

Abstract
Interferons induce a pleiotropy of responses through binding the same cell surface receptor. Here we investigated the molecular mechanism driving interferon-induced apoptosis. Using a nonbiased small interfering RNA (siRNA) screen, we show that silencing genes whose products are directly engaged in the initiation of interferon signaling completely abrogate the interferon antiproliferative response. Apoptosis-related genes such as the caspase-8, cFLIP, and DR5 genes specifically interfere with interferon-induced apoptosis, which we found to be independent of the activity of death ligands. The one gene for which silencing resulted in the strongest proapoptotic effect upon interferon signaling is the cFLIP gene, where silencing shortened the time of initiation of apoptosis from days to hours and increased dramatically the population of apoptotic cells. Thus, cFLIP serves as a regulator for interferon-induced apoptosis. A shift over time in the balance between cFLIP and caspase-8 results in downstream caspase activation and apoptosis. While gamma interferon (IFN-γ) also causes caspase-8 upregulation, we suggest that it follows a different path to apoptosis.
INTRODUCTION
Type I interferons (IFNs) are a family of homologous cytokines involved in regulatory functions by transduction of several intracellular signaling pathways, activating a pleiotropy of phenotypic responses. All type I IFNs facilitate their activity through binding the same receptor, a heterodimer composed of transmembrane proteins IFNAR1 and IFNAR2 albeit with different affinities (1–4). Following the ternary complex assembly, the interferon signal is transduced through receptor-associated Janus family kinases (JAKs), including JAK1 and TYK2, which activate signal transducer and activator of transcription (STAT) proteins. In their phosphorylated form, STATs homo- and hetero-oligomerize, followed by binding of IRF9 (ISGF3), which translocates the ternary complex into the nucleus. There, they directly regulate the transcription of IFN-stimulated genes (ISGs) by binding to specific sequences in their promoters, known as IFN-stimulated regulatory elements (ISREs) (5–7). These genes encode a large number of proteins that are responsible for antiviral, antiproliferative, and immunoregulatory processes. It is believed that specificity is achieved by the preferential binding of different STAT dimers to specific sequence elements (7).
The antiproliferative activity of IFNs was first described in 1978 (8), but the mechanism of its activation is still under debate. Antiproliferative activity is the outcome of both growth arrest and apoptosis. Several cell arrest mechanisms were described over the years, including suppression of cyclins resulting in G0/G1 arrest (9, 10) as well as transcriptional repression of the growth-promoting factor E2F-1 (11–14).
The TRAIL gene is one of the early genes induced by IFN-β in apoptosis-sensitive cell lines (15). In melanomas, IFN-β was more potent in inducing a proapoptotic effect than IFN-α2, yet the same melanoma cell lines were resistant to recombinant TRAIL protein, with no significant role identified for apoptosis inhibitors such as cFLIP, survivin, or cIAPs. An alternative signaling pathway through phosphatidylinositol 3-kinase (PI3K) and mTOR was previously suggested to drive interferon-induced apoptosis, with ISG activation being insufficient for apoptosis induction (16–19). Although the hypotheses are not contradictory, the underlining molecular basis of the antiproliferative activity is still debatable.
The robust antiviral activity of IFNs induces a state of resistance against viral attack, which is observed as early as 4 h after continuous IFN introduction (20). As opposed to the antiviral activity, the nonreversible induction of the antiproliferative response requires prolonged continuous administration of high-dose or tight-binding IFN for as long as 36 to 72 h before the effect is expressed (21). We identified an IFN-α2 mutant that binds more tightly to IFNAR1, termed IFN-YNS, which confers 5- and 100-fold decreases in 50% effective concentration (EC50) values for antiproliferative activity relative to values for IFN-β and IFN-α2, respectively, with antiviral potency hardly being affected (22). IFN-YNS confers an antiproliferative phenotype with the activation of the same transcriptional fingerprint and apoptotic biomarkers as IFN-β (23) and was used in this study.
Extrinsic apoptosis is induced by tumor necrosis factor (TNF), Fas (TNF superfamily, member 6), or TRAIL (TNF-related apoptosis-inducing ligand) binding the cell surface death receptors (DRs) (24). Binding results in the clustering of DRs, which leads to a conformational change in the receptor's intracellular domain, exposing the death domain (DD) to FADD (Fas-associated death domain) binding. This results in a conformational change in FADD, which exposes it to caspase-8 recruitment, forming the death-inducing signaling complex (DISC) (25–27). DISC assembly mediates autocatalysis of the initiator caspases (caspase-8 and -10), resulting in the activation of executioner caspase-3, -6, and -7. cFLIP (CFLAR) is an inhibitor of extrinsic apoptosis with a short half-life but tight binding to FADD (28). The cFLIP gene encodes two isoforms, long and short (29). The cFLIP long isoform (cFLIPL) contains both nuclear localization and export signals in the caspase-like domain, promoting its translocation into and out of the nucleus (28, 30, 31). Nuclear translocation of cFLIP was previously described to modulate Wnt signaling, but the apoptotic effect was not addressed. While cFLIP inhibition of caspase-8 on the DISC is well studied, cFLIP nuclear function is still under investigation.
Here we show that caspase-8 is the initiator caspase required to trigger IFN-induced apoptosis, independently of death ligand, while cFLIP serves as its inhibitor and attenuator by controlling its activation in magnitude and in time. The delicate caspase-8/cFLIP balance on the DISC eventually gravitates toward the formation of a caspase-8-dominated complex, leading to the execution of apoptosis following IFN treatment. From our results, we propose an alternative, more general mechanism of apoptosis activation by type I interferons.
MATERIALS AND METHODS
Functional siRNA screening.
WISH cells were transfected with siGENOME apoptosis (329 genes), endocytosis (58 genes), E3 ubiquitin ligase (239 genes), and protein kinase (779 genes) libraries (Dharmacon) using 0.25 μl/well INTERFERin (Polyplus Transfection) for 24 h prior to stimulation with 1 nM IFN-YNS, in a 96-well plate format. Cell density was measured by crystal violet staining. The screen was done in three biological replicates for statistical analysis. Z factor was used as the statistical score and calculated for each well (m), plate (k), and replicate (j), according to equation 1:
| (1) |
where Zjkm is Z transformations per well, Xijkm is the assay's value of a given well, Ymedjk is the median value of the whole plate, and MADjk is the median absolute deviation per plate. The average Z value was calculated as the mean Z value of the three biological replicates. For hit validation, OnTargetPlus small interfering RNA (siRNA) oligonucleotides were used as 4 separate sequences and in combination.
Antibodies and reagents.
Whole-cell lysates (radioimmunoprecipitation assay [RIPA] buffer) were separated by SDS-PAGE under reducing conditions. Mouse anti-caspase-8, rabbit anti-cFLIP/FLIP, mouse anti-caspase-3, rabbit anti-Bid (Cell Signaling), mouse anti-FADD (BD Pharmingen), rabbit monoclonal anti-caspase-8 (Millipore), mouse anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH), and antitubulin (Sigma) antibodies were used as the primary antibodies, and horseradish peroxidase (HRP)-conjugated goat anti-rabbit and goat anti-mouse antibodies were used as the secondary antibodies. Immunoreactive protein complexes were visualized with enhanced chemiluminescence (ECL) reagents (Biological Industries, Israel). RIPK1 inhibitor nectrostatin-1 (Nec-1) and caspase-8-specific inihibitor Z-IETD were purchased from ENZO. Authophagy inhibitor 3-methyladenine (3-MA) was purchased from Sigma.
Tissue culture and cell lines.
WISH (human cervical carcinoma) cells were transfected with OnTargetPlus siRNA (Dharmacon) by using INTERFERin (Polyplus Transfection) in serum-free minimal essential medium (MEM) for the indicated times and followed treatment in complete MEM for 72 h, unless indicated otherwise. MDA-MB-321 cells were grown in complete Dulbecco MEM medium, and T47D, HS578T, and BT549 cells were grown in complete RPMI medium. Transfection was performed in serum-free medium.
Lentivirus transfection.
Stable transfection was performed on WISH cells with recombinant lentivirus (a kind gift from A. Chen) carrying the FLAG-tagged FADD dominant negative (FADD-DN) recombinant vector. Positive clones (FLAG positive) were verified by using Western blotting.
