

Abstract
SurA is the primary periplasmic molecular chaperone that facilitates the folding and assembling of outer membrane proteins (OMPs) in Gram-negative bacteria. Deletion of the surA gene in Escherichia coli leads to a decrease in outer membrane density and an increase in bacterial drug susceptibility. Here, we conducted mutational studies on SurA to identify residues that are critical for function. One mutant, SurAV37G, significantly reduced the activity of SurA. Further characterization indicated that SurAV37G was structurally similar to, but less stable than, the wild-type protein. The loss of activity in SurAV37G could be restored through the introduction of a pair of Cys residues and the subsequent formation of a disulfide bond. Inspired by this success, we created three additional SurA constructs, each containing a disulfide bond at different regions of the protein between two rigid secondary structural elements. The formation of disulfide bond in these mutants has no observable detrimental effect on protein activity, indicating that SurA does not undergo large-scale conformational change while performing its function.
INTRODUCTION
The cellular envelope of Gram-negative bacteria contains two layers of membranes. Integral membrane proteins are present in both membrane layers and play important functional roles. There are more than 60 outer membrane proteins (OMPs) in Gram-negative bacteria, most of which form transportation channels called porins (1). All porins share a β-barrel structure motif in which multiple antiparallel β-strands surround a hydrophilic pore in the center. Porins are considered to be the major channels for the translocation of small hydrophilic compounds with molecular masses of less than 600 Da (2). All OMPs are first synthesized in cell cytoplasm as precursors with an N terminus signal peptide and then translocated through the inner membrane by the Sec machinery (3). After the leader peptide is digested by the N-signal peptidase, β-barrel proteins are delivered through the periplasmic space and finally inserted into the outer membrane. Periplasmic molecular chaperones are proteins in the periplasm that assist the folding and membrane insertion of OMPs (4).
To date, three major chaperones have been identified in the periplasm of Escherichia coli that are involved in the biogenesis of OMPs, including SurA, Skp, and DegP (4, 5). They share redundant chaperone activities. SurA is the primary chaperone that facilitates membrane insertion of most OMPs, while Skp and DegP scavenge OMP folding intermediates that fall off the SurA pathway and either break them down or prepare them for membrane insertion (5–7). Recently, Zhao and coworkers have shown that unfolded OMPs interact with SurA and Skp with much faster kinetics than with DegP, suggesting that SurA and Skp function at the early stage of OMP biogenesis while DegP acts subsequently (5). Depletion of SurA leads to drastically decreased density of OMPs in the outer membrane, but deletion of Skp or DegP does not have a similar effect (6–8). Two explanations have been proposed for the drastic change of OMP density in the absence of SurA. First, in a SurA-deficient strain, σE transcription factor is induced, which subsequently downregulates the mRNA level of several OMPs and leads to a decrease of OMP synthesis rate (6, 8–10). Second, without the SurA chaperone activity, nascent OMPs in the periplasm aggregate and fail to reach the outer membrane (7, 11). Furthermore, SurA-deficient cells are more susceptible to hydrophobic drugs and detergents, although a direct correlation between a change of OMP density and outer membrane permeability has not been established (12, 13).
SurA was first identified as an essential gene in Escherichia coli for stationary-phase survival (10). SurA contains 4 domains following a signal peptide: an amino terminus domain with the first 150 amino acids, two peptidyl prolyl isomerase (PPIase) domains of approximately 100 residues each, and a carboxyl terminus domain. The crystallographic structure shows that the N- and C-terminal (NCt) domains and the first PPIase domain constitute a core module, while the second PPIase segment forms a satellite domain about 30 Å away (14). The chaperone activity of SurA was found to be independent from its PPIase domains. A truncated SurA construct containing only the NCt domains is sufficient to restore SurA activity in a surA gene knockout strain in vivo (Fig. 1A) (15). The mechanism of interaction between SurA and OMPs has been the subject of many studies (16–19). However, questions remain in terms of how SurA recognizes and binds to nascent OMPs. McKay and coworkers found that SurA preferentially interacts with peptide containing an A-X-A fragment, in which A is an aromatic and X is a random residue. Such fragments appear to be prevalent in OMPs. Several peptides that bind with SurA have been identified through a phage display study. These peptides compete with unfolded OMPs to bind with SurA. However, when the structures of two SurA-peptide complexes are determined, both peptides are found to interact with the first PPIase segment of SurA, which is known to be dispensable to chaperone function (19).
Fig 1.
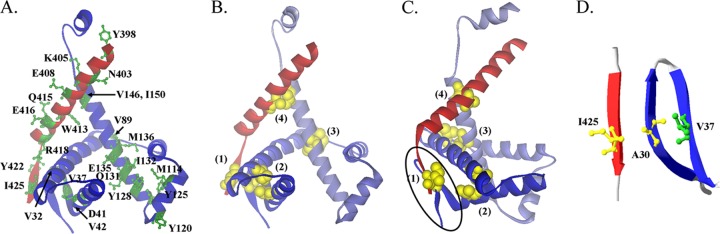
Structure of SurA N (blue) and C domain (red) (created from 1M5Y.pdb [14]). (A) Residues mutated in this study are labeled and shown in a ball-and-stick model in green. (B) Residues that are replaced by Cys to introduce a disulfide bond are highlighted: A30-I425 (1), V42-I70 (2), V89-I137 (3), and V146-A410 (4). (C) The same structure as that shown in panel B but rotated 90 degree along a vertical axis. The black oval indicates the location of a three-stranded antiparallel β-sheet formed by two strands from the N terminus and one strand from the C terminus. (D) A zoomed-in view of the β-sheet. Side chains of A30, V37, and I425 are shown in ball-and-stick models.
