

Abstract
Cells can undergo two alternative fates following exposure to environmental stress: they either induce apoptosis or inhibit apoptosis and then repair the stress-induced alterations. These processes minimize cell loss and prevent the survival of cells with aberrant DNA and protein alterations. These two alternative fates are partly controlled by stress granules (SGs). While arsenite, hypoxia, and heat shock induce the formation of SGs that inhibit apoptosis, X-ray irradiation and genotoxic drugs do not induce SGs, and they are more prone to trigger apoptosis. However, it is unclear precisely how SGs control apoptosis. This study found that SGs suppress the elevation of reactive oxygen species (ROS), and this suppression is essential for inhibiting ROS-dependent apoptosis. This antioxidant activity of SGs is controlled by two SG components, GTPase-activating protein SH3 domain binding protein 1 (G3BP1) and ubiquitin-specific protease 10 (USP10). G3BP1 elevates the steady-state ROS level by inhibiting the antioxidant activity of USP10. However, following exposure to arsenite, G3BP1 and USP10 induce the formation of SGs, which uncovers the antioxidant activity of USP10. We also found that the antioxidant activity of USP10 requires the protein kinase activity of ataxia telangiectasia mutated (ATM). This work reveals that SGs are critical redox regulators that control cell fate under stress conditions.
INTRODUCTION
Upon exposure to environmental stress, cells select two distinct fates: they either induce apoptosis or inhibit apoptosis and repair any stress-induced alterations. These processes prevent the survival of cells with DNA and protein aberrations and simultaneously minimize cell loss. These cell fate decisions are partly dependent on the type of stress. While arsenite, hypoxia, and heat shock induce the formation of stress granules (SGs) that inhibit apoptosis, genotoxins and X-ray irradiation do not induce SGs, thereby making cells more prone to undergo apoptosis (1, 2). Thus, SGs are a crucial defense mechanism against environmental stress. However, the precise mechanism underlying how SGs inhibit apoptosis has not been elucidated.
SGs are cytoplasmic RNA granules, and their formation is associated with the inhibition of translation initiation and the disassembly of polysomes (3). During stress, SGs act as storage sites of nontranslating mRNAs separated from disassembled polysomes. The mRNA composition of SGs is selective; they contain mRNAs encoding housekeeping genes but exclude those encoding stress-induced genes, such as the genes encoding heat shock proteins (4). This selective storage of mRNAs by SGs promotes the translation of stress-responsive genes, thereby driving recovery from a stress.
In addition to RNAs, SGs contain various proteins, including GTPase-activating protein SH3 domain binding protein 1 (G3BP1) (5), T-cell-restricted intracellular antigens 1 (TIA-1), T-cell-restricted intracellular antigen-related protein (TIAR) (6), poly(A)-binding protein (PABP) (6), RACK1 (1), and histone deacetylase 6 (HDAC6) (7). Although the respective roles of these proteins in SG-associated functions have not yet been fully elucidated, G3BP1 has been shown to play a critical role in the assembly of SGs (5, 7, 8). G3BP1 is an RNA-binding protein, and it is localized at polysomes under steady-state conditions. Upon exposure to stress, G3BP1 forms a multimer, which initiates the assembly of SGs.
G3BP1 has been shown to regulate the stability and translation of several mRNAs. For instance, G3BP1 inhibits the translation of the mitochondrial H+-ATP synthase subunit beta by interacting with the 3′ untranslated region of RNA (9). In addition, G3BP1 has been reported to have an endoribonuclease activity to a subset of mRNAs, such as the c-myc gene, through direct binding (10, 11). It remains unclear, however, precisely how these activities of G3BP1 are related to the SG-associated functions.
Ubiquitin-specific protease 10 (USP10) was originally identified as a binding partner for G3BP1 (12). It is ubiquitously expressed and is also recruited into SGs (3). USP10 is a deubiquitinase, and the substrates include tumor suppressor p53 (13). Following DNA damage, a fraction of USP10 translocates into the nucleus and then deubiquitinates and stabilizes p53. Such translocation of USP10 is regulated via phosphorylation by ataxia telangiectasia mutated (ATM) protein kinase. USP10, by deubiquitinating p53, suppresses tumor cell growth. Consistently with the activation of p53, the USP10 expression is downregulated in certain carcinomas without p53 mutations.
Using knockout and/or knockdown strategies against USP10 and G3BP1, we examined what roles G3BP1 and USP10 play in the stress response. We found that SGs inhibit apoptosis by reducing reactive oxygen species (ROS) production under stress conditions and that the formation of such functional SGs requires both G3BP1 and USP10. The overexpression and knockdown experiments indicate that USP10 possesses an antioxidant activity; however, the activity under steady-state conditions is suppressed by G3BP1, which is expressed at an excess amount relative to USP10. However, upon exposure to stress, SGs suppress the inhibitory activity of G3BP1 against USP10 to uncover the antioxidant activity of USP10. In addition to DNA damage, ATM is activated by its oxidation under oxidative stress and initiates antioxidant signaling by phosphorylating downstream substrates (14). The present study suggests that ATM transmits antioxidant signals partly through USP10. Collectively, the present study indicates that SGs act as components of a crucial antioxidant machinery protecting against harmful ROS-induced outcomes to mammalian cells.
MATERIALS AND METHODS
Generation of USP10 knockout mice.
The details of USP10 knockout mice (RIKEN Center for Developmental Biology accession no. CDB0605K) that lack exon 3 of the USP10 gene will be reported elsewhere.
Cell lines and culture conditions.
293T, HeLa, U2OS, Saos2, SW13, and C33A cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 4 mM l-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin (DMEM-FBS). Jurkat cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 4 mM l-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin.
Establishment of immortalized MEFs.
Embryonic tissues were isolated, washed with PBS, digested with 0.25% trypsin, and cultured in DMEM-FBS supplemented with 55 μM 2-mercaptoethanol. Mouse embryonic fibroblasts (MEFs) were immortalized by serial passages as described previously (15), and the cells within 40 passages were used for the experiments.
Plasmid constructs.
All murine USP10 (mUSP10WT), human USP10 (hUSP10WT), and their mutant cDNAs were subcloned into pCMV-HA, a mammalian expression vector encoding a protein with an N-terminal hemagglutinin (HA) epitope tag (Clontech). Mutant mUSP10s (mUSP1077–792, mUSP1095–792, mUSP10F89A, mUSP10C418A, mUSP101–114, and mUSP101–76) and mutant hUSP10s (hUSP1078–798, hUSP1096–798, hUSP10F90A, hUSP101–116, and hUSP101–77) were constructed using PCR-based mutagenesis. G3BP1WT cDNA was subcloned into pFLAG-CMV-2 (Sigma-Aldrich) to construct an expression vector encoding G3BP1 with an N-terminal FLAG epitope tag. N-terminal deletion mutants of G3BP1, G3BP147–466, G3BP168–466, and G3BP1105–466 were constructed by the PCR method and subcloned into pFLAG-CMV-2. Lentiviral expression vectors for mUSP10 and its mutants (mUSP1077–792, mUSP1095–792, mUSP10F89A, mUSP10C418A, and mUSP101–114) were generated by subcloning the respective cDNAs into the lentiviral vector plasmid CSII-EF-RfA with a blasticidin resistance gene (16). Lentiviral short hairpin RNA (shRNA) expression plasmids targeting human USP10 (sh-USP10-1 and sh-USP10-3) with a puromycin resistance gene were purchased from Sigma-Aldrich.
Establishment of stable cell lines by lentiviral transduction.
Vesicular stomatitis virus G (VSV-G)-pseudotyped HIV-1-based viruses were produced by the cotransfection of three plasmids (lentiviral plasmid [1.3 μg], pCAG-HIVgp [0.87 μg], and pCMV-VSV-G-RSV-Rev [0.87 μg]) into 293T cells (1.0 × 106) on a 60-mm dish by using FuGENE 6 reagent according to the manufacturer's instructions (Roche). Seventy-two hours after transfection, culture supernatants were harvested and used to infect 293T cells or MEFs in the presence of 8 μg/μl Polybrene. These cells were cultured in the selection medium containing 2 μg/ml puromycin or 5 μg/ml blasticidin, respectively, for 10 days.
