

Abstract
To determine talin1's role in osteoclasts, we mated TLN1fl/fl mice with those expressing cathepsin K-Cre (CtsK-TLN1) to delete the gene in mature osteoclasts or with lysozyme M-Cre (LysM-TLN1) mice to delete TLN1 in all osteoclast lineage cells. Absence of TLN1 impairs macrophage colony-stimulating factor (M-CSF)-stimulated inside-out integrin activation and cytoskeleton organization in mature osteoclasts. Talin1-deficient precursors normally express osteoclast differentiation markers when exposed to M-CSF and receptor activator of nuclear factor κB (RANK) ligand but attach to substrate and migrate poorly, arresting their development into mature resorptive cells. In keeping with inhibited resorption, CtsK-TLN1 mice exhibit an ∼5-fold increase in bone mass. Osteoclast-specific deletion of Rap1 (CtsK-Rap1), which promotes talin/β integrin recognition, yields similar osteopetrotic mice. The fact that the osteopetrosis of CtsK-TLN1 and CtsK-Rap1 mice is substantially more severe than that of those lacking αvβ3 is likely due to added failed activation of β1 integrins. In keeping with osteoclast dysfunction, mice in whom talin is deleted late in the course of osteoclastogenesis are substantially protected from ovariectomy-induced osteoporosis and the periarticular osteolysis attending inflammatory arthritis. Thus, talin1 and Rap1 are critical for resorptive function, and their selective inhibition in mature osteoclasts retards pathological bone loss.
INTRODUCTION
The osteoclast is the principal, if not exclusive, skeletal resorptive cell whose activity is initiated by contact with bone. In all cells, including osteoclasts, integrins recognize substrate, participate in cytoskeletal organization, and transmit matrix-derived signals, events essential to the resorptive process (1, 2).
αvβ3 is the principal integrin mediating the osteoclast's capacity to resorb bone. This heterodimer remodels the cell's cytoskeleton via recruitment of a complex including c-Src, Syk, Dap12, SLP-76, and Vav3. In consequence, the cytoskeleton-organizing GTPase Rac transits from its inactive GDP- to its active GTP-bound form. Absence of any component of this signaling pathway yields a common osteoclast phenotype with a deranged cytoskeleton and inability to spread (3–8). Macrophage colony-stimulating factor (M-CSF), which promotes osteoclastogenesis, also organizes the cytoskeleton of the mature bone resorptive cell by activating a signaling complex strikingly similar to that stimulated by αvβ3 (9, 10).
In osteoclasts, αvβ3 mediates bone matrix recognition by binding ligands containing the RGD motif, but its ability to do so depends upon its configuration. Like integrins in other cells, αvβ3 in the osteoclast is maintained in a default low-affinity state by interactions between the α and β subunits (11, 12). In its inactive conformation, the integrin heterodimer assumes a “bent” configuration in which the α and β intracellular domains interact via a membrane-proximal salt bridge. Upon integrin activation (i.e., increased affinity), the salt bridge is disrupted, the cytoplasmic tails separate, and the extracellular domain “straightens” to achieve a configuration permitting it to effectively bind ligand (13).
Integrins are activated by two basic mechanisms (13). Outside-in activation is achieved by initial ligand binding, which causes secondary conformational modifications, further enhancing the integrin's affinity. Outside-in conformational change can also be induced by chemicals or monoclonal antibodies (MAbs) prior to ligand binding. Inside-out activation, on the other hand, is an indirect process in which signals derived from an occupied receptor, typically that of a cytokine or growth factor, target the intracellular domain of an integrin. Such targeting of αvβ3 disrupts its salt bridge, separates its tails, and straightens the bent conformation of its external domain. The integrin is now positioned to bind ligand with high affinity and transmit matrix-derived signals, including those that organize the cytoskeleton.
Virtually all studies of integrin function in the osteoclast have focused on outside-in signaling. These experiments typically entail parachuting suspended, preterminally differentiated osteoclastic cells onto an αvβ3 ligand. This approach, however, does not mirror the resorptive process, in which osteoclasts are attached to the bone surface and constantly remodel their cytoskeleton as they move and degrade skeletal tissue. Nor does it mimic the physiological setting, in which cells are exposed to an array of cytokines and growth factors with the potential to alter integrin conformation.
Talin is a 270-kDa cytosolic adaptor protein that links β integrins to the actin cytoskeleton and in so doing regulates multiple biological events such as macrophage participation in innate immunity (14). In all cells studied, the interaction between talin and the β-integrin cytoplasmic domain is a final common and necessary step to inside-out integrin activation (15). The high-affinity heterodimer now avidly binds extracellular ligand, which stimulates the transmission of intracellular signals, including those that organize the cytoskeleton. We find that M-CSF, which activates αvβ3 in osteoclasts, promotes the association of integrin with talin via Y747 in the β3 tail. Likely reflecting the inhibition of multiple integrins, we also note that conditional deletion of talin or its activator Rap1, in mature osteoclasts, arrests the resorptive function and increases bone mass substantially more than does absence of αvβ3. Additionally, osteoclast-specific talin deletion protects against estrogen-deficient osteoporosis and inflammatory osteolysis. Our observation that the pronounced bone-sparing effect of deleting talin or Rap1 occurs only in committed bone resorbing cells gives credence to a therapeutic strategy that avoids targeting early osteoclast precursors such as macrophages. Hence, the Rap1/talin signaling complex is a necessary component of physiological and pathological bone resorption.
MATERIALS AND METHODS
Mice.
Generation of Tln1fl/fl, β3Y747A, and Rap1Afl/fl Rap1Bfl/fl mice has been described (16–18). CtsKcre/+ mice were provided by S. Kato (University of Tokyo, Tokyo, Japan) (19). LysMCre-expressing mice were purchased from the Jackson laboratory. To obtain mice with talin1-deficient macrophages (LysM-TLN), Tln1fl/fl LysMCre+/WT males were bred with Tln1fl/fl LysMCre+/WT females. In all experiments, Tln1fl/fl Cre+ or Rap1A/Bfl/fl Cre+ mice were compared with Tln1fl/fl Cre− or Rap1A/Bfl/fl Cre− sex-matched littermates.
Reagents.
Recombinant murine M-CSF was obtained from R&D Systems (Minneapolis, MN). Glutathione S-transferase-receptor activator of nuclear factor κB ligand (GST-RANKL) was expressed in our laboratory as described previously (6). The sources of antibodies are as follows: mouse antitalin MAbs and antiactin antibodies were from Sigma (St. Louis, MO); antiphosphotyrosine MAb 4G10 was from Upstate (Charlottesville, VA); MAb 327, directed against the c-Src protein, was a gift of A. Shaw (Department of Pathology, Washington University School of Medicine, St. Louis, MO); rabbit anti-Src p-Y416 antibody and rabbit anti-β3 integrin antibody were from Cell Signaling (Beverly, MA). Anti-NFATc1, anti-Rap1A, and anti-Rap1B antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). WOW1 ligand mimetic, which recognizes activated αvβ3 integrin, was provided by Sanford Shattil (University of California at San Diego). All other chemicals were obtained from Sigma.
Macrophage isolation and osteoclast culture.
Primary bone marrow macrophages (BMMs) were prepared as described previously (6), with slight modification. Marrow was extracted from femora and tibiae of 6- to 8-week-old mice with α-minimal essential medium (α-MEM) and cultured in α-MEM containing 10% inactivated fetal bovine serum (FBS), 100 IU/ml penicillin, and 100 μg/ml streptomycin (α-10 medium) with 1:10 CMG medium (conditioned medium containing 100 ng/ml mouse M-CSF) (20) on bacterial plastic dishes. Cells were incubated at 37°C in 6% CO2 for 3 days and then washed with phosphate-buffered saline (PBS) and lifted with 1× trypsin/EDTA (Invitrogen, Carlsbad, CA) in PBS. A total of 5 × 103 cells were cultured in 200 μl α-MEM containing 10% heat-inactivated FBS with 100 ng/ml GST-RANKL and 30 ng/ml of mouse recombinant M-CSF in 96-well tissue culture plates, some containing sterile bone slices. Cells were fixed and stained for tartrate-resistant acid phosphatase (TRAP) activity after 6 days in culture, using a commercial kit (Sigma 387-A, St. Louis, MO). For preosteoclast generation, 1.5 × 106 BMMs were plated per 10-cm tissue culture dish and maintained in 30 ng/ml M-CSF and 100 ng/ml GST-RANKL for 3 days.
