

Abstract
Adr1 and Cat8 are nutrient-regulated transcription factors in Saccharomyces cerevisiae that coactivate genes necessary for growth in the absence of a fermentable carbon source. Transcriptional activation by Adr1 is dependent on the AMP-activated protein kinase Snf1 and is inhibited by binding of Bmh, yeast 14-3-3 proteins, to the phosphorylated Adr1 regulatory domain. We show here that Bmh inhibits transcription by binding to Adr1 at promoters that contain a preinitiation complex, demonstrating that Bmh-mediated inhibition is not due to nuclear exclusion, inhibition of DNA binding, or RNA polymerase II (Pol II) recruitment. Adr1-dependent mRNA levels under repressing growth conditions are synergistically enhanced in a mutant lacking Bmh and the two major histone deacetylases (HDACs), suggesting that Bmh and HDACs inhibit gene expression independently. The synergism requires Snf1 and Adr1 but not Cat8. Inactivating Bmh or preventing it from binding to Adr1 suppresses the normal requirement for Cat8 at codependent promoters, suggesting that Bmh modulates combinatorial control of gene expression in addition to having an inhibitory role in transcription. Activating Snf1 by deleting Reg1, a Glc7 protein phosphatase regulatory subunit, is lethal in combination with defective Bmh in strain W303, suggesting that Bmh and Snf1 have opposing roles in an essential cellular process.
INTRODUCTION
The 14-3-3 proteins are important and ubiquitous components of diverse signal transduction pathways in which they bind to a phosphorylated peptide motif in substrate proteins (reviewed in reference 1). Phosphorylation-dependent cytoplasmic retention is a common theme in 14-3-3-mediated regulation but is only one of many mechanisms used to control the activity of its binding partners. 14-3-3 proteins also regulate function by altering the enzyme activity of a protein and by promoting or preventing its degradation.
Saccharomyces cerevisiae has two redundant 14-3-3 isoforms, Bmh1 and Bmh2, which are required for viability in most common laboratory strains (2). Bmh proteins function in numerous signal transduction pathways, including glucose repression, pseudohyphal differentiation, cell cycle regulation, DNA damage response, vesicular transport, TOR-mediated growth control, and trehalose synthesis (3–5).
A recent genome-wide analysis identified 271 putative Bmh-binding partners (4, 6), including 18 transcription factors. A few of these proteins have been confirmed, or were already known Bmh targets, such as Msn2, Msn4, and Mks1 (7–9). Adr1, a transcription factor regulated by protein kinase A and AMP-activated protein kinase (Snf1) (reviewed in reference 10), was identified in an earlier systematic search for Bmh-interacting proteins (11). We recently showed that Adr1 is directly regulated by Bmh binding to its regulatory domain (12).
How 14-3-3 proteins affect the activity of transcription factors has been investigated in only a few cases. 14-3-3 proteins inhibit the activity of the FoxO family of transcriptional regulators (reviewed in references 13 and 14) and may also regulate, and may be regulated by, the p53 oncoprotein (15–17). In yeast, there are only a few confirmed Bmh interactions with transcription factors. Bmh inhibits the retrograde signaling (RS) pathway (18) by binding and cytoplasmic sequestering of Mks1 and Rtg3 (8). The stress-responsive transcription factors Msn2 and Msn4 bind Bmh (7) and were also identified in the global analysis as possible Bmh interactors. However, the importance of their interaction with Bmh is unclear (19).
In the examples just cited, transcription factor activity is inhibited in different ways, including cytosolic retention, DNA binding, and protein stability. The possibility that 14-3-3 have a role in modulating transcription at the promoter is suggested by their interactions with chromatin-associated histone acetyltransferases (HATs), histone deacetylases (HDACs), and acetylated histone H3 (20–22). Bmh1 has been shown to interact with the GAL1 promoter when the gene is induced, and this interaction is decreased when histone H3 cannot be acetylated on Lys14 or phosphorylated on Ser10 (22). The interaction may be important because deletion of BMH1 and BMH2 decreases GAL1 transcription.
We recently showed that Bmh proteins directly bind to a phosphorylated regulatory domain (RD) in the carbon source-regulated transcription factors Adr1 and Mxr1 (12, 23). Loss of Bmh activity is associated with constitutive activation of target genes that are bound by Adr1. Importantly, activating mutations in the Adr1 RD, such as a change from Ser230 to Ala, are immune to Bmh-mediated inhibition, demonstrating the importance of direct binding to Adr1. In Pichia pastoris, the analogous mutation in Mxr1 (Ser215 to Ala) blocks binding to the P. pastoris 14-3-3 protein and leads to constitutive activation of methanol-inducible, ethanol- and glucose-repressed Mxr1-dependent genes (23). Thus, both yeast 14-3-3 proteins act as inhibitors of transcription by directly binding to a regulatory domain in a transcriptional activator of the affected genes.
Bmh-mediated inhibition of Adr1 activity could occur at any one of several steps: nuclear entry, DNA binding, or a post-promoter binding step. In this work, we show that Bmh binding at the promoter can inhibit activation of gene expression after Adr1 has bound the promoter and formed a preinitiation complex (PIC), suggesting that Bmh neither excludes Adr1 from the nucleus nor inhibits DNA binding or RNA polymerase II (Pol II) recruitment. We further show that Bmh does not act exclusively through histone deacetylases. Surprisingly, loss of Bmh function and loss of Bmh binding to Adr1 suppress the requirement for a coregulatory transcription factor at promoters regulated by Adr1 and a second transcription factor, either Cat8 or Oaf1/Pip2. Thus, one function of Bmh at active Adr1-dependent promoters may be to modulate combinatorial control of gene expression.
MATERIALS AND METHODS
Yeast strains and culture conditions.
The S. cerevisiae strains used in this study were derived from W303 and are listed in Table 1. Deletions and epitope tags were introduced using standard procedures. Yeasts were grown at 30°C with shaking in yeast extract-peptone (YP) or synthetic medium containing 5% glucose for repressing (R) conditions or 0.05% glucose for derepressing (DR) conditions. YP containing 2% glucose (YPD) containing 2% glucose was used for experiments performed with the GBD-Adr1 fusion proteins. To maintain selection of plasmids with TRP1 or URA3 markers, synthetic medium lacking tryptophan or uracil and containing 0.1% Casamino Acids was used.
Table 1.
Strains used in this study
| Straina | Relevant genotype | Reference or source |
|---|---|---|
| W303-CH1a (also called W303-1a) | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 ssd1-d2 trp1-1 ura3-1 rho+ | 24 |
| CHY35a | MATa reg1Δ::natMX | This study |
| CKY13 | MATa adr1Δ::kanMX | This study |
| CKY23 | MATa adr1Δ::natMX cat8Δ::kanMX | 25 |
| CTY-TY44 | MATα hda1Δ::natMX rpd3Δ::LEU2 | 26 |
| KBY3 | MATa bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX hda1Δ::natMX rpd3Δ::LEU2 | This study |
| KBY4 | MATα bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX rpd3Δ::LEU2 | This study |
| KBY5 | MATα bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX hda1Δ::natMX | This study |
| KBY7 | MATα bmh2Δ::kanMX hda1Δ::natMX rpd3Δ::LEU2 | This study |
| KBY15 | MATα cat8Δ::hphMX4 | This study |
| KBY18 | MATa cat8Δ::hphMX4 hda1Δ::natMX rpd3Δ::LEU2 | This study |
| KBY20 | MATa cat8Δ::hphMX4 bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX | This study |
| KBY22 | MATa cat8Δ::hphMX4 bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX hda1Δ::natMX rpd3Δ::LEU2 | This study |
| KBY26 | MATα adr1Δ::natMX | This study |
| KBY30 | MATα adr1Δ::natMX bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX | This study |
| KBY31 | MATa adr1Δ::natMX hda1Δ::natMX rpd3Δ::LEU2 | This study |
| KBY34 | MATα adr1Δ::natMX bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX hda1Δ::natMX rpd3Δ::LEU2 | This study |
| KBY35 | MATa snf1Δ::kanMX | This study |
| KBY39 | MATa snf1Δ::kanMX bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX | This study |
| KBY41 | MATa snf1Δ::kanMX hda1Δ::natMX rpd3Δ::LEU2 | This study |
| KBY44 | MATα snf1Δ::kanMX bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX hda1Δ::natMX rpd3Δ::LEU2 | This study |
| KBY48 | MATα bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX | This study |
| KBY57 | MATa BMH1-9MYC::klTRP1 bmh2Δ::kanMX hda1Δ::natMX rpd3Δ::LEU2 | This study |
| KBY67 | MATa gcn5Δ::URA3 | This study |
| KBY76 | MATα gcn5Δ::URA3 hda1Δ::TRP1 rpd3Δ::LEU2 bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX | This study |
| KBY83 | MATα ura3::SNF1as::URA3 snf1Δ::kanMX bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX hda1Δ::natMX rpd3Δ::LEU2 | This study |
| KBY88 | MATa BMH1-9MYC::klTRP1 ADR1-3FLAG::hphMX bmh2Δ::kanMX hda1Δ::natMX rpd3Δ::LEU2 | This study |
| PPY6 | MATa ADR1-13MYC::natMX bmh2Δ::kanMX | This study |
| PPY13 | MATa ADR1-13MYC::natMX bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX | This study |
| YLL908 | MATa bmh2Δ::kanMX | 27 |
| YLL1087 | MATa bmh1Δ::HIS3::bmh1-170(ts)::LEU2 bmh2Δ::kanMX | 27 |
All strains are based on W303.
