

Abstract
Centromeres, the sites of spindle attachment during mitosis and meiosis, are located in specific positions in the human genome, normally coincident with diverse subsets of alpha satellite DNA. While there is strong evidence supporting the association of some subfamilies of alpha satellite with centromere function, the basis for establishing whether a given alpha satellite sequence is or is not designated a functional centromere is unknown, and attempts to understand the role of particular sequence features in establishing centromere identity have been limited by the near identity and repetitive nature of satellite sequences. Utilizing a broadly applicable experimental approach to test sequence competency for centromere specification, we have carried out a genomic and epigenetic functional analysis of endogenous human centromere sequences available in the current human genome assembly. The data support a model in which functionally competent sequences confer an opportunity for centromere specification, integrating genomic and epigenetic signals and promoting the concept of context-dependent centromere inheritance.
INTRODUCTION
Centromeres are essential for proper chromosome segregation during mitosis and meiosis. All normal human centromeres are defined by the presence of a predominant satellite DNA family called alpha satellite (1); however, the functional interplay between genome sequences and the epigenetic network involved in kinetochore assembly is poorly understood (2–5). Efforts to explore the nature of such genomic signals have relied on the ability to study representative functional centromere sequences that colocalize with kinetochore proteins (6–8), combined with assessment of de novo centromere formation in artificial chromosome assays (7, 9–11). Previous studies of particular alpha satellite sequence DNAs have supported a sequence-based model of centromere identity (7, 12). However, such studies have been limited to a small number of well-characterized alpha satellite families, and the vast majority of such sequences in the genome have not been evaluated.
The human genome assembly (13) provides the largest available collection of alpha satellite sequences assigned to individual chromosomes and, in concert with extensive experimental evidence, contributes to current models of human centromere sequence organization (7, 14, 15). Well-characterized and assembled alpha satellite DNAs are defined by a highly divergent 171-bp monomer repeat unit, with pairwise sequence identities on the order of 60 to 80% within and between chromosomal subsets (14, 16, 17). This level of sequence divergence within the genome-wide collection of alpha satellite sequences provides an inventory of sequence features for studying CENP-A association and centromere function. Nonetheless, our understanding of the range of sequences capable of de novo centromere formation is limited to a small number of highly characterized alpha satellite DNAs (18, 19), restricting the opportunity to discern genome-wide signals of centromere competency within the majority of assembled alpha satellite sequences.
In this study, to overcome these limitations, we apply a novel strategy for extracting functional satellite sequence information from assembled human centromeric regions. To achieve this, we provide an annotation of all assembled alpha satellite sequences, reporting sites of intra- and interchromosomal homogenization patterns among assembled monomers. These alpha satellite sequence features are evaluated in the context of a global alpha satellite database from a single individual genome (20), resulting in an informed centromere mappability track from which we are able to monitor epigenetic cell line-matched CENP-A enrichment patterns in endogenous human assembled regions. From this combined analysis, we are able to classify human centromeric regions as either functioning or nonfunctioning alpha satellite sequences. Next, to evaluate alpha satellite monomers that are not enriched for CENP-A in the genome, yet have similar monomer content and organization as satellite sequences classified as functioning, we selected collections of alpha satellite DNA (in total, comprising ∼1 Mb) to test for de novo centromere formation in human artificial chromosome assays, thus identifying sequences that, while not currently functioning in the particular genome tested, might be competent for centromere function in other settings.
This combination of genomic and functional strategies has allowed us to develop an initial epigenomic and functionally annotated map of human assembled centromeric regions, which provides a genetic and epigenetic foundation for further study of these regions of the human genome, their variation, and their underlying biology and function.
MATERIALS AND METHODS
Assembled alpha satellite annotation.
Assembled alpha satellite sequences in the UCSC GRCh37/hg19 human reference genome were previously determined by RepeatMasker annotation (RepBase library, version 15.10) (21, 22). These assembled satellite sequences were partitioned into full-length monomers by utilizing both hidden Markov models (HMMER, version 2.0) (23) and local alignments (Smith-Waterman/BLAST) relative to the consensus alpha satellite sequence (16). As our analysis is sensitive to incorrectly parsed monomers, special attention was given to the intermonomer transitions in an effort to monitor and correct incorrect spacing and monomer start and end assignments (correcting an estimated 3.2% of monomers characterized in our data set). Global Needleman-Wunsch alignments (EMBOSS Needle software [http://www.ebi.ac.uk/Tools/psa/emboss_needle/nucleotide.html], with a gap penalty of 1 and gap extension of 0.5, optimized using 100 diverse collections of test monomers [described as ≤60% shared identity with the alpha satellite consensus]) were performed to determine monomer homology or pairwise sequence identity estimates. In preliminary analyses, all monomers with pairwise identity thresholds of >90%, 95%, and 98% were characterized. Here, we report analyses using a homology cutoff of ≥95%, a threshold that provided the best prediction of higher-order repeat (HOR) arrangements in the reference assembly (data not shown), along with the corresponding best inter- and intrachromosome alignment scores. From these data, we monitored repeating high-homology, monomer homology patterns within a 50-kb sliding window to detect and characterize HORs in the hg19 assembly.
All assembled sequences in the human genome (alpha satellite and all other sequences) were evaluated for sites of homology to the reported CENP-B box motif (in forward and reverse complement orientations), as defined by a regular expression that allowed for mismatches only outside the nine biologically assigned functional bases (*TTCG****A**CGGG*) (24). All identified CENP-B boxes were characterized relative to their distance from the alpha satellite consensus CENP-B motif (CTTCGTTGGAAACGGGA) (24, 25) and estimated density per 10-kb sliding window. A total of 2,018 CENP-B boxes were initially defined; however, 35% appeared to be highly divergent (defined as more than five differences from the CENP-B box consensus, with all nine critical bases correctly identified) and largely associated with non-alpha satellite regions of the genome (802 divergent CENP-B boxes were detected outside alpha satellite regions in low density, defined as a single motif identified in a 20-kb window). Therefore, in this study, we considered only those CENP-B boxes that are present in alpha satellite sequences and that are not considered divergent and provide an estimate of both CENP-B density and similarity in all assembled alpha satellite sequences.
Visualization of all monomer homology relationships is provided as files interpreted by the UCSC genome browser (bed files) (22) and presented using Circos software (26).
Global alpha satellite sequence reference and determination of satellite sequence mappability.