Actin ring and bone resorptive pit visualization.
For actin ring staining, cells were cultured on bovine bone slices in the presence of M-CSF and RANKL for 6 days, after which cells were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100, rinsed in PBS, and immunostained with AlexaFluor 488-phalloidin (Molecule Probes). To quantitate resorption lacunae, cells were removed from bone slices with mechanical agitation. Bone slices were incubated with peroxidase-conjugated wheat germ agglutinin (WGA) (Sigma) for 1 h and stained with 3,3′-diaminobenzidine (Sigma).
Plasmids and retroviral transduction.
Talin head constructs containing amino acids 1 to 433 were inserted into a pMX retrovirus vector in which the puromycin resistance sequence was replaced with one coding for blasticidin resistance. Generation of the pMX Turbo-Cre construct has been described (21). It was transfected transiently into Plat-E packaging cells using FuGENE 6 transfection reagent (Roche). Virus was collected 48 h after transfection. BMMs were infected with virus for 24 h in the presence of 100 ng/ml M-CSF and 4 μg/ml Polybrene (Sigma). Cells were selected in the presence of M-CSF and 1 μg/ml blasticidin (Calbiochem) for 3 days prior to use as osteoclast precursors.
RNA preparation and RT-PCR analyses.
Total RNA from cultured cells was isolated using the Total RNA Isolation kit (Sigma). For reverse transcription-PCR (RT-PCR) analysis, cDNAs were synthesized from 1 μg of total RNA using reverse transcriptase and oligo(dT) primers in a volume of 20 μl. The reaction mixture was adjusted to 100 μl with Tris-EDTA (TE) buffer for PCR analysis. PCR was performed with 1 μl of cDNA reaction mixture by using Platinum Pfx polymerase (Invitrogen, Carlsbad, CA) and appropriate primers in a volume of 50 μl. The following primers were used: for Tln1, 5′-CAGAGGCCGTAGAAGACCTG and 3′-GGCTGGTGTTTGACTTGGTT; for Tln2, 5′-TGCAATGTGGTGAAGACCAT and 3′-GTGTCCGACCTGCTTCTAGC; and for b3, 5′-TTACCCCGTGGACATCTACTA and 3′-AGTCTTCCATCCAGGGCAATA.
Western blotting and immunoprecipitation.
Cultured cells were washed twice with ice-cold PBS and lysed in radioimmunoprecipitation assay (RIPA) buffer containing 20 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM NaF, and 1× protease inhibitor mixture (Roche). After incubation on ice for 10 min, cell lysates were clarified by centrifugation at 21.1 × 103 g for 10 min. Forty micrograms of total lysates was subjected to 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes. Filters were blocked in 0.1% casein in PBS for 1 h and incubated with primary antibodies at 4°C overnight followed by probing with fluorescence-labeled secondary antibodies (Jackson Lab). Proteins were detected with the Odyssey Infrared Imaging system (LI-COR Biosciences).
Rac1 and Rap1 activation assay.
Preosteoclasts cultured on plastic dish or bone powder were stimulated with M-CSF and lysed, and Rac or Rap1 activity was determined by an effector domain, GST-fusion pulldown protocol using the EZ-detect Rac1 or Rap1activation kit (Pierce Biotechnology Inc.).
Medium CTx-1 assay.
Macrophages were cultured on bone with 100 ng/ml RANKL and 1/100 volume CMG14-12 mouse M-CSF-producing cell line (20) supernatant for 6 days. The medium (α-10) was changed 1 day before harvesting. Medium CTx-1 concentration was determined using the CrossLaps for Culture enzyme-linked immunosorbent assay (ELISA) kit (Nordic Bioscience Diagnosis A/S).
Phagocytosis assay.
Thymocytes isolated from 4-week-old C57BL/6 mice were γ-irradiated (1,400 rads) and cultured for 4 h to induce apoptosis. Thymocytes were 50 to 60% apoptotic as assessed by fluorescein isothiocyanate (FITC)-annexin V (BD Pharmingen) and 7-ADD (Calbiochem) staining (annexin V+, 7-ADD−). CFSE (5,6-carboxyfluorescein diacetate succinimidyl ester) (5 μM) was used to label thymocytes for 10 min at room temperature. Labeled thymocytes were cocultured with macrophages at a ratio of 5:1 at 37°C. Cells were harvested with PBS-EDTA, stained with CD11b-allophycocyanin (CD11b-APC), and analyzed by flow cytometry. The percentage of phagocytosis was calculated as the fraction of CD11b+ cells that were positive in the FL1 channel.
Serum osteocalcin measurements.
Blood was collected retro-orbitally under anesthesia immediately prior to sacrifice. Serum osteocalcin levels were measured by ELISA (Biomedical Technologies Inc.). Plasma was obtained using plasma separator tubes with lithium heparin (Becton, Dickinson).
Microcomputed tomography (μCT).
The trabecular volume in the distal femoral metaphysis or proximal tibia was measured using a Scanco μCT40 scanner (Scanco Medical AG, Bassersdorf, Switzerland). A threshold of 250 was used for evaluation of all scans. Forty to 100 slices were analyzed, starting with the first slice in which condyles and primary spongiosa were no longer visible.
Calcein labeling.
We injected calcein (Sigma) intraperitoneally at 7.5 mg/kg of body weight on days 7 and 2 before killing and then sectioned tibiae in methyl-methacrylate, which were analyzed using BioQuant OsteoII (BioQuant Image Analysis Corporation, Nashville, TN).
Serum transfer arthritis and histological analysis.
Arthrogenic serum was obtained from K/B × N mice (6 to 12 weeks old). Serum (100 μl per mouse) was injected intraperitoneally on days 7 and 2 prior to sacrifice, whereas 100 μl of PBS was administered to control animals. Five or more mice were used for each group. Paw thickness was measured daily.
Histology and histomorphometry.
The tibiae of 2-month-old mice were fixed with 10% neutral buffered formalin, followed by decalcification in 14% EDTA for 10 days, paraffin embedding, and TRAP staining. Osteoclastic perimeters were measured and analyzed using BioQuant OsteoII (BioQuant Image Analysis Corporation, Nashville, TN) in a blinded fashion. Pit areas were measured with ImageJ.
Statistics.
Statistical significance was determined using Student's t test. All experiments were performed at least twice.
RESULTS
M-CSF stimulates inside-out activation of the αvβ3 integrin in osteoclasts.
The fact that the signaling events by which M-CSF alters the osteoclast cytoskeleton are similar to those mediated by activated αvβ3 suggests a collaborative association. Confirming cytokine dependence on the integrin, M-CSF activation of Rac, the currently established most distal component of a canonical osteoclast cytoskeleton organizing complex, requires αvβ3 (Fig. 1A). Establishing that the collaboration between αvβ3 and M-CSF reflects inside-out stimulation, the cytokine approximately doubles binding of WOW1, an αvβ3 ligand mimetic recognizing the activated integrin (Fig. 1B). Furthermore, αvβ3 progressively associates with talin under the influence of M-CSF (Fig. 1C), which also promotes talin migration from the podosome belt to cytokine-induced fillipodia (Fig. 1D).
Fig 1.