β-Galactosidase assays.
The activity of the lacZ reporters was determined using β-galactosidase assays as previously described (28). The results from at least three transformants were averaged, and the values in Miller units were plotted.
mRNA isolation and reverse transcription-quantitative PCR (RT-qPCR).
RNA was purified from cells using the hot-phenol method (29). Contaminating DNA was removed using DNase I treatment (Ambion). cDNA was synthesized from mRNA using Superscript III and oligo(dT) primers (Invitrogen). cDNA was diluted 1:300 and assayed by quantitative PCR using SsoFast EvaGreen Supermix (Bio-Rad) and a PTC-200 thermocycler coupled to a Chromo 4 continuous-fluorescence detector (MJ-Research). Opticon 3 software (MJ-Research) was used for the data analysis. A standard curve was generated for each primer pair to determine the efficiency, and the fold increase of cDNA relative to ACT1 was calculated using the Pfaffl method (30). Primer sequences used for qPCR are listed in Table S1 in the supplemental material.
mRNA expression profiling using microarrays.
Total RNA was purified from triplicate cultures of W303-1a (wild type [WT]), YLL1087 (bmh1-ts bmh2Δ), CTY-TY44 (hda1Δ rpd3Δ), and KBY3 (bmh1-ts bmh2Δ hda1Δ rpd3Δ) cells exponentially growing in YP broth containing 5% glucose. mRNA profiling was carried out using oligonucleotide arrays and equipment from Affymetrix (Santa Clara, CA). Labeled cRNAs were prepared from 250 ng of each total RNA, fragmented and hybridized to Yeast 2.0 arrays using a 3′ IVT Express kit, and stained and washed using a Fluidics Station 450 according to the manufacturer's instructions. The stained arrays were scanned using a GeneChip scanner to generate the CEL files containing the raw fluorescent intensities for the probes on the arrays. Gene expression values were generated and analyzed using the Bioconductor suite of programs (http://www.bioconductor.org/) for the R statistical programming environment (http://cran.r-project.org/; http://www.bioconductor.org/packages/release/bioc/html/affy.html) and EBarrays (http://www.bioconductor.org/packages/release/bioc/html/EBarrays.html). First, each probe whose sequence falls on a single nucleotide polymorphism in the W303 genome sequence compared to the S288C reference sequence was removed from the data. Then, the intensities for the remaining probes were summarized by RMA as implemented in the affy package. Next, genes in the mutant strains with expression values less than 2-fold different from the wild-type value were filtered from the data. Finally, pairwise differential analysis of gene expression was performed on the filtered data using the EBarrays package. Genes that were significantly changed in at least one of the mutants were clustered using a decision tree based on the 2-fold change combinations listed in Fig. 2C. The gplots R package was used to plot a heat map of the scaled data.
Fig 2.

Bmh proteins and HDACs repress transcription of Adr1-dependent genes in glucose. (A) Heat map of clustered bmh1-ts bmh2Δ (bmh [YLL1087]), hda1Δ rpd3Δ (hdacΔ [CTY-TY44]), and bmh1-ts bmh2Δ hda1Δ rpd3Δ (bmh hdacΔ [KBY3]) microarray data. Genes were clustered using a decision tree based on the direction of 2-fold differences in expression for each mutant compared to the BMH1 BMH2 HDA1 RPD3 wild-type strain (W303-1a). (B) Genes exhibiting the strongest bmh-hdacΔ synergism from cluster 8. The top 52 of 72 cluster 8 genes are listed in order of the magnitude of bmh-hdacΔ synergism. Expression values listed for each mutant are the log2 ratio to the wild-type strain. The synergism value was calculated as (bmh hdacΔ/BMH HDAC)/[(bmh/BMH) + (hdacΔ/HDAC)]. Values greater than 1 indicate a higher-than-additive increase in expression of the bmh hdacΔ mutant compared to the sum of the individual bmh and hdacΔ mutants. Genes highlighted in yellow are Adr1 dependent (45), and the genes in bold have promoters bound by Adr1 (38). (C) Enrichment of Adr1, Cat8, Snf1, and glucose-regulated genes in gene clusters. The P value represents the enrichment of the indicated gene sets in each cluster over the yeast transcriptome which was considered to be the genes represented on the Affymetrix Yeast 2.0 arrays. Bold values indicate significant enrichment based on the Fisher's exact test P value. The source of Adr1 targets was YEASTRACT (46). The source of Cat8 targets was described in reference 38. Snf1-dependent and glucose-repressible genes were those identified by Young et al. (45). (D) RT-qPCR. Duplicate cultures of YLL908 (WT bmh2Δ), YLL1087 (bmh1-ts bmh2Δ), CTY-TY44 (hda1Δ rpd3Δ), KBY8 (bmh2Δ hda1Δ rpd3Δ), and KBY3 (bmh1-ts bmh2Δ hda1Δ rpd3Δ) were grown in YP plus 5% glucose, and repressed samples were collected for RT-qPCR. Cells from the repressed cultures were pelleted and resuspended in YP plus 0.05% glucose and grown for 6 h, and derepressed samples were collected for RT-qPCR. The levels of ADH2 mRNA relative to ACT1 mRNA were measured for repressed and derepressed samples. The values represent the means and SDs of three biological replicates.
ChIPs.
Chromatin immunoprecipitations (ChIPs) were performed as previously described (31). Briefly, cells were crossed-linked with ethylene glycol-bis(succinimidylsuccinate) (EGS; catalog no. 21565; Thermo Scientific) and formaldehyde. Whole-cell extracts were generated by bead beating, and the chromatin was sheared by sonication. Immunoprecipitations (IP) were performed with 1 mg of protein extract and anti-Gal4 DNA-binding domain (GBD) polyclonal antibody (sc-577 X; Santa Cruz) for GBD and GBD-Adr1 fusion proteins, anti-c-Myc monoclonal antibody (9E10, sc-40 X; Santa Cruz) for Bmh1-Myc or Adr1-Myc, and anti-RNA Pol II CTD monoclonal antibody (8WG16; Abcam ChIP grade ab817) in combination with protein A magnetic beads (protein A Dynabeads [catalog no. 100.02D; Invitrogen] or Protein A Mag Sepharose Xtra [catalog no. 28-9670-62; GE Healthcare]) or anti-Flag M2 magnetic beads (catalog no. M8823; Sigma-Aldrich) for Adr1-Flag. The cross-linking was reversed and the DNA purified using the QIAquick PCR purification kit (catalog no. 28106; Qiagen). The amount of precipitated DNA was quantitated using qPCR as described above. The results are expressed as a ratio of the ChIP threshold cycle (CT) − input CT value for the region of interest relative to the telomere (TEL-VI-R), with the values corrected for the efficiency of each primer set. Sequential ChIPs were performed as described by Geisberg and Struhl (32). In the first IP, either Adr1-Flag or RNA Pol II was precipitated from 4 mg of protein extract with either anti-Flag M2 magnetic beads or anti-RNA Pol II CTD and protein A magnetic beads, respectively. The DNA purified from the first IP was combined with bovine serum albumin (BSA), lambda phage DNA, and Escherichia coli tRNA, and Bmh1-Myc was precipitated with anti-c-Myc and protein A magnetic beads. The cross-linking was reversed, and the DNA from the second IP was purified and analyzed by qPCR as described above.
Microarray data accession number.
The Affymetrix CEL files and the RMA-summarized probe intensities have been deposited with Gene Expression Omnibus under the accession number GSE40116.
RESULTS
Bmh inhibits promoter-bound Gal4-Adr1 fusion proteins.
GAL4 DNA-binding domain (GBD)-Adr1 fusion proteins are inactive in a strain with wild-type (WT) Bmh function (BMH1 bmh2Δ) but are active in a bmh mutant (bmh1-ts bmh2Δ) strain because Bmh binds to the Adr1 regulatory domain (RD; amino acids [aa] 215 to 260) and inhibits a nearby cryptic activation domain (cAD; aa 255 to 360) (12). The mechanism of inhibition of fusion protein activity by Bmh is unknown. Bmh could inhibit a step before or after promoter binding. The use of GBD-Adr1 fusions provides a simpler system in which to determine whether Bmh regulates promoter occupancy than using WT Adr1 because Adr1 is regulated at multiple steps, including promoter binding (10). By using fusion proteins with the strong ADH1 promoter driving expression of GBD-Adr1 fusion proteins, we expected overexpressed GBD to overcome Mig1-mediated inhibition of DNA binding of Gal4 fusion proteins.