To facilitate an annotation pipeline for alpha satellite sequences in the hg19 assembly, we generated a comprehensive alpha satellite sequence database from a single genome (20), representing an estimated 2.6% of the HuRef genome and consisting of 826,394 alpha satellite-containing reads, as determined by RepeatMasker (RepBase library, version 15.10) (21), were involved in the annotation pipeline for alpha satellite sequences in the hg19 assembly. Confident alignments of near-identical satellite sequences to the human assembly are sensitive to parameterization and misrepresentation of all possible sequence variation in the assembled reference sequence. The following steps were taken to account for this level of uncertainty: (i) initial alignments of all quality-masked alpha satellite reads (masking all bases with a phred score of ≤20) to the hg19 assembly with default alignment parameters in BWA (27, 28) (bwasw default parameters tolerate an estimated 10% error for 1,000-bp or longer alignments [http://bio-bwa.sourceforge.net]) were performed, (ii) all read assignments were further evaluated by global alignment estimates of the entire sequence with read trimming of low-quality bases to optimize the alignment, and (iii) 315,071 HuRef alpha satellite whole-genome sequencing (WGS) read global alignments (accounting for 38.1% of all alpha satellite sequences) were detected to have ≥95% sequence identity to the reference assembly. As a result, each alpha satellite monomer in the hg19 assembly could be referenced by both the number of reads that align to those sites as well as all sequences that differ from the underlying reference sequence. To account for redundancy, hg19 monomers that were previously shown to have ≥95% homology were “collapsed,” or combined to present additive read depth and underlying sequence variation.
This final data set of assigned alpha satellite reads provides an estimate of monomer copy number, as estimated from HuRef WGS base coverage relative to a background estimate of single-copy sites in the genome. Read depth estimates were used in this study to identify those regions in the genome suspected to be multicopy, as demonstrated by the increased read depth and coverage estimates associated with higher-order repeat arrays at the edge of the centromere gaps.
To account for “mappability,” the hg19 reference sequence was reformatted to represent sliding windows of k-mers (where k equals 24, 36, 40, and 50 bp with 1-bp overlaps), reporting results from those k-mers of 50 bp, as this data set maximized the sequence-based information within our enrichment search while limiting edge effects observed when mapping exact matches in our short-read chromatin immunoprecipitation-sequencing database (Illumina 72-bp reads [discussed below]). These hg19 alpha satellite sequences were then evaluated for the ratio of those matches that were identified in the assigned read alignments to the total number of matches detected in all HuRef WGS reads (involving all 31 million reads in the HuRef genome, subtracting those read alignments that are provided in the referenced alignment). hg19 reference k-mers that are estimated to have full representation by HuRef global alignments are deemed “mappable,” while those that are shared with reads that are not contained within the region of interest are labeled “nonmappable” and were ignored in this study. From this analysis, 50-mers in the hg19 assembly were determined to be uniquely mappable if they were determined to be single copy by read depth estimates. Single-copy estimates are determined by being <2 standard deviations away from the mean of a single-copy distribution (representing the HuRef frequency for 1 million randomly selected 50-mers from the UCSC mappability track for nonsatellite sequences) for both autosomes (estimated mean, 6.8 reads) and sex chromosomes (mean, 2.9 reads). Conversely, those remaining mappable 50-mers represent sequences that are strictly defined within provided annotation on the human assembly yet are shared within multiple copies of the corresponding repeat.
Identification of sites of CENP-A enrichment in the HuRef cell line.
Native chromatin immunoprecipitation (N-ChIP) analysis was performed as follows. Chromatin was prepared by micrococcal nuclease (30 U; Worthington) digestion of HuRef cell nuclei to predominantly mono- and dinucleosomes. Immunoprecipitation was performed using 5 μg of antibodies against human CENP-A (mouse monoclonal; Stressgen) and normal mouse IgG (Upstate) to control for nonspecific binding. One-tenth of the starting material was reserved as an input DNA control. Immunoprecipitated DNA was extracted with phenol-chloroform and precipitated with ethanol. The extracted DNA was resuspended in 10 mM Tris–1 mM EDTA (pH 8.0) supplemented with 10 μg/ml RNase A. Sequencing was performed at the Duke IGSP Genome Sequencing and Analysis Core Facility (Illumina GAII, with 72-bp single-end reads; 42,172,690 high-quality CENP-A IP reads; and 39,496,123 mock, or IgG background, reads).
To test for general enrichment patterns, providing an opportunity to identify any potential CENP-A sites not associated with alpha satellite sequences, we initially processed the CENP-A ChIP-seq data set using a well-established data-processing pipeline, involving (i) short-read alignment (BWA and Bowtie) to the human genome assembly (Human Feb. 2009 [GRCh37/hg19]) and (ii) enrichment peak calling (QuEST and MACS). We characterized 26.9 million (63.9%) CENP-A and 36.8 million (93.4%) mock ChIP-seq read alignments to the human reference genome using either BWA (28) (allowing 4 mismatches per 72-bp read) or Bowtie aligner (29) (removing the first 7 low-quality bases and allowing 2 mismatches per 65-bp read). Unmapped CENP-A ChIP reads largely represent alpha satellite sequence variation missing from the current genome assembly, as an estimated 80% of unmapped immunoprecipitation (IP) reads are detected as alpha satellite reads using RepeatMasker libraries; all remaining sequences that remained uncharacterized by RepeatMasker were not included in further analyses. We used two methods for enrichment peak estimates with an estimated false discovery rate of <0.08: (i) QuEST (30) (parameter of histone-type ChIP resulting in wide regions of enrichment [bandwidth = 100 bp; region_size = 1,000 bp] and permissive peak-calling parameter [ChIP_threshold = 0.272198860714286; enrichment fold = 3; rescue fold = 3]) and (ii) MACS (31) (bandwidth = 300 bp; tag size = 25 [in addition to default parameters]). All peaks were annotated relative to the current UCSC hg19 database (22). These data were used to determine the absence of detectable CENP-A outside alpha satellite domains and directed our analysis to further investigate those enrichment profiles of alpha satellite sequences with reference to those mappable k-mer enrichments.