Antiproliferative and antiviral viability assays using crystal violet staining.
Live-cell staining was done as previously described (22). Briefly, cells were treated for the indicated times and then soaked in a crystal violet solution for 1 min, washed extensively in a basin with tap water, dried, and resuspended with a sodium citrate solution (50 mM sodium citrate [pH 4.2], 50% ethanol [EtOH]). Optical density was measured with an Infinite 200 plate reader (Tecan) at a 540-nm wavelength. IFN-γ was provided by M. Rubinstein (Weizmann Institute of Science, Israel).
Death ligand elimination assay.
Cells were treated with IFN-YNS (1 nM) and anti-Fas and anti-TRAIL (Axxora) recombinant fused proteins and/or anti-TNF-α polyclonal antibody (pAb) (1 μg/ml) (Peprotech) for 72 h. TNF-α (25 ng/ml), TRAIL (25 ng/ml) (Peprotech), and 1% Fas ligand (a kind gift from D. Wallach) were used as positive controls, and Z-VAD (100 μM) (Axxora) was used as an apoptosis control. Crystal violet staining was used to assess cell density.
Caspase activity and apoptosis assays.
Caspase-8, caspase-9, or caspase-3/7 activity was assayed by using the Caspase-Glo 8, 9, or 3/7 assay kit (Promega), according to the manufacturer's protocol. Luminescence was measured 30 min after applying substrate in a Modulus microplate luminometer (Turner Biosystems).
Annexin V staining.
Methods for determining the phosphatidylserine content on the outer leaflet of the cell membrane were previously described (22). In short, cells were detached with 5 mM EDTA in phosphate-buffered saline (PBS) and labeled with annexin V (Ax) and propidium iodide (PI) according to the kit manufacturer's instructions. Samples were analyzed with a FACSCalibur flow cytometer (BD Biosciences).
DNA fragmentation by TUNEL assay.
For the measurement of DNA fragmentation, a TUNEL (terminal deoxynucleotide transferase-mediated dUTP-biotin nick end labeling) detection kit, the ApoDIRECT In SituDNA Fragmentation assay kit (BioVision Research Products, Mountain View, CA), was used. Briefly, siRNA-transfected and untransfected WISH cells were treated with either 1 nM YNS or 100 μM etoposide. At 24, 48, and 72 h after treatment, cells were fixed with 1% paraformaldehyde in PBS. TUNEL staining was performed according to the manufacturer's instructions. All cells were counterstained by using a PI-RNase solution, and the cells containing fragmented DNA were analyzed by flow cytometry (LSRII).
Immunofluorescence.
WISH cells were plated onto glass coverslips and transfected with the indicated siRNAs. At 48 h after transfection, cells were treated with 1 nM YNS. After the indicated times, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 for 15 min at room temperature, and blocked with 5% bovine serum albumin (BSA) for 30 min at room temperature. Cells were incubated with the following antibodies for 1 h at room temperature: rabbit anti-cFLIP, mouse anti-caspase-8, and mouse antitubulin antibodies. After three washes with PBS, secondary anti-rabbit Cy3-conjugated IgG (1:500 dilution) and anti-mouse Alexa-conjugated IgG (1:500 dilution) were used for 1 h at room temperature. Cells were mounted onto glass slides with 4′,6-diamidino-2-phenylindole (DAPI)-containing mounting medium, and fluorescence was visualized with a Delta Vision inverted microscope. Photomicrographs were obtained by using a red, green, or blue (RGB) triple-filter cube. Analysis of fluorescence intensities was done with FIJI software. The intensity (nuclei and cytoplasm) was analyzed for ∼100 cells, and averages and standard deviations were calculated.
Proximity ligation assay.
Cells were treated as indicated above and fixed in 2.5% paraformaldehyde (PFA) on ice for 30 min. Thereafter, they were subjected to an in situ proximity ligation assay (PLA) using a Duolink detection kit (Olink Bioscience, Uppsala, Sweden) according to the manufacturer's instructions for Duolink blocking solution and the detection protocol. Briefly, slides were blocked, incubated with rabbit anti-cFLIP (Cell Signaling) and mouse anti-FADD (BD Pharmingen) antibodies or with rabbit anti-caspase-8 (N terminus, clone E6, rabbit monoclonal; Millipore) and mouse anti-FADD (BD Pharmingen) antibodies, and thereafter incubated with PLA probes, which are secondary antibodies (anti-mouse and anti-rabbit antibodies) conjugated to unique oligonucleotides. Circularization and ligation of the oligonucleotides were followed by an amplification step. The products were detected by a complementary fluorescently labeled probe. The nuclei were counterstained with DAPI. Slides were mounted, and the PLA signals were visualized with a fluorescence microscope at a ×60 magnification (Olympus). Statistical analysis of images was done for ∼10 images for each treatment (∼200 cells) by using a Matlab script, which counts the number of interactions (spots) within and outside the nucleus.
RESULTS
Molecular basis for interferon-induced antiproliferative activity.
To identify genes required for the interferon-induced antiproliferative response, we executed a functional siRNA screen of 329 apoptosis-, 58 endocytosis-, 239 E3 ubiquitin ligase-, and 779 protein kinase-related genes for their ability to alter interferon growth inhibition in WISH cells. Cells were transfected with siRNA following treatment with IFN-YNS (YNS) for 72 h. Cell density was evaluated by using crystal violet staining, with IFNAR1 and IFNAR2 serving as positive controls for knockdown (K/D) efficiency. IFNAR1 K/D results in complete rescue, while IFNAR2 K/D provides partial rescue (Fig. 1A and B). Results were evaluated for each complete screen separately. Z values lower than −3 or higher than 3 were considered significant. Most knockdowns had no significant effect on IFN-YNS-induced antiproliferative activity, testifying to the quality of the screen. K/D of genes involved in the first steps of interferon signaling, such as the TYK2, JAK1, STAT2, and IRF9 genes, confers rescue (Fig. 1B). In addition, significant Z values were determined for CUL3 and cFLIP, which decreased and increased, respectively, the antiproliferative effect. cFLIP inhibits the activation of caspase-8/10, whereas CUL3 induces the polyubiquitination and posttranslational stabilization of caspase-8 (32). To validate the role of apoptosis in interferon-induced cell death, the siRNA assay was repeated using siRNA oligonucleotides with decreased off-target effects. The results in Fig. 1B show that caspase-8, CUL3, and DR5 partially rescue antiproliferation, while cFLIP strongly and DR4 K/D partially enhance antiproliferation. Since cFLIP K/D had the strongest effect on cell death, we tested the combined effect of other gene K/Ds on the increased antiproliferative effect upon cFLIP silencing. As expected, caspase-8 K/D reversed the increased antiproliferative effect of cFLIP K/D (Fig. 1B). To validate the K/D efficiency, we confirmed the reduction in mRNA and protein levels. The procaspase-8 level was decreased by ∼70% after 24 h (Fig. 1C) and by ∼80% after 48 h of transfection. The levels of the long and short isoforms of cFLIP decreased by ∼40% and ∼75%, respectively, at 24 h posttransfection. Cells transfected with FADD siRNA showed ∼30% and ∼70% decreases in protein levels following 24 and 48 h of transfection, respectively (Fig. 1C). The screen and subsequent validation clearly placed the genes involved in interferon-induced antiproliferative activity into two functional groups. One group includes the genes participating in the activation of the canonical interferon-induced signaling cascade, and a second set of hits includes genes involved in apoptosis. Whereas K/D of genes involved in the early stages of interferon signaling resulted in complete rescue of the antiproliferative response, K/D of the caspase-8 and CUL3 genes resulted in only partial rescue.
Fig 1.

siRNA screen of apoptosis-related genes. (A) The gene count was plotted as a function of the average Z score of the YNS-treated group. cFLIP and PLK1 had a Z score of <−3, indicating enhanced antiproliferative activity, while IFNARs, CUL3, and DAP3 had a Z score of >3, indicating a rescue effect. (B) WISH cells were transfected with OnTargetPlus siRNA oligonucleotides as single K/Ds or with cFLIP oligonucleotides as double K/Ds for 48 h following treatment with 1 nM IFN-YNS for 72 h. Cell density was normalized to values for nontransfected (Non trx), nontreated cells. Data are represented as means ± standard errors of the means. (C) Western blot analysis validating downregulation upon siRNA transfection.