To determine whether Bmh regulates the occupancy of GAL promoters by Gal4-Adr1 fusion proteins, ChIP was performed using extracts prepared from WT and bmh mutant strains carrying GBD alone or GBD-Adr1 (RD-cAD; aa 154 to 424) fusion proteins. The GBD-Adr1 fusion protein occupied two different GAL promoters in both the WT and the bmh mutant strains (Fig. 1A), as shown by the high level of promoter DNA compared to the control telomeric region in the ChIP samples. There was a 2-fold-higher level of occupancy in the bmh mutant strain, but this difference was also observed for GBD alone, so the increase is not related to binding of Bmh to the Adr1 RD. The levels of GBD and GBD-Adr1 protein were similar in both strains, with slightly more in the WT strain than in the bmh mutant strain (data not shown). In related studies, we found that GBD-Adr1 binding was unaffected by the presence of either the cAD or the RD (see Table S2A in the supplemental material).
Fig 1.

GBD-Adr1 binds to GAL genes and recruits RNA Pol II in the presence of Bmh1 activity. (A) GBD ChIP. YLL908 (BMH1 bmh2Δ) and YLL1087 (bmh1-ts bmh2Δ) were transformed with a plasmid expressing either GBD (pOBD2) or GBD-Adr1 (154–424) (pGBDA4) under the control of the ADH1 promoter. Triplicate cultures were grown in selective medium plus 2% glucose to maintain the TRP1 GBD plasmids. Samples were processed for a GBD ChIP, and GBD binding to the GAL1-10 and GAL7 promoters relative to the telomere (TEL) was determined by qPCR. (B) RT-qPCR. Triplicate samples for RT-qPCR were collected from the cultures in panel A. GAL1 and GAL7 mRNA levels relative to ACT1 mRNA were determined. (C) RNA Pol II ChIP. Duplicate cultures of the strains listed in panel A were grown in selective medium plus 2% glucose. Samples were collected for RNA Pol II ChIP, and the levels of RNA Pol II binding to the GAL1-10 and GAL7 promoters relative to the telomere were determined by qPCR. The values represent the means and SDs of three biological replicates.
We confirmed that the cAD was inhibited in the WT strain by analyzing GAL mRNA by RT-qPCR. The fusion protein was highly active in the bmh mutant strain but was inactive in the WT strain, as shown by the high GAL mRNA levels in the bmh mutant and very low mRNA levels in the WT strain (Fig. 1B). Thus, despite showing a high level of promoter occupancy in the presence or absence of Bmh, the GBD-Adr1 fusion protein was inactive in the WT strain. Therefore, Bmh can inhibit the cAD at a step subsequent to binding of GBD-Adr1 to the GAL promoters, indicating that inactivity of the GBD-Adr1 fusion protein is not due to nuclear exclusion or failure to bind DNA.
Bmh inhibits a step after recruitment of a preinitiation complex.
To investigate the step at which inhibition of gene expression occurs ChIP was performed for RNA polymerase II (RNA Pol II) at the inactive GAL promoters in the WT strain carrying GBD-Adr1 fusion proteins or GBD alone. As shown in Fig. 1C, RNA Pol II was present at both promoters, and its occupancy was dependent on the Adr1 portion of the fusion protein. The level of RNA Pol II at these promoters was not significantly increased in the bmh-ts strain (data not shown). Thus, RNA Pol II is present but apparently inactive at promoters where Bmh inhibits GBD-Adr1.
Bmh and HDACs independently inhibit transcription of ADH2 under repressing growth conditions.
To investigate the possibility that Bmh also inhibits the activity of WT Adr1 at a step subsequent to promoter binding, we utilized strains containing a poised preinitiation complex (PIC). In a strain lacking the two major histone deacetylases, Hda1 and Rpd3 (hdacΔ), Adr1 is bound to its target promoters but fails to activate transcription (26, 33). An inactive PIC containing Adr1, RNA Pol II, and some general transcription factors is present in the hdacΔ mutant, and mRNA levels are derepressed 100-fold when the cells are starved for glucose. Importantly, a Ser230-to-Ala constitutive ADR1 mutation (ADR1c) in the Adr1 RD partially activated the PIC in the hdacΔ mutant (26), suggesting that Bmh might play an important role in inhibiting PIC activation. The PIC was referred to as poised as opposed to paused or stalled because we could not detect a promoter-proximal transcript.
To determine whether Bmh activity inhibits the inactive PIC in the hdacΔ mutant, strains were constructed that contained all the different combinations of WT and mutant alleles of RPD3, HDA1, BMH1, and BMH2. Gene expression in these strains grown in high-glucose medium was analyzed by measuring mRNA levels using both 3′ in vitro transcription (IVT) expression microarrays and RT-qPCR. If Bmh inhibits the inactive PIC, then the combination of the hdacΔ and bmh1-ts bmh2Δ alleles should synergistically activate gene expression in repressed cells; i.e., the increase in expression due to the combination of the hdacΔ and bmh1-ts bmh2Δ sets of mutations should be larger than the sum of the increase in expression from each individual set of mutations: (bmh hdacΔ/BMH HDAC)/[(bmh/BMH) + (hdacΔ/HDAC)]. This was observed for many Adr1-dependent genes (Fig. 2A and B). Nine gene clusters exhibiting at least a 2-fold increase or decrease in expression in at least one of the mutant strains compared to the WT strain were identified in the microarray data. Cluster 8 contained the genes whose expression was synergistically higher in the bmh1-ts bmh2Δ hdacΔ strain than in either the bmh1-ts bmh2Δ or hdacΔ strain. This cluster of 74 genes was significantly enriched (P value = 1.6 × 10−15) in genes directly regulated by Adr1 (Fig. 2C) and had Adr1-dependent genes as 56% of the top 52 genes in this cluster when ranked by the magnitude of the synergistic increase in expression (Fig. 2B). Consistent with the enrichment for Adr1-dependent genes was a coenrichment for Cat8-dependent, Snf1-dependent, and glucose-repressible genes having Fisher's exact test P values of 7.8 × 10−4, 5.0 × 10−14, and 3.0 × 10−22, respectively (Fig. 2C). RT-qPCR measurements of selected Adr1-dependent mRNAs confirmed these observations. The level of ADH2 mRNA was 26-fold higher in the bmh1-ts bmh2Δ hdacΔ strain than in the hdacΔ strain and 6-fold higher than in the bmh1-ts bmh2Δ strain (Fig. 2D). Other ADR1-dependent genes, such as ACS1 and ADY2, showed similar synergistic increases in expression, although at a lower level (see Table S2B in the supplemental material). The synergism in mRNA levels suggests that 14-3-3 proteins and histone deacetylases inhibit Adr1-dependent gene expression through different pathways.
Not all Adr1-dependent genes were corepressed by 14-3-3 proteins and histone deacetylases. Repression by the HDAC appears to be the more important regulatory theme for the group of Cat8 target, Snf1-dependent, and glucose-repressible genes in cluster 6 (Fig. 2C), while corepression by Bmh and the HDAC appears to be more important for the group of Adr1 target genes that includes the prototypical Adr1-dependent gene ADH2 in cluster 8.
Bmh may play a role in limiting the maximal level of expression in derepressed cells. Under derepressing growth conditions, the hdacΔ, bmh1-ts bmh2Δ, and bmh1-ts bmh2Δ hdacΔ strains all had 2-fold-higher levels of ADH2 mRNA than the BMH1 bmh2Δ strains (Fig. 2D). When derepressed expression of other Adr1-dependent genes was assayed, an even more dramatic increase was observed. In particular, expression of ADY2, ATO3, FDH, and POX1 was dramatically enhanced when Bmh was nonfunctional (see Table S2E in the supplemental material). Thus, Bmh also inhibits derepressed gene expression.
To determine the roles of Rpd3 and Hda1 in repressing gene expression, we tested the activity of Adr1 under repressing conditions in bmh1-ts bmh2Δ strains lacking either RPD3 or HDA1 individually. The rpd3Δ mutant caused a greater increase in ADH2 expression than the hda1Δ mutant (see Table S2B in the supplemental material), suggesting that the major effect in the double hda1Δ rpd3Δ mutant is due to loss of RPD3. Rpd3 targets histones H2A, H2B, H3, and H4, whereas Hda1 targets only histones H2B and H3 (reviewed in reference 34), suggesting that acetylation of histone H2A and/or H4 may play a more important role in inhibiting Adr1 binding than acetylation of other histones.