In terms of SurA-OMP interaction, neither the binding site(s) nor the potential structural rearrangement during the interaction has been elucidated. Here, we conducted extensive mutagenesis studies to identify mutations that affect SurA function. In addition, we limited the structural flexibility of SurA through the introduction of 4 individual disulfide bonds. We found that SurA was highly tolerant of mutations, and its function was not affected by the introduction of a disulfide bond.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
An E. coli surA gene knockout strain (BW25113ΔsurA) was used as the host for protein expression and activity assays. Cells were grown at 37°C in Luria-Bertani (LB) broth supplemented with 100 μg/ml ampicillin and 50 μg/ml kanamycin when needed.
Plasmid construction.
The surA gene lacking the signal peptide sequence was amplified from the genomic DNA of E. coli strain K-12 and cloned into vector pET22b in frame between NdeI and XhoI sites. For periplasmic SurA, the full-length surA gene was amplified and cloned into vector pMal-pIII in frame between NdeI and EcoRI sites. A polyhistidine tag was introduced at the C terminus of both proteins to facilitate purification. Site-directed mutagenesis was conducted on pMal-surA by following the manufacturer's manual using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). All sequences were confirmed by DNA sequencing (Operon).
Protein purification.
SurA has been expressed in both periplasm and cytoplasm. Cytoplasmic SurA was purified using nickel-nitrilotriacetic acid (Ni-NTA) beads (Qiagen, Chatsworth, CA) as described previously (5). For periplasmic SurA, the periplasmic fraction was prepared by osmotic shock. Cells were cultured in LB medium at 37°C overnight before being harvested by centrifugation at 4,000 × g for 20 min. Cell pellets were resuspended in buffer A (30 mM Tris-HCl, 20% sucrose, 1 mM EDTA, pH 8.0) and incubated at room temperature with shaking for 10 min. Cells were collected through centrifugation at 8,000 × g for 20 min at 4°C and then resuspended in 80 ml ice-cold solution containing 5 mM MgSO4 and 1 mM phenylmethylsulfonyl fluoride (PMSF). The mixture was stirred for 10 min on ice. The supernatant, which contained the periplasmic fluid, was collected through centrifugation. Tris-HCl (pH 7.4) was then added to the supernatant to a final concentration of 20 mM. The osmotic shock solution was subjected to Western blot analysis or further purification using metal affinity chromatography as described elsewhere (pMal Protein Fusion and Purification System manual and Qiagen literature; Qiagen Inc., Chatsworth, CA). A phosphate buffer (25 mM Na-phosphate, 10% glycerol, 200 mM NaC1, pH 7.5) supplemented with 20 or 250 mM imidazole was used in the washing and elution steps, respectively. Purified SurA was dialyzed against the phosphate buffer to remove excess imidazole. The same phosphate buffer was used throughout the study unless otherwise noted.
Purified proteins were analyzed using SDS-PAGE on a 12% homogeneous polyacrylamide gel and visualized after Coomassie blue staining. Protein concentrations were determined using the bicinchoninic acid (BCA) protein assay.
Expression level of SurA.
Freshly transformed colonies of BW25113ΔsurA containing plasmid-encoded SurA were used to inoculate LB medium supplemented with ampicillin and kanamycin. After being cultured at 37°C for 6 h, cells were harvested and subjected to osmosis shock as mentioned above. The supernatant containing periplasmic SurA was analyzed using SDS-PAGE on a 12% gel and then transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA) for Western blot analysis using a polyclonal rabbit anti-SurA primary antibody and an alkaline phosphatase-conjugated anti-rabbit secondary antibody (Abcam, Cambridge, MA). The protein-antibody conjugates were detected using nitroblue tetrazolium chloride and 5-bromo-4-chloro-3′-indoyl phosphate p-toluidine (Sigma-Aldrich, St. Louis, MO) as substrates.
SurA activity assay.
BW25113ΔsurA strains harboring plasmids encoding different SurA constructs were used in activity assays. BW25113ΔsurA containing wild-type (WT) surA (pMal-surA) or the empty vector pMal-pIII was used as the positive or negative control, respectively. SurA activities in different strains were tested using two methods, a drug susceptibility assay and an OMP (FimD and OmpA) expression level assay.
Drug susceptibility assay.
A single colony was used to inoculate LB media supplemented with 100 μg/ml ampicillin and 50 μg/ml kanamycin. Exponential-phase cultures of different strains were diluted to an optical density at 600 nm (OD600) of 0.1 using LB broth. Ten μl of this culture was used to inoculate 1 ml LB media or LB agar plate containing the indicated concentration of novobiocin. The cultures or plates were incubated overnight at 37°C. The next morning, the minimum concentration of drug that fully inhibits the growth of the bacteria (MIC) was recorded. Each experiment was repeated at least three times.
OMP expression level assay.