Validation of IFN-induced apoptosis in WISH cells.
Cell death can be a result of various mechanisms, which are distinguished by the action of the specific inhibitors Z-IETD (caspase-8), Z-VAD (general caspase), necrostatin-1 (necrosis), and 3-MA (autophagy). Rescue of interferon-induced antiproliferative activity was observed to some extent with Z-IETD treatment and more so with Z-VAD treatment (Fig. 2A and B) but not upon necrostatin-1 or 3-MA treatment. Furthermore, the combination of Z-VAD and other cell death inhibitors had no effect above that measured for Z-VAD alone. To confirm apoptosis, IFN-YNS-treated cells were stained with annexin V (Ax) and PI. Upon YNS treatment, the percentage of Ax−/PI− cells decreased from 70% to 30% and further decreased to ∼5% with cFLIP K/D. Caspase-8 K/D resulted in a complete rescue of cells from apoptosis (Fig. 2C). The results clearly show the opposing effects of caspase-8 and cFLIP on interferon-induced apoptosis. To further validate these results, IFN-YNS-treated cells were subjected to a TUNEL assay, which probes late apoptosis. Positive TUNEL staining was observed after 72 h of YNS treatment, while in cFLIP K/D cells, it was already observed after 24 h, reaching ∼80% after 72 h of treatment (Fig. 2D). Etoposide-treated cells were used as an apoptosis control. Interferon-induced apoptosis was observed in ∼60% of the cells, a percentage that was increased to almost 100% upon cFLIP K/D.
Fig 2.

Cell death inhibitors and apoptosis markers. (A) WISH cells were treated with Z-VAD at 100 μM, Z-IETD at 50 μM, Nec-1 at 63 μM, 3-MA at 1 mM, or their combination with or without (not treated [NT]) 1 nM IFN-YNS for 72 h. Cell density was normalized to values for nontreated cells. Data are represented as means ± standard errors of the means. (B) WISH cells were transfected with OnTargetPlus siRNA following treatment with 1 nM IFN-YNS and/or 100 μM Z-VAD. Cell density was normalized to values for nontransfected, nontreated cells. (C) WISH cells were transfected with siRNA targeting IFNAR1, cFLIP, or caspase-8 or with nontargeting siRNA (control) prior to treatment with 1 nM IFN-YNS for 48 h. At the end of the process, the cells were stained with annexin V (Ax) and PI to determine apoptosis. (D) Similar to panel C, but apoptosis was analyzed by TUNEL staining.
Cell cycle arrest is not affected by apoptosis inhibitors.
The antiproliferative activity of interferon is a result of apoptosis and cell cycle arrest. The total live/dead cell count showed a decrease in the number of live cells upon treatment with IFN-YNS. Z-VAD increased the number of live cells but not to the same level as the nontreated control (Fig. 2B and 3A). Blocking of interferon-induced apoptosis by Z-VAD or caspase-8 K/D did not alter the induced S-phase arrest (Fig. 3B). Our data indicate that cell cycle arrest and initiation of apoptosis are two separate mechanisms induced by interferon. Only the K/D of genes located early in the interferon-induced signaling cascade, such as the IFNAR1, STAT2, JAK1, TYK2, and IRF9 genes, completely rescued the antiproliferative response of interferon (Fig. 1B).
Fig 3.

Cell cycle arrest in antiproliferative activity of IFN. (A) WISH cells were transfected with cFLIP or caspase-8 siRNA followed by treatment with 1 nM YNS with or without Z-VAD for the indicated hours. Trypan blue staining was used to count live cells. (B) Cell cycle analysis was performed on WISH cells treated with 100 μM Z-VAD and/or 1 nM YNS for 48 h or left untreated (not treated [NT]).
cFLIP knockdown accelerates IFN-induced apoptosis.
The interferon-induced antiproliferative response typically requires 2 to 3 days to develop (22), while 4 h of IFN treatment is sufficient to induce an antiviral state (20). cFLIP K/D significantly accelerates the initiation of the antiproliferative response (Fig. 4A), in line with the accelerated accumulation of active (cleaved) caspase-3 (Fig. 5C). However, even in the cFLIP K/D background, cell density started to decline only after 8 h of interferon treatment. To determine when interferon triggers cell death, the experimental setup was altered. IFN was added for the indicated times for pulse treatment. After removal of IFN from the medium, cells were grown to a completion of 72 h from the addition of IFN (Fig. 4B). In this setup, cFLIP K/D already resulted in a 50% reduction in cell density after 8 h of treatment, while without the K/D, the cell density was not altered. Twenty-four hours of IFN treatment was sufficient to eliminate >90% of cFLIP K/D cells, while in control cells, the density was decreased by only ∼25%. Seventy-two hours of continuous IFN treatment without cFLIP K/D eliminated ∼60% of the cells (Fig. 4A and B).
Fig 4.
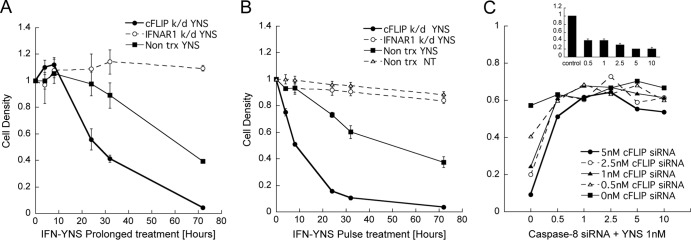
cFLIP knockdown enhances sensitivity to interferon. (A and B) Continuous IFN-YNS treatment (A) and pulse treatment (B) were performed on WISH cells transfected with siRNA targeting cFLIP or IFNAR1 following treatment with 1 nM YNS for the indicated hours. (A) The cells were treated with YNS for the indicated times. (B) All the cells were treated at time zero, and after the indicated times, the medium was replaced with fresh medium without YNS but containing 200 nM extracellular domain of IFNAR2, to exclude residual IFN, for the remaining 72 h. Data are represented as means ± standard errors of the means. (C) WISH cells were transfected with up to 10 nM siRNA targeting cFLIP and/or caspase-8 for 48 h prior to treatment with 1 nM YNS for 72 h. Caspase-8 silencing was measured by Western blotting using 0, 0.5, 1, 2.5, 5, and 10 nM siRNA (inset). The protein level was calculated compared to the level in control cells. Data are represented as means ± standard errors of the means.
Fig 5.

Caspase-3/7, -8, and -9 activity. (A) WISH cells were transfected with siRNA targeting cFLIP or caspase-8 following 24 h of treatment with 1 nM YNS. Cotreatment with the pancaspase inhibitor Z-VAD at 100 μM was used to inhibit the caspase activity. Data are represented as means ± standard errors of the means. (B) Caspase kinetics was measured in WISH cells transfected with siRNA targeting cFLIP or mock transfected following treatment with 1 nM YNS for the indicated hours. Data are represented as means ± standard errors of the means. (C) Protein cleavage was tested in nontransfected WISH cells and cells transfected with siRNA targeting cFLIP before treatment with 1 nM YNS. Fas was used as a positive control for caspase-3 and -8 cleavage. Cotreatment with 100 μM Z-VAD was used to inhibit the caspase activity. (D) WISH cells were transfected with siRNA targeting the indicated genes following treatment with 1 nM YNS for 48 and 72 h. Cells were stained with annexin V and PI to determine apoptosis. Data are represented as means. Multiple repeats and experiments have shown an average error of ±10% for the results.
The timeline of initiation of antiproliferative activity by interferon is in line with the activation of caspase-8 and -3, with and without cFLIP K/D. This suggests that cFLIP is protecting the cells from rapid apoptosis upon encountering interferon. Interferon activates the antiproliferative machinery within hours, while cFLIP inhibits its prosecution, leading to the requirement for prolonged exposure to interferon to induce apoptosis. Similar results were obtained with MDA-MB-231 breast cancer cells.
To study the balance between cFLIP and caspase-8 leading to apoptosis induction, we performed a dose-response knockdown of either cFLIP, caspase-8, or both by varying the concentration of their specific siRNAs (33). Figure 4C shows that gradual K/D of cFLIP siRNA had a partial effect on the magnitude of antiproliferative activity, which peaked at 5 nM siRNA. In contrast, even 0.5 nM caspase-8 siRNA, which was sufficient to decrease the protein level to ∼40%, sequestered interferon-induced antiproliferative activity in cFLIP K/D cells.
