

Abstract
The BAL1 macrodomain-containing protein and its partner E3 ligase, BBAP, are overexpressed in chemotherapy-resistant lymphomas. BBAP selectively ubiquitylates histone H4 and indirectly promotes early 53BP1 recruitment to DNA damage sites. However, neither BBAP nor BAL1 has been directly associated with a DNA damage response (DDR), and the function of BAL1 remains undefined. Herein, we describe a direct link between rapid and short-lived poly(ADP-ribose) (PAR) polymerase 1 (PARP1) activation and PARylation at DNA damage sites, PAR-dependent recruitment of the BAL1 macrodomain-containing protein and its partner E3 ligase, local BBAP-mediated ubiquitylation, and subsequent recruitment of the checkpoint mediators 53BP1 and BRCA1. The PARP1-dependent localization of BAL1-BBAP functionally limits both early and delayed DNA damage and enhances cellular viability independent of ATM, MDC1, and RNF8. These data establish that BAL1 and BBAP are bona fide members of a DNA damage response pathway and are directly associated with PARP1 activation, BRCA1 recruitment, and double-strand break repair.
INTRODUCTION
DNA damage initiates a tightly regulated signaling cascade and the orderly recruitment of repair factors to sites of damage. The chromatin substrate for DNA damage repair, DNA-encircling nucleosomes comprised of core histone proteins, can be modulated in multiple ways, such as incorporating histone variants, posttranslationally modifying select histones, repositioning nucleosomes, and generating DNA repair foci (1). Cells utilize specific factors to detect and repair DNA single-strand breaks (SSB) and complementary pathways, homologous recombination (HR) and nonhomologous end joining (NHEJ), to address double-strand breaks (DSB) (1).
One of the earliest responses to single-strand and double-strand DNA breaks is the activation and recruitment of poly(ADP-ribose) polymerase protein (PARP) family members. Although the PARP family includes 16 proteins, only PARP1 and PARP2 have thus far been linked to DNA damage responses (DDRs) (2). Upon activation, PARP1 catalyzes the NAD+-dependent addition of poly(ADP-ribose) (PAR) chains to target proteins, including certain histones and PARP1 itself. PARP1 activation and associated PAR synthesis occur within seconds of DNA damage and persist for minutes (1). The rapid and short-lived PARylation at DNA damage sites is thought to promote a more relaxed chromatin structure, which facilitates DNA repair (2–4).
One recently described ADP-ribose–PAR-binding motif is the macrodomain, an evolutionarily conserved sequence of ≈130 to 190 amino acids (aa) found in the variant histones, macro-H2A1 (splice forms H2A1.1 and H2A1.2) and macro-H2A2, and at least 8 additional human proteins, including ALC1 and BAL family members (3, 5–9). Both histone macro-H2A1.1 and the chromatin-remodeling factor ALC1 are recruited in a macrodomain- and PAR-dependent manner to DNA damage sites, where the proteins participate in chromatin reorganization and nucleosome sliding (2, 3, 9, 10).
We originally identified the most abundant BAL family member, BAL1, as an overexpressed gene product in treatment-resistant diffuse large B-cell lymphomas (DLBCLs) (5). BAL1 and two additional family members, BAL2 and BAL3, are the only known proteins with multiple N-terminal macrodomains (6). Of note, these hybrid molecules also include C-terminal regions with similarities to the PARP catalytic domain, and BAL2 and -3, but not BAL1, catalyze ADP ribosylation (6).
In subsequent studies, we identified the BAL1 binding partner, B-lymphoma and BAL-associated protein (BBAP), an E3 ligase with C-terminal identity to Deltex (DTX) family members (11). The BBAP and BAL1 genes are located in a head-to-head orientation on chromosome 3q21 and are regulated by the same bidirectional gamma interferon (IFN-γ)-responsive promoter (12). Of interest, BBAP and BAL1 are most abundant in DLBCLs with a prominent immune/inflammatory infiltrate and increased IFN-γ production (12, 13).
Because BAL1 contains structural motifs potentially associated with chromatin remodeling (6), we previously assessed the role of the BAL1 binding partner, BBAP, in these processes. The BBAP E3 ligase monoubiquitylated histone H4 lysine 91 and selectively modulated the kinetics of 53BP1 accumulation at DNA damage sites (14). Disruption of BBAP-mediated histone H4K91 ubiquitylation was associated with a loss of chromatin-associated histone H4K20 methylase and methylated H4K20, notable because 53BP1 localizes to DNA damage sites by binding to methylated H4K20 (14). Although these studies implicated BBAP in the ubiquitylation and additional posttranslational modification of histones, neither BBAP nor its partner protein, BAL1, was directly associated with a DDR, and the function of BAL1 remained undefined.
Current studies suggest that DDR proteins assemble in a coordinated sequential manner at sites of DNA breaks (1). The initial recruitment phase is rapid and transient and depends upon PARylation at DNA damage sites (1). A second phase, which also begins within seconds but lasts for hours, includes the sequential phosphorylation and ubiquitylation of multiple DSB repair factors. Following the initial recruitment of the Mre11-RAD50-Nbs1 (MRN) complex, HR DSB repair involves ATM localization and phosphorylation of γH2AX and MDC1 (1). ATM-mediated phosphorylation of MDC1 promotes the recruitment of the RNF8 E3 ligase, which targets H2A histones (1, 15, 16). A second E3 ligase, RNF168, interacts with ubiquitylated H2A-type histones in an RNF8-dependent manner and amplifies the local concentration of ubiquitin conjugates (1, 17). Of note, RNF8 and RNF168 also regulate the retention of the checkpoint mediators 53BP1 and BRCA1 at sites of DNA damage (15, 16, 18–22). The mechanisms of RNF8- and RNF168-mediated recruitment of 53BP1 remain undefined, whereas BRCA1 localizes to DNA breaks via RAP80, an adaptor protein with ubiquitin-interacting motifs (UIM) (17, 19, 22–24).
Although the components and dynamics of ATM-, γH2AX-, and MDC1-dependent DDR have been extensively studied, the initial PARP-dependent recruitment phase is less well characterized (1). Herein, we define a functionally significant direct link between PARP1 activation and DSB repair via recruitment of the BAL1 macrodomain to PARylated proteins, local BBAP-dependent ubiquitylation, and subsequent recruitment of 53BP1 and BRCA1.
MATERIALS AND METHODS
Generation of BAL1, BBAP, and PARP1 constructs.
The specific oligonucleotides used to generate green fluorescent protein (GFP)-tagged BAL1, BBAP, and PARP1 constructs are listed in Table S1 in the supplemental material. Human PARP1 cDNA (product no. SC119157) was purchased from OriGene Technologies (Rockville, MD). The cDNAs for human BAL1, BBAP (NCBI accession no. NM_031458.2) (5, 11), and PARP1 were inserted by PCR into pcDNA3.1/NT-GFP-TOPO (Invitrogen, Carlsbad, CA) to generate the respective N-terminally tagged proteins GFP-BAL1, GFP-BBAP, and GFP-PARP1. The GFP-BAL1 domain constructs, Macros-BBD and Macro1 (see Fig. 2), were generated by introducing a stop codon (nucleotide [nt] 2454, C→A; nt 1202, C→T) into the GFP-BAL1 plasmid with appropriate DNA oligonucleotides and the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Macro2-BBD and Macro2Δ were generated with appropriate PCR products cloned into pcDNA3.1/NT-GFP-TOPO; Macro2 was constructed by introducing a stop codon (nt 1913, C→A) into Macro2-BBD. D126A and IE326,327AA were produced with the appropriate oligonucleotides (nt 704, A→C; nt 1303, nt 1304, and nt 1307, AT, A→GC, and C) and site-directed mutagenesis. The double-mutation BAL1 construct (BAL1-DM) was generated by introducing the IE326,327AA mutation into the D126A construct.
Fig 2.