RNA interference.
Stealth Select RNAi small interfering RNAs (siRNAs) specific to human G3BP1 (oligonucleotide identification no. HSS115446) and human USP10 (oligonucleotide identification no. HSS113446) and a Stealth RNAi negative-control duplex were purchased from Invitrogen. MISSION siRNA specific to the 3′ untranslated region of human G3BP1 (siRNA identification no. SASI_Hs01_00045804) and MISSION siRNA universal negative control were purchased from Sigma-Aldrich. Transfection was carried out with 100 to 150 pmol siRNA using Lipofectamine 2000 or Lipofectamine RNAiMAX reagents according to the manufacturer's protocol (Invitrogen).
Reagents and antibodies.
The following reagents were purchased from the indicated companies: sodium arsenite (Wako Pure Chemical Industries), hydrogen peroxide (Sigma-Aldrich), puromycin (Calbiochem), blasticidin (Sigma-Aldrich), cycloheximide (Sigma-Aldrich), RNase (Wako Pure Chemical Industries), KU-55933 (Calbiochem), and N-acetylcysteine (Sigma-Aldrich). The following antibodies (at the indicated dilutions) were used in this study: anti-HA (1:2,000) (Cell Signaling Technology), anti-FLAG (1:1,000) (Sigma-Aldrich), anti-G3BP1 (1:2,000) (BD Transduction Laboratories), anti-PABP1 (1:1,000 and 1:500) (Santa Cruz Biotechnology and Abcam, respectively), anti-RACK1 (1:1,000) (Santa Cruz Biotechnology), anti-TIA-1/TIAR (1:1,000) (Santa Cruz Biotechnology), anti-ATM (1:1,000) (Calbiochem), anti-phospho-ATM (1:1,000) (Rockland Immunochemicals), and anti-α-tubulin (1:1,000) (Oncogene Research Products). The anti-USP10-C antibody (catalog no. A300-901A; Bethyl Laboratories) recognizes the central region of human USP10 and was used to detect human USP10 at a dilution of 1:2,000. The anti-USP10-N (catalog no. A300-900A; Bethyl Laboratories) and anti-USP10-163 (1:200 to 1:1,000; Cell Signaling Technology) antibodies recognize amino acids 50 to 100 and the amino acid regions surrounding Leu-163 of human USP10, respectively, and were used to detect murine USP10.
RT-PCR.
Total RNA was isolated using the NucleoSpin RNA II kit (Macherey-Nagel) and reverse transcribed using the PrimeScript reverse transcription (RT) reagent kit (TaKaRa) according to the manufacturers' instructions. Then, cDNAs were amplified with 30 cycles of PCR under the following conditions: 94°C for 30 s, 55°C for 30 s, and 72°C for 20 s. The primers 5′-ACCCACAGTATATCTTTGGC-3′ and 5′-CTGTAGCTAGGAGTTGGCGG-3′ were used for PCR to detect murine USP10 cDNA.
Coimmunoprecipitation assay.
The immunoprecipitation assay was performed as described previously (16). The cells (1.0 × 107) were lysed with ice-cold lysis buffer (1% Nonidet P-40, 25 mM Tris-HCl [pH 7.2], 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml aprotinin) and incubated with the indicated antibodies. The immune complexes were precipitated by protein G-Sepharose beads (GE Healthcare). Next, the beads were washed, boiled in sodium dodecyl sulfate (SDS) lysis buffer (62.5 mM Tris-HCl [pH 6.8], 2% SDS, 1 mM phenylmethylsulfonyl fluoride, and 20 μg/ml aprotinin), and subjected to a Western blot analysis. To evaluate the RNA-dependent protein-protein interactions, the cell lysates were pretreated with 100 μg/ml RNase at 16°C for 1 h prior to incubation with the primary antibodies.
Western blot analysis.
A Western blot analysis was performed as described previously (16). Briefly, the cells were lysed with SDS lysis buffer, and cell lysates (20 μg of proteins) were separated by SDS-PAGE, electrophoretically transferred onto an Immobilon polyvinylidene difluoride (PVDF) membrane (Millipore), and incubated with the indicated antibodies. Immunoreactive bands were visualized with an enhanced chemiluminescence (ECL) detection system (Amersham Pharmacia Biotech).
Immunofluorescence analysis.
The cells on glass coverslips in 6-well culture plates were washed with phosphate-buffered saline (PBS), fixed with 4% formaldehyde in PBS, and permeabilized with 0.1% Triton X-100 in PBS. After being washed twice with PBS containing 3% bovine serum albumin (BSA), the cells were incubated with the first antibodies and further incubated with either Alexa Fluor 488-conjugated anti-rabbit immunoglobulin antibody (Molecular Probes) for the anti-USP10 and anti-PABP1 antibodies, Alexa Fluor 488-conjugated anti-mouse immunoglobulin antibody (Molecular Probes) for the anti-G3BP1 antibody, Alexa Fluor 594-conjugated anti-rabbit immunoglobulin antibody (Molecular Probes) for the anti-FLAG antibody, or Alexa Fluor 594-conjugated anti-mouse immunoglobulin antibody (Molecular Probes) for the anti-G3BP1, anti-PABP1, anti-RACK1, anti-HA, and anti-FLAG antibodies. Cell nuclei were stained with Hoechst 33258. The samples were then mounted in Fluoromount/Plus (Diagnostic Biosystems), and the images were analyzed with a fluorescence microscope (model BZ-8000; Keyence). More than 300 cells in three random fields were analyzed by the staining of SG markers, PABP1, and G3BP1, and the SG percentage was calculated as the ratio of SG-positive cells to the total number of cells. The intensities of anti-PABP1 staining in 60 stress granules in 6 randomly selected cells were measured using a fluorescence analysis software package (BZ-II analyzer; Keyence) to determine the level of SG formation (PABP-F). Jurkat cells were washed with PBS, cultured on poly-l-lysine-coated glass slides in 6-well plates, and fixed, and the cells with SGs were visualized by a fluorescence microscope.
Quantitative determination of apoptosis.
The level of apoptosis was measured by three methods. First, cells were stained with propidium iodide, and sub-G1 DNA content was measured by flow cytometry. Second, apoptotic cells were detected with a terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay according to the manufacturer's instructions (ApoAlert DNA fragmentation assay kit; Clontech). Third, the cells cultured on coverslips in 6-well plates were fixed with 4% formaldehyde in PBS and permeabilized with 0.1% Triton X-100 in PBS. Fixed cells were stained with Hoechst 33258. After the TUNEL assay and Hoechst 33258 staining, the numbers of cells that exhibited TUNEL-positive and condensed nuclei were counted with a fluorescence microscope. More than 300 cells in three random fields were analyzed per sample.
Detection of ROS.
The cells were treated with 0.5 to 1.0 mM sodium arsenite for the indicated time, washed twice with PBS, and then incubated with 5 μM 5-(and -6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) (Molecular Probes) for 5 min at 37°C. The cells were washed twice with PBS and fixed, and CM-H2DCFDA fluorescence in the cells was quantified using the fluorescence analysis software package (BZ-II analyzer). More than 400 cells in four random fields were analyzed, and the data are presented as the mean fluorescence intensity (DCFDA-F). ROS production in G3BP1-expressing cells was assessed by transfecting 293T cells with plasmids encoding FLAG-G3BP1WT or its deletion mutants (G3BP147–466, G3BP168–466, and G3BP1105–466). The transfected cells were incubated with 5 μM CM-H2DCFDA for 5 min at 37°C, fixed, and incubated with anti-FLAG antibody and Hoechst 33258 stain. The fluorescence intensities of 20 cells in randomly selected fields were quantified using the fluorescence analysis software package (BZ-II analyzer).
Statistical analysis.
Data were analyzed with an unpaired Student t test, and results are presented as means plus or minus standard deviations (SD).
RESULTS
USP10 interacts with G3BP1 and PABP1.