E. coli cells were grown in LB media at 37°C to an OD600 of 0.8. Outer membrane fractions were extracted as described previously, with some modifications (5). Briefly, cells were collected by centrifugation and lysed using a French press, followed by ultracentrifugation at 100,000 × g for 1.5 h. The pellet was resuspended in buffer B (50 mM Na-phosphate, 100 mM NaCl, 1% N-lauroyl sarcosinate, pH 7.5) and incubated at 4°C for 1 h. The outer membrane fraction was isolated by ultracentrifugation at 100,000 × g for 1.5 h. The pellet was then resuspended in denaturing buffer C (4 M urea, 2% SDS, 50 mM Na-phosphate, 100 mM NaCl, pH 7.5, 10 mM EDTA) and sonicated for 5 min. All samples were centrifuged for 5 min at 16,000 × g, and the soluble component was analyzed using SDS-PAGE. Proteins on the gel were transferred to a PVDF membrane and detected using Western blotting as described above, except that either an anti-OmpA antibody or an anti-FimD antibody was used as the primary antibody.
CD spectroscopy.
Circular dichroism (CD) spectra were collected using a JASCO J-810 spectrometer (JASCO, United Kingdom) with a 1-nm bandwidth. Blank scans were performed using phosphate buffer. Spectra were then corrected for background by subtracting the blank scan.
Fluorescence spectroscopy.
Fluorescence emission spectra were collected using a PerkinElmer LS-55 fluorescence spectrometer (PerkinElmer, Waltham, MA) at 4°C. Tryptophan emission of SurA was monitored at an excitation wavelength of 280 nm. Blank scans were collected and subtracted from the spectra.
Protease accessibility assay.
A trypsin-to-SurA molar ratio of 1:200 was used in the experiments. The reaction was performed in phosphate buffer at room temperature for the indicated period of time, and then PMSF was added to a final concentration of 2 mM to stop the reaction. SDS loading dye was immediately added, and samples were heated at 95°C for 5 min. Samples were stored on ice before being analyzed using SDS-PAGE.
Quantification of levels of disulfide bond formation.
The extent of disulfide formation was quantified after fluorescent labeling as described previously, with slight modifications (21). SurA mutants containing a pair of Cys residues were freshly expressed in BW25113ΔsurA as described above. Cells were harvested by centrifugation at 4,000 × g for 20 min and resuspended in buffer A. After being incubated with shaking for 10 min, cells were collected using centrifugation and resuspended in an ice-cold solution containing 5 mM MgSO4 and 1 mM PMSF. Iodoacetamide (IAM) was added to a final concentration of 10 mM to block all exposed free thiol groups. The mixture was stirred for 10 min on ice before the periplasmic extraction was separated from the cell debris through centrifugation. Proteins were then purified as described above. All subsequent wash and elution buffers contained 5 mM IAM. After purification, maleimide (MAL) and SDS were immediately added to protein samples to final concentrations of 50 mM and 4% (wt/vol), respectively.
Protein concentrations of different SurA mutant samples were adjusted to the same level by measuring their absorbance at 280 nm. Dithiothreitol (DTT) was added to the samples at a final concentration of 50 mM at 37°C for 1 h. Proteins were precipitated using 15% trichloroacetic acid (TCA). After centrifugation at 14,000 × g for 5 min, the protein pellet was washed using ice-cold acetone. This step separated the reduced proteins from the extra DTT in the solution. Finally, a buffer containing 4% SDS, 50 mM Tris, pH 8.0, and 5 mM N-(5-fluoresceinyl) maleimide (FMAL) was added immediately to the protein pellet to label the newly reduced free thiol groups. The pellet was resuspended using a pipette tip and incubated at room temperature for 30 min. After the incubation, 10 mM DTT was added to quench the labeling reaction.
For each mutant, we have also conducted positive- and negative-control experiments in parallel. For the positive control, all steps were the same, except that IAM and MAL treatment were omitted from the procedure. Therefore, all thiols in the protein would be reduced by DTT and subsequently labeled by fluorescein after denaturation. For the negative control, all steps were the same, except that no DTT was added to reduce the disulfide bond. Therefore, no thiol-specific fluorescent labeling would occur. The labeled sample as well as positive and negative controls were analyzed using SDS-PAGE, and the fluorescence image was taken using the MiniVisionary gel documentation system (FOTODYNE Inc., Hartland, WI) under UV light. The same gel was then subjected to Coomassie blue staining to confirm that the same amount of protein was loaded in each lane. The fluorescent intensity of labeled SurA in the gel was quantified using ImageJ (22), and the percent disulfide bond formation (DS%) was calculated through the equation DS% = [(Is − In)/(Ip − In)] × 100, where Is, In, and Ip are the fluorescent intensities of the sample, negative control, and positive control, respectively.
RESULTS AND DISCUSSION
Identification of residues critical for SurA function.
SurA is highly conserved in most Gram-negative bacteria (20, 23–25). In an attempt to identify key amino acids for SurA chaperone function, we conducted site-directed mutagenesis at 23 different sites. Since deletion of P1 and P2 domains has been shown to have little effect on SurA activities, we focused our mutational study on conserved residues in the N- and C-terminal (NCt) domain, which contains 170 residues. The sites of mutation were chosen based on two considerations: (i) conserved residues identified through sequence alignment of E. coli SurA with its homologues, and (ii) surface-exposed residues that could play a role in OMP binding. In addition, we have also mutated all aromatic residues in the NCt domain. Aromatic residues have bulky hydrophobic moieties and usually play critical roles at sites of low binding specificity (26). One Trp (W413), five Tyr (Y120, Y125, Y128, Y398, and Y422), and one Phe (F406) residue can be found in the NCt domain. Figure 1A illustrates the structure of the NCt domain and locations of mutated residues. Most residues were mutated to Ala or Gly (Table 1). D41 was mutated to Lys for a change of charge on the side chain.
Table 1.