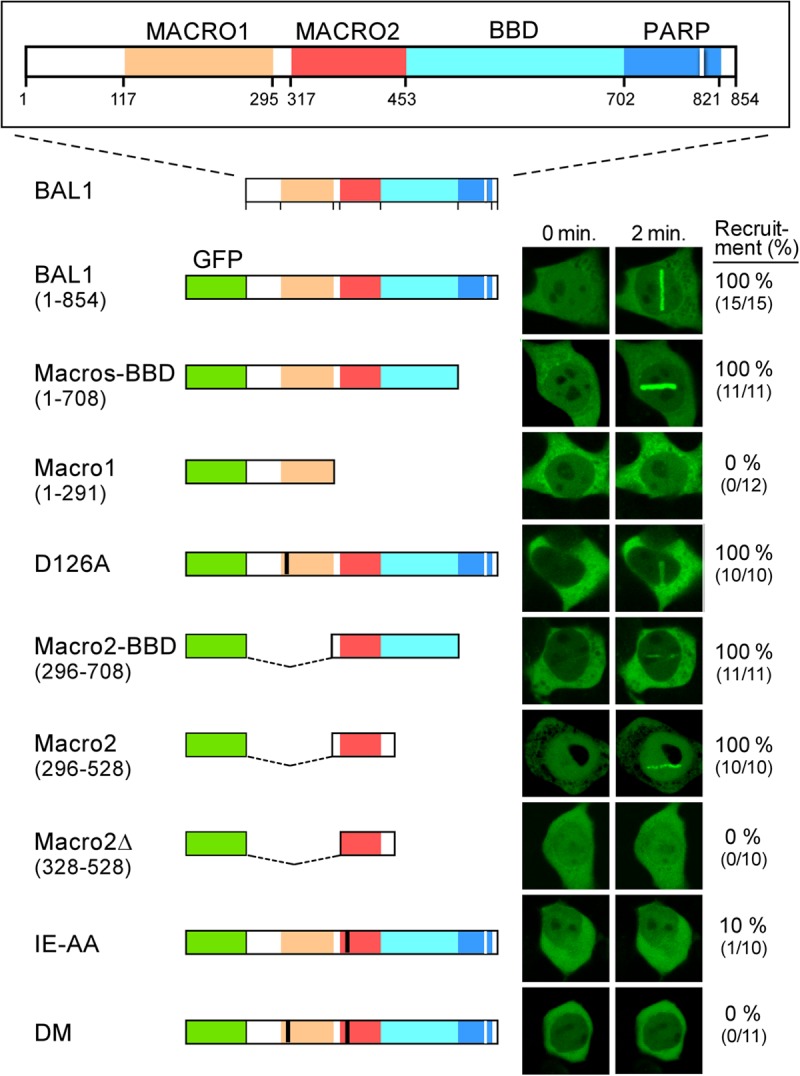
BAL1 macrodomain 2 is required for recruitment to DNA damage sites. Top, BAL1 protein functional domains, including macrodomains 1 and 2 (orange and red), BBAP binding domain (BBD) (light blue in BAL1) (2), and a region with partial sequence homology to the PARP catalytic domain (dark blue) (domain sizes and amino acids below). GFP-BAL1 constructs are labeled and represented below the BAL1 diagram. Mutations in macrodomain 1 (D126A) and macrodomain 2 (IE 326,327AA [IE-AA]) are shown in black. Representative images of GFP-BAL1 (293T cell) transfectants at baseline and 2 min following laser microirradiation are shown on the right, and the percentages of cells with GFP-BAL1 recruitment to laser-induced DNA breaks are shown on the far right.
Cell culture and in vivo expression of GFP-tagged proteins.
HeLa and 293T human embryonic kidney cells (ATCC) were grown in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum (Invitrogen). HeLa or 293T cells were transiently transfected with the indicated expression plasmids (GFP-BAL1, BAL1 domains, BBAP, or PARP1) for 24 h prior to further analysis.
In additional experiments, 5′ untranslated region (UTR)-specific BAL1 (see Table S2 in the supplemental material) or control small interfering RNA (siRNA)-treated HeLa cells were transfected with GFP vector, GFP-BAL1, or GFP-BAL1-DM constructs; 24 h later, these cells were cultured in the presence or absence of doxorubicin (Dox) (50 ng/ml) for an additional 24 h, and their viabilities were analyzed thereafter with anti-annexin V and propidium iodide (PI).
Protein depletion by siRNA.
All siRNA oligonucleotides targeting DDR factors (see Table S2 in the supplemental material) (14) were synthesized by Integrated DNA Technologies (Coralville, IA). Thereafter, the siRNAs and the control nontargeting siRNA (10 μM) were transfected into HeLa cells by using X-tremeGENE siRNA transfection reagent (Roche, Basel, Switzerland) according to the manufacturer's instructions. Cells were cultured for 48 h, lysed, and subjected to NuPAGE and Western blotting as described previously (14). The efficiency of protein depletion was assessed by immunoblotting with the following individual antibodies: BAL1 (11) or rabbit polyclonal antibody (product no. ab53796; Abcam, Cambridge, MA), PARP1 (mouse monoclonal antibody, product no. 51-6639GR; BD Biosciences, Franklin Lakes, NJ), BBAP (mouse monoclonal antibody) (11, 14), ATM (mouse monoclonal antibody, product no. 23922; Santa Cruz Biotechnology, Santa Cruz, CA), and MDC1 (rabbit polyclonal antibody) (product no. A300-051A; Bethyl Laboratories, Montgomery, TX).
Laser microirradiation.
Cells were initially seeded on coverslips and sensitized with 10 μM 5-bromo-2′-deoxyuridine (BrdU) (Roche) in phenol red-free medium (Invitrogen) for 24 h at 37°C. In selected experiments, the PARP inhibitor PJ34 (5 μM) (Sigma, St. Louis, MO) was added to the culture medium for 1 h prior to laser microirradiation. Laser microirradiation was carried out on a Zeiss LSM 510 NLO confocal microscope (Carl Zeiss MicroImaging, LLC, Thornwood, NY) equipped with a Coherent Chameleon pulse laser focused through an ×40 LD C-Apochromat 0.9-numerical-aperture water immersion objective to yield a spot size of 0.5 to 1 μm. Cells were exposed to the Ti-Sapphire pulse laser (740 nm) for ≈200 ms (fast scanning mode) at 30% laser power for 5 to 10 iterations. These settings generated a detectable damage response restricted to the laser path without noticeable cytotoxicity to the cells.
To ensure that cells with GFP proteins were assayed, cells with moderate levels of GFP were systematically chosen using identical argon laser (488-nm) settings. The association and dissociation kinetics of GFP proteins at sites of laser microirradiation were monitored on the same microscope by measuring GFP fluorescence over time in the damaged region using the 488-nm argon laser. The images were detected and stored using Carl Zeiss AIM software (Carl Zeiss MicroImaging). Variations in fluorescence intensity (I) were plotted as a function of time (t) using the Zeiss AIM software. Data were normalized against the fluorescence intensity at the right time before microirradiation (I0). Deviations in the GFP level at each time point and condition were determined by averaging values from ≥10 cells from a representative experiment.
Analysis of DNA damage sites of laser microirradiation by confocal microscopy.
HeLa cells were grown on coverslips and subjected to laser microirradiation as described above. Indirect immunofluorescence was performed as described previously (14) with minor modifications. Primary antibodies included BAL1 (product no. ab53796; Abcam), PARP1 (rabbit monoclonal antibody, 9532 [Cell Signaling Technology, Danvers, MA], or mouse monoclonal antibody, 51-6639 [BD Biosciences]), anti-PAR (4335; Trevigen, Gaithersburg, MD), γH2AX (05-636; Millipore, Billerica, MA), BBAP (mouse monoclonal antibody [14] or rabbit polyclonal antibody, A300-833A [Bethyl Laboratories]), FK2 (multiubiquitin monoclonal antibody, SPA-205; Enzo Life Sciences International, Plymouth Meeting, PA), ATM (mouse monoclonal antibody, 23922; Santa Cruz Biotechnology), MDC1 (rabbit polyclonal antibody, A300-051A; Bethyl Laboratories), RAP80 (rabbit polyclonal antibody, A300-763A; Bethyl Laboratories), BRCA1 (07-434; Millipore), 53BP1 (22760; Santa Cruz Biotechnology), ATM-P-1981 (clone 10H11.E12; Millipore), and RNF8 (rabbit polyclonal antibody, product no. ab4183; Abcam). After multiple phosphate-buffered saline (PBS) washes, coverslips were incubated with fluorescein isothiocyanate (FITC)- or Cy5-conjugated anti-mouse or anti-rabbit antibodies (Alexa Fluor; Molecular Probes, Invitrogen) for 45 min and stained with PBS containing 4-diamidino-2-phenylindole (DAPI) (counterstain for DNA). Slides were mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA) after additional PBS washes. Confocal images were acquired on an LSM 510 (Carl Zeiss MicroImaging) mounted on a Zeiss-Axiovert 100 M equipped with a Plan-Neofluar 403/1.3 oil immersion objective as reported previously (14). All images for a given condition and immunostain over time were obtained with the same image acquisition settings.
Low-dose irradiation and focus formation.
The γ-irradiation was performed as described previously (14) with minor modifications. In brief, HeLa cells grown on coverslips were treated with low-dose irradiation (100 cGy) and analyzed at serial time points for repair foci by immunostaining with the following antibodies: anti-BAL1 (product no. ab53796; Abcam), anti-PAR (4335; Trevigen, Gaithersburg, MD), anti-53BP1 (22760; Santa Cruz Biotechnology), and anti-γH2AX (05-636; Millipore, Billerica, MA).