We attempted to examine whether a decrease in histone acetylation would prevent activation of transcription in the absence of Bmh activity. However, we found that the combination of a deletion of the HAT, GCN5, and bmh1-ts bmh2Δ was synthetic lethal and that deletion of RPD3 and HDA1 rescued that lethality (K. Braun, unpublished data). It has previously been shown that a temperature-sensitive allele of the other major HAT, ESA1, is also synthetically lethal with bmh1-ts bmh2Δ and that the synthetic lethality was rescued by a deletion of RPD3 (20). These results suggest that Bmh may regulate transcription by helping to maintain the proper balance of histone acetylation. However, the strong synergism in gene expression when both Bmh and histone deacetylase activities are reduced suggests that Bmh-mediated inhibition can also function independently of histone acetylation.
Bmh inactivation enhances transcription without significantly increasing Adr1 promoter binding.
Glucose repression of ADH2 expression is partially relieved in bmh1-ts bmh2Δ mutant strains (12) (Fig. 2), as well as in a bmh1Δ bmh2Δ strain in the Σ1278 background (35). Whether this partial relief from glucose repression is due to a low level of promoter occupancy by Adr1 and thus a low level of transcription activation or to full promoter occupancy and incomplete transcription activation has not been determined.
To determine the level of promoter occupancy of Adr1 in the absence of Bmh-mediated inhibition, we performed ChIP for Adr1-Myc under repressing and derepressing conditions in both a bmh2Δ ADR1-MYC and a bmh1-ts bmh2Δ ADR1-MYC strain. The results indicate that partial release from glucose repression due to inactivating Bmh is associated with a low level of promoter binding (Fig. 3A). In fact, we do not consider Adr1 occupancy at the ADH2, ACS1, and POX1 promoters under repressing conditions to be significantly higher in the bmh1-ts bmh2Δ strain than in the WT strain (P values equal 0.01, 0.09, and 0.03, respectively). However, ADH2 mRNA levels were elevated about 24-fold in the bmh1-ts bmh2Δ strain, suggesting that Adr1 was present and active at the promoter (Fig. 3B). Adr1 occupancy was significantly increased at the ADH2 promoter upon derepression, and this was accompanied by 630- and 35-fold increases in ADH2 mRNA levels in the WT and bmh1-ts bmh2Δ strains, respectively. These results suggest that the small amount of Adr1 that is presumably bound at the ADH2 promoter under repressing conditions in the absence of Bmh activity may be fully active. Weak Adr1 binding and enhanced gene expression were also observed for ACS1 and POX1 (Fig. 3). Thus, Bmh inhibition may act primarily after Adr1 has occupied the promoter, as was observed in the bmh1-ts bmh2Δ hdacΔ mutant.
Fig 3.

Bmh inactivation enhances transcription without significantly increasing Adr1 promoter binding. (A) Adr1-Myc ChIP. Triplicate cultures of PPY6 (BMH1 bmh2Δ ADR1-MYC) and PPY13 (bmh1-ts bmh2Δ ADR1-MYC) were grown in YP plus 5% glucose (repressed) or YP plus 0.05% glucose (derepressed). Samples were processed for Adr1-Myc ChIP, and Adr1 binding to the ADH2, ACS1, and POX1 promoters relative to the telomere was determined by qPCR. The data are expressed as binding (ChIP/input) at ADH2, ACS1, and POX1 relative to binding (ChIP/input) at the telomere. (B) RT-qPCR. mRNA was extracted from the strains used for panel A after growth under repressed (5% glucose) and derepressed (0.05% glucose) conditions. RT-qPCR was performed to measure the levels of mRNA, and the levels of ADH2, ACS1, and POX1 mRNA were normalized to ACT1 mRNA. For panels A and B, the error bars represent the means of three biological replicates assayed in triplicate.
Adr1 is necessary and sufficient for transcription activation in the absence of Bmh activity.
The data presented in the previous section suggest that a low level of promoter-bound Adr1 suffices for significant levels of constitutive gene expression in the absence of Bmh activity. Although transcriptional activators are generally a prerequisite for transcription in vivo, there are situations where they are apparently dispensable (36). Therefore, it was important to determine whether Adr1 has a role in glucose-resistant gene expression in the bmh1-ts bmh2Δ hdacΔ strain. We did this in two ways. First, we asked whether UAS1, the Adr1 binding site, was sufficient to cause activation of transcription of two different reporters under repressing conditions in the bmh1-ts bmh2Δ hdacΔ mutant (Fig. 4A). Reporter activity in the bmh1-ts bmh2Δ hdacΔ mutant was enhanced 13- to 1,400-fold under repressing conditions compared to the WT strain (bmh2Δ). In either of the single bmh1-ts bmh2Δ and hdacΔ mutants, there was a low level of activity associated with the reporters under repressing conditions, whereas in the bmh1-ts bmh2Δ hdacΔ double mutant, their activity approached the value measured under derepressing conditions. Therefore, UAS1 is sufficient to synergistically activate reporter gene expression in the bmh1-ts bmh2Δ hdacΔ strain, and other elements of this Adr1-dependent promoter are not essential.
Fig 4.

Adr1 is required for transcriptional activation of ADH2 in the bmh hdac strain in glucose. (A) β-Galactosidase assay. YLL908 (WT bmh2Δ), YLL1087 (bmh1-ts bmh2Δ), CTY-TY44 (hda1Δ rpd3Δ), and KBY3 (bmh1-ts bmh2Δ hda1Δ rpd3Δ) were transformed with one of the ADH2-lacZ reporters (pLGADH2-lacZ or pHDY10). Three or more transformants of each strain were grown in selective medium plus 5% glucose to maintain the URA3 reporter plasmids, and repressed samples were collected for β-galactosidase assays. Cells from the repressed YLL908 culture were pelleted and resuspended in selective medium plus 0.05% glucose and grown overnight for derepression. Derepressed (DR) samples were collected for β-galactosidase assays. The level of reporter activity is reported in Miller units and represents the mean of at least three transformants. (B) RT-qPCR. Triplicate cultures of the adr1Δ strains KBY26 (adr1Δ), KBY30 (adr1Δ bmh1-ts bmh2Δ), KBY31 (adr1Δ hda1Δ rpd3Δ), and KBY34 (adr1Δ bmh1-ts bmh2Δ hda1Δ rpd3Δ) and the cat8Δ strains KBY15 (cat8Δ), KBY20 (cat8Δ bmh1-ts bmh2Δ), KBY18 (cat8Δ hda1Δ rpd3Δ), and KBY22 (cat8Δ bmh1-ts bmh2Δ hda1Δ rpd3Δ) were grown in YP plus 5% glucose. Repressed samples were collected, and ADH2 mRNA levels were assayed by RT-qPCR and plotted relative to ACT1 mRNA. The values represent the means and SDs of three biological replicates. (C) RT-qPCR. The adr1Δ strains KBY26 (adr1Δ), KBY30 (adr1Δ bmh1-ts bmh2Δ), KBY31 (adr1Δ hda1Δ rpd3Δ), and KBY34 (adr1Δ bmh1-ts bmh2Δ hda1Δ rpd3Δ) were transformed with a plasmid expressing either WT Adr1 (pKD16) or Adr1c (S230A) (pKD14) from the ADR1 promoter. Triplicate cultures were grown in selective medium plus 5% glucose to maintain the TRP1 plasmid. Repressed samples were collected for RT-qPCR, and the levels of ADH2 mRNA relative to ACT1 mRNA were determined. The values represent the means and SDs of three biological replicates.
To confirm the importance of Adr1 for activation of gene expression in the absence of Bmh and/or Hdac activity, we combined a deletion of ADR1, adr1Δ::kanMX with bmh1-ts bmh2Δ, hdacΔ, and both mutations and measured Adr1-dependent gene expression. The absence of Adr1 reduced ADH2 expression 10-, 30-, 5-, and 100-fold in the WT and the bmh1-ts bmh2Δ, hdacΔ, and bmh1-ts bmh2Δ hdacΔ mutants, respectively, under repressing conditions (Fig. 4B). Other Adr1-dependent genes were affected similarly by the absence of Adr1 (see Table S2C in the supplemental material). These results together with the UAS1-reporter analysis demonstrate that Adr1 is necessary and sufficient to activate transcription in the absence of Bmh activity in the hdacΔ strain.
Adr1 and Cat8 bind cooperatively to the ADH2 promoter to recruit coactivators, remodel chromatin, and activate transcription (31, 37, 38). Therefore, it was important to determine whether Cat8 also has a role in activating the inactive PIC in the bmh1-ts bmh2Δ hdacΔ strain. To accomplish this objective, a cat8 deletion was introduced into the WT, bmh1-ts bmh2Δ, hdacΔ, and bmh1-ts bmh2Δ hdacΔ strains and gene expression was measured. The results indicate that Cat8 does not play an important role in activating transcription under repressing conditions in any of the mutant strains (Fig. 4B; see also Table S2C in the supplemental material). Genes whose expression is strictly dependent on Cat8, such as FBP1, were not activated by the bmh1-ts bmh2Δ allele in the hdacΔ strain (see Table S2C).
Loss of Bmh activity is not equivalent to loss of Bmh binding to the regulatory domain of Adr1.