The antiproliferative responses (EC50 values) for IFN-treated cells with and without cFLIP K/D were similar (cFLIP K/D, 0.10 nM; nontransfected control, 0.19 nM), suggesting that cFLIP affects the magnitude and time of activation but not the pathway leading to IFN-induced apoptosis. IFN-induced apoptosis through caspase-8 is highly tunable, with the cFLIP/caspase-8 ratio determining apoptosis initiation.
Caspase activity and apoptosis markers.
Apoptotic programmed cell death is characterized by initiator caspase cleavage, which further cleaves executioner caspase-3 and -7. Figure 5A and B show the activities of caspase-3/7, caspase-8, and caspase-9 following YNS treatment. In the absence of cFLIP K/D, caspase-3 activity increased with time until 48 h, reaching ∼3-fold the basal activity. Caspase-8 and -9 activities did not change significantly. cFLIP K/D had a dramatic effect both on shortening the time of initiation and on increasing the magnitude of caspase activity. Caspase-8 and -9 activities peaked within 16 h of IFN treatment, while caspase-3 activity peaked at 20 h (Fig. 5B) and was even higher than that observed with etoposide. Z-VAD completely inhibited caspase activity independent of cFLIP K/D. Monitoring cleavage of caspase-8 and -3 by Western blotting showed maximum cleavage at 48 h of IFN treatment (Fig. 5C). cFLIP K/D cells exhibited a much higher percentage of cleaved caspases as early as 8 h. Bid cleavage was observed in IFN-treated cells as opposed to Fas-treated cells. This correlates with the low level of caspase-9 activation following IFN treatment (Fig. 5B). Since caspase-9 silencing did not affect the IFN antiproliferative activity (Fig. 1B) or annexin V staining (Fig. 5D), we assume that caspase-8 is the initiator caspase that facilitates caspase-9 amplification through Bid cleavage (Fig. 5C). The partial rescue effect of DR5 K/D was also shown in the annexin V assay, as opposed to DR4 K/D, which increased the population of apoptotic cells.
IFN-induced apoptosis is death ligand independent.
The common mechanism leading to caspase-8 cleavage on the DISC involves the binding of an external ligand (Fas, TRAIL, or TNF-α [24]). It was suggested previously that interferon-induced upregulation of TRAIL induces apoptosis through an autocrine and paracrine effect (18, 34). However, as also shown previously for melanoma cells (15), the addition of TRAIL to WISH cells did not induce apoptosis (Fig. 6A and B). To investigate whether either Fas, TRAIL, TNF-α, or their combination is the mediator of interferon-induced apoptosis, we sequestered the specific activities of these ligands using antiligand proteins fused to the human Fc domain (Fig. 6A and B). As a control, WISH cells were treated with the purified ligands. Treatment of nontransfected cells (mock) with death ligands alone showed that WISH cells are resilient to TRAIL but sensitive to Fas and TNF-α. cFLIP K/D cells were highly sensitive to all three ligands (Fig. 6A). Next, we added IFN-YNS and either one or a combination of the inhibitors. The death ligand inhibitors had no effect on the interferon-induced antiproliferative activity, either alone or in combination (Fig. 6, YNS+ α-all). To determine whether the observed antiproliferative activity of interferon in the presence of the inhibitors was indeed apoptosis, we monitored caspase-3/7 activity (Fig. 6B) and annexin V staining (Fig. 6C) following treatment with IFN-YNS with or without the inhibitors. Neither inhibitor alone nor their combination altered YNS-induced caspase-3/7 activity, while Z-VAD eliminated caspase-3/7 activity even in a cFLIP K/D background.
Fig 6.

Death ligand elimination assay. (A and B) WISH cells were transfected with cFLIP siRNA or mock and treated with YNS at 1 nM, TNF-α at 25 ng/ml, 1% Fas ligand, TRAIL at 25 ng/ml, Z-VAD at 100 μM, and anti-TNF-α/Fas TRAIL fused protein at 1 μg/ml for 72 h prior to determination of cell density (A) and for 24 h for the caspase-3/7 activity assay (B). Data are represented as means ± standard errors of the means. (C and D) WISH cells were transfected with siRNA targeting cFLIP, RIPK1, cIAP1, or cIAP2 or not transfected (control) following treatment with 1 nM YNS with or without Z-VAD, 1 μg/ml anti-death ligand mix, or 63 μM necrostatin-1 (Nec-1) for 48 and 72 h. Cells were stained with annexin V and PI to determine apoptosis. Data are represented as means. Multiple repeats and experiments have shown an average error of ±10% for the results. (E and F) WISH cells were transfected with siRNA targeting the indicated genes following treatment with 1 nM YNS for an additional 72 h to determine cell density (E) or 24 h for caspase-3/7 activity (F). Caspase activity is relative to untreated cells.
It has been shown that caspase-8 activation can be facilitated by the ripoptosome, a cytoplasmic RIPK1-dependent platform utilizing FADD as an adaptor protein (35, 36). A RIPK1 inhibitor, nerostatin-1, did not inhibit cell death in either control or cFLIP K/D cells (Fig. 6C). To further validate this result, we monitored the effect of RIPK1 K/D. Even without IFN, RIPK1 K/D decreased the live-cell population by ∼40%, with the cell death rate being increased to close to 100% following 48 h of IFN-YNS treatment (Fig. 6D and E) and with a sharp increase in the apoptotic population. cIAP1/2 were reported to destabilize the ripoptosome by facilitating ubiquitin-dependent proteasomal degradation. cIAP1/2 K/D enhanced cell death even without IFN, which was increased significantly by IFN-YNS (Fig. 6D and E). This was further verified by the caspase-3/7 activity in RIPK1 and cIAP K/D upon IFN-YNS treatment (Fig. 6F). Following IFN-YNS treatment, the RIPK1 protein level increased 2- to 4-fold (Fig. 5C), and the gene transcription levels of cIAP1 and -2 were upregulated by 4-fold within 24 h and 5- to 10-fold after 48 h of induction. Considering these data, we conclude that IFN-induced apoptosis is not dependent on RIPK1 and its kinase activity, which is required for ripoptosome-induced caspase-8 activation. It seems that interferon is utilizing the antiapoptotic role of cIAPs to inhibit rapid cell death (by upregulating the expression of these genes), and thus, the proapoptotic effect of interferon is independent of cIAPs.
FADD dominant negative overexpression rescues cells from IFN-induced apoptosis.
The FADD dominant negative (FADD-DN) recombinant protein is a truncated form of FADD harboring the death domain (DD) alone. Its overexpression rescues cells from FADD-dependent apoptosis (37). Stably transfected FADD-DN WISH cells were generated by lentiviral infection, and their effectiveness was validated by the lack of sensitivity to Fas-induced cell death (Fig. 7A and B). The IFN-induced antiviral response of FADD-DN cells was similar to that of wild-type WISH cells, with the same EC50 and magnitude of response, suggesting that the canonical JAK-STAT pathway was intact. Gene K/D in these cells showed the antiproliferative activity to be dependent on IFNAR1, with the magnitude of the antiproliferative activity in the FADD-DN clone being reduced to that observed with the caspase-8 K/D (Fig. 7C). Indeed, caspase-3/7 activation was suppressed in the FADD-DN clone, even in a cFLIP K/D background (Fig. 7D), clearly indicating a loss of the apoptotic potency of interferon in these cells. The inhibition of activation of caspase-3/7 suggests that the competition between cFLIP and caspase-8 is FADD dependent. DR5 K/D resulted in decreased caspase-3/7 activity and increased cell density, showing its proapoptotic role in interferon signaling (Fig. 7C and D). Overall, our data support the involvement of DR5 and FADD in caspase-8 activation upon interferon induction, although it is death ligand independent.
Fig 7.