53BP1 and γH2AX repair foci were identified and counted with an Image J macro program. In brief, DAPI staining was utilized to create a nuclear mask. The numbers and intensities of repair foci within the nuclear mask were captured by applying a 3-μm rolling-ball background reduction and maximum entropy threshold algorithm to both Cy5 (γH2AX) and FITC (53BP1) channels (details available upon request).
Coimmunoprecipitation assays.
HeLa cells were incubated with PJ-34 or medium alone for 1 h, treated with 50 ng/ml of doxorubicin (Dox) for 10 min, or left untreated; subsequently, cells were harvested and lysed in Triton buffer (TBS) (1% Triton X-100 with protein inhibitors). Thereafter, cell lysates were incubated with anti-PARP1 (rabbit) antibody, anti-BAL1 (rabbit) antibody, or control rabbit IgG overnight at 4°C; protein A-Sepharose beads were subsequently added to bind the IgG. After multiple washes in Triton buffer, antibody-associated proteins were released by boiling in protein sample buffer, resolved by NuPAGE, and immunoblotted with anti-PARP1 (mouse), anti-PAR, or anti-BBAP (mouse) followed by donkey anti-mouse horseradish peroxidase (HRP)-conjugated antibody or anti-BAL1 antibody, followed by protein A-labeled HRP. In input samples, antiactin antibody was used as a loading control.
Analysis of cellular proliferation and apoptosis following BAL1 depletion and Dox treatment.
HeLa cells, which have high levels of endogenous BAL1, were first transfected with scrambled control (SC) or BAL1 siRNAs. Thereafter, siRNA-transfected or parental cells were seeded onto 96-well plates at 1 × 104 cells/well, left untreated, or treated with 50 ng/ml or 200 ng/ml Dox for 1 to 96 h and were subsequently evaluated by a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (CellTiter 96 aqueous nonradioactive assay; Promega Corporation, Madison, WI). Assays were read at an absorbance of 490 nm using a SpectraMax 190 microplate reader (Molecular Devices, Sunnyvale, CA). All assays were performed in triplicate.
In additional studies, parental or BAL1- or control siRNA-treated HeLa cells were cultured in the presence or absence of Dox (50 or 200 ng/ml) for 24 h. Thereafter, the cells were detached with trypsin, washed with PBS, and incubated with anti-annexin V antibody and PI in 100 μl of binding buffer (10 mM HEPES-NaOH [pH 7.4], 140 mM NaCl, 2.5 mM CaCl) for 15 min in the dark. Following the addition of 400 μl more binding buffer, the samples were analyzed by flow cytometry (Cytomics FC 500; Beckman Coulter, Fullerton, CA).
Comet assay.
DNA damage was evaluated using the alkaline comet assay according to the manufacturer's instructions (Trevigen). PJ-34-treated and siRNA-transfected cells were subjected to low-dose irradiation (200 cGy) and subsequently incubated in normal medium for 15 min, 60 min, and 24 h before processing. Harvested cells (3 × 103) were mixed with 0.8% low-melting-point agarose and layered onto agarose-coated slides. Slides were then submerged in lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris [pH 10.0], and 1% Triton X-100) overnight at 4°C. After lysis, slides were incubated for 30 min in electrophoresis buffer (200 mM NaOH and 1 mM EDTA [pH > 13]). After electrophoresis (25 min, 21 V, 300 mA), slides were washed with 2× water and 70% ethanol and then were air dried. Slides were stained with 2 μg/ml SYBR green. The average comet tail moment was scored (50 to 100 cells/slide) from the confocal images documented on the LSM 510 (Carl Zeiss MicroImaging). The tail moment (% DNA in tail × tail length) was generated by using the software TriTek CometScore.
RESULTS
BAL1 and BBAP recruit to laser microirradiation sites.
Although BBAP protects cells exposed to DNA-damaging agents (14), neither BBAP nor its partner protein, BAL1, has been directly associated with a DDR, and the function of BAL1 remains undefined. For these reasons, we expressed GFP-tagged BAL1 and BBAP in HeLa cells and evaluated their recruitment to sites of DNA damage induced by laser microirradiation. Both GFP-BAL1 and BBAP localized to the laser-induced DNA breaks in less than 1 min with maximum recruitment for ≈10 min and subsequent release thereafter (Fig. 1A and B). Similar results were obtained when HeLa cells were laser microirradiated and immunostained for endogenous BAL1 (Fig. 1C). The macrodomain-containing protein rapidly localized to DNA damage sites, with peak BAL1 recruitment within several minutes and dispersal within the nuclear compartment in less than 60 min (Fig. 1C). Endogenous BBAP was recruited to sites of laser-induced DNA breaks with similar kinetics (Fig. 1D and data not shown).
Fig 1.

BAL1 and BBAP are recruited to DNA damage sites. GFP-BAL1 (A) and GFP-BBAP (B) recruitment to sites of laser microirradiation. HeLa cells were transfected with GFP-BAL1 or BBAP, laser microirradiated, and subsequently analyzed at serial time points for GFP fluorescence in the DNA damage site (left panels, representative photos; right panels, kinetics of recruitment). Variations in fluorescence intensity (I) were plotted as a function of time (t). GFP levels at each time point were determined by averaging values from 10 cells (±the standard error) from a representative experiment. (C) Kinetics of endogenous BAL1 recruitment to laser-induced DNA breaks. Images were obtained at baseline (0) and at serial time points (0.25 to 60 min) following laser microirradiation. (D) Endogenous BBAP localization to laser-induced DNA breaks (0, 1, and 4 min). (E) Kinetics of BAL1 focus formation following γ-irradiation. HeLa cells were treated with low-dose (100 cGy) irradiation and analyzed for BAL1 foci at baseline and at serial time points (0.5 to 60 min) thereafter.
In additional studies, HeLa cells were subjected to an alternative source of DNA damage—low-dose γ-irradiation (100 cGy)—and immunostained for BAL1 (Fig. 1E). BAL1 foci were detectable in less than 1 min, were most prominent at 4 min, and decreased in number and intensity by 30 min (Fig. 1E). Taken together, these data directly implicate the macrodomain-containing BAL1 protein and its partner E3 ligase, BBAP, in the early stages of a DDR.
BAL1 macrodomain 2 is required for recruitment to DNA damage sites.
To assess the role of the BAL1 tandem macrodomains in localization to DNA damage sites, we generated a series of GFP-BAL1 constructs that encoded both macrodomains (Macro1 and Macro2), the respective single macrodomains, or mutated versions lacking critical residues in one or both macroregions (Fig. 2). The individual GFP-BAL1 constructs were transfected into 293T cells and confirmed to be expressed in the nucleus and cytoplasm (see Fig. S1 in the supplemental material). Thereafter, the transfected 293T cells were subjected to laser microirradiation and analyzed by confocal microscopy (Fig. 2; see also Fig. S1).
GFP-BAL1 proteins containing both macrodomains (BAL1 [1 to 854] and Macros-BBD [1 to 708]), the complete macrodomain 2 (Macro2-BBD [(296 to 708)] or Macro2 [296 to 528]) efficiently localized to sites of laser-induced DNA breaks (Fig. 2). In contrast, GFP-BAL1 proteins, including a truncated macrodomain 2 (Macro2Δ [328 to 528]) or macrodomain 1 alone (Macro1 [1 to 291]) did not localize to DNA damage sites (Fig. 2). Consistent with these findings, targeted mutation of critical amino acids in macrodomain 2 (IE326,327AA) markedly reduced BAL1 recruitment, whereas a macrodomain 1 mutation (D126A) had no effect (Fig. 2). Therefore, BAL1 recruitment to DNA damage sites depends upon its macrodomains, and macrodomain 2 plays a nonredundant and essential role.
The BAL1-BBAP complex colocalizes with PARP1 and PAR and physically associates with PARylated proteins following DNA damage.
The kinetics of BAL1 recruitment to DNA damage sites were similar to those reported for PARP1 and its product, PAR (3, 9). We postulated that BAL1 interacted with PARP1 and/or PAR at sites of DNA damage, and we analyzed BAL1, PARP1, and PAR recruitment in HeLa cells following laser microirradiation (Fig. 3A). PARP1 colocalized with its product, PAR, and with BAL1 and related family members in laser-induced DNA breaks (Fig. 3A; also see Fig. S2 in the supplemental material); in addition, BBAP colocalized with PARP1 and PAR in DNA damage sites (Fig. 3B). We also subjected HeLa cells to low-dose irradiation and observed early colocalization of BAL1 and PAR repair foci (Fig. 3C).
Fig 3.