To determine CENP-A enrichment in the assembled alpha satellite sequences, IP and mock frequency estimates were provided for all 50-mers represented on the hg19 mappability track. Enrichment estimates were determined by taking log-transformed estimates of this normalized ratio, identifying those sites that show a 2-fold or greater enrichment to provide evidence for CENP-A nucleosome occupancy. CENP-A 50-mer enrichment was designated “single copy” or “multicopy” based on comparisons to whole-genome estimates, described above for the generation of the mappability data set.
Human artificial chromosome assay.
Bacterial artificial chromosomes (BACs) from seven human pericentromeric regions of the genome (outlined below) were validated for alpha satellite sequence content and organization by restriction mapping and pulsed-field gel electrophoresis (PFGE). BACs containing monomeric and higher-order repeat alpha satellite DNA were chosen to maximize the percentage of higher-order repeat arrays. In order to transform these newly created BAC-based human artificial chromosome (HAC) vectors, a Kan/Neo cassette was transposed into the vector region of each construct using the Epicentre Biotechnologies EZ-Tn5 pMOD-2⟨MCS⟩ transposon construction vector with a Kan/Neo resistance cassette cloned between the EcoRI and KpnI sites. To select for insertion of the cassette into the vector region and to thus avoid interruptions in alpha satellite DNA in our BAC-based constructs, a duplex long-range PCR assay was designed to screen the vector region from positive transformants for an increase in vector size corresponding to the Tn5 cassette. Final constructs were further confirmed by direct junction-site sequencing and PFGE insert size estimates. Transfection of human artificial chromosome test constructs into HT1080 cells was performed as previously described (9). Stable drug-resistant colonies (after 10 to 14 days) were screened for artificial chromosomes by fluorescence in situ hybridization (FISH) according to standard procedures (9, 32). Immunostaining procedures were performed as previously described (9), using a monoclonal CENP-A antibody (catalog number ADI-KAM-CC006; Enzo). Cell lines containing HAC constructs with colocalization of alpha satellite DNA and CENP-A were grown without selection for 30 days and then reexamined for these structures in order to determine mitotic stability.
RESULTS
Assessment of alpha satellite sequence variation in an individual genome.
The relative abundance of alpha satellite sequence and the level and pattern of sequence variation between individuals are highly polymorphic (33–35), thereby limiting the ability to compare individual genomic data sets to the incomplete reference genome assembly. To overcome this, we evaluated centromere sequences within the genome of a single individual (HuRef) (20) relative to annotated alpha satellite sequences in the reference assembly (see Materials and Methods). By studying patterns of intra- and/or interchromosomal sequence homology within the current human chromosome assemblies (GRCh37/hg19), we observed three patterns of homogenization: type 1, local homogenization, where either single monomers or multimeric blocks of monomers have at least one near-identical tandem repeat unit (4,407 monomers; 42.9% of all homogenized monomers); type 2, monomers that share high intrachromosomal identity but that are not in tandem (4,468 monomers; 43.4% of all homogenized monomers); and type 3, monomers that share high interchromosomal identity (2,920 monomers; 28.4% of all homogenized monomers), of which roughly half (1,521 monomers) are observed to also show intrachromosomal homology (Fig. 1; see also Tables S1 to S3 and Fig. S1 in the supplemental material). These analyses generated a genomic reference annotation of alpha satellite regional and long-distance homology, enabling an opportunity to contextualize sequence variation within the HuRef genome (Fig. 2; see also Tables S1 to S4 in the supplemental material).
Fig 1.

Schematic overview of alpha satellite organization, assembly, and read depth analysis. The assembled alpha satellite is organized by sequence similarity. (a) Alpha satellite monomers from hg19 assembled chromosomes were evaluated for the presence of local homogenization in tandem, intrachromosomal homogenization patterns that are regional but not in tandem, and interchromosomal or interarray patterns (defined by pairwise identities of ≥95%). (b) In this study, homogenized monomers are then clustered, or “purposely collapsed,” as illustrated. As a result, a single region on the human assembly could represent a cluster of more than one monomer. (c) These multimonomer sites represent multicopy regions, as demonstrated by the elevated read depth estimates.
Fig 2.

Alpha satellite annotation in the human genome assembly. (a) As illustrated for the centromeric region on the X chromosome, all full-length alpha satellite monomers are characterized by sequence identity to all other assembled monomers, resulting in a database of homogenization relationships (intra- and/or interchromosomal), local multimonomer homogenization patterns (typical of higher-order repeats [HORs]), the presence and density of CENP-B boxes, and the estimated read depth from HuRef WGS global alignments. Monomers are represented as single-tick boxes and are linearly arrayed relative to the order on the chromosome assembly spanning either side of the centromere gap (shown as the compressed bar). Pericentromeric regions (defined operationally as 2 Mb around the centromere-assigned clone gap) (28, 46) are highlighted in gray. Homogenized alpha satellite monomers (defined as ≥95% identical to another assembled monomer) are shown in black. Local, intrachromosomal homogenization patterns are represented in red (previously known HORs), blue (newly defined HORs containing >3 monomers), and yellow (newly defined HORs containing 2 or 3 monomers). CENP-B boxes are indicated in green, with darker greens signifying an identical match to the published alpha satellite CENP-B box consensus motif and lighter colors indicating relative divergence. (b) Monomers that show interchromosomal homogenization patterns are shown as connections, with the color indicating the number of monomers (or “block” of monomers) that share identity across chromosomes (purple indicates ≥5 monomers within the 1-kb window, light purple indicates ≥4, yellow indicates ≥3, and black indicates <3).
Global alignments (see Materials and Methods) of HuRef alpha satellite WGS reads (20) were used to evaluate the annotated alpha satellite in the reference assembly (GRCh37/hg19). Sequence coverage over near-identical monomers (types 1 to 3) was pooled, providing both an estimate of sequence abundance and a summary of inherent alpha satellite sequence variation in the HuRef genome (Fig. 1 and 3a). By these criteria, we estimate that 6.6 Mb of assembled alpha satellite sequence in the reference assembly is “single copy” in HuRef (defined operationally here as being sufficiently low in copy number that it cannot be distinguished in terms of read depth from the bulk of unique sequences in the euchromatic genome) (see Fig. S2 in the supplemental material). Only 19.6% of the alpha satellite annotated in the GRCh37/hg19 reference assembly is demonstrably multicopy in the HuRef genome, presumably indicating that most homogenized alpha satellite is missing from the reference assembly (14, 15).
Fig 3.