FADD-DN overexpression rescues cells from IFN-induced apoptosis. (A) Extrinsic death signal blocking was validated in FADD-DN-expressing cells versus wild-type cells (w.t.). WISH cells were treated using cross-linked anti-Fas monoclonal antibody, following crystal violet staining. (B) FLAG-tagged FADD-DN expression was monitored following treatment with TNF-α (25 ng/ml) or 1 nM YNS for 24 h in wild-type and FADD-DN WISH cells using anti-FLAG antibody. (C and D) Wild-type and FADD-DN cells were transfected with siRNA targeting the indicated genes following treatment with 1 nM YNS for 72 h, after which cell density was monitored (C), or were treated for 24 h with YNS before caspase-3/7 activity was monitored (D). Data are represented as means ± standard errors of the means.
Balance between cFLIP and caspase-8 on the DISC determines cell fate.
It was suggested previously that increased caspase-8 and decreased cFLIP levels are sufficient to induce apoptosis independent of death ligands (38, 39). Examination of the relative amounts of caspase-8 upon interferon treatment showed an increase in caspase-8 protein levels from 8 h onwards, reaching a maximum of ∼4-fold the basal level at 48 h (Fig. 5C). This is in line with previous reports showing that IFN-α and IFN-γ upregulate caspase-8 through STAT1 in TRAIL-resistant cells (40, 41). Monitoring of cFLIP cellular localization by immunostaining showed that without treatment, cFLIP was concentrated in the nucleus and that cFLIP K/D reduces its level (Fig. 8A). Upon IFN-YNS treatment, cFLIP was already seen in the cytoplasm at 11 h. At later time points, its level in the cytoplasm increased dramatically (Fig. 8A). Like IFN-YNS, IFN-γ is known to increase caspase-8 levels (Fig. 8A and 9D). Treatment with IFN-γ also increased the levels of cFLIP. However, contrary to IFN-YNS, it was concentrated predominantly in the nucleus, which may suggest a different mechanism of apoptosis induction by these two interferons.
Fig 8.

In situ quantification of caspase-8 and cFLIP interactions with FADD following YNS treatment. (A) cFLIP and caspase-8 localization and levels were monitored with immunofluorescence using specific antibodies. (B and C) cFLIP-FADD (B) and caspase-8–FADD (C) interactions visualized by an in situ proximity ligation assay. WISH cells were transfected or not with cFLIP or caspase-8 siRNA following treatment with 1 nM YNS, as indicated, or were left untreated (not treated [NT]). (D) Quantification of PLA interactions per cell (shown in panels B and C). (E) Same as panels B and C. WISH cells were transfected or not with DR4 or DR5 siRNA following treatment with 1 nM YNS, as indicated.
Fig 9.

Interferon-induced caspase-8 upregulation in interferon-sensitive and -resistant cell lines. (A) MDA-MB-321, T47D, and HS578T cells were treated with 1 nM YNS for 3 days following crystal violet staining for cell density evaluation. (B) Caspase-3/7 activity was measured in T47D and HS578T IFN-resistant cell lines following treatment with 1 nM YNS for 48 h. (C) WISH, MDA-MB-321, T47D, BT549T, and HS578T cells were treated with 1 nM YNS for the indicated hours following protein extraction for Western blotting. Error bars represent the standard errors of 2 to 4 independent replicates. The dashed line marks a 3-fold increase in the caspase-8 level. (D) Representative Western blotting for WISH cells treated with 1 nM YNS and/or 1,000 U/ml IFN-γ for the indicated times. (E and F) WISH cells were transfected with siRNA targeting the indicated genes following treatment with 1 nM YNS and/or 1,000 U/ml IFN-γ for 72 h (E) or 24 h (F), after which after cell density (E) or caspase-3/7 activity (F) was monitored. For panels A to C, E, and F, data are represented as means ± standard errors of the means.
To test whether caspase-8 increasingly binds FADD following IFN treatment, we used PLA (proximity ligation assay) (42–44). This method detects the proximity of endogenous proteins in individual cells, suggesting either direct or indirect interactions between them. The signal is viewed as high-intensity spots in the microscope image that can be quantified. Its advantages over classical coimmunoprecipitation are that PLA can also be used if only a small portion of the population is in complex and that it provides quantitative spatial information. Probing of cFLIP-FADD interactions using specific primary antibodies detected increasing amounts of interactions upon IFN treatment from 8 to 48 h (Fig. 8B and D). As expected, cFLIP K/D decreased the number of interactions with FADD. Probing of the caspase-8–FADD interaction showed few spots in untreated cells, while IFN treatment increased the number of interactions at times longer than 11 h but not at 2 to 8 h (Fig. 8C and D). In cFLIP K/D cells, the number of caspase-8–FADD interactions increased, peaking at 4 h. This correlates with the faster initiation of apoptosis upon cFLIP K/D. Importantly, the increased cFLIP-FADD and caspase-8–FADD interactions following IFN-YNS treatment were abolished when DR5 but not DR4 was knocked down (Fig. 8E). This correlates with the decrease in IFN-induced apoptosis in DR5 K/D cells. The results suggest that caspase-8 activation is governed by the ratio between caspase-8 and cFLIP on FADD and DR5.
Interferon induces caspase-8 upregulation in interferon-sensitive and -resistant cell lines.
While the antiviral response is a robust feature common to all cell lines, the antiproliferative response differs greatly. WISH and MDA-MB-231 cells are sensitive to interferon-induced antiproliferation, while T47D, BT549T, and HS578T cells are resistant (Fig. 9A) (33). We monitored caspase-8 accumulation in these five cell lines, to evaluate whether caspase-8 accumulation upon interferon treatment predicts cell responsiveness (Fig. 9C). Indeed, the caspase-8 level increased >3-fold in WISH and MDA-MB231 cells, while only minor accumulation was observed in T47D and BT549 cells. HS578T cells did show some accumulation of caspase-8, despite a lack of interferon-induced apoptosis. Therefore, we measured caspase-3/7 activity, which did not increase by interferon treatment in HS578T or T47D cells (Fig. 9B).
Type I versus type II interferons.
Gamma interferon upregulates caspase-8 expression in many cell lines (45, 46). Figure 9D shows that in WISH cells, IFN-γ increases caspase-8 levels comparably to IFN-YNS, reaching ∼6-fold within 48 h. The addition of both IFNs simultaneously further increased procaspase-8 protein levels. Indeed, cotreatment of both interferons resulted in reduced cell viability to an extent seen only upon cFLIP K/D (Fig. 9E). As expected, IFNAR1 K/D inhibited only IFN-YNS signaling. Interestingly, cFLIP silencing had a much-reduced prodeath effect upon IFN-γ treatment, and caspase-8 K/D seemed to have no effect. These results are in line with the results of the caspase-3/7 activity assay (Fig. 9F) showing that caspase-8 K/D cells still activate caspase-3/7 upon IFN-γ but not IFN-YNS treatment. It was shown previously that IFN-γ induces apoptosis through the intrinsic pathway (46), which is not crucially dependent on caspase-8. To further investigate the difference in mechanisms of type I and type II interferon-induced cell death, we monitored caspase-8 and cFLIP levels and localization in cells using immunofluorescence. Figure 7A shows that while IFN-YNS resulted in a dramatic increase in cFLIP, particularly in the cytoplasm, IFN-γ increased cFLIP levels but not in the cytoplasm. Both interferons increased the levels of caspase-8, with the increase being more pronounced when using IFN-γ (in line with the Western blot results). Together, these results suggest that the difference in K/D of cFLIP observed between IFN-YNS and IFN-γ is a result of its different localization, suggesting that not only increased levels of caspase-8 but also the localization of cFLIP drives type I-induced apoptosis.
DISCUSSION
The cellular response to interferon is broad, with the expression of over 1,000 genes being affected (23). This stimulates a pleiotropy of biological responses, of which antiviral activity is the most studied. In addition, interferon induces a strong antiproliferative response in many cell lines, which is a combination of cell cycle arrest (particularly S-phase arrest [47, 48]) and initiation of apoptosis (49). To activate interferon-induced apoptosis, an intact JAK-STAT pathway is required (50). The mechanism of how the JAK-STAT pathway leads to caspase activation and apoptosis is still under debate. The most obvious mechanism would be through the activation of TNF receptors by Fas (51), TRAIL (38, 52), or TNF. Conversely, Panaretakis et al. (17) suggested previously that interferon induces apoptosis through PI3K, which activates the downstream extracellular signal-regulated kinase 1/2 (ERK1/2), protein kinase Cδ (PKCδ), and JNK genes, leading to intrinsic apoptosis.
