

Abstract
A variety of external perturbations can induce endoplasmic reticulum (ER) stress, followed by stimulation of epithelial cells to produce an innate immune element, the cathelicidin antimicrobial peptide (CAMP). ER stress also increases production of the proapoptotic lipid ceramide and its antiapoptotic metabolite, sphingosine-1-phosphate (S1P). We demonstrate here that S1P mediates ER stress-induced CAMP generation. Cellular ceramide and S1P levels rose in parallel with CAMP levels following addition of either exogenous cell-permeating ceramide (C2Cer), which increases S1P production, or thapsigargin (an ER stressor), applied to cultured human skin keratinocytes or topically to mouse skin. Knockdown of S1P lyase, which catabolizes S1P, enhanced ER stress-induced CAMP production in cultured cells and mouse skin. These and additional inhibitor studies show that S1P is responsible for ER stress-induced upregulation of CAMP expression. Increased CAMP expression is likely mediated via S1P-dependent NF-κB–C/EBPα activation. Finally, lysates of both ER-stressed and S1P-stimulated cells blocked growth of virulent Staphylococcus aureus in vitro, and topical C2Cer and LL-37 inhibited invasion of Staphylococcus aureus into murine skin. These studies suggest that S1P generation resulting in increased CAMP production comprises a novel regulatory mechanism of epithelial innate immune responses to external perturbations, pointing to a new therapeutic approach to enhance antimicrobial defense.
INTRODUCTION
The 37-amino-acid carboxy-terminal peptide (LL-37) of the cathelicidin antimicrobial protein (CAMP) mediates innate immune responses to external pathogen challenges and also modulates cytokine production, angiogenesis, and downstream adaptive immune responses (1, 2). Prior studies demonstrated that various unrelated external perturbations, including UV B (UVB) irradiation, wounding, and permeability barrier abrogation, induce endoplasmic reticulum (ER) stress (3), which in turn stimulates a set of responses that rescue cells from apoptosis (4). These responses include increased expression of CAMP in epithelial tissues such as mammalian epidermis (5–8). We showed recently that comparable external perturbations also stimulate CAMP production in epithelial but not myeloid cells, via a novel NF-κB- and C/EBPα-mediated pathway independent of the well-known, vitamin D receptor (VDR)-mediated mechanism (9). Yet these studies left unanswered the key question of how ER stress signals the downstream production of CAMP.
Prior studies (10, 11) showed that ER stress, like external perturbations, stimulates the production of ceramide (Cer), the backbone of all sphingolipids (12). Not only is Cer a component of mammalian cell membranes, but Cer itself, as well as its hydrolytic metabolite, sphingosine, is also proapoptotic and antimitogenic (13). After exposure to ER stressors, either an accumulation of unfolded protein or an overaccumulation of folded protein occurs in cells (14). Thapsigargin (Tg), which is a specific inhibitor of intracellular sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA)-type Ca2+ pumps, disrupts ER calcium homeostasis, which causes ER stress, including accumulation of unfolded or misfolded proteins in the ER. Yet while subtoxic levels of ER stress mediate an unfolded protein response (UPR) that restores normal ER function (4), excessive ER stress can overwhelm this mechanism (15), triggering Cer-induced apoptosis (12). We identified two protective mechanisms that allow cells exposed to subtoxic levels of ER stress to escape from Cer-induced apoptosis (11): (i) enhanced metabolic conversion of Cer to glucosylceramide (16); and (ii) accelerated ceramidase-mediated hydrolysis of Cer to sphingosine, followed by its phosphorylation to sphingosine-1-phosphate (S1P) (10). While S1P generated by the sphingosine kinase 1 isoform (SPHK1) is antiapoptotic (17), S1P generated by the SPHK2 isoform can increase apoptosis and induce cell growth arrest (17, 18). S1P-mediated signaling of cellular functions is often dependent upon prior binding to one or more of five known G-protein-coupled receptors (S1P1 to S1P5), located on the outer surface of the plasma membrane (19). S1P generated in the nucleus can also directly modulate histone acetylation (20), potentially regulating gene expression. S1P likely mediates additional cellular functions via S1P receptor-independent mechanisms, including a recently identified novel role of S1P as a cofactor in the RelA pathway, leading to polyubiquitination of receptor interacting protein 1 (RIP1) and activation of the classical NF-κB pathway (21).
We show here, first, that subtoxic levels of ER stress, which are known to increase Cer production, are followed by downstream conversion of Cer to S1P, and then that S1P is the specific signal that stimulates CAMP expression under stressful conditions, via a recently identified NF-κB–C/EBPα-dependent but VDR-independent mechanism (9). We further show that the S1P signal operates independently of the S1P receptors. We show that this stress-initiated mechanism stimulates sufficient CAMP production to inhibit in vitro growth of virulent, exogenous Staphylococcus aureus and, finally, that topical applications of the S1P precursor, cell-permeative C2Cer, attenuate S. aureus invasion in murine skin, while coapplications of a specific sphingosine kinase inhibitor diminish this effect. Together, these studies identify and characterize a new ER stress-induced, lipid-mediated rescue mechanism that protects epithelial cells from apoptosis while simultaneously stimulating epithelial innate immunity to improve/enhance the antimicrobial defense barrier.
MATERIALS AND METHODS
Cell culture.