M-CSF activates αvβ3 in osteoclasts. (A) Cytokine-starved, bone-residing WT and β3−/− prefusion osteoclasts were M-CSF treated over time. Total and GTP-bound Rac levels were determined. (B) Prefusion osteoclasts were cytokine starved, lifted, and treated with M-CSF or carrier for 30 min. WOW1 binding was analyzed by fluorescence-activated cell sorting (FACS). Control binding is normalized to 100% (*, P < 0.05). (C) Cytokine-starved β3−/− prefusion osteoclasts transduced with WT human β3 were treated with M-CSF over time. β3 immunoprecipitates were immunoblotted for talin, and the blot was densitometrically analyzed. (D) Cytokine-starved osteoclasts generated from WT BMMs were exposed to M-CSF or carrier for 30 min. The cells were stained for F-actin (green) and talin (red). Arrows indicate fillipodia induced by the cytokine. Bar, 50 μm. Data are representative of 3 independent experiments for panels A to C and 2 experiments for panel D. Numbers next to the gels represent molecular weights (in thousands).
β3Y747 regulates talin-mediated cytoskeletal organization in osteoclasts.
In other cells, optimal integrin activation requires talin recognition of β3Y747 (17). To determine if the same is true regarding the organization of the osteoclast cytoskeleton, bone marrow macrophages (BMMs) of knock-in mice bearing an alanine mutation of the residue were differentiated into osteoclasts in vitro. These cells, while normal in number, develop the same nonspread, crenated appearance extant in osteoclasts lacking the β3 integrin or its cytoskeleton-organizing effectors (Fig. 2A and B) (3–8). The abnormal appearance of these mutant cells does not represent arrested differentiation (Fig. 2C). Like cultured β3-deficient osteoclasts, β3Y747A cells generate actin rings, but their bone-resorptive activity is diminished (Fig. 2D to G).
Fig 2.

β3Y747 regulates osteoclast function. (A) WT or β3Y747A (YA) BMMs were cultured in M-CSF and RANKL for 5 days and stained for TRAP activity. Bar, 200 μm. (B) Quantification of the osteoclasts (Ocs) shown in panel A. (C) WT and β3Y747A BMMs were cultured in M-CSF and RANKL. The osteoclast differentiation markers, β3 integrin subunit, NFATc1, and c-Src, were immunoblotted with time. Actin served as loading control. (D) WT and β3Y747A BMMs were cultured in M-CSF and RANKL on bone slices for 6 days. Rhodamine-phalloidin-stained actin rings were visualized by fluorescence microscopy. Bar, 100 μm. (E) After 6 days, the cells were removed and resorption pits visualized by peroxidase-labeled wheat germ agglutinin staining. Bar, 100 μm. (F) Quantitation of resorption pits visualized in panel E. (G) Medium collected from cultures shown in panel E was analyzed for CTx content. (H) Cytokine-starved β3−/− prefusion osteoclasts transduced with WT or Y747A human β3 were treated with M-CSF or carrier for 30 min. β3 immunoprecipitates were probed for talin. β3 integrin served as loading control. (I) Osteoclasts generated from β3−/− BMMs transduced with WT, Y747A, or Y747F (YF) human β3 or empty vector were stained for TRAP activity (upper panels; bar, 400 μm). Expression of β3 constructs (lower panel) with actin as loading control. (J) μCT image of distal femoral metaphysis of 6-month-old WT and β3Y474A knock-in mice. (K) μCT images of distal femur of WT and β3Y747A mice 4 weeks after ovariectomy. (L) μCT determination of BV/TV of WT and β3Y747A mice 4 weeks after ovariectomy. n = 3 per group (*, P < 0.05; **, P < 0.01). Data are representative of at least 3 independent experiments (panels A through F and J through L) or 2 experiments (panels G and H). Numbers next to the gels represent molecular weights (in thousands).
Immunoblots of β3 immunoprecipitates document that M-CSF promotes talin association with wild-type (WT) β3, but not β3Y747A (Fig. 2H). In contrast to β3Y747A osteoclasts, those expressing nonphosphorylatable β3Y747F are essentially normal (22). Like WT, β3Y747F completely rescues β3−/− osteoclasts, whereas β3Y747A is unable to do so (Fig. 2I). Thus, the mechanism by which β3Y747A disrupts the cytoskeleton does not reflect arrested phosphorylation. Unlike what is seen in β3−/− mice, no change in bone mass is evident in those bearing the β3Y747A mutation at 6 months of age (Fig. 2J) (23), but they are protected from postmenopausal bone loss (Fig. 2K and L) (24). Hence, β3Y747 is essential for talin recognition by the integrin in osteoclasts.
Talin1-deficient osteoclasts are dysfunctional in vivo.
In some cell types, talin2 can compensate for loss of talin1, but only talin1 is expressed by osteoclasts and their precursors (Fig. 3A) (25, 26). Therefore, we crossed mice with a conditional TLN1 allele to those expressing cathepsin K Cre (CtsK-Cre), which is active only in mature osteoclasts (19) (mice and osteoclasts with conditional deletion of TLN1 by CtsK-Cre are here referred to as CtsK-TLN1). In keeping with diminished resorptive activity, CtsK-TLN1 tibias and femurs are more radiodense than control tibias and femurs (Fig. 3B). Similarly, trabecular bone mass and connectivity, determined by μCT, is substantially increased in CtsK-TLN1 mice, as are volumetric and whole-body bone mineral density (BMD) (Fig. 3C to E). Histological analysis confirms enhanced trabecular bone volume (BV/TV) in the mutant mice, despite normal numbers of osteoclasts (Fig. 3F and G). In contrast, histomorphometrically measured bone formation rates in Cre+ and Cre− mice are indistinguishable, as is the circulating osteoblast function marker osteocalcin (Fig. 4A to D). In keeping with the skeletal consequences of talin deletion likely representing targeting of integrins in addition to αvβ3, CtsK-mediated deletion of only the latter does not impact bone mass in 8-week-old mice (Fig. 4E and F).
Fig 3.

TLN1 deletion in mature osteoclasts causes osteopetrosis. (A) TLN1 and TLN2 mRNA expression in WT BMMs (Mϕ) and osteoclasts (OC) assessed by RT-PCR. The β3 integrin subunit serves as a marker of osteoclast differentiation and actin as loading control (left panel). To validate TLN2 primers, mRNA expression by macrophages (Mϕ), osteoclasts, and osteoblasts (OB) was determined by quantitative PCR (right panel). (B) Radiographs of femurs and tibias/fibulas of 8-week-old control (−) and CtsK-TLN1 (+) mice. (C) μCT images of distal femoral metaphysis (middle and lower panel) and proximal tibia (upper panels) of control (−) and CtsK-TLN1 (+) mice. (D) μCT determination of BV/TV, trabecular connectivity, and volumetric bone mineral density (BMD) of distal femoral metaphysis of control (−) and CtsK-TLN1 (+) mice. (E) Whole-body dual-energy X-ray absorptiometry (DEXA) analysis of areal BMD of control (−) and CtsK-TLN1 (+) mice. (F) TRAP-stained histological sections of proximal tibia of 8-week-old control (−) and CtsK-TLN1 (+) mice. Bars: 500 μm (upper panel), 50 μm (lower panel). (G) The total number of osteoclasts normalized to bone surface and the percentage of bone surface to which osteoclasts are juxtaposed in panel F were histomorphometrically determined (*, P < 0.05; ***, P < 0.001). Data are representative of 2 independent experiments in panel A. At least 10 animals were included in each group in panels B to F and 5 animals in panel G.
Fig 4.

TLN1 deletion in mature osteoclasts does not affect bone formation. (A to D) CtsK-TLN1 (+) and control (−) mice were administered two time-spaced doses of calcein. (A) Fluorescence microscopic images of proximal tibia. Bars: 400 μm (upper panel), 100 μm (lower panel). Calculation of bone formation rates (BFR) (B) and mineral apposition rates (MAR) (C). (D) Circulating osteocalcin of CtsK-TLN1 (+) and control (−) mice. (E and F) β3fl/fl mice (generated by Katherine Weilbaecher, Washington University School of Medicine) were crossed to those expressing Ctsk-Cre. (E) β3 subunit and c-Src immunoblots of lysates of Cre+ and Cre− BMMs exposed to M-CSF (Mϕ), or M-CSF + RANKL (OC) for 6 days. Actin served as loading control. (F) μCT images of 8-week-old Cre+ and Cre− progeny. n = 5 in each group in panels A to D; n = 3 in panel F. Data are representative of at least 2 independent experiments. Numbers next to the gel represent molecular weights (in thousands).