Novobiocin MIC of BW25113ΔsurA expressing surA mutants encoded by plasmids
| Protein | MIC (μg/ml) | CSc | Mutated residue(s) | MIC (μg/ml) | CS |
|---|---|---|---|---|---|
| BW25113a | 128 | N403A | 128 | 6 | |
| pMal-pIIIb | 16 | K405A | 128 | 7 | |
| Wild-type SurA | 128 | F406A | 128 | 2 | |
| V32G | 64 | 5 | E408A | 64 | 5 |
| V37G | 16 | 7 | W413G | 128 | 7 |
| D41K | 128 | 5 | Q415A | 128 | 5 |
| V42A | 128 | 7 | E416A | 128 | 2 |
| M114G | 128 | 4 | R418G | 128 | 8 |
| Y120A | 128 | 3 | Y422A | 128 | 4 |
| Y125A | 128 | 3 | I425A | 128 | 4 |
| Y128A | 128 | 5 | Y120A/Y125A/Y128A | 128 | |
| Q131G | 128 | 3 | A30C/I425C | 256 | |
| I132G | 128 | 4 | V37G/A30C/I425C | 256 | |
| E135G | 128 | 6 | V89C/I137C | 128 | |
| M136G | 128 | 9 | V42C/I70C | 128 | |
| Y398A | 128 | 2 | V146C/A410C | 128 |
The parent strain is shown as a positive control.
BW25113ΔsurA containing vector pMal-pIII is shown as the negative control.
CS, conservation score (20).
To examine the effect of mutations on the function of SurA, we monitored the drug susceptibility levels of BW25113ΔsurA containing plasmids encoding different SurA mutants (Table 1). The same strains containing plasmid encoding WT SurA or the empty vector were used as the positive and negative controls, respectively. Among the 23 single mutants tested, V37G showed the most dramatic decrease of SurA activity. V32G and E408A also showed a 2-fold decrease in MIC. In addition, we have mutated Y120, Y125, and Y128 simultaneously to create a triple mutant, SurAY120A/Y125A/Y128A. This triple mutant has activity similar to that of WT SurA as well.
Through sequence alignment, Behrens-Kneip assigned a conservation score (CS) to each residue in SurA (20). The conservation score ranges from 0 to 10, with 10 indicating the highest level of conservation. The conservation scores of the residues mutated are listed in Table 1. Although all three residues whose mutation affected the MIC were conserved, overall the conservation score was not directly correlated with the level of activity change. Residues with the highest level of conservation, including M136, R418, K405, and W413, could be replaced with either Gly or Ala without affecting the measured MIC.
A nonfunctional mutant, SurAV37G, had structure similar to that of wild-type SurA but was less stable.
Among the conserved residues tested in this study, V37 was the only one showing a drastic effect on SurA activity when mutated. To determine how the Val37-to-Gly mutation reduced the activity of SurA, we expressed and purified both WT SurA and SurAV37G without the signal peptide. SurA contains no intrinsic Cys or disulfide bonds. Studies have shown that SurA purified from the cytoplasm has the same structure as SurA purified from the periplasm (14). We examined the structure of SurAV37G and WT SurA by CD. Far-UV CD spectra of SurAV37G and WT SurA superimposed well, suggesting the proteins had similar secondary structures, and they were approximately 50% alpha-helical (Fig. 2A). The intrinsic fluorescence of WT SurA and the mutant was also measured (Fig. 2B). We found that SurAV37G had slightly higher fluorescence than WT SurA, suggesting that certain local structural rearrangements occurred as a result of mutation. Overall, the data from CD and fluorescence spectroscopy suggested that while the secondary-structure composition of the two proteins was very similar, there was some global structure rearrangement in SurAV37G that caused a change of the microenvironment of some aromatic residues.
Fig 2.
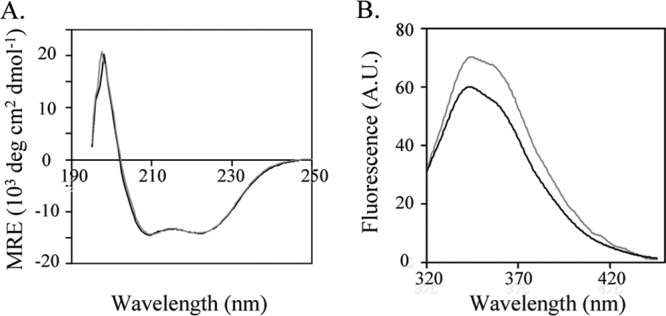
Characterization of SurAV37G (gray) and WT SurA (black). (A) Far-UV CD spectra. (B) Fluorescence emission spectra. The excitation wavelength was 280 nm.
We next examined the structural stability of WT SurA and SurAV37G by using limited protease digestion. SurAV37G and WT SurA were treated with trypsin, and the progress of digestion was monitored (Fig. 3A). Approximately 90% of SurAV37G was digested within 5 min of digestion, while 60% of WT SurA remained intact after 30 min of trypsin treatment. The mutation did have a drastic effect on SurA stability.
Fig 3.