Characterization of mappable sites of CENP-A enrichment at endogenous human centromeres. (a) HuRef WGS read alignments provide an estimate of read depth, or copy number, as well as provide a database of sequences that are assigned to a particular hg19 annotation. Mappability is illustrated as the comparison of those hg19 annotated 50-mers relative to the assigned HuRef WGS sequence library. This is most clearly demonstrated for those sites suspected to be single copy by read depth estimates, as a given 50-mer is expected to be represented completely by those sequence reads that are assigned to that region. Multicopy sites are defined as mappable if the 50-mer is also identified outside a given location. (b and c) Mapping of CENP-A-enriched 50-mers to these specific mappable sites (shown in teal) can be divided into single-copy and multicopy sites, as defined by read depth. As an example, in DXZ1, we observed both multicopy and single-copy sites within each copy of the higher-order repeat (b) and throughout the assembled portions of the array (c).
Extending such analyses to genomes sequenced with next-generation methods will require adapting this approach to short-read data sets. To do this, we created a mappability index across alpha satellite sequences in the hg19 annotated reference assembly (developed to match the existing mappability index for nonsatellite DNA, representing 24, 36, 40, and 50 bp). Conceivably, short stretches of high sequence homology might be shared between divergent (yet related) alpha satellite monomers; this could apply to sequences represented multiple times in the current assembly and/or to those found in the remaining, unmapped alpha satellite sequences representing sequence types currently absent from the human reference assembly. To identify such unmappable regions, we evaluated the alpha satellite reference sequences relative to the library of sequence variation derived from the HuRef WGS read alignments to determine if each site is capable of mapping to annotated satellite sequence features (e.g., either to precise locations in uniquely mapping alpha satellite sequences or to features describing in aggregate a collection of homogenized, annotated hg19 monomers). If a given reference sequence was represented at the same frequency among reads characterized by a particular annotation as in the global context of all WGS reads, it was considered mappable to that annotation; conversely, if it was observed at a higher frequency among all WGS reads than among those characterized by the annotation, it was deemed not mappable (illustrated in Fig. 3a). Of all alpha satellite reference sequences (here focusing on 50-mers to maximize the sequence-based information), 95.4% (∼6.7 million) were present in the HuRef genome and could be assessed for their mappability. Of these, 89% of the 50-mers were mappable in the HuRef genome, thus reducing the number of unmappable 50-mers to fewer than 700,000.
As expected, the vast majority (>4.2 million, or 94.1%) of 50-mers that derive from divergent monomeric alpha satellite sequences are mappable and single copy in the HuRef genome. Nonetheless, 250,085 50-mers from these sequences are not uniquely mappable to these regions, as they share stretches of homology with alpha satellite sequences elsewhere in the current assembly or in the much larger proportion of alpha satellite sequences that are present in the unassembled centromere gaps. Perhaps not unexpectedly, given reported relationships among various subfamilies of higher-order repeat alpha satellite sequences (17, 33, 36), a smaller proportion of 50-mers from homogenized alpha satellite sites are uniquely mappable to those sequences; nonetheless, we identified 832,559 50-mers that provide unique markers in these otherwise highly homogenized arrays (representing about half of the 1.5 million homogenized sites characterized above as types 1 to 3). In addition to these uniquely mappable sites, we identified 315,295 50-mers that are specific to alpha satellite monomers annotated as belonging to a particular subfamily, thus representing multicopy mappable sites. For example, the hg19 assembly contains ∼63 kb of sequences belonging to the DXZ1 higher-order repeat on the X chromosome, roughly half of which appear to be mappable, defining 13,863 single-copy sites and 14,959 multicopy sites (Fig. 3b). Similar patterns of single-copy and multicopy mappable sites were found for other homogenized subsets of alpha satellite from around the genome.
Mapping endogenous sites of CENP-A enrichment at human centromeres.
In order to derive an initial epigenomic map of CENP-A enrichment for the HuRef genome and to thus distinguish between sequences that are “functioning” and “nonfunctioning” at the endogenous centromeres in the HuRef cell line, we carried out CENP-A ChIP-seq analysis (generating 42.1 million ChIP and 39.5 million mock reads [see Materials and Methods]). Mapping of CENP-A-associated sequences to the human assembly using standard methods provided no evidence of CENP-A enrichment for sequences other than alpha satellite sequences. To further characterize the specific alpha satellite sequence features that are enriched in CENP-A, we generated a HuRef CENP-A enrichment profile (log-transformed 50-mer frequency ratio of CENP-A ChIP relative to paired mock data) anchored on the annotated and mappable regions of alpha satellite.
Of the total 5.3 million mappable alpha satellite sites, we identified only 46,488 50-mers that are CENP-A enriched in the HuRef cell line, providing evidence that the majority of the monomeric alpha satellite sequence is not implicated in centromeric chromatin, in line with previously reported observations (7, 8) (see Table S5 in the supplemental material). The vast majority of these (98.2%) were enriched at specific higher-order arrays present in the current human assembly at the borders of the large centromere gaps (Fig. 3a). We detected CENP-A enrichment at individual higher-order arrays on seven chromosomes (chromosomes 1 [D1Z7], 2 [D2Z1], 6 [D6Z1], 7 [D7Z1], 8 [D8Z2], X [DXZ1], and Y [DYZ3]) (see Table S6 in the supplemental material), consistent with previous cytological studies associating these arrays with centromere function. Some centromeric regions are known to contain more than one array (37), raising the possibility of using CENP-A enrichment to distinguish between sequences that are functioning from those that are nonfunctioning in the HuRef genome. Indeed, on chromosome 7, as indicated above, we detected CENP-A enrichment for monomers mappable to only one (D7Z1 but not D7Z2) of the two known centromeric higher-order repeat arrays on this chromosome (38, 39). In contrast to these examples, the remaining chromosomes showed no detectable CENP-A enrichment on any sequences present in the assembly. This presumably indicates that the functioning centromere sequences on those chromosomes are missing from the assembly; different approaches will be necessary to address this possibility and to fully identify, characterize, and map the relevant sequences.
Notably, we detected enrichment of CENP-A at both multicopy and single-copy mappable sites within higher-order repeat sequences. Using the example of DXZ1 at the X chromosome centromere (Fig. 3b and c), multicopy enrichment sites indicate likely CENP-A distribution across many copies of the homogenized repeat. Single-copy enrichment sites, in contrast, provide evidence for the presence of CENP-A at particular copies of the repeat that are distinguishable from the sequences of most DXZ1 repeats. Such patterns of both multicopy and single-copy enrichment were found for all examples of higher-order repeats and other homogenized sites in the current assembly.