To delineate the function of USP10, we searched for USP10-binding proteins in T cells by using a glutathione S-transferase fusion protein of USP10 and identified G3BP1 and PABP1 as the dominant interacting proteins (12, 17, 18; our unpublished observations). An immunoprecipitation analysis confirmed that endogenous USP10 formed a complex with G3BP1 and PABP1 in 293T cells (Fig. 1A). Pretreatment of the cell lysates with RNase prior to immunoprecipitation did not affect the interaction between G3BP1 and USP10; however, the interaction between G3BP1 and PABP1 was significantly reduced, indicating that the G3BP1 interaction with USP10 is RNA independent, whereas the interaction with PABP1 is RNA dependent (Fig. 1B). Plasmids encoding various murine USP10 (mUSP10) mutants were constructed to identify the domain of USP10 responsible for these interactions (Fig. 2A). mUSP10F89A has a point mutation in the PABP-interacting motif 2 (PAM2), which is a putative binding motif for PABP1 (17). mUSP10C418A has a point mutation in the cysteine protease domain, a mutation that inactivates deubiquitinating activity (12). An immunoprecipitation analysis of 293T cells showed that endogenous G3BP1 and PABP1 interacted with the amino acids 1 to 76 and 1 to 114 of mUSP10, respectively, and the PAM2 is involved in binding with PABP1 but not G3BP1 (Fig. 2B and C). The same sets of human USP10 (hUSP10) mutants showed identical binding specificities of hUSP10 to G3BP1 and PABP1 (Fig. 3).
Fig 1.
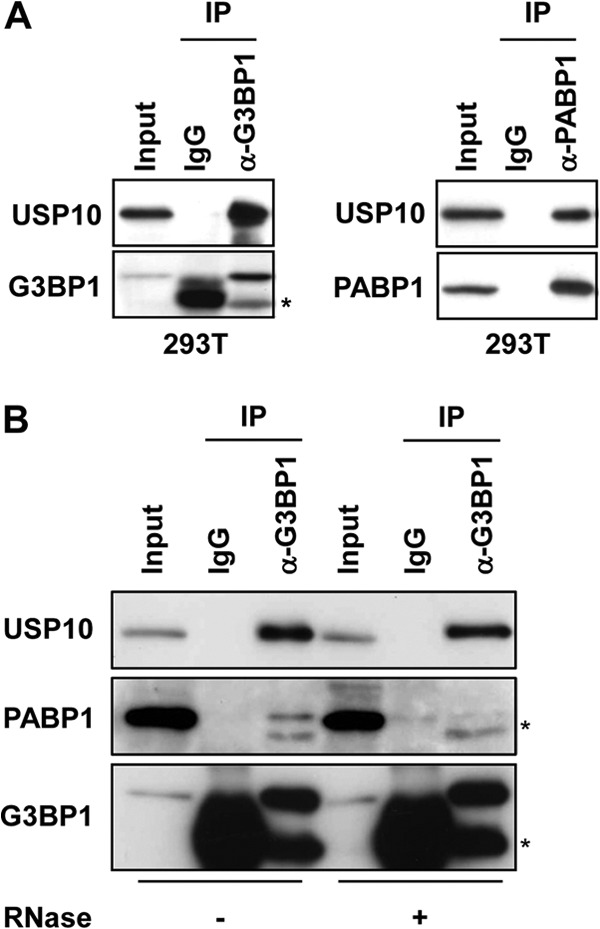
USP10 interacts with G3BP1 and PABP1. (A) Cell lysates prepared from 293T cells were immunoprecipitated with anti-G3BP1, anti-PABP1, and control antibodies. The cell lysate (input) and immunoprecipitates (IP) were characterized using a Western blot analysis with anti-USP10-C, anti-G3BP1, and anti-PABP1 antibodies. The asterisk indicates a nonspecific band. (B) Cell lysates prepared from 293T cells were treated with 100 μg/ml RNase at 16°C for 1 h and then immunoprecipitated with anti-G3BP1 antibody. The input and immunoprecipitates were characterized using a Western blot analysis with anti-USP10-C, anti-PABP1, and anti-G3BP1 antibodies. The asterisks indicate nonspecific bands.
Fig 2.
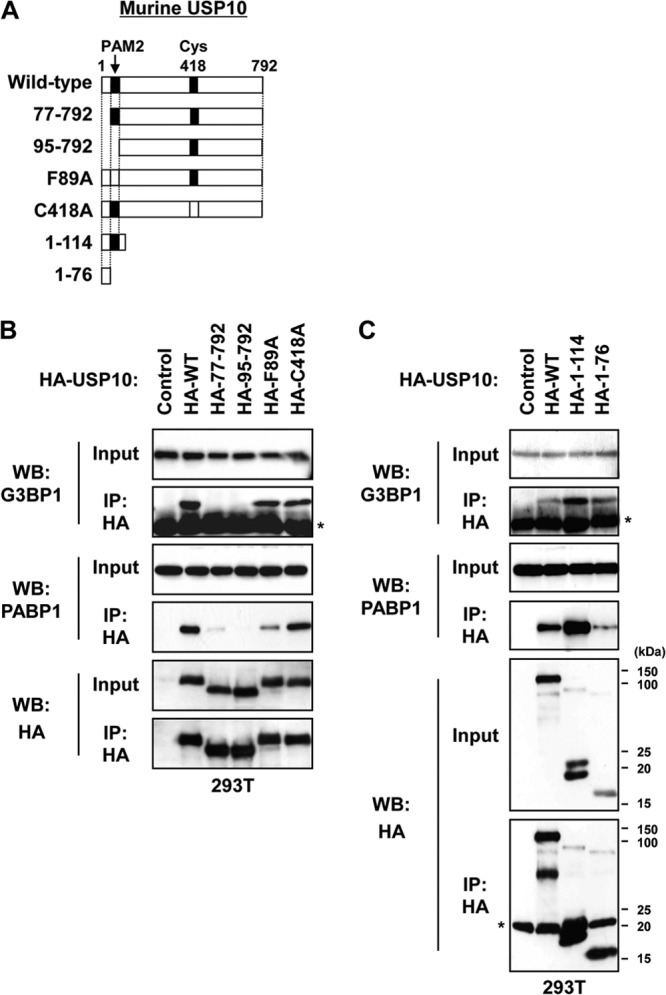
Domains of murine USP10 involved in binding with G3BP1 and PABP1. (A) Schematic representation of the murine USP10 (mUSP10) mutants used in this study. (B and C) 293T cells were transfected with plasmids encoding HA-tagged mUSP10 (HA-WT) and its mutants (HA-77-792, HA-95-792, HA-F89A, HA-C418A, HA-1-114, HA-1-76) as described for panel A. Cell lysates prepared from 293T cells were then immunoprecipitated with anti-HA antibody. The input and immunoprecipitates were characterized using a Western blot (WB) analysis with anti-G3BP1, anti-PABP1, and anti-HA antibodies. The asterisks indicate nonspecific bands.
Fig 3.