Here we used a nonbiased siRNA screen on WISH cells to systematically elucidate the interferon-induced apoptotic pathway. Silencing genes, the products of which are directly engaged in the initiation of the interferon signal, such as the two cell surface receptors, STAT2, JAK1, and TYK2, completely abrogate the interferon antiproliferative response. Conversely, genes involved in apoptosis, such as the DISC-related DR5, FADD, caspase-8, and cFLIP genes, are required for interferon-induced apoptosis but do not affect interferon-induced S-phase arrest (which also occurs after addition of Z-VAD). This indicates that cell cycle arrest and apoptosis are independently activated by interferon, downstream of ISGF3 formation.
The one gene for which silencing resulted in the strongest proapoptotic effect upon interferon signaling is the cFLIP gene. Silencing of cFLIP shortened the time of initiation of apoptosis from days to hours and increased the percentage of apoptotic cells from 60 to close to 100%. Thus, cFLIP serves as an attenuator for interferon-induced apoptosis, explaining why it takes only hours for interferon to initiate the antiviral response but days to cause apoptosis. The delay in the onset of apoptosis may well come to rescue cells from an overshoot of transient interferon production caused by an invasion of viruses. The interferon-induced antiviral response does not harm the cells but only prepares them for further viral attack (20). Apoptosis, which is induced by prolonged exposure to high levels of interferon, serves as a second line of defense to stop viral replication and to remove infected cells (53). Indeed, viruses such as the Urabe AM9 vaccine strain of mumps virus encode apoptosis inhibitors and decrease the activation of extrinsic IFN-α2-induced apoptosis (54). Interestingly, viral FLIP genes were the first members of the family to be identified as containing the death effector domain (DED), which interacts with certain caspases: caspase-8 and -10 (55). These proteins are principally composed of two homologous DED regions, which protect cells against apoptosis induced by several death receptors. This indicates the predominant role of FLIP in the arms race of viruses and human cells, which both exploit.
We were surprised to see that sequestering of either TRAIL, Fas, TNF, or their combination had no effect on interferon-induced apoptosis. Several previous studies reported apoptosis induction by cFLIP silencing on the DISC, without the involvement of an external ligand (38, 39). Of particular interest is previous work by Wilson et al. (39), who demonstrated that a higher expression level of procaspase-8 or silencing of cFLIP is sufficient to induce apoptosis with an involvement of the DR5 receptor but without external death ligand. Here we show that interferon induces the accumulation of procaspase-8 to levels similar to those previously reported by Wilson et al. (39), to initiate apoptosis and that DR5 is involved in this process. PLA confirmed that in the presence of interferon, cFLIP K/D abolishes the interactions between cFLIP and FADD and doubles the number of interactions between caspase-8 and FADD. Moreover, maximal caspase-8–FADD interactions that are observed within 11 h of interferon treatment are shortened to 4 h upon cFLIP K/D. Interestingly, interferon-induced cFLIP-FADD interactions precede caspase-8–FADD interactions (Fig. 8C), suggesting an inhibitory role of cFLIP in caspase-8 accumulation on FADD, which leads to its activation. As the increased interactions were abolished upon DR5 K/D, we suggest that DR5 serves as the receptor molecule of the DISC on which cFLIP and caspase-8 compete.
To further investigate the role of caspase-8 and cFLIP, we compared their levels of expression and cellular localization upon treatment with either type I or type II IFNs. Both interferons induced production of caspase-8, which was evenly distributed throughout the cell. Conversely, immunofluorescence clearly showed that cFLIP, which was observed mainly in the nuclei prior to the addition of IFN-YNS, was massively upregulated in the cytoplasm by IFN-YNS but not by IFN-γ. These findings may explain why induction of apoptosis by IFN-YNS is more dependent on cFLIP than is apoptosis induced by IFN-γ (see comparison upon cFLIP K/D in Fig. 9).
The results of this study suggest that the increased expression level of caspase-8 upon interferon treatment has an important role in triggering the apoptotic effect of interferon through the DR5 receptor on FADD. Indeed, T47D and BT549T cells, which do not express higher levels of caspase-8 upon IFN induction, are resistant to interferon-induced apoptosis. Conversely, the caspase-8 expression level was increased in two sensitive cell lines, WISH and MDA-MB-231. The simultaneous addition of IFN-YNS and IFN-γ resulted in an even higher increase in caspase-8 levels and a greatly enhanced apoptotic response. An outlier cell line is HS578T, where increased expression levels of caspase-8 did not activate caspase-3 or apoptosis. This suggests that in addition to the caspase-8 concentration, other factors contribute to interferon-induced apoptosis.
The role of the cFLIP long and short isoforms in inducing apoptosis is still under debate (56–58). cFLIPL can apparently be antiapoptotic at lower levels but proapoptotic at higher levels. If this is the case, perhaps the elevated cFLIP levels in the cytoplasm at later times of induction by IFN-YNS treatment are proapoptotic, while the lower levels at shorter times of IFN-YNS treatment are antiapoptotic. This would allow cFLIP to inhibit apoptosis if viral infection is transient but would allow apoptosis upon consistent interferon stimulation. While IFN-γ also causes an increase in caspase-8 levels, it does not promote a massive increase in cFLIP levels in the cytoplasm. Moreover, the effect of caspase-8 K/D and cFLIP K/D upon IFN-γ treatment is less pronounced. IFN-γ induces apoptosis through the intrinsic pathway (46), which is different from what we show here for IFN-YNS. Clearly, the balance between cFLIP and caspase-8 is of greater importance for the latter in inducing apoptosis than for the former. Further studies underlining the role and mechanism of cFLIP levels and translocation to the cytoplasm by the nuclear export and import signals on cFLIP (30, 31) are required to shed light on the nature of this mechanism.
If interferon indeed induces apoptosis through a gradual upregulation of caspase-8 on receptor-mediated DISC, cells should be rescued either by FADD K/D or in FADD dominant negative cells. While FADD K/D reduced its quantity by ∼70%, its effect on interferon-induced antiproliferative activity was marginal. This may be explained by an excessive FADD concentration in the cytoplasm (59) relative to that of the DISC-bound FADD (60). Indeed, in a stable FADD-DN cell line, which is resistant to receptor-dependent DISC activation, reduced caspase-3/7 activity was observed upon interferon treatment. The FADD-DN cell line supports the notion that caspase-8/cFLIP competition is taking place on FADD.
Figure 10 summarizes the mechanism of interferon-induced apoptosis in WISH cells determined in this work: the formation of the interferon-receptor complex induces the JAK-STAT pathway, resulting in ISGF3 formation, which in turn induces caspase-8 production. Caspase-8 is directed toward the DISC containing DR5, where it is inhibited by cFLIP, which rapidly accumulates on the DISC upon interferon induction. Due to the faster cFLIP accumulation, it takes days to reach apoptosis in the majority of cells, while STAT phosphorylation dwindles within a few hours.
Fig 10.
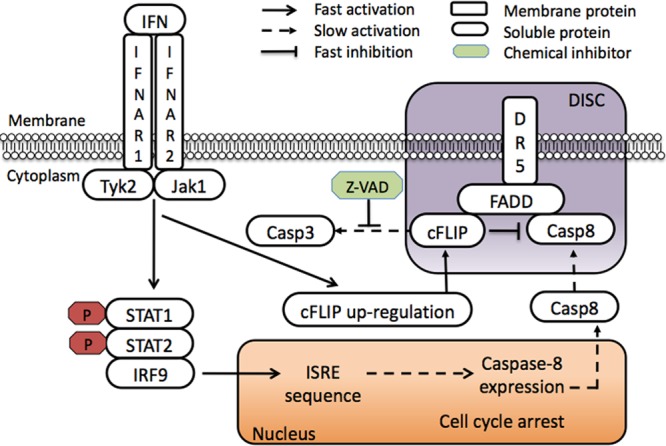
Schematic model of IFN-induced apoptosis in WISH cells. IFN binding to its receptors activates the canonical JAK-STAT pathway, leading to cell cycle arrest and expression of caspase-8, which progresses within days. Meanwhile, the cFLIP cytoplasmic concentration increases rapidly, binding FADD within hours after IFN induction to inhibit caspase-8 cleavage. This results in slow and gradual caspase-8 activation, followed by caspase-3 cleavage and apoptosis. Caspase-8–cFLIP competition is FADD dependent and requires DR5 as a platform independent of death ligands.