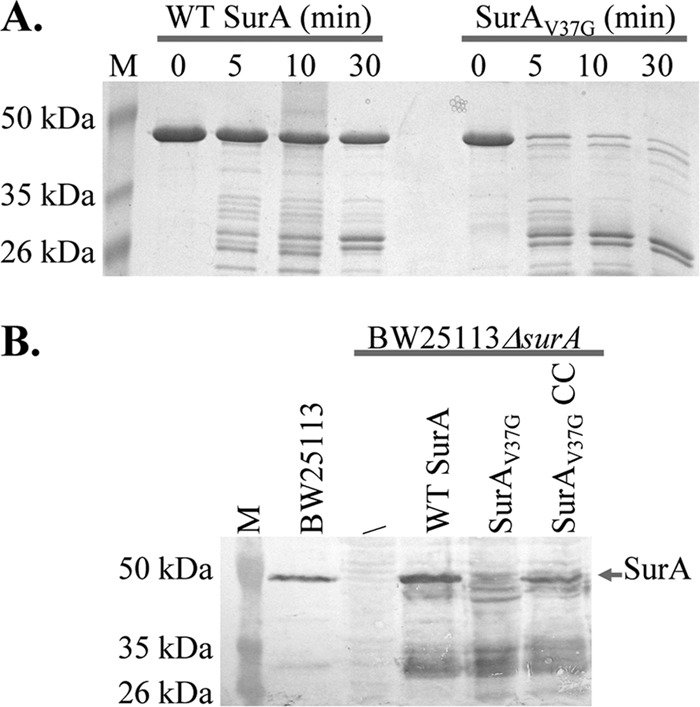
Effect of V37G mutation of SurA stability. (A) Limited trypsin digestion of purified WT SurA and SurAV37G. The molecular masses of bands in the molecular mass marker (M) are marked on the left of the gel. (B) SurA expression levels in different strains. Western blot analysis of osmotic fluid from BW25113, BW25113ΔsurA, and BW25113ΔsurA containing plasmid-encoded WT SurA, SurAV37G, and SurAV37G/A30C/I425C (SurAV37G CC). An anti-SurA antibody was used. The molecular masses of bands in the molecular mass marker are marked on the left of the gels.
Finally, to examine the stability of the mutant in vivo, we introduced plasmids coding for each protein with its signal peptide into BW25113ΔsurA and then determined the SurA level in the periplasm of each strain using anti-SurA Western blotting (Fig. 3B). For WT SurA, we detected both full-length protein and fragments, possibly due to protease digestion. In the case of SurAV37G, only fragments were observed. There was very little, if any, full-length SurA. It appeared that the Val37-to-Gly mutation drastically decreased the stability of the protein both in vivo and in vitro, which led to a very low concentration of full-length protein in the cell periplasm.
V37G mutation affects the function of SurA through destabilizing its structure.
As discussed above, the mutation destabilized the protein and reduced its concentration in the cell periplasm. The low concentration of SurA could contribute to a decrease of activity. However, it was not clear if this is the only reason for the decrease of activity. For example, Val37 could make contact with substrate OMPs, and the mutation to Gly could have weakened this interaction. To examine if stability is the major reason for the observed loss of function, we designed an experiment to partially restore the stability of the mutant. A close examination of SurA structure revealed that Val37 was located in an N-terminal β strand that was part of a three-stranded anti-parallel β-sheet involving two strands from the N terminus and one strand from the C terminus (Fig. 1B and C). The β-sheet appears to lock the N and C termini together and thus plays an important role in stabilizing SurA tertiary structure. In terms of secondary-structure preferences, Val is a strong β-sheet former and Gly is not (27). The mutation of V37 to Gly may have disrupted the formation of the β-sheet, which may subsequently affect its interaction with the neighboring strands and therefore destabilize the three-stranded β-sheet. If this is actually the case, then the introduction of a pair of cysteines to form a disulfide bond between the N-terminal and C-terminal strands may help stabilize the β-sheet and restore the function loss caused by the V37-to-Gly mutation. Specifically, we introduced two Cys residues at positions 30 and 425 (Fig. 1D). According to the crystal structure of SurA, these two Cys residues should be close enough to form a disulfide bond.
We have previously developed an effective thiol-trapping and fluorescence-labeling protocol to monitor and quantify the formation of disulfide bonds in protein structures (21). Using this method, we examined the formation of disulfide bond in a WT background in SurAA30C/I425C. We found that disulfide bond formed completely between A30C and I425C (Fig. 4A). We next introduced an additional V37G mutation into the construct to create the triple mutant SurAV37G/A30C/I425C. The extent of disulfide bond formation in the triple mutant was estimated to be approximately 50% (Fig. 4B). These results indicated that V37G mutation disrupted the local structure of the β-sheet. However, half of the proteins still formed a disulfide bond. We expect the disulfide bond would increase the stability of the mutant and therefore restore its function.
Fig 4.

Characterization of SurAV37G/A30C/I425C (SurAV37G CC). (A) Fluorescent image (F) and Coomassie blue (CB) stain of SurAA30C/I425C (SurA CC) after fluorescent labeling. Lane 1 and lane 3 were positive and negative controls, respectively, which were used to quantify the extent of disulfide bond formation (see the text). (B) Fluorescent image (F) and Coomassie blue (CB) stain of SurAA30C/I425C (SurA CC), SurAV37G CC, and WT SurA after fluorescent labeling. WT SurA was used as a control to confirm the absence of nonspecific labeling under the current experimental conditions. (C) FimD expression levels in different strains. A representative blot of anti-FimD Western blotting of outer membrane vesicles extracted from strain BW25113ΔsurA containing the empty vector (/) or plasmids encoding WT SurA, SurAV37G, or SurAV37G CC. The position of FimD is marked by an arrow.