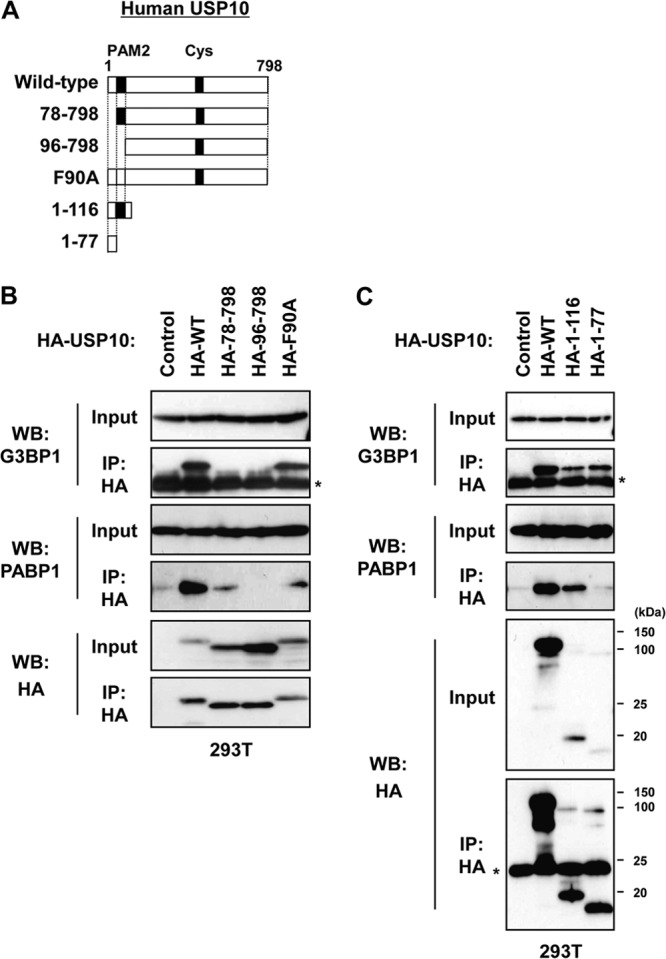
Human USP10 proteins interact with G3BP1 and PABP1. (A) Schematic representation of the human USP10 (hUSP10) mutants used in this study. (B and C) 293T cells were transfected with plasmids encoding HA-tagged hUSP10 (HA-WT) and its mutants (HA-78-798, HA-96-798, HA-F90A, HA-1-116, HA-1-77) as described for panel A. Cell lysates prepared from 293T cells were then immunoprecipitated with anti-HA antibody. The input and immunoprecipitates were characterized using a Western blot analysis with anti-G3BP1, anti-PABP1, and anti-HA antibodies. The asterisks indicate nonspecific bands.
USP10 is recruited into SGs.
G3BP1 has been shown to be essential for the assembly of SGs under various stress conditions (5, 7, 8). Therefore, we next examined whether USP10 is localized in SGs. USP10 was found to be diffusely localized in the cytoplasm of 293T cells, and the distribution was almost identical to that of G3BP1. Upon treatment with 0.5 mM arsenite, USP10 was detected predominantly in the SGs containing G3BP1 in 293T cells (Fig. 4A). The granules also recruited PABP1 and RACK1, which are other SG marker proteins (Fig. 4A). Similar recruitment of USP10 into SGs was also detected in 293T cells exposed to heat shock (Fig. 4B) and in HeLa and Jurkat cells treated with arsenite (Fig. 4C). These results show that USP10 is recruited into SGs in cells exposed to stress. This recruitment of USP10 into SGs is consistent with the findings of a previous study (3).
Fig 4.

Subcellular localization of USP10 and SG marker proteins. (A to C) 293T (A and B) and HeLa and Jurkat (C) cells were treated with 0.5 mM sodium arsenite for 60 min or heat shock at 42°C for 60 min as indicated. The cells were then fixed and stained with anti-USP10-C (green) together with anti-G3BP1 (red), anti-PABP1 (red), or anti-RACK1 (red). Nuclei were counterstained using Hoechst 33258 (blue). The bars indicate 10 μm.
USP10 inhibits arsenite-induced apoptosis.
To investigate whether USP10 is involved in SG-associated functions, USP10 knockout mice were generated, and USP10-deficient mouse embryonic fibroblasts (USP10Δ/Δ MEFs) were established (Fig. 5). The USP10Δ/Δ mice had the deletion of the USP10 exon 3 and were expected to encode the 30 N-terminal amino acids of USP10 and an additional 11 amino acids derived from the out-of-frame sequence of exon 4 (Fig. 5A). An RT-PCR analysis detected the expected size of the fragments corresponding to the USP10 mutant mRNA with a deletion of exon 3 in the USP10Δ/Δ MEFs (Fig. 5A and B). A Western blot analysis showed that two USP10 antibodies that recognize distinct N-terminal regions of USP10 (located downstream of the USP10 exon 3) detected USP10 proteins in the USP10+/+ MEFs but not in the USP10Δ/Δ MEFs (Fig. 5C). Treatment with arsenite (1 mM) induced SGs in USP10Δ/Δ MEFs, but there were fewer SGs, and they were smaller than those in USP10+/+ MEFs (Fig. 6A to C). In addition, the SGs in the USP10Δ/Δ MEFs disappeared much more rapidly than those in the USP10+/+ MEFs (Fig. 6B). USP10 deficiency in MEFs, however, had little effect on the levels of G3BP1 and PABP1 (Fig. 5C). Low SG-forming activity was recapitulated in USP10 knockdown 293T cells established by shRNA under stress conditions (arsenite, heat shock) (Fig. 6D to F). These results indicate that USP10 is not essential for the formation of SGs in MEFs, but they demonstrate that it does promote their formation.
Fig 5.
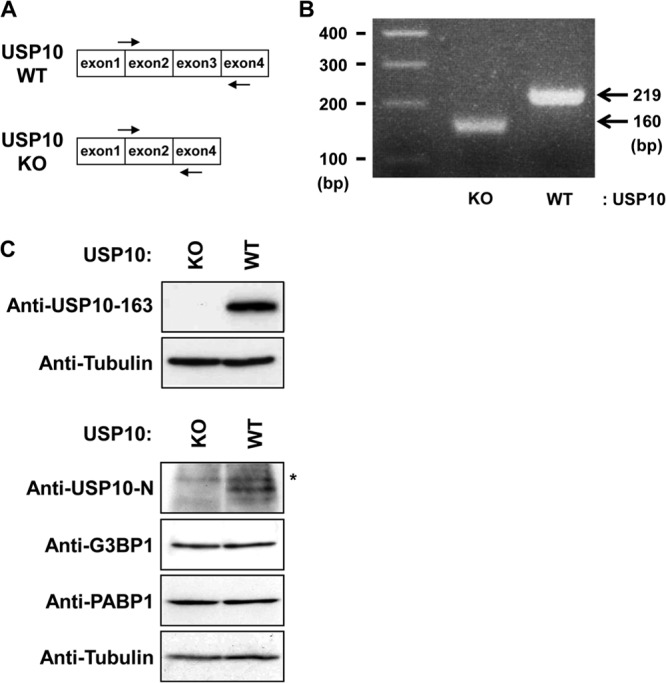
Characterization of the USP10 null allele. (A) The boxes indicate exons 1 to 4 of the murine USP10 genome. The arrows indicate the positions of the primer sets in the USP10 genome. (B) An RT-PCR analysis of murine USP10 mRNA in the USP10Δ/Δ (knockout [KO]) and USP10+/+ (wild-type [WT]) MEFs. The arrows and numbers indicate the PCR products and expected sizes, respectively. (C) Cell lysates prepared from USP10Δ/Δ (KO) and USP10+/+ (WT) MEFs were characterized using a Western blot analysis with anti-USP10, anti-G3BP1, anti-PABP1, and anti-α-tubulin antibodies. Anti-USP10-163 and anti-USP10-N antibodies (upper blots, Cell Signaling Technology; lower blots, Bethyl Laboratories) recognize the amino acid regions surrounding Leu-163 and amino acids 50 to 100 of human USP10, which are located downstream of USP10 exon 3. The asterisk indicates a nonspecific band.
Fig 6.

USP10 plays critical roles in SG formation. (A and B) USP10Δ/Δ (KO) and USP10+/+ (WT) MEFs were treated with 1 mM sodium arsenite for 60 min (A) or the indicated times (B). The cells were fixed and then stained with anti-PABP1 (green) and anti-G3BP1 (red) antibodies and Hoechst 33258 (blue). Staining was visualized with a fluorescence microscope (A). The bar indicates 20 μm. (B) The proportions (percentages) of cells containing SGs at the indicated time points. (C) Cells were treated with 1 mM sodium arsenite for 40 min, and the levels of PABP1 fluorescence in 60 SGs (PABP-F) were quantitatively determined by the fluorescence analysis software. (D) 293T cells were infected with lentiviruses encoding human USP10 shRNA (sh-USP10-1) or control nontargeting shRNA (sh-NT), and the cells were cultured in the presence of puromycin. Cell lysates were prepared from selected cultures and characterized using a Western blot analysis with anti-USP10-C, anti-G3BP1, anti-PABP1, anti-TIA-1/TIAR, and anti-α-tubulin antibodies. (E) USP10 knockdown and control cells were either treated with 0.5 mM sodium arsenite for 20 min (upper panels) or exposed to heat shock at 42°C for 60 min (lower panels). The cells were then stained with anti-USP10-C (green) and anti-G3BP1 (red) antibodies and Hoechst 33258 (blue). The bars indicate 10 μm. (F) SG percentages. In all panels, the values denote the means ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant.