While the vast majority of enriched sites are associated with higher-order repeat arrays in the genome, we were also able to track low-frequency CENP-A enrichment in nonhomogenized monomeric subsets in pericentromeric regions adjacent to the centromere gaps in the current assembly, in agreement with a previously reported observation of divergent satellite adjacent to the centromere on the mouse Y chromosome (40). For example, we found evidence of CENP-A enrichment extending 12.6 kb from the DXZ1 array over adjacent monomeric monomers in the p-arm assembly, in agreement with previous studies that described the CENP-A domain closer to the p-arm of the array (7, 41) (Fig. 3c). A similar extension of CENP-A was observed in a 2-kb DYZ3 q-arm transition zone on the Y chromosome. Additionally, we identified 34 sites of limited CENP-A enrichment within nonfunctioning higher-order repeat units and monomeric alpha satellite sites; such ectopic sites may correspond to temporally or spatially limited sites of CENP-A deposition in the population of HuRef cells (see Table S7 in the supplemental material).
We have used CENP-A ChIP-seq analysis to distinguish between alpha satellite monomers that are functioning and those that are nonfunctioning at endogenous centromeres in HuRef cells. This data set raises two questions for further analysis. First, are the assignments of functioning and nonfunctioning consistent for all human genomes, or are they relevant only to the HuRef genome (or perhaps even to just this particular cell line)? Second, are the sequences identified as nonfunctioning in HuRef actually nonfunctional, or are they potentially competent to serve as functioning sequences in a different experimental or native context? While the first question will require a more extensive analysis of multiple genomes together with a better understanding of the centromere gaps in the current reference genome assembly, the second question is amenable to exploration using human artificial chromosome assays.
Human artificial chromosome assays identify competent centromere sequences.
To combine the study of sequence characterization and CENP-A localization with functional assays, a subset of the alpha satellite sequences that were designated nonfunctioning in the HuRef genome were tested in human artificial chromosome assays for their competence to form de novo centromeres. To do this, we selected 970 kb of annotated nonfunctioning alpha satellite sequence from seven chromosomes (that is, sequences corresponding to alpha satellite types not associated with CENP-A at endogenous human centromeres in the HuRef cell line, as presented above) (see Fig. S3 in the supplemental material). Genomic sites for testing were selected to represent various patterns of monomer organization and homogenization, corresponding to (i) regions containing previously characterized higher-order repeat units from chromosomes 7, 17, and 19 (D17Z1B, D7Z2, and D19Z2/D5Z1); (ii) novel higher-order repeat units from chromosomes 4 and 11 (HOR4-13mer and HOR11-12mer); and (iii) nonhomogenized monomeric regions from chromosomes 3 and X, selected on the basis of previous high-resolution cytological studies that indicated that these sequences were physically separate from the major sites of centromere protein localization (see Fig. S4 in the supplemental material). While each of these sequences is nonfunctioning in the HuRef genome, they represent a range of density and periodicity of CENP-B boxes (24) (from ∼0.24/kb to ∼1.7/kb) (Table 1), which have previously been associated with the ability of subsets of alpha satellite to form artificial chromosomes (18, 42).
Table 1.
Characteristics and results of alpha satellite constructs tested for human artificial chromosome formationc
| Chromosome | Starting BACa | Assay construct |
Human artificial chromosome assay |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | Higher-order repeat α-satellite |
Monomeric α-satellite |
No. of colonies tested | No. (%) of colonies with HACs | CENP-A IF-FISH | Mitotic stability | ||||||
| Size (kb) | No. of CENP-B boxes | Presence of CENP-A enrichment | Size(s) (kb) | No. of CENP-B boxes | Presence of CENP-A enrichment | |||||||
| Xp | RP13-971O21 | DXZ1-9HOR | 19 | 26 | + | 59, 23b | 1 | − | 19 | 4 (21) | 2/2 | 2/2 |
| Xp | RP11-65I6 | X-monomeric | 0 | NA | NA | 129 | 14 | − | 30 | 0 | NA | NA |
| 3p | RP11-557B13 | 3-Monomeric | 0 | NA | NA | 203 | 22 | − | 24 | 0 | NA | NA |
| 4q | RP11-365H22 | D4Z2bn-9HOR | 23 | 37 | − | 9 | 0 | − | 15 | 4 (27) | 2/2 | 1/1 |
| 7q | RP11-435D24 | D7Z2-8HOR | 22 | 53 | − | 27 | 2 | − | 18 | 5 (28) | 4/4 | 2/2 |
| 11p | RP11-100E23 | D11Z1-7HOR | 14 | 9 | − | 27 | 8 | − | 17 | 4 (24) | 4/4 | 2/2 |
| 17p | RP11-458D13 | D17Z1B-7HOR | 17 | 27 | − | 132 | 0 | − | 20 | 4 (20) | 2/2 | 2/2 |
| 19p | RP11-21H14 | D19Z1-10HOR | 29 | 6 | − | 19 | 0 | − | 10 | 0 | NA | NA |
The genomic location of each BAC is indicated in Fig. S3a to h in the supplemental material.
Two versions of DXZ1-9HOR containing different amounts of monomeric α-satellite were tested. Results were indistinguishable and were pooled.
Alpha satellite arrays from different chromosomes and representing different types of arrays were chosen for their capacity to form de novo centromeres in human artificial chromosome assays. A 19-kb stretch of DXZ1, a CENP-A-associated array within the context of the HuRef genome, was chosen as a control for use of a relatively small amount of higher-order repeat (HOR) array DNA in this assay. Two monomeric arrays, from chromosomes Xp and 3p, were chosen due to their inclusion of CENP-B boxes and the absence of higher-order repeat array DNA. Five additional arrays were included to test the competency of nonfunctioning sequences in the HuRef genome. NA, not applicable.