Rap1-deficient osteoclasts are dysfunctional.
Despite the relatively robust in vivo phenotype of CtsK-TLN1 mice, osteoclasts generated in vitro, for 5 days, are normal (Fig. 5A). As seen in Fig. 5B, this unexpected phenomenon likely represents undiminished talin1 protein in these cells until 6 days of culture, the limit of their in vitro viability. To circumvent this limitation, we attempted, but failed, to delete the gene by transducing TLN1fl/fl-bearing BMMs with Cre recombinase-transduced retrovirus (not shown). Based upon our previous experience that the success of in vitro Cre-mediated deletion is gene specific (21, 27), we turned to Rap1, a Ras family GTPase, which in conjunction with Rap1 interacting adaptor molecule (RIAM) mediates talin/integrin recognition (28). Like its effect on Rac (Fig. 1A), M-CSF transits Rap1 to its active, GTP-bound form in prefusion osteoclasts (Fig. 5C).
Fig 5.

Rap1-deficient osteoclasts are dysfunctional in vitro. (A) CtsK-TLN1 (+) and control (−) BMMs maintained in M-CSF and RANKL for 5 days were stained for TRAP activity. Bar, 400 μm. (B) CtsK-TLN1 BMMs were exposed to M-CSF and RANKL for 2 to 6 days or M-CSF (M) for 6 days. Talin1 and Cre expression were determined by immunoblotting. The β3 integrin subunit serves as a marker of osteoclast differentiation and actin as loading control. (C) Cytokine-starved, WT prefusion osteoclasts were exposed to increasing amounts of M-CSF for 15 min. Lysate containing GTP-Rap1 was measured by pulldown assay. Total Rap1 served as loading control. (D) Rap1Afl/fl × Rap1Bfl/fl BMMs were transduced with Cre recombinase (+) or vector (−). Lysates were immunoblotted for Rap1A and Rap1B. Actin served as loading control. (E) Rap1Afl/fl × Rap1Bfl/fl Cre+ and Cre− BMMs were exposed to RANKL and M-CSF for 5 days and stained for TRAP activity. Bar, 100 μm. (F) The cells were removed and resorption pits (dark brown reaction product) visualized by staining with wheat germ agglutinin. Bar, 100 μm. (G) Medium was analyzed for CTx content. (H) Rap1Afl/fl × Rap1Bfl/fl Cre+ and Cre− BMMs were treated with M-CSF and RANKL for 2 to 5 days or M-CSF (M) alone for 5 days. NFATc1, β3 integrin, and c-Src were immunoblotted. Actin served as loading control. (I) Cytokine-starved Rap1Afl/fl × Rap1Bfl/fl Cre+ and Cre− prefusion osteoclasts were lifted and maintained in suspension (S) or plated on vitronectin (A) for 30 min. Vav3 immunoprecipitates were immunoblotted for phosphotyrosine (p-Y) and Vav3. (J) Cytokine-starved Rap1Afl/fl × Rap1Bfl/fl Cre+ and Cre− prefusion osteoclasts were exposed to M-CSF or carrier for 30 min. β3 integrin immunoprecipitates were immunoblotted for talin and β3 subunit. Data are representative of at least 3 independent experiments in panels A to F and H to J and 2 in panel G. Numbers next to the gels represent molecular weights (in thousands).
In contrast to our attempts to target TLN1fl/fl, Cre-mediated deletion of Rap1Afl/fl/Rap1Bfl/fl in vitro was successful (Fig. 5D). This exercise produced osteoclasts exhibiting the characteristic crenated appearance of those lacking integrin-activated, cytoskeleton-organizing molecules and with compromised bone-resorptive capacity (Fig. 5E to G). In keeping with failed integrin activation, Cre+ Rap1 BMMs normally express differentiation markers under osteoclastogenic conditions (Fig. 4H). Furthermore, liganding of αvβ3 does not phosphorylate its effector, Vav3 (Fig. 4I), and M-CSF fails to prompt talin/β3 association in the gene-deleted, prefusion osteoclasts (Fig. 4J).
Mice with Rap1-deficient osteoclasts are osteopetrotic.
To determine the physiological relevance of Rap1 in osteoclasts, we crossed Rap1Afl/fl/Rap1Bfl/fl mice to those expressing CtsK-Cre, generating animals whose in vivo phenotype mirrors those with conditional deletions of talin in that they develop a marked increase in bone mass despite an abundance of osteoclasts (Fig. 6A to E) (mice and osteoclastic cells with conditional deletions of Rap1A/B by CtsK-Cre are here referred to as CtsK-Rap1). CtsK-induced loss of Rap1A/B is apparent within 3 days of exposing CtsK-Rap1 BMMs to M-CSF and receptor activator of nuclear factor κB ligand (RANKL) (Fig. 6F). In keeping with this early deletion, CtsK-Rap1 osteoclasts so generated have the crenated appearance characteristic of cytoskeletal disarray (Fig. 6G). Thus, disruption of Rap1/talin activity arrests integrin function in osteoclasts and inhibits their resorptive activity in vitro as well as in vivo.
Fig 6.

Rap1 deletion in mature osteoclasts causes osteopetrosis. (A) X-rays of femurs of CtsK-Rap1 (+) and control (−) mice demonstrating enhanced radiodensity of the former. (B) μCT images of femurs of CtsK-Rap1 and control mice. (C) Quantitative μCT data of femurs of CtsK-Rap1 and control mice. (D) TRAP-stained (red reaction product) histological sections of tibias of CtsK-Rap1 and control mice. Bars: 500 μm (upper panel), 100 μm (lower panel). (E) Histomorphometric quantification of trabecular bone volume and osteoclast abundance shown in panel D. (F) CtsK-Rap1 BMMs were exposed to M-CSF and RANKL for 2 to 6 days or M-CSF (M) for 6 days. Rap1b expression was determined by immunoblotting. Actin served as loading control and c-Src as a marker of differentiation. (G) Control (−) and CtSk-Rap1 (+) BMMs were cultured with M-CSF and RANKL for 4 days, after which they were stained for TRAP activity. Bars: 400 μm (upper panels), 100 μm (lower panel) (**, P < 0.01; ***, P < 0.001). n = 4 in each group (A to E). Data are representative of 3 independent experiments (F and G). Numbers next to the gel represent molecular weights (in thousands).
Talin1-deficient osteoclast lineage cells are dysfunctional.
Our previous experiments indicate that talin deletion late in osteoclastogenesis produces osteopetrosis in vivo but fails to yield an abnormal phenotype ex vivo. Given the success of in vitro Rap1 targeting, however, we reasoned that deleting TLN1 in early osteoclast lineage cells in vivo would obviate this limitation. To this end, we mated TLN1fl/fl mice with those expressing Cre driven by the lysozyme M (LysM) promoter, expressed in all myeloid lineage cells (osteoclast lineage cells with conditional deletion of TLN1 by LysM-Cre are here referred to as LysM-TLN1). Talin1 is reduced by approximately 65% in LysM-TLN1 cells (Fig. 7A), while expression of kindlin 3 mRNA, whose product binds and activates integrins, is intact (29–31).
Fig 7.