GBD-Adr1 fusion proteins with ADR1c mutations, such as S230A, are not inhibited by Bmh (12). To determine whether a nonfusion Adr1c also activates gene expression independently of Bmh, we transformed congenic adr1Δ strains with and without the bmh1-ts bmh2Δ and hdacΔ alleles with low-copy-number plasmids expressing ADR1-S230A or WT ADR1 from the ADR1 promoter. mRNA levels of Adr1-dependent genes were determined by RT-qPCR.
The results indicate that inactivating Bmh is not equivalent to the ADR1-S230A mutation (Fig. 4C; see also Table S2D in the supplemental material). Loss of Bmh activity had a more pronounced effect on Adr1-dependent gene expression than did the ADR1-S230A mutation, an effect that is particularly evident in the hdacΔ strain. For example, Adr1-S230A-activated ADH2 expression in the hdacΔ strain was about 4-fold lower than expression activated by WT Adr1 in the bmh1-ts bmh2Δ hdacΔ mutant (compare columns 3 and 4 in Fig. 4C). If loss of Bmh activity had been equivalent to activation by Adr1-S230A, these activities would have been identical. The enhanced effect of the bmh1-ts bmh2Δ mutation compared to the effect of the non-Bmh-binding Adr1-S230A allele is presumably because loss of Bmh activity both directly and indirectly affects WT Adr1 activity but only indirectly affects Adr1c activity by releasing Snf1 inhibition (12, 35). In the presence of histone deacetylase activity, however, WT Adr1 in the bmh1-ts bmh2Δ strain had activity equivalent to that of Adr1-S230A in the BMH WT strain (compare columns 1 and 2 in Fig. 4C). This may be due to some residual Bmh activity in the bmh1-ts bmh2Δ strain, an explanation that is consistent with the 3-fold-increased activity of Adr1-S230A compared to WT Adr1 in that strain (Fig. 4C, column 2). Importantly, the levels of ADH2 expression in the bmh1-ts bmh2Δ hdacΔ strain were equivalent with both activators, indicating that WT Adr1 and Adr1-S230A activators have equivalent activities when Bmh is nonfunctional.
Bmh is bound to Adr1-dependent promoters under both repressed and derepressed conditions.
If Bmh plays a role in regulating promoter-bound Adr1 activity, it might act directly at the promoter. To test this possibility, we tagged Bmh1 with a Myc epitope and did ChIP for Bmh1-Myc using extracts prepared from repressed and derepressed bmh2Δ hdacΔ cultures (KBY57). There was a weak signal at several Adr1-dependent promoters, suggesting that Bmh1 might be bound to DNA indirectly. For example, it might be bound to Adr1 but cross-linked to DNA only indirectly via protein-protein interactions. To enhance the signal of Bmh1-Myc at promoter DNA, we performed sequential ChIP. The first IP was performed with either anti-Flag or anti-RNA Pol II antibodies to recover chromatin and other proteins bound to Adr1-Flag and RNA Pol II, respectively. A second IP was performed using anti-Myc antibodies to recover Bmh1-Myc that was associated with the chromatin that was present after the first IP.
Sequential ChIP significantly enhanced the recovery of DNA containing Adr1-dependent promoters (Fig. 5). ACS1 promoter DNA was the most enriched, but ADH2, ADY2, POT1, and POX1 promoter DNAs were also evident in the Bmh1-Myc ChIP (Fig. 5 and data not shown). We found Bmh1-Myc most enriched at these promoters under derepressing conditions. A possible explanation for the apparent increased level of Bmh at the promoter under derepressing conditions is that there was more Adr1 and RNA Pol II in this sample. However, previous studies indicated that the level of Adr1 and RNA Pol II at the promoter in the hdacΔ strain did not increase significantly upon derepression (26). We confirmed this result by measuring the level of promoter DNA in the ChIP for RNA Pol II and Adr1 and found a slight increase upon derepression (data not shown), but this change was lower than the increase in Bmh binding upon derepression (Fig. 5). This result appears to rule out increased chromatin levels in the derepressed sample from the first IP as being responsible for increased promoter DNA in the second Bmh ChIP. The apparent increase in Bmh occupancy under derepressing conditions could have multiple causes. Bmh might be more efficiently recruited to the promoter or more efficiently immunoprecipitated when transcription is activated. In conclusion, Bmh is associated with promoter-bound Adr1 and RNA Pol II under both repressing and derepressing conditions.
Fig 5.
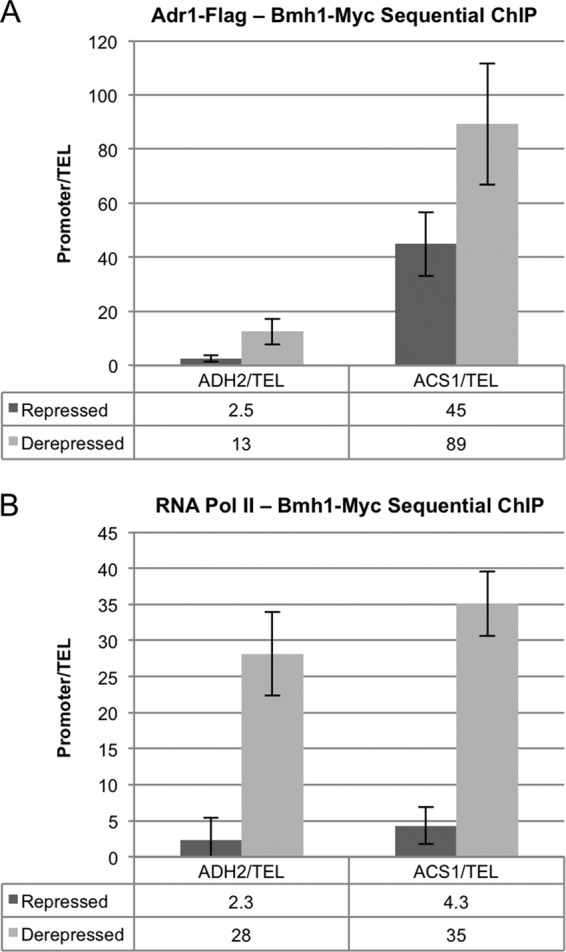
Bmh is bound to Adr1-dependent promoters in hdac strains under both repressed and derepressed conditions. Triplicate cultures of KBY88 (ADR1-FLAG BMH1-MYC bmh2Δ hda1Δ rpd3Δ) were grown in YP plus 5% glucose (repressed), and then the cells were pelleted and resuspended in YP plus 0.05% glucose and derepressed for 4.5 h. Repressed and derepressed samples were collected for sequential ChIP. The values represent the means of three biological replicates. (A) Adr1-Flag–Bmh1-Myc sequential ChIP. Adr1-Flag was immunoprecipitated from the ChIP extracts, followed by Bmh1-Myc. Bmh1 binding to the ADH2 and ACS1 promoters relative to the telomere was determined by qPCR. (B) RNA Pol II–Bmh1-Myc sequential ChIP. RNA Pol II was immunoprecipitated from the ChIP extracts, followed by Bmh1-Myc. Bmh1 binding to the ADH2 and ACS1 promoters relative to the telomere was determined by qPCR.
Constitutive activation of Snf1 is deleterious in the absence of normal Bmh activity.
Glucose-resistant, constitutive ADH2 expression is dramatically elevated in the hdacΔ strain when Bmh-mediated inhibition is relieved, either by mutating Ser230 to Ala in the Adr1 RD or by inactivating Bmh (Fig. 2 and 4). However, glucose repression is still present, as shown by increased ADH2 expression when a bmh1-ts bmh2Δ hdacΔ strain is starved of glucose (Fig. 2). In previous studies, complete relief from glucose repression was achieved by activating Snf1 in an hdacΔ strain expressing the constitutive ADR1-S230A allele (26). Activation of Snf1 was accomplished by deleting REG1, the regulatory subunit of the Glc7 protein phosphatase that dephosphorylates and inactivates Snf1. Thus, it seemed likely that the absence of fully active Snf1 might explain glucose repression in the bmh1-ts bmh2Δ hdacΔ strain. To test this possibility, we attempted to delete REG1 in the bmh1-ts bmh2Δ hdacΔ strain. However, we were unable to isolate viable transformants containing reg1Δ, suggesting that the triple mutant might be inviable. The synthetic lethality of the combination of bmh1-ts bmh2Δ and reg1Δ was confirmed by dissecting asci derived from a bmh1-ts bmh2Δ REG1/BMH1 BMH2 reg1Δ diploid strain. Tetra- and ditype tetrads had one and two spore colonies, respectively, that were inviable, and they were usually the putative bmh1-ts bmh2Δ reg1Δ spores (Fig. 6A). Similarly, we were unable to cure a bmh1-ts bmh2Δ reg1Δ strain of a plasmid-borne copy of BMH1. Thus, it appears that activating Snf1 under repressing growth conditions is lethal in the presence of low or temperature-sensitive Bmh activity.