Human artificial chromosome test constructs were prepared by modifying BACs that contain alpha satellite sequences from the seven chosen nonfunctioning regions to maximize the percentage of higher-order repeat alpha satellite sequences (when present) relative to the adjacent monomeric sequence (see Materials and Methods) (Table 1). As a positive control, we also tested constructs containing DXZ1 sequences shown previously to interact with CENP-A both at endogenous human centromeres and in previous human artificial chromosome studies (7, 43). Constructs to be tested were transfected into HT1080 cells (9), and 10 to 30 drug-resistant colonies were isolated for each construct. To identify colonies that carry putative human artificial chromosomes, we performed FISH using the relevant BAC DNA as a probe. Human artificial chromosomes were identified for five of the six higher-order repeat constructs tested (Table 1). In the cases of DXZ1 and D17Z1B, our data are consistent with previous reports (7, 43–45). The one higher-order repeat construct that did not support formation of human artificial chromosomes was a construct containing D19Z1 alpha satellite, which, perhaps notably, contains the lowest CENP-B box density with the most varied motif periodicity among the alpha satellite arrays tested here. The two constructs containing monomeric alpha satellite sequences from chromosomes 3 and X did not form detectable artificial chromosomes (Table 1), consistent with immunofluorescence-FISH (IF-FISH) experiments that indicated that these sequences map adjacent to, but not coincident with, centromere proteins at the functional centromere on these chromosomes. Although the observed CENP-B density for these monomeric regions is similar to that of the other constructs used in this study, these sequences lack the periodicity of the motifs that is evident in sequences organized into tandem higher-order repeats. The inability of these constructs to form artificial chromosomes here suggests that it is not the presence of CENP-B boxes per se but rather the higher-order periodicity and/or the pattern of specific variants within the array that contributes to centromere competency, through the ability to establish the necessary centrochromatin to recruit kinetochore assembly.
Standard criteria for validating human artificial chromosomes include association with the centromere protein CENP-A or CENP-E and mitotic stability for at least 30 days in the absence of drug selection (7, 9). To test for centromere protein associations, we performed IF-FISH to identify artificial chromosomes that hybridized to the relevant alpha satellite probe and colocalized with CENP-A (Fig. 4 and Table 1; see also Fig. S5 in the supplemental material). In addition, all artificial chromosomes tested here were mitotically stable for at least 30 days in the absence of selection, thus fulfilling the criteria for human artificial chromosomes containing functional de novo centromeres (Table 1). Notably, there was no difference in the frequency of detection or behavior of artificial chromosomes generated from higher-order repeat constructs containing alpha satellite sequences identified as functioning or nonfunctioning by CENP-A ChIP-seq analysis in the HuRef genome. Both types of constructs appeared to be competent, in contrast to constructs containing nonhomogenized monomeric alpha satellite sequences, which are not competent in this assay (Table 1).
Fig 4.

Defining nonfunctioning genomic arrays as centromere competent or not competent. Shown is an overview of the experimental strategy. CENP-A ChIP-seq analysis was utilized to map CENP-A enrichment sites in the HuRef genome. To determine if arrays determined to be functioning or nonfunctioning in HuRef are competent for centromere formation when removed from their genomic locations, BAC-based constructs harboring these classes of arrays were tested in a human artificial chromosome (HAC) assay for de novo centromere formation by IF-FISH analysis and mitotic stability (Table 1). Nonfunctioning higher-order repeat arrays with CENP-B boxes are competent to form centromeres in this assay, while nonfunctioning monomeric units are not competent. Here, we demonstrate this assay using three examples of a centromeric region, illustrated with two higher-order arrays (shown as blue and purple blocks). Sites enriched for CENP-A enrichment are shown as red circles over genomic sites indicated. (a) When testing sequences previously shown to recruit and stably maintain CENP-A in the HuRef genome in a BAC-based construct (shown as the construct containing blue sequence), we detected the de novo establishment of CENP-A domains in the human artificial chromosome assay (where CENP-A is shown in red and the BAC construct is shown in green). (b) In a similar manner, we demonstrate that those sites that do not appear enriched by CENP-A in the genomic context, yet are organized into higher-order repeats, are still competent to recruit and stably propagate CENP-A. (c) However, for those sites that both lack CENP-A enrichment in the genome as well as HOR organization, we did not detect sequence competency by the HAC assay.
DISCUSSION
To explore those genomic sequence features that are competent for centromere identity, we have functionally characterized the centromeric sequences assigned to individual chromosomes in the current human (hg19) genome assembly. This study advances two aspects of our current genomic and functional understanding of these sequences. First, we have used CENP-A ChIP-seq analysis of the HuRef genome to provide an initial epigenomic enrichment map of endogenous centromeric regions, permitting distinction between functioning and nonfunctioning alpha satellite subsets in that genome. This represents a substantial advance in the field of centromere genomics, as we demonstrate the utility of pairing existing satellite patterns in the reference assembly with sequence frequencies observed for data from individual genomes. Second, we have used human artificial chromosome assays across a number of genomic sites containing alpha satellite DNA to demonstrate that at least some sequences that are nonfunctioning in the HuRef genome are nonetheless competent for de novo centromere formation. This indicates that the particular alpha satellite sequences that are used as centromere sequences in any given genome are only a subset of the collection of potentially functional centromeric sequences, thus raising the possibility that the designation of which sequences are in fact functional differs among different genomes within the human population. From an evolutionary viewpoint, this excess of regional centromere-competent sequences may provide some security for preservation of centromere function, as a rapid gain or loss of sequence in this region may be more easily tolerated, ultimately conferring stability to centromeres and the chromosomes on which they are found.
From the results of both the CENP-A ChIP-seq analysis and the artificial chromosome assays, we present a model, illustrated in Fig. 5, in which both genomic competency and context-dependent epigenetic interactions act together to define centromere identity (Fig. 5a). Given that individuals are known to have different sizes of higher-order repeat arrays (35, 46, 47) and corresponding array sequence variation (17, 33, 48), one could hypothesize that sequence abundance and composition might have an effect on CENP-A deposition and centromere sequence use. This analysis begins to outline the concept of centromere-competent sequences, revealed through testing of those sequences that are capable of recruiting and stably propagating CENP-A in the genome and/or when tested in a de novo artificial chromosome assay. Expansion of this sequence analysis to capture this process over time, or between cell lines, is expected to further address whether such a combined genomic/epigenomic model of centromere specification is stable over time. The initial domain of CENP-A deposition could be stably maintained throughout cell division as centromeric chromatin is replenished each cell cycle; alternatively, these sites could fluctuate stochastically whereby CENP-A enrichment is a signature of a cell population (49) (Fig. 5b).
Fig 5.