SGs play an essential role in the recovery of cells from some types of stress, and the inability to form SGs induces apoptosis (1, 7). Therefore, the apoptosis sensitivity of USP10Δ/Δ MEFs was examined. Arsenite induced apoptosis in USP10Δ/Δ MEFs, and the level was much higher than that of USP10+/+ MEFs (Fig. 7). The apoptosis induced in USP10Δ/Δ MEFs was totally inhibited by N-acetylcysteine (NAC), a precursor of the ROS scavenger glutathione, indicating that USP10 inhibits ROS-dependent apoptosis in MEFs (Fig. 8A). Therefore, the kinetics of ROS production in USP10Δ/Δ MEFs treated with arsenite were monitored. The amounts of ROS in USP10Δ/Δ and USP10+/+ MEFs were equivalently reduced 30 min after arsenite exposure, but the amount of ROS in USP10Δ/Δ MEFs at 60 min was elevated more than that in USP10+/+ MEFs (Fig. 8B and C). These results indicate that arsenite reduces ROS production for 60 min in MEFs, and the ROS reduction within 30 min is independent of USP10, but the ROS reduction from 30 to 60 min and thereafter is dependent on USP10 (Fig. 8B and C). Collectively, these results indicate that USP10 plays important roles in arsenite-induced SG formation, suppression of ROS elevation, and ROS-dependent apoptosis in MEFs.
Fig 7.

Genetic ablation of USP10 enhances arsenite-induced apoptosis. (A) USP10Δ/Δ (KO) and USP10+/+ (WT) MEFs were treated with 1 mM sodium arsenite for 60 min, washed, and further cultured in fresh medium for 10 h. MEFs were stained with propidium iodide (PI), and the sub-G1 apoptotic population was analyzed by flow cytometry. (B) Proportion of the sub-G1 fraction in USP10Δ/Δ (KO) and USP10+/+ (WT) MEFs. (C to F) Apoptotic cells were also assessed by a TUNEL assay (C and D) or staining of nuclei with Hoechst 33258 (E and F) under a fluorescence microscope. In all panels, the values denote the means ± SD. ***, P < 0.001. The bars indicate 50 μm.
Fig 8.
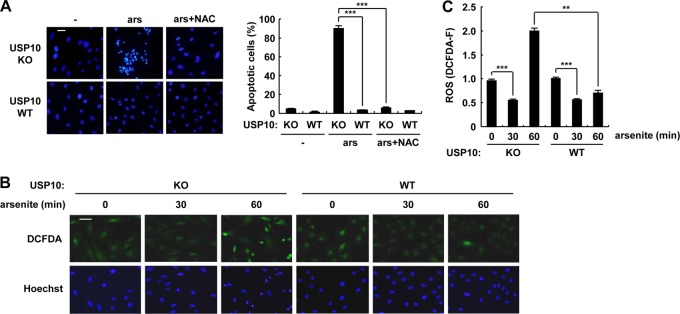
USP10 plays critical roles in ROS production and ROS-dependent apoptosis. (A) USP10Δ/Δ (KO) and USP10+/+ (WT) MEFs were pretreated with or without 5 mM N-acetylcysteine (NAC) and further treated with 1 mM sodium arsenite (ars) for 60 min. The cells were washed, cultured in fresh medium for 10 h, and then stained with Hoechst 33258. The numbers of cells containing condensed nuclei (apoptotic cells) were counted by a fluorescence microscope. The bar indicates 50 μm. (B and C) USP10Δ/Δ (KO) and USP10+/+ (WT) MEFs were treated with 1 mM sodium arsenite for 0, 30, and 60 min and stained with 5 μM CM-H2DCFDA (a redox-sensitive dye) (green) and Hoechst 33258 (blue) for 5 min at 37°C. The staining of the cells was characterized under a fluorescence microscope (B). The bar indicates 50 μm. ROS levels (DCFDA-F) were quantitatively determined by the fluorescence analysis software (C). In all panels, the values denote the means ± SD. **, P < 0.01; ***, P < 0.001.
SGs reduce ROS production.
Hydrogen peroxide (H2O2) is an oxidative stress (19) that does not induce SGs in MEFs (Fig. 9A). This study next examined the function of USP10 to a stress without inducing SGs. H2O2 also induced apoptosis in USP10Δ/Δ MEFs, but unlike the results with arsenite, the level was equivalent to that in USP10+/+ MEFs (Fig. 9B). These results suggest that USP10 inhibits arsenite-induced apoptosis as a result of the formation of SGs. USP10Δ/Δ MEFs stably expressing mUSP10s were established to test this hypothesis (Fig. 10A). The expression of mUSP10WT in USP10Δ/Δ MEFs increased SG formation and inhibited ROS production and apoptosis (Fig. 10B to D), confirming that USP10 plays a role in all three activities. mUSP10F89A and mUSP10C418A rescued all three activities, although their activities to SGs and apoptosis were slightly less than those of mUSP10WT (Fig. 10E and F). These results indicate that the PABP1 binding and deubiquitinase activity of USP10 are dispensable for these three SG-associated activities. On the other hand, mUSP1077–792, mUSP1095–792, and mUSP101–114 barely rescued any activities in USP10Δ/Δ MEFs, thus indicating that both N-terminal and C-terminal regions of mUSP10 are required for these three activities (Fig. 10E and F). Interestingly, the expression of mUSP101–114 or mUSP1095–792 in USP10Δ/Δ MEFs augmented arsenite-induced ROS production (Fig. 10F), thus indicating that these mutants might interact with the proteins negatively controlling ROS production. In this respect, mUSP101–114 might augment the arsenite-induced ROS production in USP10Δ/Δ MEFs through the interaction with G3BP1, since mUSP101–114 can interact with G3BP1 (Fig. 2C). In addition, it should be noted that arsenite-induced ROS production and apoptosis in the USP10Δ/Δ MEFs expressing USP10 mutants were correlated qualitatively but not precisely quantitatively (Fig. 10E and F). We measured the ROS production and apoptosis in these MEFs 100 min and 4 h after arsenite treatment, respectively. The difference between the two assays might explain why ROS production and apoptosis in MEFs do not quantitatively show an exact correlation.
Fig 9.
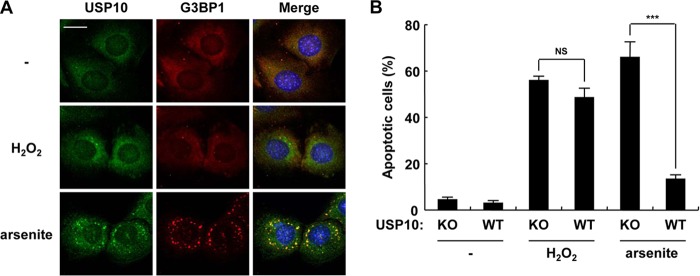
Hydrogen peroxide (H2O2) equivalently induces apoptosis in USP10Δ/Δ and USP10+/+ MEFs. (A) USP10+/+ MEFs were treated with 1 mM H2O2 for 60 min or 1 mM sodium arsenite for 60 min, fixed, and stained with anti-USP10-163 (green) and G3BP1 (red) antibodies. Nuclei were counterstained using Hoechst 33258 (blue). The staining of cells was examined by a fluorescence microscope. The bar indicates 20 μm. (B) USP10Δ/Δ (KO) and USP10+/+ (WT) MEFs were treated with 1 mM H2O2 for 180 min or 1 mM sodium arsenite for 90 min, fixed, and stained with Hoechst 33258. The numbers of cells with condensed nuclei were counted using a fluorescence microscope. The values denote the means ± SD. ***, P < 0.001; NS, not significant.