Normal human keratinocytes (KC) isolated from neonatal foreskins or immortalized, nontransformed (HaCaT) KC, derived from normal human skin (a gift from N. Fusenig, Heidelberg, Germany), were grown as described previously (9). Culture medium was switched to serum-free KC growth medium containing 0.07 mM calcium chloride and growth supplements (Invitrogen, Carlsbad, CA) 1 day prior to treatment with Tg (Sigma, St. Louis, MO), S1P, dihydro-sphingosine-1-phosphate (dhS1P), C2Cer (N-acetylsphingosine) (Avanti Polar Lipids, Inc., Alabaster, AL), 5-[4-phenyl-5-(trifluoromethyl)-2-thienyl]-3-[3-(trifluoromethyl)phenyl]-1,2,4-oxadiazole (SEW2871) (Cayman Chemical Company, Ann Arbor, MI), pertussis toxin (Ptx) (Sigma-Aldrich, St. Louis, MO), (R)-phosphoric acid mono-[2-amino-2-(3-octyl-phenylcarbamoyl)-ethyl] ester (VPC23019) (Avanti Polar Lipids), JTE-013 (Cayman Chemical Company), N-oleoylethanolamine (NOE) and/or N,N-dimethylsphingosine (DMS) (Matreya, Pleasant Gap, PA), or 4-[4-(4-chlorophenyl)-2-thiazolyl]amino phenol (SKI-II) (Enzo Life Sciences, Inc., Farmingdale, NY). Cell toxicities, including apoptosis, were determined by poly(ADP-ribose) polymerase (PARP) cleavage as well as a trypan blue dye exclusion assay (10).
Measurement of intercellular levels of Cer, sphingosine, and S1P.
To assess the levels of cellular sphingolipids, HaCaT KC were incubated with Tg or C2Cer and washed with phosphate-buffered saline (PBS), followed by extraction of total lipids as we reported previously (10). S1P, sphingosine, and Cer were derivatized with o-phthalaldehyde (OPA) reagent and then quantitated using a high-performance liquid chromatography (HPLC) system equipped with a fluorometric detector system (Jasco, Tokyo, Japan) as described previously (10, 22). Sphingolipid levels are expressed in pmol per mg protein.
qRT-PCR analysis.
Quantitative real-time PCR (qRT-PCR) was performed using cDNAs prepared from mRNA fractions of cell lysates by use of specific primer sets, as we described previously (9). mRNA expression was normalized to levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Western immunoblot analysis.
Western immunoblot analysis was performed as described previously (9). The following antibodies were used: anti-human or anti-mouse β-actin antibody (Abcam, Cambridge, MA), anti-human or anti-mouse cathelicidin (anti-CAMP or anti-mCAMP, respectively) (Santa Cruz Biotech, Santa Cruz, CA, or LifeSpan Biosciences, Seattle, WA), anti-phospho-Ser-225 SPHK1, anti-phosphoTyr-204 or anti-phospho-Thr-202 extracellular signal-regulated kinase 1/2 (ERK1/2) (ECM Biosciences, Versailles, KY), anti-human phospho-Ser-21, anti-phospho-Thr-222/226 C/EBPα (Cell Signaling, Boston, MA), and anti-PARP (BD Sciences, Franklin Lakes, NJ). Equal amounts of protein (30 μg) were loaded per lane.
Quantification of CAMP.
CAMP contents in cell lysates were quantitated using an LL-37 human enzyme-linked immunosorbent assay (ELISA) kit (Hycult Biotech Inc., Plymouth Meeting, PA).
Dual-luciferase reporter assay of NF-κB transcriptional activity.
Transcriptional activities of NF-κB were assessed using reporter kits (SABiosciences, Frederick, MD). HaCaT KC were transfected with NF-κB-responsive luciferase constructs carrying the firefly luciferase reporter gene, as well as a constitutively expressing Renilla luciferase construct, using HilyMax (Dojindo, Rockville, MD). After transfection, cells were pretreated with or without the sphingosine kinase inhibitor DMS for 1 h, followed by stimulation with Tg or S1P for 18 h. Promoter activity was assessed using a dual-luciferase assay system (Promega, Madison, WI).
siRNAs and transfections.
HaCaT KC were transfected with 20 nM small interfering RNA (siRNA) for SPHK1, SPHK2 (Santa Cruz Biotech), or S1P lyase (Qiagen) or with nontargeted, control siRNA (Dharmacon, Lafayette, CO) by use of siLentFect (Bio-Rad, Hercules, CA) as previously described (10).
Ex vivo murine experiments.
Full-thickness pieces of murine skin harvested from mice or S1P lyase-null mice (5-day-old, female C57BL/6 mice [23] used under an Institutional Animal Care and Use Committee [Children's Hospital Oakland Research Institute and San Francisco Veterans Affairs Medical Center]-approved protocol) were placed dermis side down on filter paper and maintained at the air-medium interface in KC growth medium (described above). Tg (0.1 μM) or vehicle (dimethyl sulfoxide [DMSO]) was applied epicutaneously (20 μl/cm2), followed by incubation for 24 h at 37°C in 5% CO2 in air. Skin was separated, followed by extraction of the mRNA or protein, as previously described (16).
Antimicrobial assay.
Antimicrobial activity in KC cell lysates was assessed against S. aureus (ΔmprF strain) as described previously (24). Briefly, S. aureus in 50 mM bicarbonate buffer containing 20% Trypticase soy broth (TSB) (optical density at 600 nm [OD600], 0.6 to 0.8) was incubated with LL-37 (as a positive control) or with cell lysates isolated from KC treated with C2Cer or Tg at 37°C. The growth of S. aureus was determined by measuring absorbance values at 600 nm, using a microplate spectrophotometer.
S. aureus invasion assay.
Full-thickness pieces of murine skin treated with C2Cer (0.5 mM) with or without SKI (1 mM) and/or treated with vehicle (propylene glycol-ethanol [7:3]) were harvested from hairless mice (24 weeks old, female, hr/hr; n = 5) under an Institutional Animal Care and Use Committee (San Francisco Veterans Affairs Medical Center)-approved protocol. The epidermal permeability barrier was attenuated by topical application of topical oxazolone (1%) once every other day for five applications and then treated with C2Cer and/or SKI as described above. Skin was placed dermis side down on filter paper and maintained at the air-medium interface in KC growth medium (as described above). S. aureus (ISP479C) (25) in PBS or PBS alone was applied epicutaneously (20 μl/cm2), followed by incubation for 8 or 24 h at 37°C in 5% CO2 in air. Gram staining was performed to assess S. aureus invasion.