To examine if this was actually the case, we measured the MIC of BW25113ΔsurA containing SurAV37G/A30C/I425C. As expected, the introduction of a disulfide bond between C30 and C425 restored the activity of SurA (Table 2). To further confirm that this increase of activity was actually caused by the formation of a disulfide bond, we measured the MIC of the strain in the presence of 5 mM DTT and found that the MIC was reduced by 4-fold (Table 2). Under the same conditions, 5 mM DTT had no effect on the MIC of BW25113ΔsurA containing SurAV37G or wild-type SurA.
Table 2.
Novobiocin MIC of BW25113ΔsurA expressing surA mutants encoded by plasmids in the presence or absence of DTT
| Protein | MIC (μg/ml) |
|
|---|---|---|
| No DTT | 5 mM DTT | |
| No SurA | 16 | 16 |
| Wild type | 128 | 128 |
| V37G | 16 | 16 |
| V37G/A30C/I425C | 256 | 64 |
If the disulfide bond improved the stability of SurA, there should be more full-length SurA in the periplasm. We measured the expression level of SurA in BW25113ΔsurA containing plasmid-encoded SurAV37G/A30C/I425C and SurAV37G (Fig. 3B). Similar to SurAV37G, SurAV37G/A30C/I425C was partially degraded in vivo. However, there was still a significant fraction of full-length protein in the case of SurAV37G/A30C/I425C, which was clearly sufficient to maintain the wild-type level of SurA activity.
Using quantitative Western blotting, we estimated that there were approximately 1,500 molecules of SurA per cell in strain BW25113. Figure 5 shows a representative blot. Purified SurA was diluted to the indicated concentrations and used as the standard. Band intensities were plotted against protein concentrations for the standards to obtain a calibration curve, which was used to determine the SurA level in the periplasm of BW25113. When BW25113ΔsurA was transformed using plasmid encoding wild-type SurA (pMal-III-SurA), under basal expression conditions there were ∼3,500 molecules of SurA per cell. However, this increase of SurA expression level did not further increase the MIC of the strain compared to that of BW25113. When BW25113ΔsurA was transformed with plasmid encoding SurAV37G/A30C/I425C, the copy number of full-length SurA was approximately 1,000. However, the MIC of the strain was twice the level of the wild-type strain or surA knockout strain transformed with plasmid-encoded wild-type SurA. This result revealed that E. coli needs as little as 1,000 or fewer SurA units per cell for normal SurA activity and to maintain membrane integrity. We have also tested the MIC of BW25113ΔsurA containing SurAA30C/I425C, and it was also twice as high as that of the wild-type strain (Table 1). The higher MIC of SurAA30C/I425C suggested that SurA functioned better with a more rigid structure.
Fig 5.
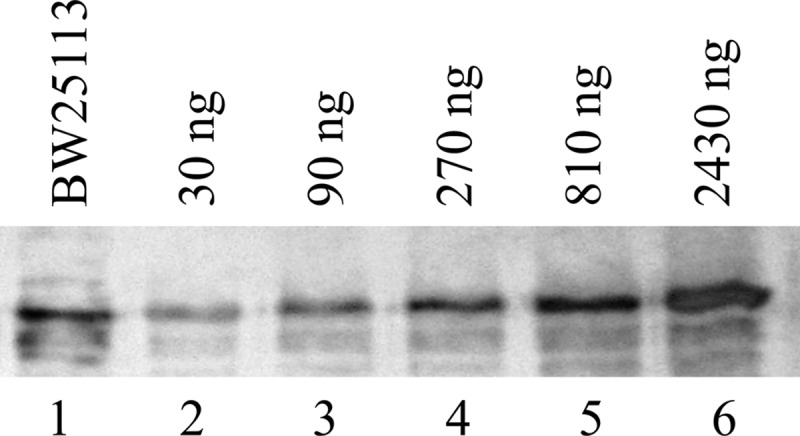
Representative blot of quantitative Western blotting. Periplasmic fraction obtained from BW25113 (lane 1) was loaded together with purified SurA diluted to different concentrations (lanes 2 to 6).
As an alternative method to examine the activity of SurA, we measured the expression level of FimD in BW25113ΔsurA expressing plasmid-encoded WT SurA, SurAV37G, or SurAA30C/I425C/V37G (Fig. 4C). Studies have shown that the FimD expression level in the outer membrane depends on the function of SurA (28). Consistent with the MIC assay, while the levels of FimD in cells containing WT SurA or SurAA30C/I425C/V37G were similar, the expression level in cells without SurA or SurAV37G were significantly lower.
OmpA is another well-established SurA substrate (11). We have measured the OmpA abundance in the outer membrane using anti-OmpA Western blot analysis (Fig. 6). BW25113ΔsurA in the absence of plasmid-encoded SurA was used as a control (lane 2). When SurAV37G was introduced, the OmpA level remained unchanged (lane 3). When wild-type SurA (lane 1) or SurAV37G/A30C/I425C (lane 4) was expressed, the OmpA level increased by 2.6- and 3.5-fold, respectively. This result is consistent with the MIC measurement.
Fig 6.
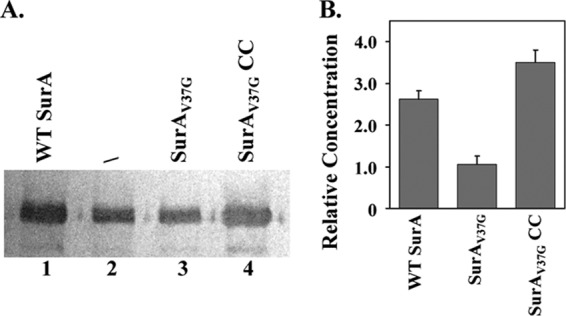
OmpA expression levels in different strains. (A) A representative blot of anti-OmpA Western blotting of outer membrane vesicles extracted from strain BW25113ΔsurA containing the empty vector (/; lane 2) or a plasmid encoding WT SurA (lane 1), SurAV37G (lane 3), or SurAV37GCC (lane 4). (B) The OmpA relative expression level was obtained from the ratio of band intensities between lanes 1 and 2 (WT SurA), lanes 3 and 2 (SurAV37G), or lanes 4 and 2 (SurAV37G CC). The average values and standard deviations from three measurements are shown.