Fig 10.

Domains of USP10 required for SG-associated functions. (A) USP10Δ/Δ (KO) and USP10+/+ (WT) MEFs expressing a series of HA-mUSP10 mutants were established. The cell lysates were prepared from these transfectants and characterized using a Western blot analysis with anti-HA and anti-USP10-N antibodies. (B) Indicated MEFs were treated with 1 mM sodium arsenite for 60 min, and cells were stained with anti-PABP1 (green) and anti-HA (red) antibodies and Hoechst 33258 (blue). The bar indicates 20 μm. (C) Indicated MEFs were treated with 1 mM sodium arsenite for 100 min. The cells were washed and cultured in fresh medium for 4 h. The cells were then fixed and stained with Hoechst 33258 to detect apoptotic cells. The bar indicates 50 μm. (D) Indicated MEFs were treated with 1 mM sodium arsenite for 100 min and stained with 5 μM CM-H2DCFDA (green) and Hoechst 33258 (blue) for 5 min at 37°C. The bar indicates 50 μm. (E) USP10Δ/Δ (KO) and USP10+/+ (WT) MEFs expressing the indicated mUSP10 mutants were treated with 1 mM sodium arsenite and stained in a manner similar to that described for panel B. Upper panel, SG formation (PABP-F) was determined. MEFs were also treated with 1 mM sodium arsenite and stained in a manner similar to that described for panel C. Lower panel, the numbers of cells containing condensed nuclei were counted. (F) USP10Δ/Δ (KO) and USP10+/+ (WT) MEFs expressing the indicated mUSP10 mutants were treated with 1 mM sodium arsenite and stained in a manner similar to that described for panel D. The ROS level (DCFDA-F) was measured. In all panels, the values denote the means ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Two different mechanisms of the arsenite-induced ROS reduction were considered: ROS reduction is specific to arsenite treatment, or the reduction is a common function of SGs. To distinguish which of these two possibilities was the case, we attempted to induce SGs without arsenite treatment (5). The expression of exogenous G3BP1 without arsenite treatment successfully induced SGs in 293T cells, and it reduced the ROS level only in the cells with SGs (Fig. 11A). All three N-terminal deletion mutants of G3BP1 that lost the ability to induce SGs simultaneously failed to suppress ROS production (Fig. 11B and C). These three inactive G3BP1 mutants did not interact with USP10 in 293T cells (27). Since SGs induced by exogenous G3BP1 collected G3BP1 into SGs and reduced the amount of G3BP1 outside SGs (Fig. 11D), exogenous G3BP1-induced SGs contained endogenous G3BP1. In addition to arsenite, we observed an ROS reduction in 293T cells exposed to heat shock, and the reduction was detected simultaneously with SG formation (Fig. 12). Collectively, these results indicate that G3BP1-induced SGs can reduce the ROS level and that this effect is not specific for exposure to arsenite.
Fig 11.

G3BP1-induced SGs reduce ROS production. (A to C) 293T cells were transfected with plasmids encoding FLAG-G3BP1WT or the indicated deletion mutants (G3BP147–466, G3BP168–466, and G3BP1105–466). The transfected cells were treated with 5 μM CM-H2DCFDA (green) for 5 min at 37°C. The cells were then fixed and stained using anti-FLAG (red) and Hoechst 33258 (blue). (A) The arrows indicate SG-positive cells, and the bar indicates 10 μm. (B) SG percentages in cells stained with anti-FLAG. (C) ROS level (DCFDA-F) in FLAG-positive cells. (D) 293T cells were transfected with plasmid encoding FLAG-G3BP1WT, fixed, and stained using anti-G3BP1 (green), anti-FLAG (red), and Hoechst 33258 (blue). The bar indicates 10 μm. In all panels, the values denote the means ± SD. ***, P < 0.001.
Fig 12.
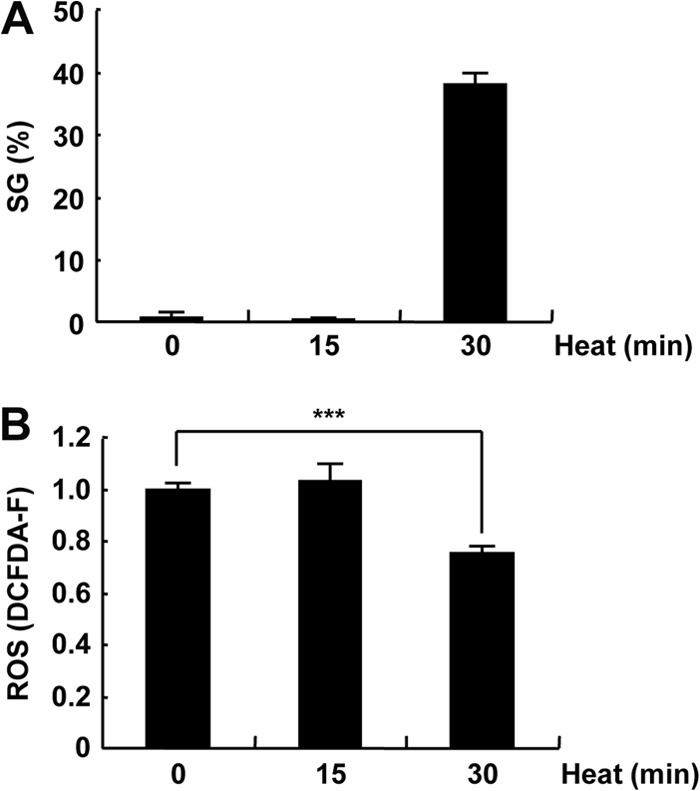
Heat shock reduces ROS production. (A and B) 293T cells were exposed to heat shock at 44°C for 0, 15, and 30 min. The SG percentages (A) and ROS levels (DCFDA-F) (B) are shown. The values denote the means ± SD. ***, P < 0.001.
Treatment of cells with a protein synthesis inhibitor, cycloheximide (CHX), has been shown to prevent SG formation (20) (Fig. 13A and B). We next examined whether CHX treatment prevents arsenite-induced ROS reduction. Pretreatment of 293T cells with CHX upregulated the ROS level under steady-state conditions (Fig. 13C), suggesting that ongoing protein synthesis is required for downregulation of the steady-state ROS level. Upon treatment with arsenite, the ROS level was reduced, even in the CHX-treated cells; however, the reduction was less than that observed in the CHX-untreated cells (Fig. 13C). These results suggest that arsenite reduces the ROS level in both SG-dependent and SG-independent manners.
Fig 13.

Cycloheximide inhibits SG formation and partially prevents arsenite-induced ROS reduction. (A to C) 293T cells were pretreated with or without 50 μg/ml cycloheximide (CHX) for 30 min, and the cells were further treated with 0.5 mM sodium arsenite for 30 min. The cells were then stained with anti-PABP1 (green) and anti-G3BP1 (red) antibodies and Hoechst 33258 (blue) (A). The bar indicates 10 μm. The SG percentages (B) and the ROS levels (DCFDA-F) (C) are shown. The values denote the means ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
G3BP1 inhibits the antioxidant role of USP10 under steady-state conditions.
We next assessed the role of endogenous G3BP1 and USP10 in ROS production (Fig. 14A). Knockdown of endogenous G3BP1 by using siRNA reduced the steady-state ROS levels in all six cell lines tested (Fig. 14B). The cellular ROS level was also reduced by another siRNA targeting the 3′ untranslated region of G3BP1 mRNA (Fig. 14C and D), and the reduction of the ROS was rescued by the expression of siRNA-resistant G3BP1 (Fig. 14E and F). It should be noted that the expression of G3BP1 in G3BP1 knockdown cells induced SGs, but the amount of SGs was lower than that observed in wild-type 293T cells (our unpublished observations). On the other hand, while knockdown of USP10 in the G3BP1 knockdown cells enhanced the ROS level, knockdown of USP10 in the G3BP1-competent cells had minimal effects on ROS production (Fig. 14G and H). Furthermore, exogenous USP10 reduced ROS production only in the G3BP1 knockdown cells and not in the G3BP1-competent cells (Fig. 14I and J). Collectively, these data demonstrate that USP10 possesses an antioxidant activity; however, the activity observed under steady-state conditions was masked by an excess amount of G3BP1 relative to USP10 in C33A cells.