Statistical analyses.
Statistical comparisons were performed using an unpaired Student t test.
RESULTS
ER stress accelerates formation of Cer and its downstream metabolites in parallel with enhanced CAMP production.
External perturbations that induce ER stress, such as UV irradiation, also upregulate murine CAMP (mCAMP; the murine homologue of human CAMP) (7), and ER stress alone stimulates production of Cer (10, 12). As in our prior studies (9), we employed cultured human KC as a model of epithelial cells to identify whether Cer and/or its distal metabolites are responsible for the ER stress-initiated increases in CAMP expression. First, we ascertained whether levels of cellular Cer and its distal metabolites, sphingosine and S1P, increase following exposure to subtoxic doses of Tg, a specific pharmacological ER stressor (26). Lipid quantification demonstrated an increase not only in cellular Cer but also in sphingosine and S1P in KC subjected to ER stress (Table 1), and the changes were comparable to those induced by subtoxic (low-dose) UVB (10). These studies show that subtoxic levels of ER stress stimulate elevation of not only Cer but also its distal metabolites, sphingosine and S1P, to levels potentially linking ER stress to signaling of CAMP production via Cer and/or its metabolite(s).
Table 1.
Sphingolipid content in human KC exposed to ER stress
| Treatment | Lipid content (pmol/mg protein)a |
||
|---|---|---|---|
| Cer | Sphingosine | S1P | |
| Vehicle | 672.14 ± 55.50 | 53.53 ± 5.61 | 12.53 ± 0.41 |
| Tg | 1,107.82 ± 38.57* | 80.38 ± 4.46* | 17.47 ± 0.24* |
| Vehicle | 711.54 ± 35.99 | 46.96 ± 2.99 | 11.93 ± 0.78 |
| C2Cer | 11,448.3 ± 953.65* | 418.63 ± 24.88* | 14.21 ± 0.51* |
Data are means ± SD. *, P < 0.01 (n = 3) versus vehicle.
To assess the direct effects of Cer and/or its metabolites on CAMP expression, we next incubated KC with subtoxic concentrations of exogenous, cell-permeating short-chain Cer, which is a precursor that leads to increased intracellular long-chain Cer (27). As expected, exogenous, cell-permeative Cer (N-acetylsphingosine [C2Cer]) (7.5 μM) increased not only cellular long-chain Cer but also sphingosine and S1P levels (Table 1), without altering cell viability or inducing apoptosis, which was assessed as PARP cleavage (Fig. 1A and B). qRT-PCR analysis revealed significant increases in CAMP mRNA levels in KC incubated not only with Tg (Fig. 1C) but also with exogenous C2Cer (Fig. 1C and D). Western immunoblot and ELISA analyses showed that CAMP protein levels in cell lysates also increased following treatment with subtoxic doses of either Tg or C2Cer (Fig. 1E and F). Together, these results show that subtoxic levels of ER stress stimulate production of cellular Cer and its downstream metabolites, suggesting that either Cer itself or one of its downstream metabolites can regulate CAMP expression in KC.
Fig 1.

S1P, but neither Cer nor sphingosine, is responsible for stimulation of CAMP expression. HaCaT KC pretreated with or without ceramidase (25 μM NOE) or SPHK inhibitors (2.5 μM DMS and 1 μM SKI) for 30 min were incubated with or without exogenous C2Cer (7.5 μM), S1P (1 μM), or Tg (0.1 μM) for 24 h. Cell viability was determined by a trypan blue dye exclusion assay (A) or PARP cleavage as a measure of apoptosis (B). CAMP mRNA and protein expression was assessed by qRT-PCR (C and D), Western immunoblotting (E), and ELISA detection of a cleaved active form of CAMP (LL-37) (F). Protein levels were assessed using cell lysates. Similar results were obtained when the experiment was repeated in triplicate, using different cell preparations. Data are means ± standard deviations (SD) (n = 3; P < 0.01 [n = 2 for panel F]).
S1P is the sphingolipid metabolite that signals the increase in CAMP.
We next assessed whether Cer itself or one or more of its distal metabolites was responsible for increased CAMP expression. Blockade of ceramidase by use of only NOE, a specific and broad pharmacological inhibitor of acidic, neutral, and alkaline ceramidases, did not alter endogenous CAMP mRNA levels in KC (Fig. 1C). NOE cotreatment with exogenous C2Cer, however, significantly attenuated the expected C2Cer-induced increase in CAMP mRNA and protein expression (Fig. 1C and D), suggesting that a Cer metabolite likely regulates CAMP expression.
To further distinguish which Cer metabolite, i.e., sphingosine and/or S1P, is the signal responsible for increased CAMP expression, we next blocked the conversion of sphingosine to S1P by using a pan-sphingosine kinase inhibitor, DMS. While exogenous C2Cer increased CAMP mRNA and protein expression, DMS treatment significantly attenuated the C2Cer-mediated increase in these CAMP expression levels (Fig. 1D and E). Moreover, SKI, another highly specific inhibitor of sphingosine kinase (28), also attenuated the expected upregulation of CAMP protein expression (Fig. 1E and F), further suggesting that S1P accounts for the stimulation of CAMP expression. Finally, the ER stress-induced increase in CAMP mRNA and protein expression was significantly attenuated in KC cotreated with either DMS or SKI (Fig. 1C, E, and F).