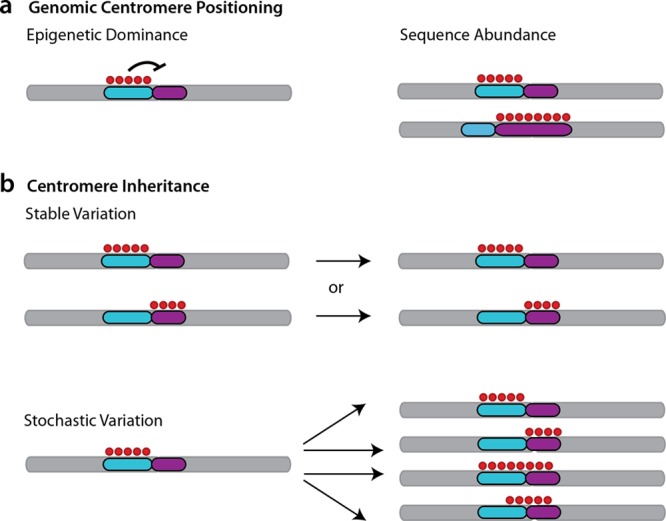
Models for designation of functioning centromere sequences. (a) Genomic features underlying the positioning of functioning centromeres. In an epigenetic model of centromere positioning, the pattern of CENP-A and other marks (red nucleosomes) can act as a positive or negative signal to act upon (and distinguish between) underlying competent genomic sequences (here, purple or teal arrays). Alternately, satellite feature abundance may be a factor contributing to CENP-A recruitment where ancient or more recent polymorphic differences in individual sizes of higher-order arrays influence centromere positioning. (b) Propagation of centromere designation. Under either model of localization shown in panel a, additional models must account for centromere inheritance (mitotic and/or meiotic) and propagation over time. Once loaded, CENP-A association might be stably maintained through mitosis and/or meiosis via nucleosome loading. Conversely or less frequently, these sites might be susceptible to stochastic variation whereby CENP-A enrichment is a signature of a given time point, cell type, or family member but is subject to change and dispersal across all regional competent sequences in other cellular, family, or population contexts.
Efforts to fully explore these centromeric regions will depend on careful and comprehensive sequence comparisons between genome-matched satellite DNA data sets derived from a number of individual genomes, as consensus sequences alone will likely be inadequate to provide the complete spectrum of information needed to make full functional inferences either within a given genome or across populations. Here, we provide a genome assessment for one individual, in one cell line, and at a single time point. It will be necessary to address whether such genome annotation, both sequence based and epigenetic, is a shared property of different genomic samples, exploring different cellular states (varying by developmental stage, tissue type, and health or disease) and different genomes (within an individual and among families or population groups). By extending this approach, a universal library of competent centromere sequences might be identified, promoting a comprehensive approach to defining parameters of human centromere and centromere sequence plasticity.
Supplementary Material
Footnotes
Published ahead of print 10 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01198-12.
REFERENCES
- 1. Manuelidis L. 1978. Chromosomal localization of complex and simple repeated human DNAs. Chromosoma 66:23–32 [DOI] [PubMed] [Google Scholar]
- 2. Henikoff S, Ahmad K, Malik HS. 2001. The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293:1098–1102 [DOI] [PubMed] [Google Scholar]
- 3. Karpen GH, Allshire RC. 1997. The case for epigenetic effects on centromere identity and function. Trends Genet. 13:489–496 [DOI] [PubMed] [Google Scholar]
- 4. Schueler MG, Sullivan BA. 2006. Structural and functional dynamics of human centromeric chromatin. Annu. Rev. Genomics Hum. Genet. 7:301–313 [DOI] [PubMed] [Google Scholar]
- 5. Ugarković DI. 2009. Centromere-competent DNA: structure and evolution. Prog. Mol. Subcell. Biol. 48:53–76 [DOI] [PubMed] [Google Scholar]
- 6. Haaf T, Ward DC. 1994. Structural analysis of alpha-satellite DNA and centromere proteins using extended chromatin and chromosomes. Hum. Mol. Genet. 3:697–709 [DOI] [PubMed] [Google Scholar]
- 7. Schueler MG, Higgins AW, Rudd MK, Gustashaw K, Willard HF. 2001. Genomic and genetic definition of a functional human centromere. Science 294:109–115 [DOI] [PubMed] [Google Scholar]
- 8. Vafa O, Sullivan KF. 1997. Chromatin containing CENP-A and alpha-satellite DNA is a major component of the inner kinetochore plate. Curr. Biol. 7:897–900 [DOI] [PubMed] [Google Scholar]
- 9. Harrington JJ, Van Bokkelen G, Mays RW, Gustashaw K, Willard HF. 1997. Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat. Genet. 15:345–355 [DOI] [PubMed] [Google Scholar]
- 10. Ikeno M, Grimes B, Okazaki T, Nakano M, Saitoh K, Hoshino H, McGill NI, Cooke H, Masumoto H. 1998. Construction of YAC-based mammalian artificial chromosomes. Nat. Biotechnol. 16:431–439 [DOI] [PubMed] [Google Scholar]
- 11. Nakano M, Okamoto Y, Ohzeki J, Masumoto H. 2003. Epigenetic assembly of centromeric chromatin at ectopic alpha-satellite sites on human chromosomes. J. Cell Sci. 116:4021–4034 [DOI] [PubMed] [Google Scholar]
- 12. Masumoto H, Ikeno M, Nakano M, Okazaki T, Grimes B, Cooke H, Suzuki N. 1998. Assay of centromere function using a human artificial chromosome. Chromosoma 107:406–416 [DOI] [PubMed] [Google Scholar]
- 13. International Human Genome Sequencing Consortium 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921 [DOI] [PubMed] [Google Scholar]
- 14. Rudd MK, Willard HF. 2004. Analysis of the centromeric regions of the human genome assembly. Trends Genet. 20:529–533 [DOI] [PubMed] [Google Scholar]
- 15. She X, Horvath JE, Jiang Z, Liu G, Furey TS, Christ L, Clark RA, Graves T, Gulden CL, Alkan C, Bailey JA, Sahinalp C, Rocchi M, Haussler D, Wilson RK, Miller W, Schwartz S, Eichler EE. 2004. The structure and evolution of centromeric transition regions within the human genome. Nature 430:857–864 [DOI] [PubMed] [Google Scholar]
- 16. Waye JS, Willard HF. 1987. Nucleotide sequence heterogeneity of alpha satellite repetitive DNA: a survey of alphoid sequences from different human chromosomes. Nucleic Acids Res. 15:7549–7569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Willard HF, Waye JS. 1987. Chromosome-specific subsets of human alpha satellite DNA: analysis of sequence divergence within and between chromosomal subsets and evidence for an ancestral pentameric repeat. J. Mol. Evol. 25:207–214 [DOI] [PubMed] [Google Scholar]