A decrease of structure flexibility does not affect SurA activity.
The observation of increased MIC of SurAA30C/I425C prompted us to investigate the relationship between SurA structure flexibility and activity. The functional NCt domain of SurA is small, containing ∼170 residues (Fig. 1B and C). We identified three additional locations scattered at different regions of the domain for the introduction of cysteine pairs (Fig. 1B and C). According to SurA crystal structure, the distances between the α carbons of each Cys pair are within 6 Å, which allows the formation of disulfide bonds (21). As shown in Fig. 7, lanes 1 and 3 were positive and negative controls, respectively, for the quantification of disulfide bond percentages. The extents of disulfide bond formation were calculated as described in Materials and Methods and are shown in Table 3. Formation of disulfide bond was close to completion in all four pairs of Cys residues. We next tested the activity of these mutants using the drug susceptibility assay and found that they had activity similar to that of WT SurA (Table 1).
Fig 7.
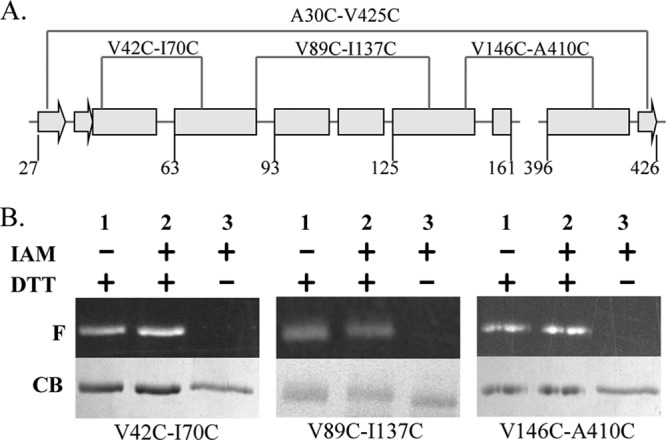
(A) Secondary structure scheme of the NCt domain of SurA with the position of disulfide bonds shown. Arrows and rectangles denote β-strands and α-helices, respectively. (B) Fluorescent image (F) and Coomassie blue (CB) stain of three SurA cysteine pairs mutants after fluorescent labeling. Lane 1 and lane 3 were positive and negative controls, respectively, which were used to quantify the extent of disulfide bond formation (see the text).
Table 3.
Percent disulfide formation in mutants containing Cys pairs
| Cysteine pair | % Disulfide bond |
|---|---|
| A30C-I425C | 95.1 ± 4.4 |
| V42C-I70C | 98.1 ± 4.9 |
| V89C-I137C | 96.5 ± 2.1 |
| V146C-A410C | 97.4 ± 6.3 |
Many chaperones, such as HSC70 and SecB, have been shown to undergo conformational changes upon binding with their substrates (29, 30). We introduced a disulfide bond at four different locations in the functionally relevant NCt domain. To our surprise, while all four disulfide bonds formed in vivo, none of them had a negative impact on SurA function. While the result from one pair of Cys residues might not be conclusive, the lack of observable effect on function for all four Cys pairs distributed throughout the tertiary structure strongly suggested that SurA does not undergo global conformational changes during its operation. Another interesting discovery was that SurA was highly tolerant to mutations. All conserved/invariable and aromatic residues that we tested, except for V37 and, to a lesser extent, V32 and E408, could be replaced without significantly affecting the activity of the protein. This plasticity might be important for SurA to bind and interact with various nascent OMPs lacking well-defined structure.
ACKNOWLEDGMENTS
We acknowledge the University of Kentucky Center for Structure Biology for the use of the CD spectrometer. We thank Luis Angel Fernandez for the kind gift of anti-FimD antibody.
We thank the Kentucky NASA EPSCoR (NNX10AV39A; Y.W.), Oak Ridge Associated Universities (ORAU) Ralph E. Powe Junior Faculty Enhancement Award (Y.W.), the University of Kentucky faculty startup fund (Y.W.), and the University of Kentucky Presidential Fellowship (M.Z.) for supporting this work.