To further investigate the role of S1P and also to identify which SPHK isoform, i.e., SPHK1 or -2, generates the S1P that regulates CAMP expression, we next transfected KC with siRNA against either SPHK1 or SPHK2. Both SPHK1 and SPHK2 mRNA expression levels were significantly suppressed (≥50% versus control scrambled siRNA) in HaCaT KC transfected with siRNA against either SPHK1 or SPHK2 (Fig. 2A). Moreover, the silencing effects of siRNA against either SPHK1 or SPHK2 were specific, i.e., siRNA against SPHK1 or SPHK2 did not suppress the other isoform of SPHK (Fig. 2A). Only silencing of SPHK1, not that of SPHK2, significantly attenuated CAMP expression in cells that were either subjected to ER stress or treated with exogenous C2Cer (Fig. 2B and C). In contrast, siRNA against SPHK2 modestly increased CAMP mRNA expression, suggesting an opposing role for SPHK2 in the downregulation of CAMP (Fig. 2B). Moreover, siRNA against SPHK2 did not suppress the expected C2Cer- or Tg-mediated increases in CAMP expression (Fig. 2B). Finally, we investigated whether ER stress regulates SPHK1 at a transcriptional level and/or whether it stimulates catalytic activity by phosphorylating SPHK1. Western immunoblot analysis revealed a transient phosphorylation of SPHK1 following ER stress, induced by treatment of KC with Tg (Fig. 2D), while in contrast, qRT-PCR analysis showed no significant changes in SPHK1 mRNA levels (1.45-fold versus vehicle control). Together, these results confirm that (i) ER stress-mediated S1P signaling accounts for CAMP upregulation in response to external perturbations known to increase ER stress, (ii) SPHK1 is the kinase isoform that generates the S1P responsible for the ER stress-mediated stimulation of CAMP expression, and (iii) posttranscriptional upregulation of SPHK1 catalytic activity likely accounts for increased S1P production.
Fig 2.

SPHK1 plays a critical role to stimulate CAMP expression. HaCaT KC cells transfected with scrambled-, SPHK1-, or SPHK2-siRNA were exposed to ER stress or exogenous C2Cer (7.5 μM) treatment as shown in Fig. 1. (A and B) The mRNA levels of SPHK and CAMP were determined by qRT-PCR. (C and D) CAMP protein and expression of CAMP and SPHK1 phosphorylation in KC following ER stress generated by Tg were assessed by Western immunoblot analysis. Fold changes compared with the zero time point are shown in parentheses. Data are means ± SD (n = 3; P < 0.01).
We next assessed the role of S1P by another approach, i.e., blockade of the irreversible catabolism of S1P by S1P lyase by use of siRNA. Initial studies showed that siRNA significantly suppressed S1P lyase mRNA expression (to 27% of that with control scrambled siRNA). In KC previously transduced with siRNA against S1P lyase, basal S1P levels were significantly increased and were further elevated following ER stress induced by either Tg or C2Cer treatment compared with the levels for control, scrambled siRNA-treated cells (Table 2). Next, qPCR and Western immunoblot analyses showed that CAMP mRNA expression was not increased in KC transfected with siRNA for S1P lyase under basal conditions (Fig. 3A), while CAMP protein levels were elevated in these cells (Fig. 3B). Exposure to either Tg or C2Cer enhanced CAMP mRNA and protein expression in KC transduced with S1P lyase siRNA versus scrambled siRNA (Fig. 3A and B). Hence, distal metabolites of S1P, such as phosphoethanolamine and hexadecenal, do not regulate CAMP expression, further pointing to the central role of cellular S1P in stimulating CAMP production. These results also suggest that feedback regulation of CAMP transcription likely occurs by accumulation of CAMP protein in cultured KC under basal conditions.
Table 2.
Blockade of S1P lyase further increases S1P content
| siRNA | Treatment | S1P content (pmol/mg protein)a |
|---|---|---|
| Scrambled (control) | Vehicle | 12.35 ± 0.60 |
| Tg | 14.81 ± 0.20* | |
| Vehicle | 10.71 ± 2.11 | |
| C2Cer | 18.54 ± 0.68* | |
| S1P lyase | Vehicle | 33.00 ± 0.83* |
| Tg | 41.73 ± 0.56*, ** | |
| Vehicle | 36.32 ± 0.39* | |
| C2Cer | 60.35 ± 3.67*, ** |
Data are means ± SD. Significance is indicated for P values of <0.01 (n ≥ 3). *, versus scrambled (control) siRNA plus vehicle; **, versus S1P lyase siRNA plus vehicle.
Fig 3.

Blockade of S1P lyase further enhances human and murine CAMP expression. HaCaT KC transfected with S1P lyase siRNA or scrambled control siRNA were incubated with or without exogenous C2Cer (7.5 μM) or Tg (0.1 μM) for 24 h. (A) Excised murine skin was exposed either to vehicle or to ER stress induced by Tg (see details in Materials and Methods). CAMP or mCAMP (a murine homologue of CAMP) mRNA (A and C) and protein (B and D) expression was determined by qRT-PCR and Western immunoblot analysis, respectively. mCathelin#, cleaved peptide of mCAMP. Data are means ± SD (n = 3). *, P < 0.01; **, P < 0.03.
To validate these in vitro studies using an in vivo setting, we next induced ER stress in wild-type mouse skin by topical applications of Tg, which increased mCAMP mRNA and protein expression in mouse skin (Fig. 3C and D) (9). Under basal conditions, mCAMP mRNA expression was significantly higher in S1P lyase-deficient mouse skin than in wild-type skin, while neither mCAMP nor its catabolite, mCathelin, accumulated (Fig. 3C and D). Moreover, topical Tg modestly but significantly increased mCAMP mRNA levels (Fig. 3C) and significantly elevated mCAMP and mCathelin peptide levels in knockout mouse skin (Fig. 3D). These additional studies show that S1P, rather than phosphoethanolamine or hexadecenal, upregulates CAMP production not only in vitro but also in vivo. Moreover, these results also suggest that despite sustained elevations of basal levels of S1P content and mCAMP mRNA, a further significant increase in mCAMP protein levels occurs only in response to ER stress in S1P lyase-deficient mouse skin. A prior study showed that synthesized CAMP/LL-37 (mCathelin) is secreted into the extracellular fraction of the stratum corneum in epidermis (29), while CAMP secretion was not evident in the undifferentiated cultured keratinocytes used in this study, which also do not contain a stratum corneum. Therefore, the pools of CAMP/LL-37 (in vitro) (Fig. 3B) versus mCathelin (ex vivo) (Fig. 3D) are likely different, i.e., reflecting the intracellular pool of CAMP/LL-37 in cultured cells versus primarily an extracellular pool in whole epidermis (further details are presented in Discussion).