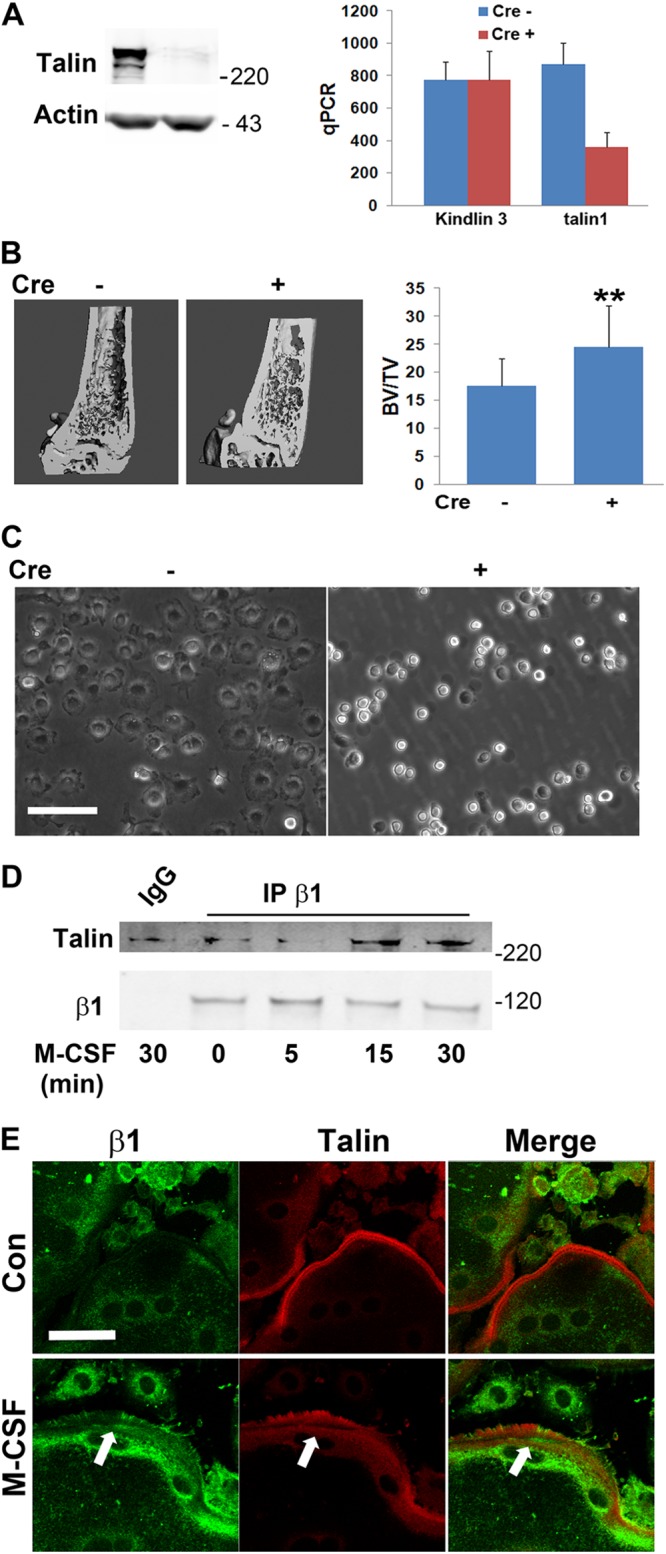
TLN1 deletion in osteoclast lineage cells disrupts BMM adhesion. (A) Talin1 protein (left panel) and quantitative PCR (qPCR)-determined talin1 and kindlin3 mRNA (right panel) expression in LysM-TLN1 (+) and control (−) BMMs. (B) μCT image and BV/TV of distal femurs of 8-week-old LysM-TLN1 (+) and control (−) mice (**, P < 0.01). (C) Phase-contrast microscopy of LysM-TLN1 (+) and control (−) BMMs. Bright cells are those that are nonadherent. Bar, 100 μm. (D) Cytokine-starved WT macrophages were exposed to M-CSF over time. β1 integrin immunoprecipitates were immunoblotted for talin and β1 subunit. IgG served as negative control. (E) Serum/cytokine-starved osteoclasts, differentially immunostained for β1 integrin subunit (green) and talin (red) were exposed to carrier or M-CSF for 5 min. Arrows indicate areas of colocalization. Bar, 50 μm. Data are representative of at least 3 independent experiments in panels A and C through E, and n = 12 in the experiment illustrated in panel B. Numbers next to the gels represent molecular weights (in thousands).
Similar to what is seen in CtsK-TLN1 mice, the number of osteoclasts in LysM-TLN1 animals is indistinguishable from that of the control (not shown). While the change is not as dramatic as that occurring in the context of CtsK-Cre, trabecular bone volume of LysM-TLN1 mice is also significantly increased (Fig. 7B). Whereas cultured CtsK-TLN1 osteoclast lineage cells appear normal, LysM-TLN1 precursors cluster and largely fail to attach to and spread on substrate, features consistent with integrin dysfunction (Fig. 7C). The impaired attachment of LysM-TLN1 BMMs does not, however, promote apoptosis (data not shown).
BMMs do not express αvβ3 until they assume the osteoclast phenotype (32). Talin1, however, also activates other integrins. In fact, M-CSF promotes talin association with the β1 subunit, which participates in osteoclast function, as documented by coimmunoprecipitation and confocal colocalization with exposure to the cytokine (30, 33) (Fig. 7D and E).
LysM-Cre-mediated TLN1 deletion impairs osteoclast formation.
To assess the phenotype of LysM-TLN1 osteoclasts, we treated Cre+ and Cre− BMMs with M-CSF and RANKL. While abundant, characteristic osteoclasts emerged in control cultures, only a few small, adherent, tartrate-resistant acid phosphatase (TRAP)-positive, talin1-deficient polykaryons appeared (Fig. 8A and B). These cells have abnormal cytoskeletons as evidenced by the presence of numerous minute, actin structures and a paucity of rings (Fig. 8C). Attesting that these features represent failed inside-out integrin activation, they are virtually normalized by the outside-in stimulator, MnCl2 (Fig. 8D). In contrast, expression of the key osteoclast differentiation markers c-Src and NFATc1 is unaltered in RANKL-treated LysM-TLN1 cells, regardless of their capacity to adhere to substrate (Fig. 8E). Similarly, LysM-Cre-mediated TLN1 deletion does not affect M-CSF-induced total tyrosine phosphorylation or signaling important for osteoclast differentiation and survival (Fig. 8F). These observations indicate that osteoclast formation requires matrix adhesion by precursors, regardless of the expression of proteins that specifically regulate the cell's differentiation and function. This conclusion is in keeping with the failure of LysM-TLN1 BMMs to undergo multinucleation when exposed to IL-4 (Fig. 8G).
Fig 8.

TLN1 deletion in osteoclast lineage cells disrupts osteoclast formation. (A) LysM-TLN1 (+) and control (−) BMMs treated with M-CSF and RANKL for 5 days were viewed by phase-contrast microscopy to visualize osteoclasts and nonadherent BMMs. Bar, 100 μm. (B) Following removal of nonadherent cells illustrated in panel A, the cells were stained for TRAP activity. Bar, 400 μm. (C) Fibrillar actin of bone-residing LysM-TLN1 (+) and control (−) osteoclasts was visualized by rhodamine-phalloidin staining. Bar, 100 μm. (D) Bone-residing LysM-TLN1 osteoclasts were exposed to MnCl2 (2 mM) or PBS for 8 h. Fibrillar actin was visualized by rhodamine-phalloidin staining. Bar, 100 μm. (E) BMMs were treated with M-CSF and RANKL (OC) or M-CSF alone (MΦ) for 4 days. LysM-TLN1 cells were separated into adherent and nonadherent components, whereas all control cells were adherent. c-Src and NFATc1 were immunoblotted. Actin served as loading control. (F) LysM-TLN1 (+) and control (−) BMMs were exposed to M-CSF over time. Lysates were immunoblotted for talin and total phosphotyrosine content (upper panel) or total and phosphorylated AKT and extracellular signal-regulated kinase (ERK) (lower panel). Actin served as loading control. (G) LysM-TLN Cre+ and Cre− BMMs were cultured in M-CSF with or without interleukin-4 (IL4) (20 ng/ml) for 24 h. The cells were stained with hematoxylin. Bar, 100 μm. Data are representative of at least 5 independent experiments (A to C) or at least 3 independent experiments (D to G). Numbers next to the gels represent molecular weights (in thousands).