- 18. Masumoto H, Nakano M, Ohzeki J. 2004. The role of CENP-B and alpha-satellite DNA: de novo assembly and epigenetic maintenance of human centromeres. Chromosome Res. 12:543–556 [DOI] [PubMed] [Google Scholar]
- 19. Warburton PE. 1999. Making CENs of mammalian artificial chromosomes. Mol. Genet. Metab. 68:152–160 [DOI] [PubMed] [Google Scholar]
- 20. Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, Lin Y, MacDonald JR, Pang AWC, Shago M, Stockwell TB, Tsiamouri A, Bafna V, Bansal V, Kravitz SA, Busam DA, Beeson KY, McIntosh TC, Remington KA, Abril JF, Gill J, Borman J, Rogers Y-H, Frazier ME, Scherer SW, Strausberg RL, Venter JC. 2007. The diploid genome sequence of an individual human. PLoS Biol. 5:e254 doi:10.1371/journal.pbio.0050254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. 2005. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110:462–467 [DOI] [PubMed] [Google Scholar]
- 22. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res. 12:996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bao Z, Eddy SR. 2002. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12:1269–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Masumoto H, Masukata H, Muro Y, Nozaki N, Okazaki T. 1989. A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J. Cell Biol. 109:1963–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alkan C, Cardone MF, Catacchio CR, Antonacci F, O'Brien SJ, Ryder OA, Purgato S, Zoli M, Della Valle G, Eichler EE, Ventura M. 2011. Genome-wide characterization of centromeric satellites from multiple mammalian genomes. Genome Res. 21:137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res. 19:1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25 doi:10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A. 2008. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods 5:829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9:R137 doi:10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grimes BR, Rhoades AA, Willard HF. 2002. Alpha-satellite DNA and vector composition influence rates of human artificial chromosome formation. Mol. Ther. 5:798–805 [DOI] [PubMed] [Google Scholar]
- 33. Durfy SJ, Willard HF. 1989. Patterns of intra- and interarray sequence variation in alpha satellite from the human X chromosome: evidence for short-range homogenization of tandemly repeated DNA sequences. Genomics 5:810–821 [DOI] [PubMed] [Google Scholar]
- 34. Warburton PE, Wevrick R, Mahtani MM, Willard HF. 1992. Pulsed-field and two-dimensional gel electrophoresis of long arrays of tandemly repeated DNA: analysis of human centromeric alpha satellite. Methods Mol. Biol. 12:299–317 [DOI] [PubMed] [Google Scholar]
- 35. Wevrick R, Willard HF. 1989. Long-range organization of tandem arrays of alpha satellite DNA at the centromeres of human chromosomes: high-frequency array-length polymorphism and meiotic stability. Proc. Natl. Acad. Sci. U. S. A. 86:9394–9398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alexandrov IA, Mitkevich SP, Yurov YB. 1988. The phylogeny of human chromosome specific alpha satellites. Chromosoma 96:443–453 [DOI] [PubMed] [Google Scholar]
- 37. Alexandrov IA, Kazakov AE, Tumeneva I, Shepelev V, Yurov Y. 2001. Alpha-satellite DNA of primates: old and new families. Chromosoma 110:253–266 [DOI] [PubMed] [Google Scholar]
- 38. Waye JS, England SB, Willard HF. 1987. Genomic organization of alpha satellite DNA on human chromosome 7: evidence for two distinct alphoid domains on a single chromosome. Mol. Cell. Biol. 7:349–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wevrick R, Willard HF. 1991. Physical map of the centromeric region of human chromosome 7: relationship between two distinct alpha satellite arrays. Nucleic Acids Res. 19:2295–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pertile MD, Graham AN, Choo KHA, Kalitsis P. 2009. Rapid evolution of mouse Y centromere repeat DNA belies recent sequence stability. Genome Res. 19:2202–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Spence JM, Fournier REK, Oshimura M, Regnier V, Farr CJ. 2005. Topoisomerase II cleavage activity within the human D11Z1 and DXZ1 alpha-satellite arrays. Chromosome Res. 13:637–648 [DOI] [PubMed] [Google Scholar]
- 42. Ohzeki J, Nakano M, Okada T, Masumoto H. 2002. CENP-B box is required for de novo centromere chromatin assembly on human alphoid DNA. J. Cell Biol. 159:765–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rudd MK, Mays RW, Schwartz S, Willard HF. 2003. Human artificial chromosomes with alpha satellite-based de novo centromeres show increased frequency of nondisjunction and anaphase lag. Mol. Cell. Biol. 23:7689–7697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maloney KA, Sullivan LL, Matheny JE, Strome ED, Merrett SL, Ferris A, Sullivan BA. 2012. Functional epialleles at an endogenous human centromere. Proc. Natl. Acad. Sci. U. S. A. 109:13704–13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rudd MK, Schueler MG, Willard HF. 2003. Sequence organization and functional annotation of human centromeres. Cold Spring Harb. Symp. Quant. Biol. 68:141–149 [DOI] [PubMed] [Google Scholar]
- 46. Mahtani MM, Willard HF. 1990. Pulsed-field gel analysis of alpha-satellite DNA at the human X chromosome centromere: high-frequency polymorphisms and array size estimate. Genomics 7:607–613 [DOI] [PubMed] [Google Scholar]
- 47. Oakey R, Tyler-Smith C. 1990. Y chromosome DNA haplotyping suggests that most European and Asian men are descended from one of two males. Genomics 7:325–330 [DOI] [PubMed] [Google Scholar]
- 48. Prades C, Laurent MA, Puechberty J, Yurov Y, Roizès G. 1996. SINE and LINE within human centromeres. J. Mol. Evol. 42:37–43 [DOI] [PubMed] [Google Scholar]
- 49. Dimitriadis EK, Weber C, Gill RK, Diekmann S, Dalal Y. 2010. Tetrameric organization of vertebrate centromeric nucleosomes. Proc. Natl. Acad. Sci. U. S. A. 107:20317–20322 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.