BAL1 and BBAP colocalize with PARP1 and PAR and physically associate with PARylated proteins following DNA damage. Colocalization of PARP1, PAR, and BAL1 (A) or PARP1, PAR, and BBAP (B) at laser-induced DNA breaks (2 min following laser microirradiation). (C) Colocalization of BAL1 and PAR foci in γ-irradiated cells. HeLa cells were treated with low-dose irradiation (100 cGy) and analyzed for BAL1 and PAR foci at baseline and at serial time points (0.5 to 60 min) thereafter. (D) Coimmunoprecipitation of PARylated proteins and BAL1. HeLa cells were untreated or treated with low-dose Dox (50 ng) for 10 min with or without PJ-34 pretreatment. Cell lysates were immunoprecipitated (IP) with anti-PARP1, anti-BAL1, or control IgG and immunoblotted with anti-PARP1, -PAR, -BAL1, or -BBAP antibodies. Input whole-cell lysates (left) were analyzed similarly and immunoblotted for actin as a loading control. Molecular weight markers are shown on the right. (E) Recruitment of GFP-PARP1, BAL1, and BBAP to laser-induced breaks in control cells or cells pretreated with the PARP inhibitor PJ-34 (2 min following laser microirradiation). (F) Kinetics of GFP-BAL1 recruitment to laser-induced DNA breaks in control or PJ-34-pretreated cells. GFP-BAL1 levels at each time point were determined by averaging values from 10 cells (±the standard error) in a representative experiment.
We next directly assessed the interactions between these proteins and protein modifications by immunoprecipitation. To define the basis of PARP1 interactions in these studies, we utilized the drug PJ-34, which inhibits PARP1 generation of PAR but leaves PARP1 DNA binding intact (9). HeLa cells were untreated or treated with low-dose Dox (50 ng/ml) for 10 min with or without PJ-34 pretreatment. Thereafter, whole-cell lysates were prepared, and PARP1 and BAL1 (and control IgG) were immunoprecipitated and immunoblotted for PARP1, PARylated proteins, BAL1, and BBAP (Fig. 3D). Input whole-cell lysates were analyzed similarly (Fig. 3D, left column).
As expected, input lysates from Dox-treated cells had a modest increase in PAR formation, which was abrogated by PJ-34 (Fig. 3D, left column). PARP1 immune complexes from Dox-treated cells contained more abundant PARylated proteins in addition to increased BAL1 and BBAP, and PJ-34 inhibited these interactions (Fig. 3D; see also Fig. S3A and B in the supplemental material). Following Dox treatment, BAL1 immune complexes also included increased PARylated pr proteins unless PARP activity was inhibited by PJ-34 (Fig. 3D). These data indicate that BAL1 binds PARylated proteins, which are more abundant following DNA damage and PARP1 activation.
To distinguish between BAL1 binding to PAR-modified proteins, including PARP1, or PARP1 itself, we performed in vitro pulldown assays using GST-tagged BAL1 proteins and FLAG-tagged PARP1 (see Fig. S3C and D in the supplemental material). We first demonstrated that FLAG-PARP1 was functionally active in vitro, catalyzing the synthesis of PARP1-associated PAR chains in the presence of NAD+ (see Fig. S3E). GST-BAL1 selectively pulled down PAR-modified PARP1 in a macrodomain-dependent manner but did not bind unmodified PARP1 protein (see Fig. S3F and G), confirming that BAL1 specifically binds to PAR.
Recruitment of BAL1-BBAP to DNA damage sites is dependent upon PARP activity.
Given the interaction between BAL1 and PARylated proteins (Fig. 3D; see also Fig. S3 in the supplemental material), we next asked whether PARP1 activity was required for BAL1 and BBAP recruitment to DNA damage sites. We first expressed GFP-PARP1, BAL1, or BBAP in HeLa cells and treated the cells with PJ-34 or vehicle alone prior to laser microirradiation. As expected, PJ-34 did not impair GFP-PARP1 binding to laser-induced DNA breaks (Fig. 3E). However, PJ-34 pretreatment inhibited the recruitment of GFP-BAL1 and BBAP to DNA damage sites (Fig. 3E and F).
Similar results were obtained when HeLa cells were preincubated with PJ-34 or vehicle alone, laser microirradiated, and immunostained for endogenous PARP1, PAR, BAL1, or BBAP (Fig. 4A). Although PJ-34 did not alter PARP1 recruitment to DNA damage sites, the compound eliminated PARP-mediated PAR formation (Fig. 4A, top row) and abrogated BAL1 and BBAP recruitment to laser-induced DNA breaks (Fig. 4A, middle and bottom rows). PJ-34 treatment also inhibited irradiation-induced PAR and BAL1 focus formation in HeLa cells (see Fig. S4A and B in the supplemental material). Taken together, these data indicate that PARP activity and PAR formation are required for BAL1 and BBAP localization to DNA damage sites.
Fig 4.

Recruitment of endogenous PARP1, PAR, BAL1, and BBAP to DNA damage sites. (A) Recruitment of endogenous PARP1, PAR, BAL1, and BBAP to laser-induced breaks in control or PJ-34-pretreated cells (2 min following laser microirradiation). (B) Depletion of PARP1, BAL1, and BBAP in HeLa cells. (C) Recruitment of BAL1, PARP1, BBAP, and PAR to laser-induced breaks in control cells or cells depleted of PARP1, BAL1, or BBAP (2 min following laser microirradiation).
To define the hierarchy of PARP1, BAL1, and BBAP interactions, we individually depleted each protein by siRNA and assessed the subsequent DDR (Fig. 4B and C). We focused on PARP1 because over 85% of in vivo PARP activity is attributed to this family member (25). PARP1 depletion (Fig. 4C, panels g to l) abrogated DNA damage-induced PARP1 localization (Fig. 4C, panel h) and PARP-mediated PAR formation (Fig. 4C, panel k). In addition, PARP1 knockdown inhibited BAL1 and BBAP recruitment to laser-induced DNA breaks (Fig. 4C, panels g and j, respectively). In BAL1- or BBAP-depleted cells, activated PARP1 and PAR were still recruited to DNA damage sites, confirming that PARP1 functions upstream of BAL1 and BBAP (Fig. 4C, panels n, q, t, and w, respectively). In BAL1-depleted cells, BBAP did not accumulate in laser-induced DNA breaks (Fig. 4C, panel p). In contrast, BAL1 localized to DNA damage sites in BBAP-depleted cells (Fig. 4C, panel s), placing BAL1 upstream of BBAP. Therefore, BAL1 is recruited to a DNA damage site via PARP1-mediated PAR formation, and BBAP localizes to the site via its interaction with BAL1.
BAL1 limits the cellular response to DNA-damaging agents.
We next assessed the function of BAL1 in tumor (HeLa) cell growth and (Dox-)induced cytotoxicity by depleting endogenous BAL1 via siRNA and treating the cells with Dox (50 to 200 ng/ml). Although BAL1 RNA interference (RNAi) reduced the growth of untreated cells, the consequences of BAL1 depletion were most striking in cells treated with low-dose Dox (50 ng/ml) (Fig. 5A) (P < 0.001, two-way analysis of variance [ANOVA]). For example, after 72 h of treatment with low-dose Dox (50 ng/ml), cellular proliferation was ≈80% lower in BAL1-depleted cells than in control RNAi or parental cells (Fig. 5A). BAL1 depletion also increased cellular apoptosis in both untreated and Dox-treated cells (Fig. 5B). These data indicate that the macrodomain-containing BAL1 protein enhances tumor cell survival and decreases Dox-induced cytotoxicity.
Fig 5.

BAL1 limits the cellular response to DNA-damaging agents. (A) BAL1 depletion augments the cellular response to DNA-damaging agents. HeLa cells were transfected with control or BAL1 siRNAs (siRNAs 1 and 2), treated with Dox at 50 ng/ml or 200 ng/ml, or left untreated for 1 to 96 h and subsequently evaluated by an MTS assay. The consequences of BAL1 depletion were most striking in cells treated with low-dose Dox (50 ng/ml) (P < 0.001, two-way ANOVA). After 72 h of treatment with low-dose Dox (50 ng/ml), cellular proliferation (as assessed by an MTS assay) was ≈70 to 80% lower in BAL1-depleted cells than in control RNAi or parental cells. OD, optical density. (B) Cellular apoptosis following BAL1 depletion and Dox treatment. Parental, control, and BAL1 siRNA-transfected HeLa cells were untreated or treated with Dox at 50 ng/ml and 200 ng/ml for 24 h and analyzed for apoptosis with anti-annexin V and PI staining. Error bars in panels A and B represent the standard deviations (SD) of the mean for three replicates in a representative experiment. (C) Recruitment of GFP-control, GFP-BAL1, or GFP-BAL1-DM to laser-induced breaks in (5′ UTR-specific) BAL1 siRNA knockdown cells. (a to c) GFP-control, GFP-BAL1, and GFP-BAL1-DM; (d to f) PARP-1 in these cells; (g to i) merged images. (D) Apoptosis of BAL1-depleted HeLa cells repleted with GFP-control, GFP-BAL1, or GFP-BAL1-DM and subsequently treated with doxorubicin (50 ng/ml) or left untreated. Apoptosis was assessed with annexin V and PI staining. Error bars represent the SD of the mean for 3 replicates in a representative experiment.