Fig 14.

G3BP1 controls steady-state ROS level. (A) The indicated cell lines were transfected with human G3BP1 siRNA (si-G3BP1) or control nontargeting siRNA (si-NT) for 48 h. The cell lysates prepared from transfected cells were subjected to a Western blot analysis with anti-G3BP1 and anti-α-tubulin antibodies. (B) The cell lines treated in panel A were assessed for ROS production (DCFDA-F). (C and D) 293T and C33A cells were transfected with human G3BP1 siRNA targeting its 3′ untranslated region (si-G3BP1/3′UTR) or control nontargeting siRNA (si-NT) for 48 h. (C) Cell lysates prepared from transfected cells were subjected to a Western blot analysis with anti-G3BP1 and anti-α-tubulin antibodies. (D) The cells were assessed for ROS production (DCFDA-F). (E and F) 293T cells transfected with the si-G3BP1/3′ UTR or si-NT were further transfected with FLAG-G3BP1 plasmid for 24 h. (E) Cell lysate prepared from transfected cells was subjected to a Western blot analysis with anti-G3BP1, anti-FLAG, and anti-α-tubulin antibodies. The upper and lower arrows indicate exogenous FLAG-G3BP1 and endogenous G3BP1, respectively, and the asterisk indicates a nonspecific band. (F) The cells were assessed for ROS production (DCFDA-F). (G) C33A cells were transfected with si-NT, si-G3BP1, USP10 siRNA (si-USP10), or si-G3BP1 plus si-USP10 for 48 h, and cell lysates were subjected to a Western blot analysis with anti-G3BP1, anti-USP10-C, and anti-α-tubulin antibodies. (H) Cells treated in panel G were assessed for ROS production (DCFDA-F). (I and J) C33A cells transfected with si-G3BP1 or si-NT were further transfected with plasmids encoding HA-hUSP10 for 24 h. (I) Cell lysates prepared from transfected cells were subjected to a Western blot analysis with anti-G3BP1, anti-HA, and anti-α-tubulin antibodies. (J) The cells were assessed for ROS production (DCFDA-F). (K) C33A cells transfected with the indicated siRNAs were treated with 0.5 mM sodium arsenite for 0, 30, and 60 min, and the cells were assessed for ROS production (DCFDA-F). (L) C33A cells transfected with the indicated siRNAs were treated with 0.5 mM sodium arsenite for 60 min. After being washed, the cells were cultured in fresh medium for 4 h. The cells were then fixed and stained with Hoechst 33258 to detect apoptotic cells. In all panels, the values denote the means ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We then examined the involvement of G3BP1 in ROS-dependent apoptosis. G3BP1 knockdown in C33A cells diminished the ROS level following treatment with arsenite, and the levels were lower than those in the control cells (Fig. 14K). G3BP1 knockdown C33A cells showed less steady-state and arsenite-induced apoptosis than the control cells, and the level of apoptosis was correlated with that of ROS (Fig. 14L). These results suggest that the prooxidant activity of G3BP1 plays a critical role in both steady-state and arsenite-induced apoptosis. These results also suggest that SGs inactivate the prooxidant activity of G3BP1 and that this is part of the mechanism by which SGs reduce the ROS level.
The antioxidant activity of USP10 requires the protein kinase activity of ATM.
Ataxia telangiectasia mutated (ATM) protein kinase is oxidated at Cys-2991 by oxidative stressors, such as H2O2, and initiates an antioxidant signaling cascade (14). Intriguingly, ATM has been shown to interact with and phosphorylate USP10 (13). To examine the involvement of ATM in USP10-induced ROS reduction, we investigated whether inhibition of ATM protein kinase blocks such USP10 activity. Treatment with KU-55933, an ATM inhibitor, upregulated arsenite-induced ROS production in USP10+/+ MEFs but not in USP10Δ/Δ MEFs, and the inability to upregulate ROS in the USP10Δ/Δ MEFs was rescued by the expression of USP10WT (Fig. 15A and B). A Western blot analysis showed that KU-55933 inhibited arsenite-induced ATM phosphorylation at Ser-1981 (Fig. 15C). On the other hand, KU-55993 did not affect SG formation in the 293T cells treated with arsenite (Fig. 15D). Collectively, these results suggest that USP10 is a downstream mediator of the antioxidant signaling of ATM. It is noteworthy that KU-55933 upregulated ROS in the USP10Δ/Δ MEFs with transduced mUSP10 more significantly than that observed in the USP10+/+ MEFs (Fig. 15A and B). We assume that ATM downregulates ROS in USP10Δ/Δ mice exposed to physiological stress, such as hypoxia, and that such a long-term contribution of ATM to ROS regulation in USP10Δ/Δ mice might therefore enhance the antioxidant role of ATM in USP10Δ/Δ MEFs.
Fig 15.

Inhibition of ATM suppresses the antioxidant activity of USP10. (A and B) The indicated MEFs were pretreated with 15 μM KU-55933 (an ATM inhibitor [ATMi]), and the cells were treated with 1 mM sodium arsenite for 100 min and stained with 5 μM CM-H2DCFDA (green) and Hoechst 33258 (blue) for 5 min at 37°C. The staining was characterized under a fluorescence microscope. The bar indicates 50 μm. The ROS levels (DCFDA-F) in panel A were quantitatively determined using fluorescence analysis software (B). (C) The indicated MEFs were treated with sodium arsenite in the absence and presence of KU-55933, and the cell lysates were characterized using a Western blot analysis with anti-phospho-ATM and anti-ATM antibodies. (D) USP10+/+ MEFs were treated with sodium arsenite in the absence and presence of KU-55933, and the cells were then stained with anti-PABP1 (green) antibody and Hoechst 33258 (blue). The bar indicates 20 μm. In all panels, the values denote the means ± SD. *, P < 0.05; **, P < 0.01; NS, not significant.
DISCUSSION
Environmental stressors, such as heat, hypoxia, arsenite, and viral infections, stimulate ROS production. Continuously high ROS production causes tremendously harmful outcomes to cells, organs, and organisms, including DNA damage, massive cell death, inflammation, cancer, and aging (19). To protect against these disasters, cells quickly activate antioxidant mechanisms. We found that SGs have antioxidant activity during these stress responses, and this activity is critical for inhibiting apoptosis and for recovery from the stress. This antioxidant activity of SGs is controlled by two SG components, G3BP1 and USP10. USP10 possesses an antioxidant activity; however, the activity observed under steady-state conditions is masked by an excess amount of G3BP1 relative to USP10. However, when cells are exposed to a stress, G3BP1 and USP10 cooperatively induce SGs, and then the SGs, possibly by altering the conformation of USP10 and/or G3BP1, disrupt G3BP1 inhibition against USP10, thereby uncovering the antioxidant activity of USP10 to reduce ROS production (Fig. 16). These findings indicate that SGs are components of a quickly inducible antioxidant machinery that protects cells from detrimental ROS-induced alterations.
Fig 16.
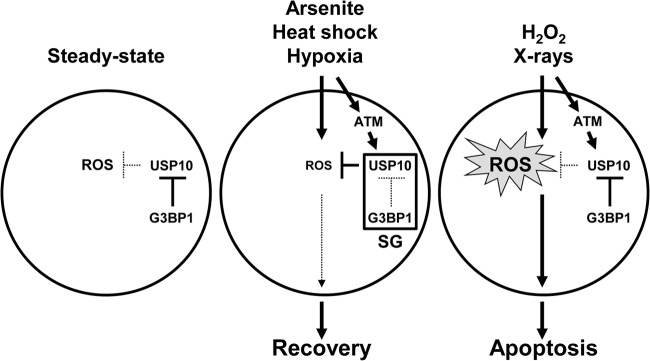
A current model for the functions of SGs, USP10, and G3BP1 in the stress response. G3BP1 masks the antioxidant function of USP10 under steady-state conditions. One type of stress, such as arsenite, heat shock, or hypoxia, induces the formation of SGs and simultaneously activates ATM. SGs inactivate the inhibitory activity of G3BP1 against USP10, and USP10 is activated by ATM or ATM-activated proteins. Next, USP10 reduces ROS production and inhibits ROS-dependent apoptosis. Other types of stress, such as H2O2 and X-ray irradiation, do not induce SG formation, and the cells are more prone to undergo apoptosis.