Stimulation of S1P signaling inhibits Staphylococcus aureus growth and invasion.
We next explored the physiological relevance of the S1P-mediated increase in CAMP production by measuring its net impact on antimicrobial defense, using two different approaches. In the first approach, a virulent S. aureus strain (ΔmprF strain) was incubated with cell lysates isolated from KC previously treated with either exogenous C2Cer, Tg, or vehicle alone. These treatments increased CAMP production 7.8-fold (by C2Cer) and 9.4-fold (by Tg), but neither C2Cer nor Tg significantly increased mRNA levels of another major epidermal AMP, i.e., human β-defensin 2 (hBD2) (for C2Cer, 0.93- ± 0.1-fold versus vehicle; and for Tg, 0.90- ± 0.2-fold versus vehicle). S. aureus growth was significantly suppressed by lysates from both C2Cer- and Tg-treated cells in comparison to lysates from vehicle-treated cells, and the inhibition of growth was comparable to growth inhibition in cells exposed to synthetic LL-37 and S. aureus (Fig. 4A).
Fig 4.

Inhibition of growth and invasion of S. aureus mediated by S1P-induced CAMP expression. (A) In vitro growth inhibition studies were performed as follows. S. aureus was incubated for the indicated times with LL-37 or HaCaT KC lysates treated with vehicle, C2Cer (7.5 μM), or Tg (0.1 μM). The growth of S. aureus was assessed by measuring absorbance values at 600 nm. Data are means ± SD (n = 3). *, P < 0.01 versus vehicle-treated cell lysates. (B and C) Ex vivo bacterial invasion studies were performed as follows. S. aureus was applied epicutaneously to full-thickness pieces of murine skin (n = 2) treated with C2Cer (0.5 mM), SKI (1 mM), and/or vehicle, followed by incubation for 24 h at 37°C. (B) CAMP mRNA expression in skin was assessed by qRT-PCR. (C) Bacterial invasion/growth into murine skin was assessed by Gram staining (counterstaining with hematoxylin and eosin [H&E]).
We next addressed whether S1P-mediated CAMP production blocks S. aureus (ISP479C) invasion into murine skin. Since normal epidermis, with its competent permeability and antimicrobial barrier, prevents S. aureus invasion, we also assessed invasions into skin whose barrier had been compromised by topical oxazolone. As expected, mCAMP mRNA expression was significantly increased in C2Cer-treated skin, and this induction was blocked by coapplications of a specific sphingosine kinase inhibitor, SKI (Fig. 4B). Gram staining revealed S. aureus invasion into vehicle- and SKI-treated skin (indicated by arrows in Fig. 4C), confirming that S. aureus invasion occurs in murine skin and that blockade of S1P production allows bacterial invasion. In contrast, S. aureus invasion was decreased in skin treated with topical C2Cer as well as in samples cotreated with synthetic LL-37 (positive control) (Fig. 4C, panels d and c, respectively). Finally, blockade of S1P production by coapplication of SKI attenuated C2Cer-mediated inhibition of bacterial invasion (Fig. 4C, panel e). Finally, as in cultured cells, neither topical C2Cer nor SKI altered mBD3 (a homologue of hBD2) mRNA expression (not shown). Since neither hBD2 nor other known epidermal antimicrobial peptides potently inhibit S. aureus growth (30), the observed suppression of S. aureus growth likely can be attributed to a S1P-signaled increase in CAMP production.
S1P regulation of CAMP expression is likely receptor independent.
We next explored whether increased S1P-mediated CAMP expression is receptor dependent. Since all five S1P receptors are expressed in KC (31) and prior studies have shown that both S1P1 and S1P2 receptor ligands activate NF-κB (32, 33), one or both of these receptors could also mediate increased CAMP expression. Hence, we next investigated whether any of the following would alter CAMP expression: (i) a pan-S1P receptor activator, sphinganine (dihydrosphingosine)-1-phospate (dhS1P); (ii) a specific S1P1 ligand (SEW2871); (iii) a specific S1P1 and S1P3 antagonist (VPC23019); (iv) pertussis toxin, an inhibitor of G-protein-coupled S1P (except S1P2) receptors (34); and (v) a specific S1P2 antagonist (JTE-013). As in other cell types (35), exogenous dhS1P and SEW2871 increased the phosphorylation of ERK1/2, which is an indicator of S1P receptor activation in cells, suggesting that these ligands activate all five S1P receptors, including S1P1, in this KC line (Fig. 5). Yet exogenous SEW2871 and dhS1P, added at the same concentration, did not alter CAMP mRNA levels (Table 3). Furthermore, neither the S1P1/S1P3 antagonist (VPC23019), the S1P1/S1P3/S1P4/S1P5 inhibitor (pertussis toxin), nor the S1P2 antagonist (JTE-013) altered ER stress-induced CAMP expression (Table 3). Together, these results strongly suggest that the S1P-induced increase in CAMP expression occurs in an S1P receptor-independent fashion.
Fig 5.

S1P1 ligand stimulates ERK1/2 phosphorylation in human KC. HaCaT KC were incubated with the S1P1 ligand SEW2871 (1 μM) (A) or with dihydrosphingosine-1-phosphate (dhS1P) (B) for the indicated times. Phosphorylated forms of ERK1/2 were assessed by Western immunoblot analysis.