Fig 6.

Snf1, but not Cat8, is required for derepression in the absence of Bmh activity. (A) Tetrad analysis. CHY35a (MATa reg1Δ) and KBY48 (MATα bmh1-ts bmh2Δ) were mated; diploids were selected, purified, and sporulated; and tetrads were dissected on YPD. Growth is shown after 5 days at 30°C. Twelve tetrads are shown vertically, with each set separated horizontally. The genotype for each spore is shown to the right of the tetrads and was determined based on the markers present in each tetrad. (B) RT-qPCR. Triplicate KBY83 (SNF1as bmh1-ts bmh2Δ hda1Δ rpd3Δ) cultures were grown in YP plus 5% glucose (repressed), and then the cells were pelleted and resuspended in YP plus 0.05% glucose for derepression. At the start of derepression, Snf1as activity was inhibited using 5 μM 2NM-PP1, and an equal volume of DMSO was used for a control. Samples were collected from the repressed culture (time zero) and 30, 60, 120, 240, and 360 min following derepression (DR) and Snf1as inhibition for RT-qPCR. The level of ADH2 mRNA relative to ACT1 mRNA was determined for each time point. The values represent the means of three biological replicates. (C) RT-qPCR. Triplicate cultures of WT, YLL1087 (bmh1-ts bmh2Δ), KBY15 (cat8Δ), and KBY20 (cat8Δ bmh1-ts bmh2Δ) were grown in YP plus 5% glucose, and then the cells were pelleted and resuspended in YP plus 0.05% glucose and derepressed for 4 h. Derepressed samples were collected for RT-qPCR, and the levels of ADH2, ACS1, and FBP1 mRNA relative to ACT1 mRNA were determined. Values were plotted as a percentage of the expression in the WT strain for each of the genes. The values represent the means of three biological replicates. (D) RT-qPCR. CKY13 (adr1Δ CAT8) and CKY23 (adr1Δ cat8Δ) were transformed with a plasmid expressing either WT Adr1 (pKD16) or Adr1c (S230A) (pKD14) from the ADR1 promoter. Triplicate cultures were grown in YP plus 5% glucose, and then the cells were pelleted and resuspended in YP plus 0.05% glucose and derepressed for 4 h. Derepressed samples were collected for RT-qPCR, and the levels of ADH2, ACS1, and POX1 mRNA relative to ACT1 mRNA were determined. Values were plotted as a percentage of the expression in the corresponding WT CAT8 strain for each of the genes and represent the means of three biological replicates assayed in duplicate.
Snf1 has a role in gene activation subsequent to PIC formation.
Because we were unable to activate Snf1 by deleting REG1 in the bmh1-ts bmh2Δ strain, we tested the possibility that Snf1 has a role in activating the poised PIC in the hdacΔ mutant using an analog-sensitive allele of SNF1, SNF1as, and the ATP analog 2NM-PP1. 2NM-PP1 specifically shuts off Snf1as-dependent gene expression by inhibiting its kinase activity (39). SNF1as bmh1-ts bmh2Δ hdacΔ (KBY83) cells were initially grown in glucose and then derepressed in low glucose in the presence of the Snf1as inhibitor or the inhibitor solvent (dimethyl sulfoxide [DMSO]) for the no-inhibitor control. As shown in Fig. 6B, addition of 2NM-PP1 to an SNF1as bmh1-ts bmh2Δ hdacΔ strain reduced the derepression of ADH2 expression 30-fold compared to the DMSO control. Repressed ADH2 expression in the same strain was also reduced by inhibiting Snf1as (data not shown). Expression of other Adr1-dependent genes was affected similarly (data not shown). Recent results in a BMH HDAC WT strain indicated that Snf1 has a role in gene expression subsequent to activation of gene expression (40). Therefore, Snf1 has a role in activating Adr1-dependent gene expression subsequent to PIC recruitment in the presence and the absence of Bmh activity.
Bmh mediates combinatorial control of Adr1-, Cat8-dependent genes.
The presence of promoter-bound Bmh1 under derepressing growth conditions suggests that Bmh might have a role in Adr1-dependent gene expression unrelated to glucose repression. To explore the possibility of a role for Bmh in combinatorial control of gene expression by Adr1 and Cat8, we assayed derepression of several genes coregulated by Adr1 and Cat8 in WT CAT8 and cat8Δ mutants with and without Bmh activity (WT, bmh1-ts bmh2Δ, cat8Δ, and bmh1-ts bmh2Δ cat8Δ strains). Remarkably, derepression of ADH2 and ACS1, two genes whose transcriptional dependence on both factors has been extensively characterized (38, 41), was unaffected by deleting CAT8 in the bmh1-ts bmh2Δ strain. In contrast, derepression of the same genes was reduced to 1.1% and 16% of WT levels by deleting CAT8 in a WT strain (Fig. 6C). Expression of genes activated only by Cat8, such as FBP1, was reduced to a low level in the absence of Cat8, independent of the BMH genotype (Fig. 6C). JEN1, another gene that is coregulated by Adr1 and Cat8, showed a similar response in the bmh1-ts bmh2Δ strain (see Table S2E in the supplemental material). Thus, loss of Bmh activity specifically and efficiently suppresses the requirement for Cat8 at genes normally codependent on both Adr1 and Cat8.
Loss of Bmh activity could suppress the deficiency of Cat8 by enhancing Snf1 activity, by not inhibiting Adr1, or by both mechanisms. To distinguish between these possibilities, the expression of Adr1-, Cat8-codependent genes was assayed when Adr1c was the activator and compared to their activation by WT Adr1. Because Adr1c is not bound and inhibited by Bmh (12), we expected it to mimic the phenotype of the bmh1-ts bmh2Δ mutant strain if the suppression of the Cat8 deficiency is due to the loss of Bmh-mediated inhibition of Adr1. Strains with a deletion of ADR1 with or without CAT8 were transformed with CEN-TRP1 plasmids carrying WT ADR1 or ADR1c expressed from the ADR1 promoter.
Adr1c efficiently suppressed the absence of Cat8 for ADH2 and ACS1 expression (Fig. 6D). For example, derepression of ADH2 in the cat8Δ strain with the Adr1c activator was 100% of the level in a CAT8 strain. In contrast, when WT Adr1 was the activator, ADH2 derepression in a cat8Δ strain was only 1% of the WT level. Adr1c enhanced the expression of 16 other genes showing different levels of Adr1 and Cat8 codependency when Cat8 was absent (see Table S3 in the supplemental material). Figure 6D also compares the relative levels of derepressed gene expression for ADH2, ACS1, and FBP1 in cat8Δ BMH1 BMH2 and cat8Δ bmh1-ts bmh2Δ strains with gene expression in CAT8 and cat8Δ strains carrying WT Adr1 and Adr1c activators. The expression of the codependent genes ACS1 and ADH2 was more efficiently suppressed by inactivating Bmh than by inhibiting its binding to Adr1. This was also true for genes only modestly dependent on Cat8 (ATO3, ADY2, POX1, and FDH), but the interpretation is complicated by the dramatic increase in derepression of these genes in the bmh1-ts bmh2Δ strain (see Tables S2E and S3 in the supplemental material). JEN1 expression is nearly identical in the same comparison, and three genes whose expression is CAT8 dependent but ADR1 independent, FBP1, MLS1 and ICL1, were not significantly affected by either bmh1-ts bmh2Δ or ADR1c (Fig. 6D; see also Tables S2E and S3). Thus, for genes whose expression is highly dependent on both Adr1 and Cat8 (ACS1 and ADH2), loss of Bmh suppresses Cat8 deficiency more effectively than the Adr1c activator. We interpret this result to indicate that loss of Bmh suppresses Cat8 deficiency both indirectly, by activating Snf1, and directly, by loss of binding and inhibition of Adr1. In conclusion, when Bmh is unable to bind and inhibit the activity of Adr1, genes normally coregulated by both Adr1 and Cat8 are derepressed efficiently in the absence of Cat8.
DISCUSSION
Bmh proteins have both a direct and an indirect role in regulating Adr1-dependent gene expression (Fig. 7). The direct role occurs via Bmh binding to the Adr1 regulatory domain and inhibits activation domain function (12). We show in this work that this occurs at a step in transcription after a PIC has been formed. Moreover, Bmh is likely to influence a step in PIC activation directly at the promoter, because Bmh can be detected at promoters together with both Adr1 and RNA Pol II.
Fig 7.