The present study detected two distinct antioxidant responses against arsenite: SG-dependent activities and SG-independent activities. SG-independent ROS reduction was detected in the MEFs pretreated with CHX for 30 min after arsenite treatment (early ROS reduction) (Fig. 13). This early ROS reduction was independent of USP10, since it was observed in the USP10Δ/Δ MEFs (Fig. 8C). The mechanism underlying this early ROS reduction is likely to include the activation of antioxidant enzymes by arsenite, including oxidation of these enzymes, as reported previously (21–23). On the other hand, the SG-dependent antioxidant activity was detected 30 to 100 min after arsenite treatment in MEFs, and the activity was dependent on USP10 (Fig. 8 and 10).
USP10 is a deubiquitinase for p53, and this stabilizes the protein (13). p53 also has an antioxidant activity under various stress conditions (24). However, p53 is not a mediator of USP10 to block ROS production in MEFs treated with arsenite since mUSP10C418A, defective for p53 deubiquitination (13), showed an antioxidant activity equivalent to that of mUSP10WT (Fig. 10), and USP10Δ/Δ MEFs express p53 protein at a level equivalent to that of USP10Δ/Δ MEFs expressing mUSP10WT (our unpublished observations).
ATM is activated by various types of oxidative stress stimuli, and accumulating evidence has shown that ATM plays a crucial role in the antioxidant response in a kinase-dependent manner (25, 26). ATM interacts with and phosphorylates human USP10 at Thr-42 and Ser-337; this interaction and phosphorylation are triggered by stress stimuli (13). An ATM inhibitor abrogated the antioxidant activity of USP10 without inhibiting SG formation, thus suggesting that ATM might control the antioxidant activity of USP10 (Fig. 15). It would be interesting, therefore, to clarify whether the USP10 phosphorylation induced by ATM plays a role in the antioxidant activity of USP10. It is unclear how USP10 exerts its antioxidant effect (Fig. 14). USP10 interacts with many proteins localized at polysomes, such as PABPs, HuR, RACK1, and YBX1 (18) (our unpublished observations). Therefore, USP10 might control the stability and/or translation of mRNA(s) involved in redox control; however, the activity might be suppressed by G3BP1 under steady-state conditions.
In summary, our study reveals that USP10 and G3BP1 play critical roles in ROS regulation under both steady-state and stress conditions. Based on our findings, we present a working model for the mechanisms underlying control of ROS and ROS-dependent apoptosis by USP10, G3BP1, and SGs (Fig. 16). The antioxidant function of USP10 is inactivated by G3BP1 under steady-state conditions. Environmental stressors such as arsenite, hypoxia, and heat shock induce the formation of SGs and simultaneously activate ATM. SGs inactivate the inhibitory activity of G3BP1 against USP10, and thus USP10 becomes ready for activation. USP10 is then activated by either ATM-induced phosphorylation or ATM-phosphorylated proteins to reduce ROS and ROS-dependent apoptosis. Other types of stress, such as H2O2 and X-ray irradiation, can activate ATM; however, ATM cannot activate USP10 without SGs, and the cells are more prone to undergo apoptosis.
ACKNOWLEDGMENTS
We thank H. Miyoshi (RIKEN Tsukuba Institute, Japan) for lentiviral packaging plasmids and M. Mishina (University of Tokyo, Japan) for TLCN-Cre mice. We also thank M. Tobimatsu for technical assistance.
This work was supported in part by a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by a grant for the promotion of Niigata University research projects.
Footnotes
Published ahead of print 10 December 2012
REFERENCES
- 1. Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. 2008. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 10:1324–1332 [DOI] [PubMed] [Google Scholar]
- 2. Buchan JR, Parker R. 2009. Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36:932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson P, Kedersha N. 2009. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat. Rev. Mol. Cell Biol. 10:430–436 [DOI] [PubMed] [Google Scholar]
- 4. Nover L, Scharf KD, Neumann D. 1989. Cytoplasmic heat shock granules are formed from precursor particles and are associated with a specific set of mRNAs. Mol. Cell. Biol. 9:1298–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tourriere H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160:823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6. Kedersha NL, Gupta M, Li W, Miller I, Anderson P. 1999. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 147:1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kwon S, Zhang Y, Matthias P. 2007. The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes Dev. 21:3381–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Solomon S, Xu Y, Wang B, David MD, Schubert P, Kennedy D, Schrader JW. 2007. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol. Cell. Biol. 27:2324–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ortega AD, Willers IM, Sala S, Cuezva JM. 2010. Human G3BP1 interacts with beta-F1-ATPase mRNA and inhibits its translation. J. Cell Sci. 123:2685–2696 [DOI] [PubMed] [Google Scholar]
- 10. Gallouzi IE, Parker F, Chebli K, Maurier F, Labourier E, Barlat I, Capony JP, Tocque B, Tazi J. 1998. A novel phosphorylation-dependent RNase activity of GAP-SH3 binding protein: a potential link between signal transduction and RNA stability. Mol. Cell. Biol. 18:3956–3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tourriere H, Gallouzi IE, Chebli K, Capony JP, Mouaikel J, van der Geer P, Tazi J. 2001. RasGAP-associated endoribonuclease G3Bp: selective RNA degradation and phosphorylation-dependent localization. Mol. Cell. Biol. 21:7747–7760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soncini C, Berdo I, Draetta G. 2001. Ras-GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene 20:3869–3879 [DOI] [PubMed] [Google Scholar]
- 13. Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. 2010. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 140:384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo Z, Deshpande R, Paull TT. 2010. ATM activation in the presence of oxidative stress. Cell Cycle 9:4805–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Todaro GJ, Green H. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17:299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Higuchi M, Tsubata C, Kondo R, Yoshida S, Takahashi M, Oie M, Tanaka Y, Mahieux R, Matsuoka M, Fujii M. 2007. Cooperation of NF-kappaB2/p100 activation and the PDZ domain binding motif signal in human T-cell leukemia virus type 1 (HTLV-1) Tax1 but not HTLV-2 Tax2 is crucial for interleukin-2-independent growth transformation of a T-cell line. J. Virol. 81:11900–11907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Albrecht M, Lengauer T. 2004. Survey on the PABC recognition motif PAM2. Biochem. Biophys. Res. Commun. 316:129–138 [DOI] [PubMed] [Google Scholar]
- 18. Sowa ME, Bennett EJ, Gygi SP, Harper JW. 2009. Defining the human deubiquitinating enzyme interaction landscape. Cell 138:389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thannickal VJ, Fanburg BL. 2000. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 279:L1005–L1028 [DOI] [PubMed] [Google Scholar]
- 20. Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. 2000. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 151:1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leiser SF, Miller RA. 2010. Nrf2 signaling, a mechanism for cellular stress resistance in long-lived mice. Mol. Cell. Biol. 30:871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ray PD, Huang BW, Tsuji Y. 2012. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 24:981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu R, Chen C, Mo YY, Hebbar V, Owuor ED, Tan TH, Kong AN. 2000. Activation of mitogen-activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2-dependent mechanism. J. Biol. Chem. 275:39907–39913 [DOI] [PubMed] [Google Scholar]
- 24. Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. 2005. The antioxidant function of the p53 tumor suppressor. Nat. Med. 11:1306–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, Walker CL. 2010. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. U. S. A. 107:4153–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cosentino C, Grieco D, Costanzo V. 2011. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 30:546–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsuki M, Takahashi M, Higuchi M, Makokha GN, Oie M, Fujii M. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells, in press [DOI] [PubMed] [Google Scholar]