To directly evaluate the role of PARP-dependent BAL1 recruitment on tumor cell viability, we depleted BAL1 by siRNA and repleted it with GFP vector only, GFP-BAL1, or GFP-BAL1-DM (which lacks the PAR binding domain [Fig. 2]). In BAL1-depleted cells, BAL1 recruitment to PARP-1-associated laser-induced DNA breaks was restored by GFP-BAL1 but not by GFP-BAL1-DM (Fig. 5C, panels b versus c and h versus i). Importantly, GFP-BAL1 repletion also limited the apoptosis of BAL1-depleted cells at baseline and following Dox treatment, unlike GFP-BAL1-DM (Fig. 5D). These data directly and specifically associate the early macrodomain- and PAR-dependent recruitment of BAL1 to DNA damage sites (Fig. 5C) with enhanced tumor cell survival (Fig. 5D).
Early ubiquitin chain formation at DNA damage sites requires PARP1, BAL1, and BBAP.
After demonstrating the functional significance of BAL1 recruitment to PARylated DNA damage sites (Fig. 5), we next assessed a potential link between PARP1 activation, localization of the BAL1 partner E3 ligase BBAP, and conjugated ubiquitin chain formation. HeLa cells were pretreated with PJ-34 or vehicle alone, laser microirradiated, and immunostained for PARP1, BBAP, and newly formed ubiquitin chains (using the FK2 antibody which recognizes conjugated ubiquitin) (Fig. 6A and B). In control (vehicle-treated) cells, FK2 immunostaining was readily apparent from the earliest analyzed time point (5 min) through the last assessment (60 min) (Fig. 6A); in contrast, BBAP recruitment was most abundant at the earlier time points (Fig. 6B). Although PJ-34 did not impair PARP1 recruitment to DNA damage sites, the PARP inhibitor abrogated BBAP localization (Fig. 6B) and early ubiquitin chain formation (FK2 immunostaining) (Fig. 6A). BAL1 depletion similarly decreased BBAP recruitment and early ubiquitin chain formation at laser-induced DNA breaks (Fig. 6C and D; see also Fig. S5 in the supplemental material). Depletion of endogenous BBAP prior to laser microirradiation also abrogated early FK2 immunostaining (Fig. 6E; see also Fig. S5). The effects of PARP inhibition or BAL1 or BBAP depletion on ubiquitin chain formation were most striking in the first few minutes following laser microirradiation (Fig. 6A, C, and E; see also Fig. S5).
Fig 6.

PARP-dependent recruitment of BBAP to DNA damage sites is required for early ubiquitin chain formation. (A) Ubiquitylation (conjugated ubiquitin, FK2 immunostaining) and PARP1 recruitment at serial time points following laser microirradiation (baseline [0] and 5 to 60 min) in control or PJ-34-treated cells. (B) BBAP and PARP1 recruitment in laser-microirradiated control or PJ-34-treated cells. (C) Ubiquitylation (FK2 immunostaining) and PARP1 recruitment in laser-microirradiated control cells or cells depleted of BAL1. (D) BBAP and PARP1 recruitment in laser-microirradiated control cells or cells depleted of BAL1. (E) Ubiquitylation (FK2 immunostaining) and PARP1 recruitment in laser-microirradiated control cells or cells depleted of BBAP. (F) Ubiquitylation (FK2 immunostaining) and BAL1 recruitment in BAL1-depleted cells replete with GFP-control or GFP-BAL1. Images were obtained 5 min after laser microirradiation. (G) RNF8 and PARP1 recruitment at serial time points following laser microirradiation in control or PJ-34-treated cells. (H) RNF8 and PARP1 recruitment in laser-microirradiated control cells or cells depleted of BAL1.
To directly assess the role of BAL1 in early ubiquitin chain formation at DNA damage sites, we depleted BAL1 by siRNA, repleted it with GFP vector only or GFP-BAL1, and assessed FK2 immunostaining at 5 min (Fig. 6F). In BAL1-depleted cells, early ubiquitin chain formation (FK2 immunostaining) was selectively restored by GFP-BAL1 (Fig. 6F). Taken together, these data indicate that early ubiquitin chain formation at DNA damage sites is dependent upon PAR formation, BAL1 and BBAP recruitment, and the BBAP E3 ligase activity.
PARP1 activation and BAL1-BBAP recruitment to DNA damage sites are independent of ATM and MDC1.
Given the recently described ATM- and MDC1-dependent ubiquitylation at DNA damage sites (16, 17), we explored the relationship between ATM and MDC1 accumulation and PARP1 activation at laser-induced DNA breaks. HeLa cells were preincubated with PJ-34 or vehicle alone, laser microirradiated, and immunostained for endogenous ATM, MDC1, and PARP1 (see Fig. S6 in the supplemental material). The chemical PARP inhibitor did not impair ATM or MDC1 recruitment to DNA damage sites (see Fig. S6A). In complementary experiments, the depletion of ATM or MDC1 by siRNA (see Fig. S6B) had no effect on PARP1 or BAL1 accumulation at laser-induced DNA breaks (see Fig. S6C). Therefore, PARP1 activation and BAL1 recruitment are independent of ATM and MDC1, suggesting that there are two separately regulated pathways of DNA damage-induced ubiquitylation. Consistent with this hypothesis, neither chemical PARP inhibition nor BAL1 depletion altered the delayed kinetics of accumulation of the ATM- and MDC1-dependent E3 ligase, RNF8, at DNA damage sites (Fig. 6G and H). RNF8 was primarily detectable 40 to 60 min following laser microirradiation (Fig. 6G and H), in contrast to BBAP, which was most prominent from the earliest evaluated time point, 5 min, through 20 min (Fig. 6B and D).
Functional analyses of PARP1-BAL1-BBAP- and MDC1-RNF8-associated DDRs.
We next compared the functional consequences of the respective PARP1-PAR-dependent BAL1-BBAP and ATM-MDC1-RNF8 DDR pathways using comet assays, which measure unrepaired DNA damage in individual cells with gel electrophoresis (26). In initial studies, HeLa cells were pretreated with PJ-34 or vehicle alone, subjected to low-dose irradiation (200 cGy) and analyzed by comet assays 15 min, 60 min, or 24 h thereafter (Fig. 7A). In control cells, the intensities of the comet tails, which reflect unrepaired DNA damage, were modestly increased at 15 min, decreased at 60 min, and near baseline by 24 h (Fig. 7A, left). In PJ-34-treated cells, comet tail intensities were markedly increased at 15 min and persistently elevated through 24 h (Fig. 7A, right). BAL1- and BBAP-depleted cells had similarly increased comet tail intensities 15 min through 24 h following irradiation (Fig. 7B and D, left and middle panels). Therefore, chemical PARP inhibition and BAL1 or BBAP depletion all markedly increase unrepaired DNA damage at early and later time points (Fig. 7A, B, and D).
Fig 7.