Table 3.
S1P receptor-independent pathway accounts for regulation of CAMP expression
| Treatment | Relative CAMP mRNA expression (vs vehicle control) |
|---|---|
| Vehicle | 1.00 ± 0.03 |
| SEW2871 (1 μM) | 0.94 ± 0.06 |
| Vehicle | 1.00 ± 0.09 |
| dhS1P (0.1 μM) | 1.03 ± 0.10 |
| Vehicle | 1.00 ± 0.04 |
| VPC23019 (0.5 μM) | 1.40 ± 0.16 |
| Tg (0.1 μM) | 3.88 ± 0.37* |
| Tg + VPC23019 | 4.10 ± 0.27* |
| Vehicle | 1.00 ± 0.04 |
| JTE-013 (1 μM) | 0.85 ± 0.07 |
| Tg | 6.71 ± 0.63* |
| Tg + JTE-013 | 7.36 ± 0.42* |
| Vehicle | 1.00 ± 0.03 |
| Ptx (12.5 ng/ml) | 1.17 ± 0.15 |
| Tg | 6.15 ± 0.25* |
| Tg + Ptx | 7.69 ± 0.43* |
| C2Cer (7.5 μM) | 6.69 ± 1.18* |
| C2Cer + Ptx | 8.16 ± 0.51* |
| Vehicle | 1.00 ± 0.04 |
| Ptx | 1.32 ± 0.04 |
| S1P (1 μM) | 2.96 ± 0.66* |
| S1P + Ptx | 3.47 ± 0.42* |
Data are means ± SD. *, P < 0.01 (n = 3) versus vehicle.
S1P stimulation of CAMP expression requires NF-κB and C/EBPα activation.
Since the studies above indicated that S1P stimulates CAMP in a receptor-independent fashion, we next asked which intracellular mechanism(s) accounts for CAMP upregulation. Our prior studies showed that the ER stress-induced increase in CAMP requires (i) phosphorylation of NF-κB transactivity and (ii) NF-κB-mediated C/EBPα phosphorylation by mitogen-activated protein (MAP) kinase (9). Accordingly, blockade of S1P generation with either DMS or SKI attenuated the expected ER stress (induced by two mechanistically different established pharmacological ER stressors, Tg and tunicamycin [36]) or showed a C2Cer-induced increase in phospho-NF-κB generation and transactivation (Fig. 6A and B). Moreover, preincubation of KC with BAY11-7082, a specific inhibitor of NF-κB, suppressed the C2Cer (→ long-chain Cer → S1P)-mediated increase in CAMP mRNA levels (Fig. 6C). Together, these results suggest that ER stress-induced increases in S1P activate NF-κB, leading to increasing CAMP expression.
Fig 6.

NF-κB–C/EBPα activation is required for S1P-induced upregulation of CAMP expression. HaCaT KC were incubated with or without C2Cer, S1P (1 μM), Tg, tunicamycin (Tm), SPHK inhibitors (DMS [2.5 μM] and SKI [1 μM]), and/or an NF-κB inhibitor (2 μM BAY11-7082) as shown in Fig. 1. (A and D) Phosphorylated forms of NF-κB or C/EBPα (either Ser-21 [Ser21] or Thr-222/226 [T222/226]) in nuclear fractions were assessed by Western immunoblot analysis. Fold changes compared with vehicle are shown in parentheses in panel A. (B) NF-κB transactivation was assessed using a luciferase reporter assay. The growth inhibition at 7 h of incubation is shown. (C) CAMP mRNA levels were determined by qRT-PCR. Similar results were obtained when the experiment was repeated (duplicated) using different cell preparations. Data are means ± SD (n = 3; P < 0.01).
Since C/EBPα phosphorylation is also required for ER stress-induced CAMP expression in KC (9), we next assessed the relationship between S1P signaling and C/EBPα phosphorylation. Western immunoblot analysis of C/EBPα revealed that S1P stimulated the phosphorylation of C/EBPα at both the Ser-21 and Thr-222/226 sites (37, 38) (Fig. 6D). In contrast, the addition of either DMS or SKI selectively decreased the phosphorylation of the Thr-222/226 sites, while the Ser-21 phosphorylation site remained unaffected. Yet Ser-21, not Thr-222/226, phosphorylation was increased by SPHK inhibition (Fig. 6D). Together, these results suggest that the ER stress-initiated S1P signal stimulates NF-κB transactivation, and also that Thr-222/226 rather than Ser-21 phosphorylation of C/EBPα is important for the S1P-dependent increase in CAMP expression (Fig. 6D).
DISCUSSION
External perturbations, such as UVB irradiation, permeability barrier perturbation, wounding, or pathogen invasions, provoke ER stress (3, 9, 39), which in turn initiates a set of rescue mechanisms that protect epithelial cells from cell death (4). Our prior studies delineated this external perturbation (ER stress)-induced Cer → S1P rescue response and its ability to protect cells against Cer-induced apoptosis (10). We showed in addition that ER stress stimulates CAMP production (9). Pertinent to our findings, one recent study also showed that ER stress-initiated XBP-1-mediated signaling stimulates innate immunity in Caenorhabditis elegans (40, 41), but the details of the pathway by which ER stress upregulates CAMP expression in that system remained unresolved. Our studies now illuminate the metabolic pathway that links the ER stress-initiated rescue mechanism to enhanced epithelial innate immunity. Specifically, we now show that S1P is the sphingolipid metabolite that stimulates CAMP production in response to ER stress. We further illuminated several details of this novel regulatory mechanism of CAMP expression, including how it operates independently of the established VDR mechanism (42). While the latter process likely predominates under basal (nonstressed) conditions, we demonstrated here that the same subtoxic levels of ER stress that stimulate CAMP production also enhance S1P signaling of CAMP, independent of the VDR. Importantly, S1P itself can induce ER stress under similar circumstances (43), but exogenous S1P did not induce ER stress in our system (assessed here by XBP1 activation in KC) (Fig. 7). Thus, the increase in S1P that occurs in response to ER stress likely activates CAMP expression via the pathway ER stress → ↑S1P → ↑CAMP rather than S1P → ↑ER stress → ↑CAMP. Because epithelial tissues (in particular skin) continuously face external perturbations, including frequent exposure to microbial pathogens that in turn induce ER stress, this rescue mechanism likely boosts innate immune defense in a fashion that is important for host survival. Indeed, enhanced CAMP/LL-37 production could rescue cells by multiple mechanisms that extend well beyond antimicrobial defense (1, 2), because LL-37 is a multifunctional peptide that also modulates cytokine production, cellular differentiation, and adaptive immunity, including differentiation of both macrophages (44) and dendritic cells (45).