A still-open question is what promotes FADD-induced proximity upon interferon induction in the absence of external death ligand. A role of DR5 (but not DR4) was suggested previously by Wilson et al. (39) to be important for ligand-independent caspase-8 activation. This fits our K/D data for DR5, which reduces apoptosis. Both DR4 and DR5 promote apoptosis by binding FADD but by a different mechanism (61). The increased apoptosis observed upon DR4 K/D suggests that it competes with DR5 on FADD, and thus, silencing of DR4 enhances interferon-induced apoptosis through DR5. Interferon may also stimulate the production of an as-yet-unknown factor that mediates the recruitment of caspase-8 and/or cFLIP to FADD and promotes its clustering. The PLA data provide support for this hypothesis, as they showed that binding of cFLIP and caspase-8 was already observed at 8 and 11 h, respectively, after the addition of interferon, while accumulation of caspase-8 was observed only at later times. Moreover, cFLIP K/D without interferon induction did not cause an increase in caspase-8–FADD interactions despite the lack of the tighter-binding cFLIP competitor.
This study provides a solid molecular explanation for interferon-induced apoptosis and suggests directions for how interferon can be combined with other inhibitors or activators to increase its efficiency as an antitumor agent. Most notably, even a partial inhibition of cFLIP or simultaneous addition of IFN-γ would dramatically increase the efficiency of type I interferon. It should also be noted that interferon activates an apoptotic response to different degrees in different cell lines, which should be taken into account in any potential treatment.
ACKNOWLEDGMENTS
This research was funded by the European Community's Seventh Framework Program (FP7/2007-2013) under grant agreement no. 223608.
We thank Inder Verma of Salk Institute, CA, for sharing the lentivirus constructs used in this study. We also thank Andreas Strasser and David Huang from the Walter and Eliza Hall Institute of Medical Research Melbourne for sharing FADD-DN constructs. Mati Cohen developed the Matlab script used for the PLA analysis. Ghil Jona helped us with the siRNA screen.
A.A., G.Y., and G.S. planned and performed the biochemical and molecular cell biological studies and wrote the manuscript; S.W. planned and performed cell cycle experiments; and D.H. planned experiments.
We declare that we have no conflict of interest.
Footnotes
Published ahead of print 10 December 2012
REFERENCES
- 1. Brierley MM, Fish EN. 2002. IFN-alpha/beta receptor interactions to biologic outcomes: understanding the circuitry. J. Interferon Cytokine Res. 22:835–845 [DOI] [PubMed] [Google Scholar]
- 2. Cohen B, Novick D, Barak S, Rubinstein M. 1995. Ligand-induced association of the type I interferon receptor components. Mol. Cell. Biol. 15:4208–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, Lee C, Yarden G, Vleck SE, Glenn JS, Nolan GP, Piehler J, Schreiber G, Garcia KC. 2011. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 146:621–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Uze G, Lutfalla G, Gresser I. 1990. Genetic transfer of a functional human interferon alpha receptor into mouse cells: cloning and expression of its cDNA. Cell 60:225–234 [DOI] [PubMed] [Google Scholar]
- 5. Cebulla CM, Miller DM, Sedmak DD. 1999. Viral inhibition of interferon signal transduction. Intervirology 42:325–330 [DOI] [PubMed] [Google Scholar]
- 6. Darnell JEJ. 1998. Studies of IFN-induced transcriptional activation uncover the Jak-Stat pathway. J. Interferon Cytokine Res. 18:549–554 [DOI] [PubMed] [Google Scholar]
- 7. Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227–264 [DOI] [PubMed] [Google Scholar]
- 8. Horoszewicz JS, Leong SS, Carter WA. 1979. Noncycling tumor cells are sensitive targets for the antiproliferative activity of human interferon. Science 206:1091–1093 [DOI] [PubMed] [Google Scholar]
- 9. Sangfelt O, Erickson S, Castro J, Heiden T, Gustafsson A, Einhorn S, Grander D. 1999. Molecular mechanisms underlying interferon-alpha-induced G0/G1 arrest: CKI-mediated regulation of G1 Cdk-complexes and activation of pocket proteins. Oncogene 18:2798–2810 [DOI] [PubMed] [Google Scholar]
- 10. Tiefenbrun N, Melamed D, Levy N, Resnitzky D, Hoffman I, Reed SI, Kimchi A. 1996. Alpha interferon suppresses the cyclin D3 and cdc25A genes, leading to a reversible G0-like arrest. Mol. Cell. Biol. 16:3934–3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Einat M, Resnitzky D, Kimchi A. 1985. Close link between reduction of c-myc expression by interferon and, G0/G1 arrest. Nature 313:597–600 [DOI] [PubMed] [Google Scholar]
- 12. Furukawa Y, Iwase S, Kikuchi J, Nakamura M, Yamada H, Matsuda M. 1999. Transcriptional repression of the E2F-1 gene by interferon-alpha is mediated through induction of E2F-4/pRB and E2F-4/p130 complexes. Oncogene 18:2003–2014 [DOI] [PubMed] [Google Scholar]
- 13. Melamed D, Tiefenbrun N, Yarden A, Kimchi A. 1993. Interferons and interleukin-6 suppress the DNA-binding activity of E2F in growth-sensitive hematopoietic cells. Mol. Cell. Biol. 13:5255–5265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Resnitzky D, Yarden A, Zipori D, Kimchi A. 1986. Autocrine beta-related interferon controls c-myc suppression and growth arrest during hematopoietic cell differentiation. Cell 46:31–40 [DOI] [PubMed] [Google Scholar]
- 15. Chawla-Sarkar M, Leaman DW, Borden EC. 2001. Preferential induction of apoptosis by interferon (IFN)-beta compared with IFN-alpha2: correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin. Cancer Res. 7:1821–1831 [PubMed] [Google Scholar]
- 16. Hjortsberg L, Lindvall C, Corcoran M, Arulampalam V, Chan D, Thyrell L, Nordenskjold M, Grander D, Pokrovskaja K. 2007. Phosphoinositide 3-kinase regulates a subset of interferon-alpha-stimulated genes. Exp. Cell Res. 313:404–414 [DOI] [PubMed] [Google Scholar]
- 17. Panaretakis T, Hjortsberg L, Tamm KP, Bjorklund AC, Joseph B, Grander D. 2008. Interferon alpha induces nucleus-independent apoptosis by activating extracellular signal-regulated kinase 1/2 and c-Jun NH2-terminal kinase downstream of phosphatidylinositol 3-kinase and mammalian target of rapamycin. Mol. Biol. Cell 19:41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pokrovskaja K, Panaretakis T, Grander D. 2005. Alternative signaling pathways regulating type I interferon-induced apoptosis. J. Interferon Cytokine Res. 25:799–810 [DOI] [PubMed] [Google Scholar]
- 19. Thyrell L, Hjortsberg L, Arulampalam V, Panaretakis T, Uhles S, Dagnell M, Zhivotovsky B, Leibiger I, Grander D, Pokrovskaja K. 2004. Interferon alpha-induced apoptosis in tumor cells is mediated through the phosphoinositide 3-kinase/mammalian target of rapamycin signaling pathway. J. Biol. Chem. 279:24152–24162 [DOI] [PubMed] [Google Scholar]
- 20. Jaitin DA, Schreiber G. 2007. Upregulation of a small subset of genes drives type I interferon-induced antiviral memory. J. Interferon Cytokine Res. 27:653–664 [DOI] [PubMed] [Google Scholar]
- 21. Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, Van der Heyden J, Uze G, Schreiber G. 2006. Inquiring into the differential action of interferons (IFNs): an IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol. Cell. Biol. 26:1888–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kalie E, Jaitin DA, Abramovich R, Schreiber G. 2007. An interferon alpha2 mutant optimized by phage display for IFNAR1 binding confers specifically enhanced antitumor activities. J. Biol. Chem. 282:11602–11611 [DOI] [PubMed] [Google Scholar]
- 23. Kalie E, Jaitin DA, Podoplelova Y, Piehler J, Schreiber G. 2008. The stability of the ternary interferon-receptor complex rather than the affinity to the individual subunits dictates differential biological activities. J. Biol. Chem. 283:32925–32936 [DOI] [PubMed] [Google Scholar]