As the adhesive properties of their precursors are compromised, one would expect mobilization of osteoclasts in response to a resorptive stimulus to be dampened in LysM-TLN1 mice. To determine if this is so, we supracalvarially injected Cre+ and Cre− mice with RANKL or PBS and stained their calvaria for TRAP activity (34). Whereas control RANKL-injected calvaria contain numerous TRAP-rich resorption areas, they are much less abundant in those of LysM-TLN1 mice (Fig. 9A and B). Furthermore, calvarial mRNAs of the osteoclast markers cathepsin K and TRAP are induced by RANKL in WT but not LysM-TLN1 animals (Fig. 9C). Thus, RANKL-enhanced osteoclast formation, in vitro and in vivo, requires progenitor cell talin expression.
Fig 9.

TLN1 deletion in osteoclast lineage cells disrupts osteoclast mobilization. (A) RANKL (50 μg/mouse/day) was administered supracalvarially to LysM-TLN1 (+) and control (−) mice for 5 days. Calvaria were fixed and stained for TRAP activity. (B) Calvaria of LysM-TLN1 (+) and control (−) mice treated as described for panel A were subjected to μCT. (C) Cathepsin K and TRAP mRNA, isolated from the calvaria shown in panels A and B, was quantitated by qPCR. (D) Cytokine-starved LysM-TLN1 (+) and control (−) prefusion osteoclasts were lifted and maintained in suspension (S) or plated on vitronectin (A) for 30 min. Lysates were immunoblotted for total phosphotyrosine (p-Y) and activated c-Src (p-Src). Actin served as loading control. (E) Preosteoclast migration was assessed using transwells in which the lower membrane surface was coated with vitronectin in the absence or presence of M-CSF as a chemoattractant. The membrane-residing cells were stained with crystal violet. Bar, 100 μm. (F) Quantification of cells shown in panel E. (G) Engulfment of apoptotic T cells by LysM-TLN1 (+) and control (−) BMMs (*, P < 0.05; **, P < 0.01). Data are representative of at least 2 independent experiments. Numbers next to the gel represent molecular weights (in thousands).
αvβ3, in the osteoclast, stimulates a cytoskeleton-organizing complex in which c-Src is a proximal effector (3, 10, 35). To address this issue in the context of LysM-TLN1, we generated Cre+ and Cre− prefusion osteoclasts. They were lifted and either maintained in suspension or plated on αvβ3 ligand, which activates c-Src in control but not talin-deficient cells (Fig. 9D). Tyrosine phosphorylation of specific components of LysM-TLN1 total cell lysate is also diminished.
To determine the impact of TLN1 deletion on integrin-directed movement, the lower surfaces of transwell filters were coated with the αvβ3 ligand vitronectin. LysM-TLN1 and control prefusion osteoclasts were added to transwells in which the lower chamber did or did not contain M-CSF. Neither genotype migrated toward αvβ3 ligand in the absence of M-CSF (Fig. 9E and F). In the presence of the activating cytokine, directed migration of both is increased but that of talin1-deficient cells is less than one-third of that of WT. Reflective of cytoskeletal dysfunction, the phagocytic capacity of LysM-TLN1 BMMs is also impaired (Fig. 9G).
The talin head domain partially rescues talin1-deficient osteoclasts.
Talin1 consists of a FERM domain-containing head linked to a long, flexible rod (36). Retroviral expression of the talin1 head (talin1–433) in LysM-TLN1 BMMs prompts them to adhere to plastic and spread (Fig. 10A). Furthermore, the tyrosine phosphorylation of adherent, head domain-transduced prefusion osteoclast lysate is more profound than that of Cre+ vector control (Fig. 10B). The talin1 head substantially rescues the appearance and number of LysM-TLN1 TRAP-stained osteoclasts (Fig. 10C) but not their cytoskeletal abnormalities, as manifested by the persistence of numerous atypical, minute actin structures and a paucity of rings (Fig. 10D).
Fig 10.

Talin1 head domain partially rescues LysM-TLN1 osteoclast formation. (A) Talin11–433 (Head) or vector was retrovirally transduced into LysM-TLN1 BMMs (+). Vector was also transduced into control BMMs (−). Phase-contrast micrograph of transduced BMMs. Bar, 100 μm. (B) Cytokine-starved, transduced prefusion osteoclasts were maintained in suspension (S) or plated on vitronectin (A) for 30 min. Lysates were immunoblotted for phosphorylated tyrosine (p-Y). Actin served as loading control. (C) Transduced BMMs cultured in M-CSF and RANKL for 5 days were stained for TRAP activity. Bar, 400 μm. (D) Bone-residing transduced BMMs cultured in M-CSF and RANKL for 5 days were stained with rhodamine-phalloidin to visualize fibrillar actin. Bar, 100 μm. Data are representative of at least 3 independent experiments. Numbers next to the gel represent molecular weights (in thousands).
Talin1-deficient osteoclasts protect against pathological bone loss.
Reflecting diminished resorption, μCT and histomorphometric analysis establishes that CtsK-TLN1 mice are substantially protected from bone loss following estrogen deprivation (Fig. 11A to C). Similarly, the osteoclasts of CtsK-TLN1 mice with inflammatory arthritis, while abundant, are generally not juxtaposed to bone and exhibit the crenated appearance of those lacking integrin-activated cytoskeleton-organizing molecules, resulting in much less periarticular erosion (Fig. 11D and E). Thus, talin regulates osteoclast function in physiological and pathological circumstances.
Fig 11.

Talin1 deficiency in osteoclasts protects against pathological bone loss. μCT image of distal femur (A) and histological images of tibia (C) of CtsK-TLN1 (+) and control (−) mice 4 weeks after sham operation or ovariectomy (OVX). Bar, 500 μm. (B) μCT-determined percentage of bone loss in CtsK-TLN1 (+) and control (−) mice 4 weeks following ovariectomy, relative to sham operation. (D) TRAP-stained histological sections of ankles of CtsK-TLN1 (+) and control (−) mice 7 days following injection of arthrogenic serum. Bars: 100 μm (top panel), 25 μm (bottom panel). (E) Percentage of surface (S) exhibiting erosive features and total number of osteoclasts normalized to bone surface, (the percentage of bone surface to which osteoclasts are juxtaposed) (*, P < 0.05; **, P < 0.01). n = 7 in each group for panels A to C, and n = 4 in panels D and E. Data are representative of at least 2 independent experiments.
DISCUSSION
There is compelling evidence that M-CSF and αvβ3 interdependently remodel the osteoclast cytoskeleton. For example, the cytokine needs αvβ3 to transit the cytoskeleton-organizing GTPase Rac to its GTP-bound state, and integrin-stimulated Rac, induced by adherence to bone, requires M-CSF. Hence, αvβ3 and M-CSF induction of their associated cytoskeleton-organizing signaling complexes are perhaps related by inside-out activation of the integrin. In fact, we previously determined that M-CSF activates a hybrid complex of murine αv and human β3 (11). We presently show that the same obtains, to a similar extent, in naïve WT cells. Having established that M-CSF is capable of activating αvβ3 in the bone-resorbing cell, we turned to the causal mechanisms and biological implications.
Talin recognizes the β3 integrin cytoplasmic domain by first binding a distal motif containing β3Y747. Alanine knock-in of this residue largely obviates talin1 recognition and prompts failure of osteoclast spreading in vitro (17). In contrast to the alanine substitution, β3Y747F has no effect on osteoclast formation and function (22). In keeping with this observation, β3Y747F rescues the β3−/− osteoclast phenotype as effectively as the WT construct. Because Y>F substitution is more conservative than Y>A, the disruptive effect of β3Y747A on talin1 recognition likely reflects the conformational change of the integrin rather than failed phosphorylation (25).
To more directly determine the role of talin1 in osteoclast formation and function, we targeted the TLN1fl/fl gene by CtsK-Cre, generating mice with a 5-fold increase in bone mass. In keeping with integrin dysfunction, CtsK-TLN1 osteoclasts are abundant but when stressed, such as in the context of inflammatory osteolysis, fail to spread or normally attach to bone.