Fig 7.
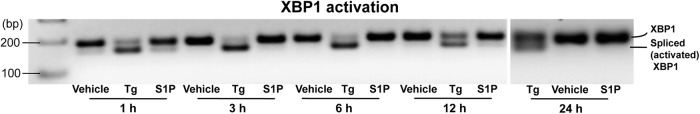
Exogenous S1P does not induce ER stress. HaCaT KC were incubated with exogenous S1P (10 μM) or Tg (0.1 μM) for 24 h. ER stress was assessed by XBP1 activation using RT-PCR.
Prior studies have shown that the ER stress-induced rescue mechanism includes a UPR that consists of three stress sensors: (i) inositol-requiring enzyme 1 (IRE1)–tumor necrosis factor receptor-associated factor 2 (TRAF2), (ii) protein kinase R-like ER kinase (PERK)–eukaryotic initiation factor 2α (eIF2α), and (iii) activating transcription factor 6 (ATF 6)-AKT (or protein kinase B). While this mechanism allows cell survival in the face of subtoxic perturbations, excessive ER stress can overwhelm these homeostatic mechanisms, leading to cell death. Both toxic and subtoxic levels of ER stress stimulate production of the proapoptotic lipid species Cer, but in the case of subtoxic stress, newly generated Cer is rapidly converted to at least two nontoxic Cer metabolites, including glucosyl-Cer and S1P (10, 16). Only the latter stimulates innate immunity, while also protecting epidermal cells from apoptosis. We showed that pharmacologically induced subtoxic ER stress, induced by using exogenous cell-permeative Cer (which first increases endogenous long-chain Cer levels and subsequently increases S1P generation), as well as exogenous S1P itself stimulates CAMP mRNA and protein expression. Moreover, using both specific pharmacological inhibitors and siRNAs against SPHKs, we showed not only that S1P is the responsible signal but also that the ER stress-mediated increase in S1P is SPHK1 dependent, while silencing of SPHK2 mRNA expression instead stimulates CAMP expression. Thus, S1P generated by SPHK2 could serve as a negative regulator of CAMP expression. Prior studies have shown that S1P generated by SPHK2 can subsequently be dephosphorylated by S1P phosphatase to generate sphingosine, followed by Cer resynthesis in the ER (17). Thus, SPHK1 and SPHK2 could regulate CAMP oppositely within different intracellular compartments.
While some S1P-mediated immune effects are S1P receptor dependent (46), our results demonstrate that regulation of innate immunity via CAMP generation likely occurs by an S1P receptor-independent mechanism. Specifically, neither an activator of all five S1P receptors (S1P1 to S1P5), dhS1P (47–49), nor a specific S1P1 ligand (SEW2871) altered the S1P-induced increase in CAMP expression. Moreover, blockade of S1P1/S1P3, S1P2, or S1P1/S1P3/S1P4/S1P5 receptors did not impede the S1P-induced increase in CAMP expression. Instead of a receptor-dependent mechanism, we show here that S1P activates NF-κB, subsequently increasing C/EBPα phosphorylation to upregulate CAMP expression (Fig. 6). Whereas the phosphorylation of two sites on C/EBPα (Ser-21 and Thr-222/226) and CAMP expression declined in cells treated with an NF-κB inhibitor (9), blockade of sphingosine-to-S1P conversion attenuated only Thr-222/226 (but not Ser-21) phosphorylation. Prior studies showed that Ser-21 phosphorylation downregulates transcriptional activities by increasing conformational changes in the C/EBPα structure (37). Thus, sphingosine may serve as a negative regulator of CAMP expression by suppressing Thr-222/226 phosphorylation (Fig. 6D).
Our studies in S1P lyase-deficient mice are relevant because they prove that (i) a sustained elevation in S1P increases basal CAMP mRNA (but not CAMP protein) levels and (ii) the same S1P-dependent mechanism is operative in epithelial tissues in vivo. Prior studies have demonstrated that an extracellular mechanism also regulates, albeit by an undefined mechanism(s), CAMP levels in vivo, i.e., CAMP is processed to the functional antimicrobial peptide LL-37 (or CRAMP, a homologue of LL-37, in mice) and cathelin by a serine protease (29). LL-37/CRAMP and cathelin are further hydrolyzed by proteases into hydrolytic products in the extracellular domains of the outermost layers of the epidermis, the stratum corneum (29). Moreover, increased epidermal turnover could affect CAMP/LL-37 levels in epidermis. In contrast to the in vivo/ex vivo system, the cultured KC employed in these studies do not recapitulate a highly differentiated epidermis, i.e., neither generating the stratum corneum nor secreting CAMP. Thus, sustained elevated S1P upregulates CAMP mRNA levels, leading to increased CAMP protein production at the basal level, but the protein and peptides can be secreted and regulated by external domains in the stratum corneum. In contrast, since CAMP is not secreted in vitro, sustained stimulation of CAMP expression by S1P results in accumulated CAMP protein in cells (Fig. 3B) and may downregulate CAMP transcription (negative-feedback regulation) at the basal level (Fig. 3A). Under stressed conditions, a feedback downregulation of CAMP transcription in response to accumulation of CAMP protein could be attenuated, i.e., increased CAMP mRNA expression, in cultured cells. The difference between in vivo (S1P lyase knockout mice) and in vitro (cultured undifferentiated KC) results is likely due to these differences in in vivo and in vitro conditions. Elucidation of the full spectrum of mechanisms that can regulate CAMP/LL-37 in vivo is important. Yet elucidation of the contribution from posttranscriptional regulatory mechanisms was not a focus in our present study to investigate the upstream signal of CAMP transcription in response to ER stress.