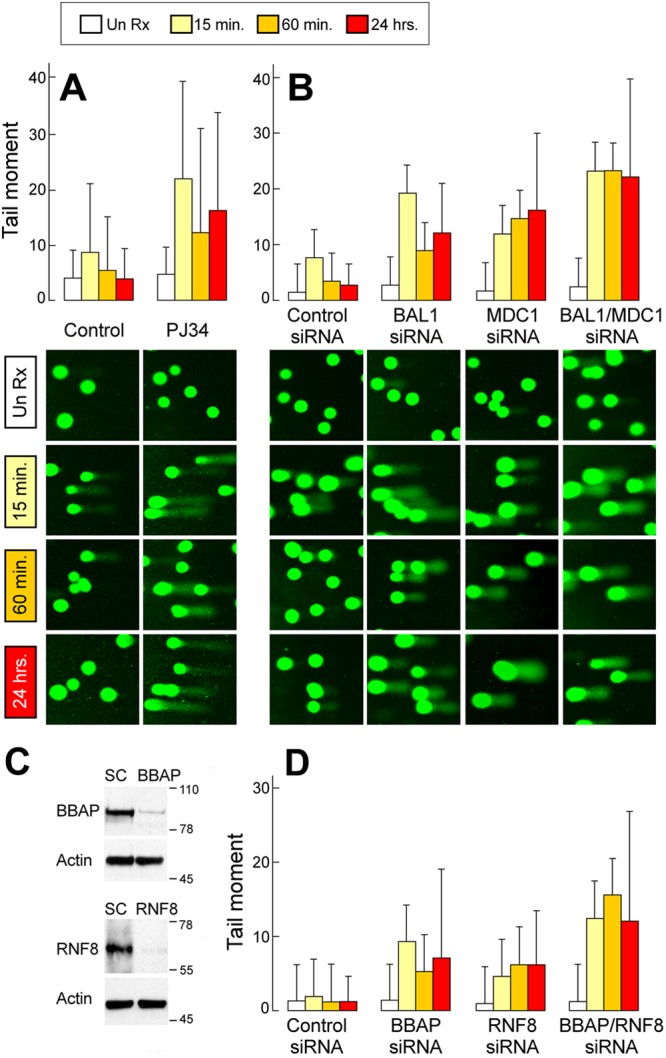
Functional analyses of PARP1-BAL1-BBAP- and MDC1-RNF8-associated DDRs by comet assay. Comet assays of HeLa cells pretreated with PJ-34 or vehicle alone (A), control, BAL1, MDC1, or BAL1 and MDC1 siRNAs (B), or control, BBAP, RNF8, or BBAP and RNF8 siRNAs (D), treated with low-dose irradiation (200 cGy) and analyzed under alkaline conditions 15 min, 60 min, or 24 h thereafter. Un Rx, untreated. (C) Depletion of BBAP or RNF8 following siRNA. HeLa cells treated with a scrambled control (SC) or BBAP or RNF8 siRNA were lysed, size fractionated, and immunoblotted with the respective antibodies and actin (as a loading control). The comet tail moment (% DNA in tail × tail length) was determined for 50 to 100 cells/condition using TriTek CometScore software. (A and B) Bar graphs (means ± SD); (C and D) representative photographs. Data are from one of three similar experiments.
The apparent independence of PARP1-BAL1-BBAP and ATM-MDC1-RNF8 recruitment and ubiquitylation at DNA damage sites (Fig. 6; see also Fig. S6 in the supplemental material) prompted us to compare these pathways using the comet assay. In contrast to BAL1-depleted cells, MDC1 knockdown cells had a more delayed pattern of unrepaired DNA damage (comet tail intensity) following irradiation (Fig. 7B). When BAL1 and MDC1 were both depleted prior to irradiation, comet tail intensity was greater than that of either single knockdown at 15 min through 24 h (Fig. 7B, right).
DNA damage was also more delayed in RNF8-depleted cells than in BBAP knockdown cells (Fig. 7C and D). When both E3 ligases, BBAP and RNF8, were depleted prior to irradiation, the comet tail intensity was greater than that of either single knockdown from early through later time points (Fig. 7D). Taken together, these data indicate that PARP1-dependent BAL1-BBAP-mediated DNA damage repair is functionally distinct and nonredundant compared to that of ATM, MDC1, and RNF8.
Early 53BP1 recruitment to DNA damage sites requires PARP1, BAL1, and BBAP.
We previously found that depletion of the BBAP E3 ligase delayed the accumulation of the checkpoint mediator 53BP1 in repair foci (14). Given the dependence of BBAP on PARP1 activation and BAL1 recruitment, we next assessed the consequences of chemical PARP1 inhibition (PJ-34 treatment) on 53BP1 accumulation at DNA damage sites (Fig. 8A). In PJ-34-treated cells, 53BP1 localization to laser-induced DNA breaks was significantly delayed (Fig. 8A). Similar results were obtained when endogenous PARP1, BAL1, or BBAP were depleted by siRNA prior to laser microirradiation (Fig. 8B [images obtained ≈20 min following microirradiation]). These data directly implicate PARP1 activation and BAL1-BBAP recruitment in the early localization of 53BP1 to DNA damage sites.
Fig 8.

Early 53BP1 recruitment to DNA damage sites requires PARP1, BAL1, and BBAP. (A) Recruitment of PARP1 and 53BP1 to laser-induced breaks in control or PJ-34-treated cells. Images were obtained at baseline (0) and 10 to 30 min following laser microirradiation. (B) PARP1 and 53BP1 recruitment to laser-induced breaks in control cells or cells depleted of PARP1, BAL1, or BBAP (via siRNA) (20 min following laser microirradiation). (C to F) Kinetics of 53BP1 and γH2AX focus formation following γ-irradiation of control or PJ-34-treated cells (C and D) or control siRNA or BAL1 siRNA-treated cells (E and F). Cells were treated with PJ-34 or vehicle alone (C and D) or control siRNA or BAL1 siRNA (E and F), subjected to low-dose (100-cGy) irradiation, and analyzed for 53BP1 and γH2AX foci at baseline and 1 to 60 min thereafter (C and E). Shown are the percentages of cells with ≥10 foci per nucleus at each time point and condition. Error bars represent the SD of the means for 3 independently stained slides for each time point and condition. At the earliest time points following irradiation (0 to 4 min), the development of repair foci (percentage of cells with ≥10 foci per nucleus) was compared in control and PJ-34-treated cells and control siRNA and BAL siRNA-treated cells with an ANOVA.
To further characterize potentially separate PARP1- and ATM-MDC1-RNF8-H2AX-dependent pathways of 53BP1 accumulation, HeLa cells were pretreated with PJ34 or vehicle alone, subjected to low-dose irradiation, and analyzed for 53BP1 and H2AX foci at 0 to 60 min thereafter. At the earliest time points following irradiation (<30 min), there were significantly more 53BP1 foci in control cells than in PJ-34-treated cells (P = 0.004) (Fig. 8C and D). In contrast, H2AX focus formation was more delayed than that of 53BP1 and unaffected by treatment with PJ-34 (P = not significant [NS]) (Fig. 8C and D). After irradiation, 53BP1 focus formation was also more rapid in control cells than in BAL1-depleted cells (P = 0.001) (Fig. 8E and F). However, H2AX focus formation was more delayed and similar in control and BAL1 knockdown cells (P = NS) (Fig. 8E and F). These data define the initial PARP1- and BAL1-dependent, H2AX-independent recruitment of 53BP1 to DNA damage sites.
Early RAP80 and BRCA1 localization to DNA damage sites requires PARP1, BAL1, and BBAP.
BRCA1 accumulates at DNA damage sites via the adaptor protein RAP80 and its ubiquitin-interacting motifs. For these reasons, we also analyzed the kinetics of BRCA1 and RAP80 accumulation at laser-induced DNA breaks following chemical PARP inhibition. In PJ-34-treated cells, the recruitment of RAP80 and BRCA1 to DNA damage sites was markedly delayed (Fig. 9A and B).
Fig 9.
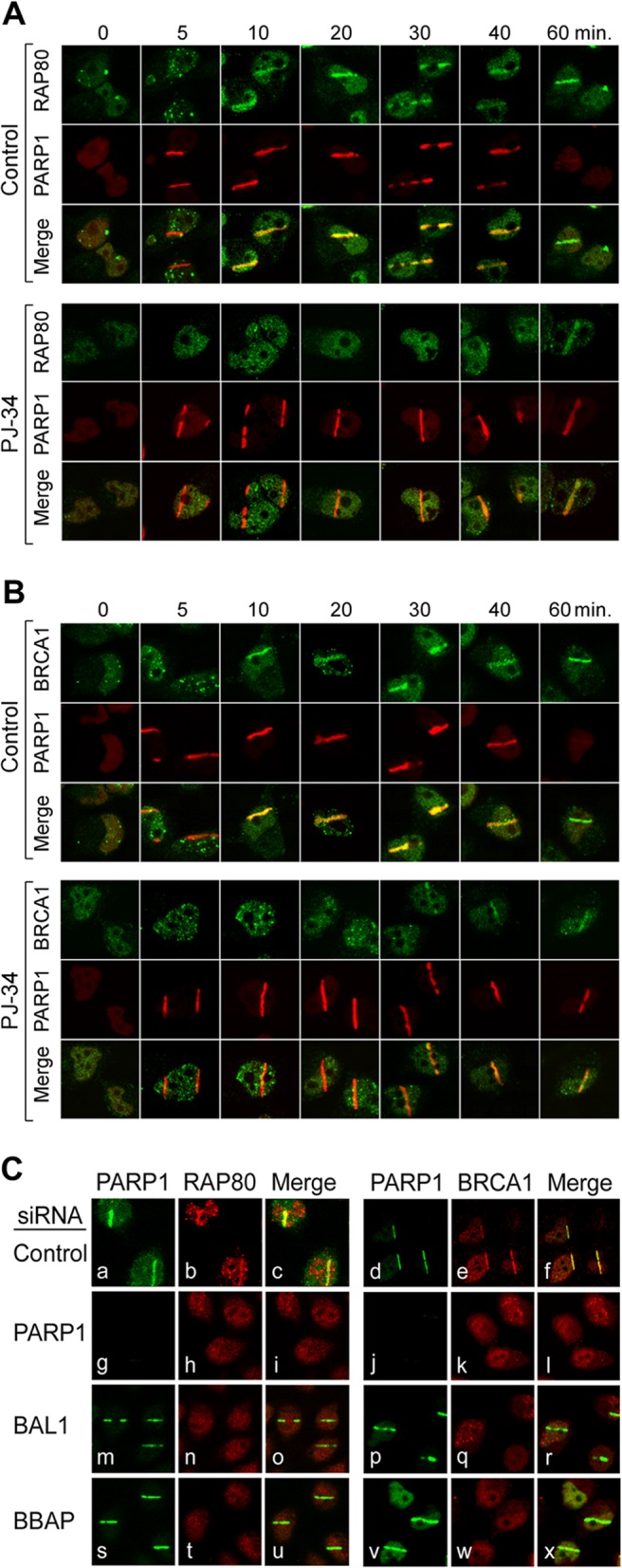
Early recruitment of RAP80-BRCA1 to DNA damage sites requires PARP1, BAL1, and BBAP. PARP1 and RAP80 (A) and BRCA1 (B) recruitment to laser-induced breaks in control or PJ-34-treated cells (0 to 60 min following laser microirradiation). (C) PARP1, RAP80, and BRCA1 recruitment to laser-induced breaks in control cells (a through f) or cells depleted of PARP1 (g to l), BAL1 (m to r), or BBAP (s to x) (via siRNA) (10 min following laser microirradiation).