Because CtsK is expressed late in osteoclastogenesis, Cre recombinase driven by the enzyme's promoter deletes target genes only in cells substantially committed to the osteoclast phenotype. To assess the impact of talin deletion throughout the osteoclastogenic process, we turned to mice expressing LysM-Cre, which is active in all myeloid cells. LysM-TLN1 BMMs and prefusion osteoclasts fail to attach to substrate, spread, or activate cytoskeleton-organizing molecules. LysM-TLN1 prefusion osteoclasts also do not migrate normally to αvβ3 ligand, another manifestation of defective integrin and cytoskeletal function. Failure of LysM-TLN1 cells to spread on substrate or migrate is not due to inadequate outside-in integrin activation, since the defect is rescued by MnCl2. Thus, the functional abnormalities of LysM-TLN1 osteoclast lineage cells likely reflect failed inside-out integrin activation.
Despite the robust in vivo skeletal phenotype of CtsK-TLN1 mice, osteoclasts generated in vitro from these animals are normal, reflecting the persistence of talin protein to the limits of cultured osteoclast viability. To circumvent this limitation, we attempted to delete TLN1fl/fl in vitro with transduced Cre recombinase. While we have used this strategy successfully in other circumstances (21, 27), we find it to be gene specific and were unsuccessful in the context of talin.
Talin contains two principal regions, the head and rod domains, which interact to autoinhibit the protein (13). Rap1 and its binding partner, RIAM, activate talin1 by exposing its head, which then associates with cytoplasmic tails of β integrins to disrupt their inactivating salt bridge and increase ligand affinity. The talin rod interacts with the cytoskeletal proteins, actin and vinculin. Given that Rap1 is essential for talin-mediated integrin activation, in other circumstances, we attempted to delete it in vitro by Cre transduction and were successful in generating osteoclasts with disorganized cytoskeletons. With the realization that Rap1 exerts effects other than talin activation, the identical skeletal appearance of mice lacking either gene provides compelling evidence that the normal appearance of osteoclasts derived from CtsK-TLN1 BMMs reflects failed deletion of the target protein.
The magnitude of skeletal resorption is reflective of the number of osteoclasts and the capacity of the individual cell to degrade bone. We have shown that the absence of αvβ3 or of members of its activated cytoskeleton-organizing complex arrests osteoclast function but not differentiation. In keeping with the essential role of talin in stimulating this complex, RANKL- and M-CSF-treated LysM-TLN1 BMMs express normal osteoclastogenic markers and activate relevant signaling molecules. On the other hand, mirroring the absence of αvβ3 (37), the number of osteoclasts derived from LysM-TLN1 BMMs in vitro is substantially reduced, a likely consequence of impaired motility and capacity to multinucleate. It is also reflected by impairment of LysM-TLN1 mice to mobilize osteoclasts in response to RANKL in vivo. The fact that two strains of poorly adherent precursors yield low numbers of osteoclasts in culture is consistent with the postulate that despite expression of normal differentiation markers, acquisition of the osteoclast phenotype requires matrix adherence and migration and is therefore impaired in the face of compromised integrin function.
Notwithstanding substantial deficiencies of osteoclast formation and function in vitro, the basal bone mass of LysM-TLN1 mice, albeit significantly increased, is not as abundant as that of CtsK-TLN1 animals. This difference, while enigmatic, may represent relative efficiencies of TLN1 deletion. Temporal differences in Cre expression may also contribute to this discrepancy, as we have observed similar inconsistencies in other circumstances involving the same two Cre-expressing mice (8). Regardless of this conundrum, LysM-TLN1 mice provided us the means of assessing the role of talin's integrin-activating domain in osteoclasts. We find that expression of the talin1 head in LysM-TLN1 BMMs normalizes their capacity to adhere to matrix and spread. The transduced head also substantially increases the number of osteoclasts recruited by M-CSF and RANKL. However, consistent with the essential role of the talin1 rod in binding to cytoskeletal proteins (25), neither osteoclast abundance nor appearance is completely rescued in LysM-TLN1 cells expressing the talin head alone. Thus, intact talin is required to functionally link the integrin to the osteoclast cytoskeleton.
The pragmatic aspect of our study relates to the fact that αvβ3 is the only integrin that has served as a clinically tested therapeutic target in the context of osteoporosis (38). The bone-sparing effects of a small-molecule αvβ3 inhibitor are relatively modest and thus have not been further pursued. Because the osteopetrotic phenotypes of CtsK-TLN1, CtsK-Rap1, and kindlin3−/− mice are more profound than of those of mice lacking the β3 integrin (23) or β3Y747A, the therapeutic failure of αvβ3 inhibition likely reflects compensation by other integrins, such as those of the β1 family. Hence, targeting common motifs of numerous integrins that recognize essential activating molecules, such as talin, Rap1, or kindlin3, should be a more promising strategy for preventing and treating pathological osteolysis. Furthermore, our data establish that therapeutic targeting needs not be extant throughout osteoclastogenesis, which would impact other myeloid cells such as inflammatory macrophages. The fact that inhibition of integrin function late in the course of osteoclast development yields healthy mice substantially resistant to inflammatory osteolysis and estrogen-deficient osteoporosis buttresses this argument.
Thus, we propose the following model of M-CSF-mediated organization of the osteoclast cytoskeleton (Fig. 12). Occupancy of c-Fms changes Rap1-GDP to its active, GTP-bound form. Rap1-GTP induces talin to associate with a variety of β subunit cytoplasmic domains, thus transiting the integrins to their active, high-affinity conformation. Upon binding bone matrix-residing ligand, the integrin activates a canonical signaling complex, the most distal established member being Rac, thus organizing the osteoclast's cytoskeleton and enabling the cell to resorb bone.
Fig 12.

Proposed model of M-CSF-mediated organization of the osteoclast cytoskeleton. In the current study, we find that ligand occupancy of the M-CSF receptor, c-Fms, transits GDP-bound Rap1 to its active, GTP-bound state. Active Rap1 promotes association of talin with the cytoplasmic domain of αvβ3 integrin, leading to induction of its high-affinity conformation. Upon ligand occupancy, the integrin activates a canonical signaling pathway, which we previously demonstrated consists of c-Src, Syk, Dap 12, and Vav3. The last is a relatively osteoclast-specific Rac guanine nucleotide exchange factor. GTP-Rac organizes the cell's cytoskeleton via Arp2/3 (not shown), including actin ring formation, thus permitting it to resorb bone.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants AR032788, AR046523, AR054618, AR057037, and AR057235 (S.L.T.) and AR027214 (M.H.G.).
The contents are solely our responsibility and do not necessarily represent the official views of the NIH.
We declare that we have no financial conflicts of interest.