- 24. Ashkenazi A, Dixit VM. 1999. Apoptosis control by death and decoy receptors. Curr. Opin. Cell Biol. 11:255–260 [DOI] [PubMed] [Google Scholar]
- 25. Scaffidi C, Schmitz I, Krammer PH, Peter ME. 1999. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 274:1541–1548 [DOI] [PubMed] [Google Scholar]
- 26. Wang L, Yang JK, Kabaleeswaran V, Rice AJ, Cruz AC, Park AY, Yin Q, Damko E, Jang SB, Raunser S, Robinson CV, Siegel RM, Walz T, Wu H. 2010. The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat. Struct. Mol. Biol. 17:1324–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilson NS, Dixit V, Ashkenazi A. 2009. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat. Immunol. 10:348–355 [DOI] [PubMed] [Google Scholar]
- 28. Hyer ML, Samuel T, Reed JC. 2006. The FLIP-side of Fas signaling. Clin. Cancer Res. 12:5929–5931 [DOI] [PubMed] [Google Scholar]
- 29. Yu JW, Shi Y. 2008. FLIP and the death effector domain family. Oncogene 27:6216–6227 [DOI] [PubMed] [Google Scholar]
- 30. Katayama R, Ishioka T, Takada S, Takada R, Fujita N, Tsuruo T, Naito M. 2010. Modulation of Wnt signaling by the nuclear localization of cellular FLIP-L. J. Cell Sci. 123:23–28 [DOI] [PubMed] [Google Scholar]
- 31. Zhang J, Chen Y, Huang Q, Cheng W, Kang Y, Shu L, Yin W, Hua ZC. 2009. Nuclear localization of c-FLIP-L and its regulation of AP-1 activity. Int. J. Biochem. Cell Biol. 41:1678–1684 [DOI] [PubMed] [Google Scholar]
- 32. Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. 2009. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 137:721–735 [DOI] [PubMed] [Google Scholar]
- 33. Levin D, Harari D, Schreiber G. 2011. Stochastic receptor expression determines cell fate upon interferon treatment. Mol. Cell. Biol. 31:3252–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC. 2003. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 8:237–249 [DOI] [PubMed] [Google Scholar]
- 35. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M. 2011. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43:449–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P. 2011. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43:432–448 [DOI] [PubMed] [Google Scholar]
- 37. Newton K, Harris AW, Bath ML, Smith KG, Strasser A. 1998. A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J. 17:706–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kitahara Y, Kawane K, Nagata S. 2010. Interferon-induced TRAIL-independent cell death in DNase II−/− embryos. Eur. J. Immunol. 40:2590–2598 [DOI] [PubMed] [Google Scholar]
- 39. Wilson TR, Redmond KM, McLaughlin KM, Crawford N, Gately K, O'Byrne K, Le-Clorrenec C, Holohan C, Fennell DA, Johnston PG, Longley DB. 2009. Procaspase 8 overexpression in non-small-cell lung cancer promotes apoptosis induced by FLIP silencing. Cell Death Differ. 16:1352–1361 [DOI] [PubMed] [Google Scholar]
- 40. Fulda S, Debatin KM. 2002. IFNgamma sensitizes for apoptosis by upregulating caspase-8 expression through the Stat1 pathway. Oncogene 21:2295–2308 [DOI] [PubMed] [Google Scholar]
- 41. Liedtke C, Groger N, Manns MP, Trautwein C. 2006. Interferon-alpha enhances TRAIL-mediated apoptosis by up-regulating caspase-8 transcription in human hepatoma cells. J. Hepatol. 44:342–349 [DOI] [PubMed] [Google Scholar]
- 42. Greenberg JI, Shields DJ, Barillas SG, Acevedo LM, Murphy E, Huang J, Scheppke L, Stockmann C, Johnson RS, Angle N, Cheresh DA. 2008. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature 456:809–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reinhardt C, Bergentall M, Greiner TU, Schaffner F, Ostergren-Lunden G, Petersen LC, Ruf W, Backhed F. 2012. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature 483:627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. 2006. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3:995–1000 [DOI] [PubMed] [Google Scholar]
- 45. Ahn EY, Pan G, Vickers SM, McDonald JM. 2002. IFN-gamma upregulates apoptosis-related molecules and enhances Fas-mediated apoptosis in human cholangiocarcinoma. Int. J. Cancer 100:445–451 [DOI] [PubMed] [Google Scholar]
- 46. Ruiz-Ruiz C, Munoz-Pinedo C, Lopez-Rivas A. 2000. Interferon-gamma treatment elevates caspase-8 expression and sensitizes human breast tumor cells to a death receptor-induced mitochondria-operated apoptotic program. Cancer Res. 60:5673–5680 [PubMed] [Google Scholar]
- 47. Vannucchi S, Chiantore MV, Fiorucci G, Percario ZA, Leone S, Affabris E, Romeo G. 2005. TRAIL is a key target in S-phase slowing-dependent apoptosis induced by interferon-beta in cervical carcinoma cells. Oncogene 24:2536–2546 [DOI] [PubMed] [Google Scholar]
- 48. Vannucchi S, Percario ZA, Chiantore MV, Matarrese P, Chelbi-Alix MK, Fagioli M, Pelicci PG, Malorni W, Fiorucci G, Romeo G, Affabris E. 2000. Interferon-beta induces S phase slowing via up-regulated expression of PML in squamous carcinoma cells. Oncogene 19:5041–5053 [DOI] [PubMed] [Google Scholar]
- 49. Bekisz J, Baron S, Balinsky C, Morrow A, Zoon KC. 2010. Antiproliferative properties of type I and type II interferon. Pharmaceuticals (Basel) 3:994–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Arulampalam V, Kolosenko I, Hjortsberg L, Bjorklund AC, Grander D, Tamm KP. 2011. Activation of STAT1 is required for interferon-alpha-mediated cell death. Exp. Cell Res. 317:9–19 [DOI] [PubMed] [Google Scholar]
- 51. Moraga I, Harari D, Schreiber G, Uze G, Pellegrini S. 2009. Receptor density is key to the alpha2/beta interferon differential activities. Mol. Cell. Biol. 29:4778–4787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rani MR, Pandalai S, Shrock J, Almasan A, Ransohoff RM. 2007. Requirement of catalytically active Tyk2 and accessory signals for the induction of TRAIL mRNA by IFN-beta. J. Interferon Cytokine Res. 27:767–779 [DOI] [PubMed] [Google Scholar]
- 53. Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 [DOI] [PubMed] [Google Scholar]
- 54. Rosas-Murrieta NH, Santos-Lopez G, Reyes-Leyva J, Jurado FS, Herrera-Camacho I. 2011. Modulation of apoptosis by V protein mumps virus. Virol. J. 8:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hofmann K. 1999. The modular nature of apoptotic signaling proteins. Cell. Mol. Life Sci. 55:1113–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fricker N, Beaudouin J, Richter P, Eils R, Krammer PH, Lavrik IN. 2010. Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J. Cell Biol. 190:377–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kavuri SM, Geserick P, Berg D, Dimitrova DP, Feoktistova M, Siegmund D, Gollnick H, Neumann M, Wajant H, Leverkus M. 2011. Cellular FLICE-inhibitory protein (cFLIP) isoforms block CD95- and TRAIL death receptor-induced gene induction irrespective of processing of caspase-8 or cFLIP in the death-inducing signaling complex. J. Biol. Chem. 286:16631–16646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Micheau O. 2003. Cellular FLICE-inhibitory protein: an attractive therapeutic target? Expert Opin. Ther. Targets 7:559–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schleich K, Warnken U, Fricker N, Ozturk S, Richter P, Kammerer K, Schnolzer M, Krammer PH, Lavrik IN. 2012. Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol. Cell 47:306–319 [DOI] [PubMed] [Google Scholar]
- 60. Dickens LS, Boyd RS, Jukes-Jones R, Hughes MA, Robinson GL, Fairall L, Schwabe JW, Cain K, Macfarlane M. 2012. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 47:291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Henry RE, Andrysik Z, Paris R, Galbraith MD, Espinosa JM. 2012. A DR4:tBID axis drives the p53 apoptotic response by promoting oligomerization of poised BAX. EMBO J. 31:1266–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]