Localization of RAP80 and BRCA1 to DNA damage sites was also delayed in BAL1-depleted cells (see Fig. S7A to D in the supplemental material). Furthermore, PARP1, BAL1, or BBAP knockdown all reduced early RAP80 and BRCA1 localization to laser-induced DNA breaks (Fig. 9C [PARP1, panels h and k; BAL1, panels n and q; and BBAP, panels t and w, respectively]). Consistent with these findings, BBAP ubiquitylated the RAP80 residues K63 (27) and K48 (see Fig. S8 in the supplemental material).
Taken together, these data indicate that PARP1-, BAL1-, and BBAP-dependent ubiquitylation provides initial access to the downstream mediators RAP80 and BRCA1 at DNA damage sites (Fig. 10). These studies define separate temporally and functionally distinct mechanisms of DNA damage-induced ubiquitylation and recruitment of 53BP1 and BRCA1—an early PARP1-, BAL1-, and BBAP-dependent pathway and a later phosphorylation-dependent ATM-MDC1-RNF8-associated route (Fig. 10).
Fig 10.
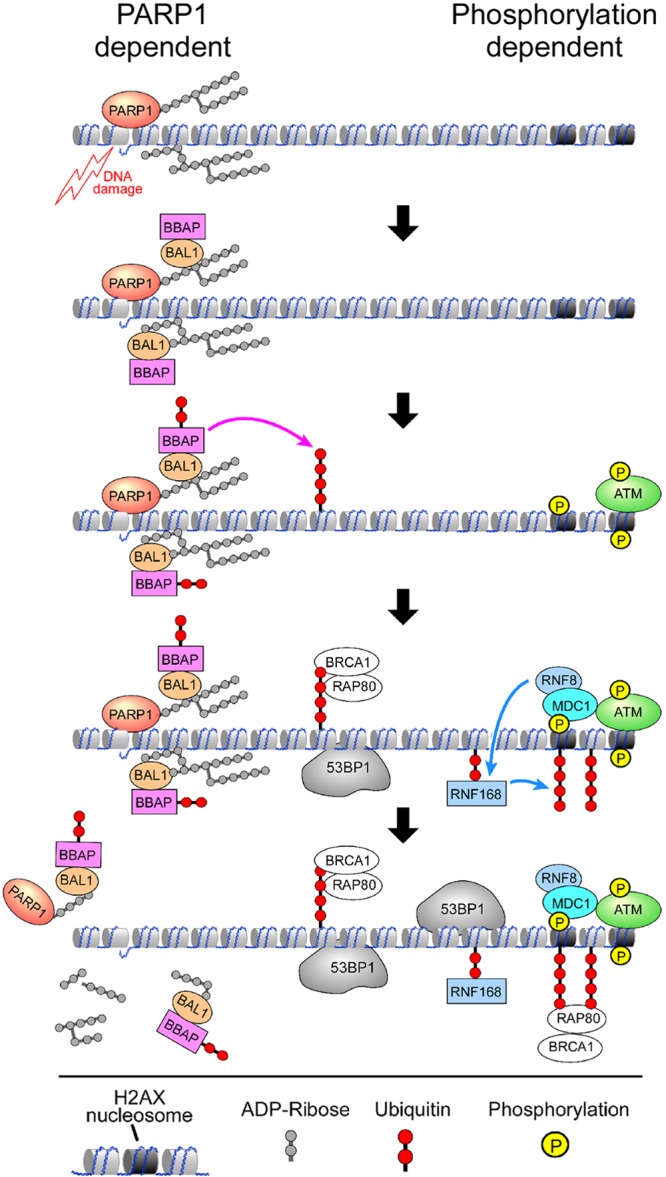
DNA damage-induced ubiquitylation and recruitment of 53BP1 and RAP80-BRCA1 occurs via an early PARP1-, BAL1-, and BBAP-dependent pathway and a later phosphorylation-dependent ATM-MDC1-RNF8-associated route.
DISCUSSION
In this study, we have defined a direct link between the initial rapid and short-lived PARylation at DNA damage sites, PAR-dependent recruitment of the BAL1 macrodomain-containing protein and its partner E3 ligase BBAP, local BBAP-mediated ubiquitylation, and subsequent recruitment of the checkpoint mediators 53BP1 and BRCA1. The PARP1-dependent localization of BAL1 and BBAP functionally limits early and delayed DNA damage and enhances cellular viability independently of ATM, MDC1, and RNF8. These data firmly establish BAL1 and BBAP as bona fide DDR pathway members and provide new insights into PARP-mediated DNA repair.
The kinetics of BAL1 and BBAP recruitment to laser-induced breaks reflect their early PAR-dependent localization at these sites. These findings provide a mechanism for the previously described delay in 53BP1 focus formation in BBAP-depleted cells (14). In the earlier studies, BBAP knockdown selectively decreased 53BP1 recruitment at early time points following DNA damage induced by low-dose doxorubicin (50 ng/ml) or γ-irradiation (100 cGy); however, at later time points, the numbers of 53BP1 repair foci were similar in BBAP-depleted cells and controls (14). These findings likely reflect selective impairment of early PAR-dependent BAL1-BBAP-mediated 53BP1 recruitment with intact delayed ATM, MDC1, and RNF8-dependent events (Fig. 10).
Although two additional macrodomain-containing proteins bind PAR and participate in chromatin reorganization, histone macro-H2A1.1 (9) and nucleosome sliding (ALC1) (3), BAL1 plays a unique role in PAR-dependent DDRs by localizing its partner E3 ligase to DNA breaks. BAL1 interacts with BBAP via the unique BAL1 C terminus (11) and binds PAR through the BAL1 N-terminal macrodomains. Of interest, BAL family members are the only described proteins with multiple macrodomains (6, 8). Previous phylogenetic analyses suggested that there might be functional distinctions between the respective macroregions in a given BAL1 protein (6). Consistent with this hypothesis, only BAL1 macrodomain 2 was required for recruitment to DNA damage sites.
In addition to having multiple N-terminal macrodomains, BAL family members contain C-terminal regions with similarities to known ADP-ribosyltransferase catalytic sites (6). However, BAL1 lacks the required catalytic glutamic acid and additional donor and acceptor sequences which are conserved in BAL2B and BAL3 (6). Consistent with these findings, BAL2B and BAL3, but not BAL1, catalyze ADP ribosylation (6). Although all BAL family members localize to laser-induced DNA breaks (Fig. 1 and 2; see also Fig. S2 in the supplemental material), the specific roles of BAL family members which catalyze ADP ribosylation, BAL2B and BAL3, remain to be defined.
The degree of PARP1-mediated poly(ADP)-ribosyl)ation is thought to reflect the severity of genotoxic stress (28). Under conditions of mild to moderate DNA damage, poly(ADP-ribosyl)ation supports repair at DNA damage sites. In contrast, severe stress conditions lead to the overactivation of PARP1, unregulated poly(ADP-ribose) synthesis, NAD+ and ATP+ depletion, and massive cell death (28). BAL1 and BBAP are most effective in limiting the DNA damage and cytotoxicity associated with low- rather than high-dose doxorubicin or γ-irradiation. These observations are consistent with the newly identified roles of BAL1 and BBAP in PARP1-dependent DNA damage repair.