Footnotes
Published ahead of print 28 December 2012
REFERENCES
- 1. Bos MP, Tommassen J. 2004. Biogenesis of the Gram-negative bacterial outer membrane. Curr. Opin. Microbiol. 7:610–616 [DOI] [PubMed] [Google Scholar]
- 2. Nikaido H. 2001. Preventing drug access to targets: cell surface permeability barriers and active efflux in bacteria. Semin. Cell Dev. Biol. 12:215–223 [DOI] [PubMed] [Google Scholar]
- 3. Pugsley AP. 1993. The complete general secretory pathway in Gram-negative bacteria. Microbiol. Rev. 57:50–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rizzitello AE, Harper JR, Silhavy TJ. 2001. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J. Bacteriol. 183:6794–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu S, Ge X, Lv ZY, Zhi ZY, Chang ZY, Zhao XS. 2011. Interaction between bacterial outer membrane proteins and periplasmic quality control factors: a kinetic partitioning mechanism. Biochem. J. 438:505–511 [DOI] [PubMed] [Google Scholar]
- 6. Leverrier P, Vertommen D, Collet J. 2010. Contribution of proteomics toward solving the fascinating mysteries of the biogenesis of the envelope of Escherichia coli. Proteomics 10:771–784 [DOI] [PubMed] [Google Scholar]
- 7. Sklar JG, Wu T, Kahne D, Silhavy TJ. 2007. Defining the roles of periplasmic chaperones SurA, Skp and DegP in Escherichia coli. Genes Dev. 21:2473–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vertommen D, Ruiz N, Leverrier P, Silhavy TJ, Collet J. 2009. Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 9:2432–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lazar SW, Almiron M, Tormo A, Kolter R. 1998. Role of the Escherichia coli SurA protein in stationary phase survival. J. Bacteriol. 180:5704–5711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tormo A, Almiron M, Kolter R. 1990. SurA, an Escherichia coli gene essential for survival in stationary phase. J. Bacteriol. 172:4339–4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lazar SW, Kolter R. 1996. SurA, assists the folding of Escherichia coli outer membrane proteins. J. Bacteriol. 178:1770–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Missiakas D, Betton JM, Raina S. 1996. New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol. Microbiol. 21:871–884 [DOI] [PubMed] [Google Scholar]
- 13. Tamae C, Liu A, Kim K, Sitz D, Hong J, Becket E, Bui A, Solaimani P, Tran KP, Yang HJ, Miller JH. 2008. Determination of antibiotic hypersensitivity among 4,000 single-gene-knockout mutants of Escherichia coli. J. Bacteriol. 190:5981–5988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bitto E, McKay DB. 2002. Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane protein. Structure 10:1489–1498 [DOI] [PubMed] [Google Scholar]
- 15. Behrens S, Maier R, de Cock H, Schmid FX, Gross CA. 2001. The SurA periplasmic PPIase lacking its parvulin domain function in vivo and has chaperone activity. EMBO J. 20:285–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bitto E, McKay DB. 2003. The periplasmic molecular chaperone SurA binds a peptide motif that is characteristic of integral outer membrane proteins. J. Biol. Chem. 278:49316–49322 [DOI] [PubMed] [Google Scholar]
- 17. Hennecke G, Nolte J, Volkmer-Engert R, Schneider-Mergener J, Behrens S. 2005. The periplasmic chaperone SurA exploits two features characteristic of integral outer membrane proteins for selective substrate recognition. J. Biol. Chem. 280:23540–23548 [DOI] [PubMed] [Google Scholar]
- 18. Webb HW, Ruddock LW, Marchant RJ, Jonas K, Klappa P. 2001. Interaction of the periplasmic peptidylprolyl cis-trans isomerase SurA with model peptides. J. Biol. Chem. 276:45622–45627 [DOI] [PubMed] [Google Scholar]
- 19. Xu XH, Wang SY, Hu YX, McKay DB. 2007. The periplasmic bacterial molecular chaperone SurA adapts its structure to bind peptides in different conformations to assert a sequence preference for aromatic residues. J. Mol. Biol. 373:367–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Behrens-Kneip S. 2010. The role of SurA factor in outer membrane protein transport and virulence. Int. J. Med. Microbiol. 300:421–428 [DOI] [PubMed] [Google Scholar]
- 21. Lu W, Zhong M, Wei Y. 2011. A reporter platform for the monitoring of in vivo conformational changes in AcrB. Protein Pept. Lett. 18:863–871 [DOI] [PubMed] [Google Scholar]
- 22. Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alcock FH, Grossmann JG, Gentle IE, Likic VA, Lithgow T, Tokatlidis K. 2008. Conserved substrate binding by chaperones in the bacterial periplasm and the mitochondrial inter-membrane space. Biochem. J. 409:377–387 [DOI] [PubMed] [Google Scholar]
- 24. Muller M, Koch HG, Beck K, Schafer U. 2001. Protein traffic in bacteria: multiple routes from the ribosome to and across the membrane. Prog. Nucleic Acid Res. Mol. Biol. 66:107–157 [DOI] [PubMed] [Google Scholar]
- 25. Sydenham M, Douce G, Bowe F, Ahmed S, Chatfield S, Dougan G. 2000. Salmonella enterica serovar Typhimurium surA mutants are attenuated and effective live oral vaccines. Infect. Immun. 68:1109–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dougherty DA. 1996. Cation-π interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science 271:163–168 [DOI] [PubMed] [Google Scholar]
- 27. Chou PY, Fasman GD. 1974. Prediction of protein conformation. Biochemistry 13:222–245 [DOI] [PubMed] [Google Scholar]
- 28. Palomino C, Marin E, Fernandez LA. 2011. The fimbrial usher FimD follows the SurA-BamB pathway for its assembly in the outer membrane of Escherichia coli. J. Bacteriol. 193:5222–5230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haimann MM, Akdogan Y, Philipp R, Varadarajan R, Hinderberger D, Trommer WE. 2011. Conformational changes of the chaperone SecB upon binding to a model substrate–bovine pancreatic trypsin inhibitor (BPTI). Biol. Chem. 392:849–858 [DOI] [PubMed] [Google Scholar]
- 30. Park K, Flynn GC, Rothman JE, Fasman GD. 1993. Conformational change of chaperone Hsc70 upon binding to a decapeptide: a circular dichroism study. Protein Sci. 2:325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]