Model illustrating the activation of a glucose-repressed gene in the presence and the absence of Bmh. In low glucose, transcriptional activators, Adr1 (purple) and Cat8 (green), activate transcription. They bind the upstream activating sequences, UAS1 and UAS2, when the histones (gray) are hyperacetylated by the HATs (red). Subsequently, Adr1 and Cat8 each recruit a subset of coactivators and RNA Pol II (blue). Bmh (orange) binds to the regulatory domain of Adr1 at the promoter under derepressed conditions and weakly inhibits transcription. When Bmh is deleted, Adr1 is sufficient to activate transcription in the absence of Cat8. We propose that Bmh inhibits the cryptic activation domain (cAD) of Adr1, which leaves only the major activation domain (TADIII) available for recruiting coactivators and RNA Pol II. Therefore, Cat8 must provide the second activation domain to recruit the additional coactivators for optimal transcription. In the absence of Bmh, both the cAD and TADIII are available to activate transcription and are sufficient to recruit all the coactivators and RNA Pol II in the absence of Cat8.
The indirect role of Bmh proteins prevents the inappropriate activation of Snf1. When Snf1 is partially activated in the bmh1-ts bmh2Δ strain (unpublished data), there is a low level of Adr1 binding and a low level of activation of gene expression relative to the expression in derepressing growth conditions (Fig. 3). By helping to keep Snf1 in an inactive state, Bmh could prevent the Snf1-dependent histone hyperacetylation (42) that promotes nucleosome mobility and Adr1 binding (33, 43, 44). Bmh has been shown to interact with both HATs and HDACs (20), and thus, it could also have an Snf1-dependent role in maintaining promoter nucleosomes in a hypoacetylated state.
We observed strong Adr1-dependent synergistic activation of gene expression in a bmh1-ts bmh2Δ hdacΔ mutant in the presence of glucose. This suggests that histone hypoacetylation and Bmh inhibit Adr1 by different pathways. In previous work, deletion of BMH1 and BMH2 in a Σ1278 strain in combination with a REG1 deletion synergistically activated ADH2 expression under repressing growth conditions, suggesting that Bmh and Reg1 regulate Adr1 activity by different pathways (35). The two cases of synergism are likely to arise for the same two reasons. First, HDAC mutations and activation of Snf1 (by deleting REG1 or inactivating Bmh) each create a promoter that is permissive for Adr1 binding. Second, there is an Adr1-dependent function that normally occurs post-DNA binding and post-PIC recruitment that is inhibited by Bmh and requires Snf1 (Fig. 6) (40). The combination of these effects leads to strong synergism of Adr1-dependent gene expression (Fig. 2).
An important and novel finding is that Bmh inhibits a step in transcription activation that occurs after PIC recruitment. Bmh may inhibit a step in transcription initiation, elongation, nucleosome remodeling, promoter escape, or a posttranscriptional process (40). Even though the mechanism of Bmh-mediated inhibition of Adr1 is unknown, nuclear exclusion or inhibition of DNA binding appears to be ruled out.
Bmh is found in association with Adr1 under both repressing and derepressing growth conditions (12), and our current work shows that Bmh associates with Adr1 at the promoter in an hdacΔ mutant under both conditions (Fig. 5). Thus, Bmh could exert an inhibitory effect on Adr1 activity under both conditions. However, under repressing growth conditions, hypoacetylated nucleosomes prevent Adr1 from binding at the promoter. Thus, Bmh may not play an important direct role in inhibiting Adr1-dependent gene expression in the presence of abundant glucose. Consistent with this prediction, ADR1c mutations cause only a low level of Adr1 activity under repressing conditions, and this effect is restricted to a few promoters (see Table S3 in the supplemental material). From these observations, we conclude that Bmh has a minor direct role in preventing Adr1-dependent gene expression in the presence of glucose. In our model, HDACs provide the primary mode of repression of DNA binding and Bmh inhibits a post-DNA binding step in gene activation. Chromatin-mediated repression and Bmh-mediated inhibition acting independently provide tight regulation of transcriptional activation.
Most genes have multiple activators that integrate diverse signals at the promoter. Adr1-dependent target genes encode a multitude of enzymes catalyzing steps in interdependent metabolic pathways (38, 45). Most of the genes encoding these activities have numerous transcription factor binding sites in their promoter in addition to UAS1. Particularly interesting in the present context are the genes that are bound and regulated by both Adr1 and Cat8, and the genes of β-oxidation and peroxisome biogenesis that are bound and regulated by both Adr1 and Oaf1/Pip2. An Adr1c allele relieves the Oaf1/Pip2 requirement for derepression of the β-oxidation genes (25), just as it relieves the Cat8 requirement for the genes of ethanol metabolism (Fig. 6D; see also Table S3 in the supplemental material). One interesting possibility is that Bmh has a direct modulatory role at promoters, serving to integrate diverse signals that allow a greater or lesser dependence on a coregulating transcription factor. The evidence presented in Fig. 6C and D and in Tables S2E and S3 in the supplemental material strongly implicates Bmh in such a role at promoters codependent on Adr1 and Cat8.
What could be the mechanism whereby Bmh modulates the activity of Adr1 and Cat8 to influence combinatorial control of gene expression? Our observations suggest that Bmh inhibits a function that is normally performed by Cat8 (or Oaf1/Pip2) but that can be performed by Adr1 in the absence of Bmh. In the WT situation chromatin remodeling, coactivator recruitment and gene expression were codependent on both Adr1 and Cat8 for several promoters (37, 38). Although a unique function for Adr1 and Cat8 at codependent promoters was not identified, gene expression activated by Adr1c was less dependent on several coactivators than expression activated by WT Adr1. The requirement for the HAT activity of SAGA was dramatically reduced when Adr1c was the activator (25). This observation suggests that Bmh may have a role opposing the HAT activity of SAGA, for example, by recruiting an HDAC.
Our hypothesis to explain a modulatory role at codependent promoters invokes Bmh-mediated inhibition of the cryptic AD, as illustrated in the model depicted in Fig. 7. Our hypothesis assumes that the cryptic AD has the potential to perform a unique role, either quantitatively or qualitatively, that is normally performed by a second transcription factor, such as Cat8 or Oaf1/Pip2, at promoters codependent on Adr1 and another activator. If Bmh is inactive or is unable to bind Adr1, recruitment or some other function in gene expression that is normally dependent on the coregulatory transcription factor is dispensable because Adr1 can perform the role itself.
We describe a new role for 14-3-3 proteins in which their binding at a promoter regulates the activity of a DNA-bound transcription factor. These results expand the molecular mechanisms that 14-3-3 proteins use to modulate the activity of signal transduction pathways. In addition, they suggest that 14-3-3 proteins can have an important and dynamic role while complexed with a promoter-bound transcription factor. One important and apparently novel role for 14-3-3 proteins in yeast is to modulate the requirement for multiple activators at promoters showing codependent gene regulation.
Supplementary Material
ACKNOWLEDGMENTS
Chao Zhang generously provided the inhibitor, 2NM-PP1, for these studies, and Jim Broach provided the SNF1as allele (SNF1-I132G).
The research was supported by Public Health Service grant GM26079 from the National Institutes of Health.
Footnotes
Published ahead of print 3 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01226-12.