The current studies also define an early wave of ubiquitylation at DNA damage sites that is dependent upon PARP1 activation, BAL1-BBAP recruitment, and BBAP E3 ligase activity and independent of ATM-MDC1-H2AX and RNF8. These findings are consistent with earlier reports which suggested that the initial detection of DNA lesions occurs independently of H2AX phosphorylation (29). In these earlier studies, the initial recruitment of 53BP1 and BRCA1 to DNA damage sites was intact in H2AX-deficient mice (29).
PARP1-dependent BAL1-BBAP-mediated ubiquitylation promotes the rapid and specific recruitment of 53BP1, RAP80, and BRCA1 to DNA damage sites. BBAP ubiquitylates histone H4 lysine 91 and increases the accessibility of H4K20 to methylation and 53BP1 recruitment via its tandem tudor domain (14). BBAP-mediated ubiquitylation likely fosters RAP80-BRCA1 localization via RAP80 ubiquitin-interacting motifs (UIMs). Given the time course and kinetics of PARP1 activation and ATM-MDC1 phosphorylation, these studies define separate and complementary pathways of BBAP- and RNF8-RNF168-mediated ubiquitylation and recruitment of 53BP1 and BRCA1 to DNA damage sites. Functional analyses confirm the nonredundant role of PARP1-dependent BAL1-BBAP recruitment and ubiquitylation on DDR (Fig. 5 and 7).
The roles of BAL1 and BBAP in PARP1-dependent DNA damage repair have additional clinical implications. The BAL1 macroprotein and its partner E3 ligase were originally identified in a screen for genes that were overexpressed in treatment-resistant lymphomas. The current studies provide a mechanism for the earlier observations and suggest that targeted inhibition of BAL1 and/or BBAP might increase the efficacy of chemotherapy (doxorubicin) or radiation treatment.
Recent studies have highlighted the utility of combining DNA-damaging chemotherapy with PARP inhibition (30–32). PARP inhibitors were initially evaluated in BRCA-deficient breast cancers based on the predicted “synthetic lethality” between PARP-dependent largely single-strand break repair and BRCA-associated DSB repair (31). More recent studies of combined PARP inhibition and chemotherapy were based on the notion of overwhelming cellular DNA repair pathways in tumors with intact BRCA1 (31, 32). Our studies provide new evidence of a direct link between PARP1 activation and BRCA1 recruitment and implicate the BAL1 macroprotein and BBAP E3 ligase in these processes. For these reasons, additional analyses of the mechanisms and consequences of BAL1 and BBAP overexpression and their targeted inhibition are warranted.
Supplementary Material
ACKNOWLEDGMENTS
We thank Lai Ding and Daniel Tom of the Harvard NeuroDiscovery Center for their assistance with laser microirradiation and confocal microscopy and Daniel Silver for his helpful comments.
We acknowledge support from NIH grants PO1CA092625 (M.A.S.) and CA133781 (J.M.).
Footnotes
Published ahead of print 10 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00990-12.
REFERENCES
- 1. Polo SE, Jackson SP. 2011. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 25:409–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol. Cell 40:179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahel D, Horejsi Z, Wieckens N, Polo SE, Garcia-Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP, Owen-Hughes T, Boulton SJ. 2009. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325:1240–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krishnakumar R, Kraus WL. 2010. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol. Cell 39:8–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aguiar R, Yakushijin Y, Kharbanda S, Salgia R, Fletcher J, Shipp M. 2000. BAL is a novel risk-related gene in diffuse large B-cell lymphomas which enhances cellular migration. Blood 96:4328–4334 [PubMed] [Google Scholar]
- 6. Aguiar RCT, Takeyama K, He C, Kreinbrink K, Shipp M. 2005. B-aggressive lymphoma (BAL) family proteins have unique domains which modulate transcription and exhibit PARP activity. J. Biol. Chem. 280:33756–33765 [DOI] [PubMed] [Google Scholar]
- 7. Han W, Li X, Fu X. 2011. The macro domain protein family: structure, functions, and their potential therapeutic implications. Mutat. Res. 727:86–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. 2010. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 35:208–219 [DOI] [PubMed] [Google Scholar]
- 9. Timinszky G, Till S, Hassa PO, Hothorn M, Kustatscher G, Nijmeijer B, Colombelli J, Altmeyer M, Stelzer EH, Scheffzek K, Hottiger MO, Ladurner AG. 2009. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 16:923–929 [DOI] [PubMed] [Google Scholar]
- 10. Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai YSSK, Washburn MP, Florens L, Ladumer AG, Conaway RC. 2009. Poly(ADP-ribosyl)ation directs recruitment and activation to an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. U. S. A. 106:13770–13774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takeyama K, Aguiar RCT, Gu L, He C, Freeman GJ, Kutok JL, Aster JC, Shipp MA. 2003. The BAL-binding protein, BBAP, and related Deltex family members exhibit E3 ubiquitin ligase activity. J. Biol. Chem. 278:21930–21937 [DOI] [PubMed] [Google Scholar]
- 12. Juszczynski P, Kutok JL, Li C, Mitra J, Aguiar RC, Shipp MA. 2006. BAL1 and BBAP are regulated by a gamma interferon-responsive bidirectional promoter and are overexpressed in diffuse large B-cell lymphomas with a prominent inflammatory infiltrate. Mol. Cell. Biol. 26:5348–5359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, Wu B, Pasqualucci L, Neuberg D, Aguiar RC, Dal Cin P, Ladd C, Pinkus GS, Salles G, Harris NL, Dalla-Favera R, Habermann TM, Aster JC, Golub TR, Shipp MA. 2005. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 105:1851–1861 [DOI] [PubMed] [Google Scholar]
- 14. Yan Q, Dutt S, Xu R, Graves K, Juszczynski P, Manis JP, Shipp MA. 2009. BBAP monoubiquitylates histone H4 at lysine 91 and selectively modulates the DNA damage response. Mol. Cell 36:110–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. 2007. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131:901–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. 2007. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131:887–900 [DOI] [PubMed] [Google Scholar]
- 17. Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, Lukas J, Lukas C. 2009. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 136:435–446 [DOI] [PubMed] [Google Scholar]
- 18. Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, Pelletier L, Jackson SP, Durocher D. 2007. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318:1637–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. 2007. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316:1198–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, Oldreive C, Wildenhain J, Tagliaferro A, Pelletier L, Taubenheim N, Durandy A, Byrd PJ, Stankovic T, Taylor AM, Durocher D. 2009. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136:420–434 [DOI] [PubMed] [Google Scholar]
- 21. Wang B, Elledge SJ. 2007. Ubc13/Rnf8 ubiqutin ligases control foci formation of the Rap80/Abraxas/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. U. S. A. 104:20759–20763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. 2007. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316:1194–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. 2011. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 25:685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim H, Chen J, Yu X. 2007. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 316:1202–1205 [DOI] [PubMed] [Google Scholar]
- 25. Yelamos J, Monreal Y, Saenz L, Aguado E, Schreiber V, Mota R, Fuente T, Minguela A, Parrilla P, de Murcia G, Almarza E, Aparicio P, Menissier-de Murcia J. 2006. PARP-2 deficiency affects the survival of CD4+CD8+ double-positive thymocytes. EMBO J. 25:4350–4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olive PI, Banath JP. 2006. The comet assay: a method to measure DNA damage in individual cells. Nat. Protoc. 1:23–29 [DOI] [PubMed] [Google Scholar]
- 27. Lok GT, Sy SM, Dong SS, Ching YP, Tsao SW, Thomson TM, Huen MS. 2012. Differential regulation of RNF8-mediated Lys48- and Lys63-based poly-ubiquitylation. Nucleic Acids Res. 40:196–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hassa PO. 2009. The molecular “Jekyll and Hyde” duality of PARP1 in cell death and cell survival. Front. Biosci. 14:72–111 [DOI] [PubMed] [Google Scholar]
- 29. Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. 2003. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 5:675–679 [DOI] [PubMed] [Google Scholar]
- 30. Balmana J, Domchek SM, Tutt A, Garber JE. 2011. Stumbling blocks on the path to personalized medicine in breast cancer: the case of PARP inhibitors for BRCA1/2-associated cancers. Cancer Discov. 1:29–34 [DOI] [PubMed] [Google Scholar]
- 31. Ellisen LW. 2011. PARP inhibitors in cancer therapy: promise, progress, and puzzles. Cancer Cell 19:165–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, Koo IC, Sherman BM, Bradley C. 2011. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 364:205–214 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.