Footnotes
Published ahead of print 10 December 2012
REFERENCES
- 1. Hynes RO. 1992. Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69:11–25 [DOI] [PubMed] [Google Scholar]
- 2. DeMali KA, Wennerberg K, Burridge K. 2003. Integrin signaling to the actin cytoskeleton. Curr. Opin. Cell Biol. 15:572–582 [DOI] [PubMed] [Google Scholar]
- 3. Zou W, Kitaura H, Reeve J, Long F, Tybulewicz VLJ, Shattil SJ, Ginsberg MH, Ross FP, Teitelbaum SL. 2007. Syk, c-Src, the αvβ3 integrin, and ITAM immunoreceptors, in concert, regulate osteoclastic bone resorption. J. Cell Biol. 176:877–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reeve JL, Zou W, Liu Y, Maltzman JS, Ross FP, Teitelbaum SL. 2009. SLP-76 couples Syk to the osteoclast cytoskeleton. J. Immunol. 183:1804–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zou W, Zhu T, Craft CS, Broekelmann TJ, Mecham RP, Teitelbaum SL. 2010. Cytoskeletal dysfunction dominates in DAP12-deficient osteoclasts. J. Cell Sci. 123:2955–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Faccio R, Zou W, Colaianni G, Teitelbaum SL, Ross FP. 2003. High dose M-CSF partially rescues the Dap12-/- osteoclast phenotype. J. Cell. Biochem. 90:871–883 [DOI] [PubMed] [Google Scholar]
- 7. Faccio R, Teitelbaum SL, Fujikawa K, Chappel J, Zallone A, Tybulewicz VL, Ross FP, Swat W. 2005. Vav3 regulates osteoclast function and bone mass. Nat. Med. 11:284–290 [DOI] [PubMed] [Google Scholar]
- 8. Croke M, Ross FP, Korhonen M, Williams DA, Zou W, Teitelbaum SL. 2011. Rac deletion in osteoclasts causes severe osteopetrosis. J. Cell Sci. 124:3811–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Faccio R, Takeshita S, Colaianni G, Chappel JC, Zallone A, Teitelbaum SL, Ross FP. 2007. M-CSF regulates the cytoskeleton via recruitment of a multimeric signaling complex to c-Fms Tyr-Y559/697/721. J. Biol. Chem. 282:18991–18999 [DOI] [PubMed] [Google Scholar]
- 10. Zou W, Reeve JL, Liu Y, Teitelbaum SL, Ross FP. 2008. DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol. Cell 31:422–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Faccio R, Novack DV, Zallone A, Ross FP, Teitelbaum SL. 2003. Dynamic changes in the osteoclast cytoskeleton in response to growth factors and cell attachment are controlled by β3 integrin. J. Cell Biol. 162:499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Faccio R, Grano M, Colucci S, Villa A, Giannelli G, Quaranta V, Zallone A. 2002. Localization and possible role of two different αvβ3 integrin conformations in resting and resorbing osteoclasts. J. Cell Sci. 115:2919–2929 [DOI] [PubMed] [Google Scholar]
- 13. Shattil SJ, Kim C, Ginsberg MH. 2010. The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 11:288–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lim J, Wiedemann A, Tzircotis G, Monkley SJ, Critchley DR, Caron E. 2007. An essential role for talin during alpha(M)beta(2)-mediated phagocytosis. Mol. Biol. Cell 18:976–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA. 2003. Talin binding to integrin β tails: a final common step in integrin activation. Science 302:103–106 [DOI] [PubMed] [Google Scholar]
- 16. Petrich BG, Marchese P, Ruggeri ZM, Spiess S, Weichert RA, Ye F, Tiedt R, Skoda RC, Monkley SJ, Critchley DR, Ginsberg MH. 2007. Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. J. Exp. Med. 204:3103–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Petrich BG, Fogelstrand P, Partridge AW, Yousefi N, Ablooglu AJ, Shattil SJ, Ginsberg MH. 2007. The antithrombotic potential of selective blockade of talin-dependent integrin alpha IIb beta 3 (platelet GPIIb-IIIa) activation. J. Clin. Invest. 117:2250–2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pan BX, Vautier F, Ito W, Bolshakov VY, Morozov A. 2008. Enhanced cortico-amygdala efficacy and suppressed fear in absence of Rap1. J. Neurosci. 28:2089–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, Nishina H, Takeda S, Takayanagi H, Metzger D, Kanno J, Takaoka K, Martin TJ, Chambon P, Kato S. 2007. Estrogen prevents bone loss via estrogen receptor α and induction of Fas ligand in osteoclasts. Cell 130:811–823 [DOI] [PubMed] [Google Scholar]
- 20. Takeshita S, Kaji K, Kudo A. 2000. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J. Bone Miner. Res. 15:1477–1488 [DOI] [PubMed] [Google Scholar]
- 21. Bai S, Kopan R, Zou W, Hilton MJ, Ong CT, Long F, Ross FP, Teitelbaum SL. 2008. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J. Biol. Chem. 283:6509–6518 [DOI] [PubMed] [Google Scholar]
- 22. Feng X, Novack DV, Faccio R, Ory DS, Aya K, Boyer MI, McHugh KP, Ross FP, Teitelbaum SL. 2001. A Glanzmann's mutation in β3 integrin specifically impairs osteoclast function. J. Clin. Invest. 107:1137–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McHugh KP, Hodivala-Dilke K, Zheng MH, Namba N, Lam J, Novack D, Feng X, Ross FP, Hynes RO, Teitelbaum SL. 2000. Mice lacking β3 integrins are osteosclerotic because of dysfunctional osteoclasts. J. Clin. Invest. 105:433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao H, Kitaura H, Sands MS, Ross FP, Teitelbaum SL, Novack DV. 2005. Critical role of β3 integrin in experimental postmenopausal osteoporosis. J. Bone Miner. Res. 20:2116–2123 [DOI] [PubMed] [Google Scholar]
- 25. Critchley DR. 2009. Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Annu. Rev. Biophys. 38:235–254 [DOI] [PubMed] [Google Scholar]
- 26. Zhang X, Jiang G, Cai Y, Monkley SJ, Critchley DR, Sheetz MP. 2008. Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat. Cell Biol. 10:1062–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ito Y, Teitelbaum SL, Zou W, Zheng Y, Johnson JF, Chappel J, Ross FP, Zhao H. 2010. Cdc42 regulates bone modeling and remodeling in mice by modulating RANKL/M-CSF signaling and osteoclast polarization. J. Clin. Invest. 120:1981–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA, Puzon-McLaughlin W, Lafuente EM, Boussiotis VA, Shattil SJ, Ginsberg MH. 2006. Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Curr. Biol. 16:1796–1806 [DOI] [PubMed] [Google Scholar]
- 29. Moser M, Legate KR, Zent R, Fassler R. 2009. The tail of integrins, talin, and kindlins. Science 324:895–899 [DOI] [PubMed] [Google Scholar]
- 30. Schmidt S, Nakchbandi I, Ruppert R, Kawelke N, Hess MW, Pfaller K, Jurdic P, Fassler R, Moser M. 2011. Kindlin-3-mediated signaling from multiple integrin classes is required for osteoclast-mediated bone resorption. J. Cell Biol. 192:883–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moser M, Bauer M, Schmid S, Ruppert R, Schmidt S, Sixt M, Wang HV, Sperandio M, Fassler R. 2009. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 15:300–305 [DOI] [PubMed] [Google Scholar]
- 32. Inoue M, Namba N, Chappel J, Teitelbaum SL, Ross FP. 1998. Granulocyte macrophage-colony stimulating factor reciprocally regulates alphav-associated integrins on murine osteoclast precursors. Mol. Endocrinol. 12:1955–1962 [DOI] [PubMed] [Google Scholar]
- 33. Helfrich MH, Nesbitt SA, Lakkakorpi PT, Barnes MJ, Bodary SC, Shankar G, Mason WT, Mendrick DL, Vaananen HK, Horton MA. 1996. Beta 1 integrins and osteoclast function: involvement in collagen recognition and bone resorption. Bone 19:317–328 [DOI] [PubMed] [Google Scholar]
- 34. Zhao B, Takami M, Yamada A, Wang X, Koga T, Hu X, Tamura T, Ozato K, Choi Y, Ivashkiv LB, Takayanagi H, Kamijo R. 2009. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat. Med. 15:1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Horne WC, Sanjay A, Bruzzaniti A, Baron R. 2005. The role (s) of Src kinase and Cbl proteins in the regulation of osteoclast differentiation and function. Immunol. Rev. 208:106–125 [DOI] [PubMed] [Google Scholar]
- 36. Campbell ID. 2010. The talin FERM domain is not so FERM. Structure 18:1222–1223 [DOI] [PubMed] [Google Scholar]
- 37. Faccio R, Takeshita S, Zallone A, Ross FP, Teitelbaum SL. 2003. c-Fms and the αvβ3 integrin collaborate during osteoclast differentiation. J. Clin. Invest. 111:749–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murphy MG, Cerchio K, Stoch SA, Gottesdiener K, Wu M, Recker R, for the L-000845704 Study Group. 2005. Effect of L-000845704, an αvβ3 integrin antagonist, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women. J. Clin. Endocrinol. Metab. 90:2022–2028 [DOI] [PubMed] [Google Scholar]