REFERENCES
- 1. Morrison DK. 2009. The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 19:16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Heusden GP, Griffiths DJ, Ford JC, Chin AWTF, Schrader PA, Carr AM, Steensma HY. 1995. The 14-3-3 proteins encoded by the BMH1 and BMH2 genes are essential in the yeast Saccharomyces cerevisiae and can be replaced by a plant homologue. Eur. J. Biochem. 229:45–53 [PubMed] [Google Scholar]
- 3. Panni S, Landgraf C, Volkmer-Engert R, Cesareni G, Castagnoli L. 2008. Role of 14-3-3 proteins in the regulation of neutral trehalase in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 8:53–63 [DOI] [PubMed] [Google Scholar]
- 4. van Heusden GP. 2009. 14-3-3 proteins: insights from genome-wide studies in yeast. Genomics 94:287–293 [DOI] [PubMed] [Google Scholar]
- 5. van Heusden GP, Steensma HY. 2006. Yeast 14-3-3 proteins. Yeast 23:159–171 [DOI] [PubMed] [Google Scholar]
- 6. Kakiuchi K, Yamauchi Y, Taoka M, Iwago M, Fujita T, Ito T, Song SY, Sakai A, Isobe T, Ichimura T. 2007. Proteomic analysis of in vivo 14-3-3 interactions in the yeast Saccharomyces cerevisiae. Biochemistry 46:7781–7792 [DOI] [PubMed] [Google Scholar]
- 7. Beck T, Hall MN. 1999. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402:689–692 [DOI] [PubMed] [Google Scholar]
- 8. Liu Z, Sekito T, Spirek M, Thornton J, Butow RA. 2003. Retrograde signaling is regulated by the dynamic interaction between Rtg2p and Mks1p. Mol. Cell 12:401–411 [DOI] [PubMed] [Google Scholar]
- 9. van Heusden GP, Steensma HY. 2001. 14-3-3 proteins are essential for regulation of RTG3-dependent transcription in Saccharomyces cerevisiae. Yeast 18:1479–1491 [DOI] [PubMed] [Google Scholar]
- 10. Hahn S, Young ET. 2011. Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 189:705–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C, Dewar D, Lin Z, Michalickova K, Willems AR, Sassi H, Nielsen PA, Rasmussen KJ, Andersen JR, Johansen LE, Hansen LH, Jespersen H, Podtelejnikov A, Nielsen E, Crawford J, Poulsen V, Sorensen BD, Matthiesen J, Hendrickson RC, Gleeson F, Pawson T, Moran MF, Durocher D, Mann M, Hogue CW, Figeys D, Tyers M. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415:180–183 [DOI] [PubMed] [Google Scholar]
- 12. Parua PK, Ratnakumar S, Braun KA, Dombek KM, Arms E, Ryan PM, Young ET. 2010. 14-3-3 (Bmh) proteins inhibit transcription activation by Adr1 through direct binding to its regulatory domain. Mol. Cell. Biol. 30:5273–5283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Calnan DR and Brunet A. 2008. The FoxO code. Oncogene 27:2276–2288 [DOI] [PubMed] [Google Scholar]
- 14. Tzivion G, Dobson M, Ramakrishnan G. 2011. FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 1813:1938–1945 [DOI] [PubMed] [Google Scholar]
- 15. Radhakrishnan VM, Putnam CW, Qi W, Martinez JD. 2011. p53 suppresses expression of the 14-3-3 gamma oncogene. BMC Cancer 11:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rajagopalan S, Jaulent AM, Wells M, Veprintsev DB, Fersht AR. 2008. 14-3-3 activation of DNA binding of p53 by enhancing its association into tetramers. Nucleic Acids Res. 36:5983–5991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang HY, Wen YY, Chen CH, Lozano G, Lee MH. 2003. 14-3-3 sigma positively regulates p53 and suppresses tumor growth. Mol. Cell. Biol. 23:7096–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Butow RA, Avadhani NG. 2004. Mitochondrial signaling: the retrograde response. Mol. Cell 14:1–15 [DOI] [PubMed] [Google Scholar]
- 19. Santhanam A, Hartley A, Duvel K, Broach JR, Garrett S. 2004. PP2A phosphatase activity is required for stress and Tor kinase regulation of yeast stress response factor Msn2p. Eukaryot. Cell 3:1261–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lottersberger F, Panza A, Lucchini G, Longhese MP. 2007. Functional and physical interactions between yeast 14-3-3 proteins, acetyltransferases, and deacetylases in response to DNA replication perturbations. Mol. Cell. Biol. 27:3266–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Macdonald N, Welburn JP, Noble ME, Nguyen A, Yaffe MB, Clynes D, Moggs JG, Orphanides G, Thomson S, Edmunds JW, Clayton AL, Endicott JA, Mahadevan LC. 2005. Molecular basis for the recognition of phosphorylated and phosphoacetylated histone h3 by 14-3-3. Mol. Cell 20:199–211 [DOI] [PubMed] [Google Scholar]
- 22. Walter W, Clynes D, Tang Y, Marmorstein R, Mellor J, Berger SL. 2008. 14-3-3 interaction with histone H3 involves a dual modification pattern of phosphoacetylation. Mol. Cell. Biol. 28:2840–2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parua PK, Ryan PM, Trang K, Young ET. 2012. Pichia pastoris 14-3-3 regulates transcriptional activity of the methanol inducible transcription factor Mxr1 by direct interaction. Mol. Microbiol. 85:282–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Infante JJ, Law GL, Wang IT, Chang HW, Young ET. 2011. Activator-independent transcription of Snf1-dependent genes in mutants lacking histone tails. Mol. Microbiol. 80:407–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ratnakumar S, Young ET. 2010. Snf1 dependence of peroxisomal gene expression is mediated by Adr1. J. Biol. Chem. 285:10703–10714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tachibana C, Biddick R, Law GL, Young ET. 2007. A poised initiation complex is activated by SNF1. J. Biol. Chem. 282:37308–37315 [DOI] [PubMed] [Google Scholar]
- 27. Lottersberger F, Rubert F, Baldo V, Lucchini G, Longhese MP. 2003. Functions of Saccharomyces cerevisiae 14-3-3 proteins in response to DNA damage and to DNA replication stress. Genetics 165:1717–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guarente L. 1983. Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 101:181–191 [DOI] [PubMed] [Google Scholar]
- 29. Collart MA, Oliviero S. 2001. Preparation of yeast RNA. Curr. Protoc. Mol. Biol. 23:13.12.1–13.12.5 doi:10.1002/0471142727.mb1312s23 [DOI] [PubMed] [Google Scholar]
- 30. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45 doi:10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Biddick RK, Law GL, Chin KK, Young ET. 2008. The transcriptional coactivators SAGA, SWI/SNF, and mediator make distinct contributions to activation of glucose-repressed genes. J. Biol. Chem. 283:33101–33109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Geisberg JV, Struhl K. 2005. Analysis of protein co-occupancy by quantitative sequential chromatin immunoprecipitation. Curr. Protoc. Mol. Biol. 70:21.8.1–21.8.7 doi:10.1002/0471142727.mb2108s70 [DOI] [PubMed] [Google Scholar]
- 33. Verdone L, Wu J, van Riper K, Kacherovsky N, Vogelauer M, Young ET, Grunstein M, Di Mauro E, Caserta M. 2002. Hyperacetylation of chromatin at the ADH2 promoter allows Adr1 to bind in repressed conditions. EMBO J. 21:1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Millar CB, Grunstein M. 2006. Genome-wide patterns of histone modifications in yeast. Nat. Rev. Mol. Cell Biol. 7:657–666 [DOI] [PubMed] [Google Scholar]
- 35. Dombek KM, Kacherovsky N, Young ET. 2004. The Reg1-interacting proteins, Bmh1, Bmh2, Ssb1, and Ssb2, have roles in maintaining glucose repression in Saccharomyces cerevisiae. J. Biol. Chem. 279:39165–39174 [DOI] [PubMed] [Google Scholar]
- 36. Adkins MW, Tyler JK. 2006. Transcriptional activators are dispensable for transcription in the absence of Spt6-mediated chromatin reassembly of promoter regions. Mol. Cell 21:405–416 [DOI] [PubMed] [Google Scholar]
- 37. Biddick RK, Law GL, Young ET. 2008. Adr1 and Cat8 mediate coactivator recruitment and chromatin remodeling at glucose-regulated genes. PLoS One 3:e1436 doi:10.1371/journal.pone.0001436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tachibana C, Yoo JY, Tagne JB, Kacherovsky N, Lee TI, Young ET. 2005. Combined global localization analysis and transcriptome data identify genes that are directly coregulated by Adr1 and Cat8. Mol. Cell. Biol. 25:2138–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shirra MK, McCartney RR, Zhang C, Shokat KM, Schmidt MC, Arndt KM. 2008. A chemical genomics study identifies Snf1 as a repressor of GCN4 translation. J. Biol. Chem. 283:35889–35898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Young ET, Zhang C, Shokat KM, Parua PK, Braun KA. 2012. The AMP-activated protein kinase Snf1 regulates transcription factor binding, RNA polymerase II activity, and mRNA stability of glucose-repressed genes in Saccharomyces cerevisiae. J. Biol. Chem. 287:29021–29034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walther K, Schuller HJ. 2001. Adr1 and Cat8 synergistically activate the glucose-regulated alcohol dehydrogenase gene ADH2 of the yeast Saccharomyces cerevisiae. Microbiology 147:2037–2044 [DOI] [PubMed] [Google Scholar]
- 42. Abate G, Bastonini E, Braun KA, Verdone L, Young ET, Caserta M. 2012. Snf1/AMPK regulates Gcn5 occupancy, H3 acetylation and chromatin remodelling at S. cerevisiae ADY2 promoter. Biochim. Biophys. Acta 1819:419–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Di Mauro E, Verdone L, Chiappini B, Caserta M. 2002. In vivo changes of nucleosome positioning in the pretranscription state. J. Biol. Chem. 277:7002–7009 [DOI] [PubMed] [Google Scholar]
- 44. Young ET, Kacherovsky N, Van Riper K. 2002. Snf1 protein kinase regulates Adr1 binding to chromatin but not transcription activation. J. Biol. Chem. 277:38095–38103 [DOI] [PubMed] [Google Scholar]
- 45. Young ET, Dombek KM, Tachibana C, Ideker T. 2003. Multiple pathways are co-regulated by the protein kinase Snf1 and the transcription factors Adr1 and Cat8. J. Biol. Chem. 278:26146–26158 [DOI] [PubMed] [Google Scholar]
- 46. Abdulrehman D, Monteiro PT, Teixeira MC, Mira NP, Lourenco AB, dos Santos SC, Cabrito TR, Francisco AP, Madeira SC, Aires RS, Oliveira AL, Sa-Correia I, Freitas AT. 2011. YEASTRACT: providing a programmatic access to curated transcriptional regulatory associations in Saccharomyces cerevisiae through a web services interface. Nucleic Acids Res. 39:D136–D140 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.