While multiple biological functions have been attributed to S1P (50), the present study illuminates an entirely new and potentially important role for S1P in modulating innate immune function through the regulation of CAMP production. Because CAMP/LL-37 is a multifunctional antimicrobial peptide, tight regulation of CAMP production could be important for the maintenance of epithelial functions under normal (basal) conditions. Since CAMP reportedly displays many of the same biological activities, e.g., modulation of cytokine secretion/production, angiogenesis, and adaptive immunity, it is pertinent to ask whether many of the functions currently ascribed to CAMP should instead be attributed, at least in part, to S1P (Fig. 8). Recent studies have demonstrated that ER stress signals are required for normal human KC differentiation (51), while S1P stimulates differentiation in KC (31). S1P might alter KC differentiation through CAMP/LL-37 production. Thus, ER stress-mediated increases in S1P might help to maintain KC differentiation in parallel with enhanced antimicrobial defense. These studies also suggest that stimulation of the S1P signal by nonapoptotic levels of external stress, ER stress, and/or metabolic modulation of S1P, i.e., by cell-permeative Cer, could be a therapeutic approach to enhance the endogenous antimicrobial defense in epithelia, including lung and gut epithelia as well as the skin.
Fig 8.
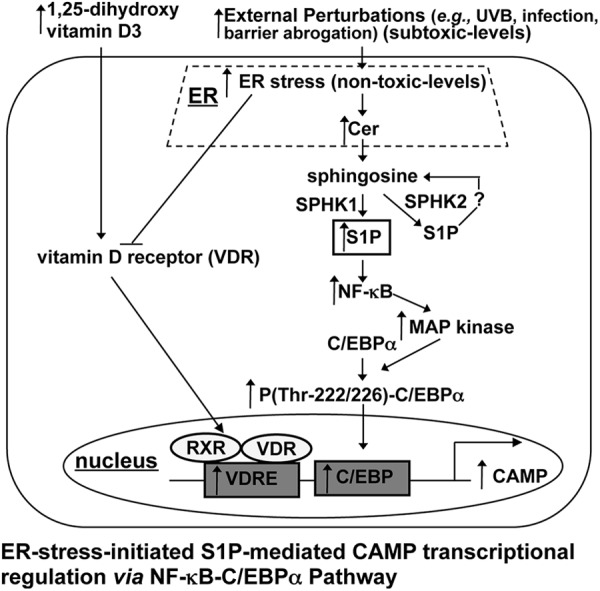
Proposed mechanism of S1P-mediated induction of CAMP in human KC.
ACKNOWLEDGMENTS
We are indebted to Sally Pennypacker (Northern California Institute for Research and Education and Veterans Affairs Medical Center, San Francisco, CA) and Meng Zhang (Children's Hospital Oakland Research Institute) for technical support in cell culture and in animal studies, respectively. We thank Paul M. Sullam and Ho Seong Seo (Northern California Institute for Research and Education and Veterans Affairs Medical Center, San Francisco, CA) for technical support and advice on bacterial invasion studies. We gratefully acknowledge Sarah Spiegel for numerous critical discussions and advice. We thank Joan Wakefield for superb editorial assistance.
We gratefully acknowledge the support of the Medical Research Services of the Veterans Affairs Medical Center, San Francisco (merit review grant to P.M.E.). This study was supported by a REAC award from the University of California, San Francisco, by the National Rosacea Society, and by National Institutes of Health grants AR051077 and AR062025 (the National Institute of Arthritis and Musculoskeletal and Skin Diseases) to Y.U. and CA77528 and CA129438 to J.S.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We declare that no competing interests exist.
Footnotes
Published ahead of print 10 December 2012
REFERENCES
- 1. Nijnik A, Hancock RE. 2009. The roles of cathelicidin LL-37 in immune defences and novel clinical applications. Curr. Opin. Hematol. 16:41–47 [DOI] [PubMed] [Google Scholar]
- 2. Schauber J, Gallo RL. 2009. Antimicrobial peptides and the skin immune defense system. J. Allergy Clin. Immunol. 124:R13–R18 [DOI] [PubMed] [Google Scholar]
- 3. Celli A, Mackenzie DS, Crumrine DS, Tu CL, Hupe M, Bikle DD, Elias PM, Mauro TM. 2011. Endoplasmic reticulum Ca(2+) depletion activates XBP1 and controls terminal differentiation in keratinocytes and epidermis. Br. J. Dermatol. 164:16–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mera K, Kawahara K, Tada K, Kawai K, Hashiguchi T, Maruyama I, Kanekura T. 2010. ER signaling is activated to protect human HaCaT keratinocytes from ER stress induced by environmental doses of UVB. Biochem. Biophys. Res. Commun. 397:350–354 [DOI] [PubMed] [Google Scholar]
- 5. Aberg KM, Man MQ, Gallo RL, Ganz T, Crumrine D, Brown BE, Choi EH, Kim DK, Schroder JM, Feingold KR, Elias PM. 2008. Co-regulation and interdependence of the mammalian epidermal permeability and antimicrobial barriers. J. Invest. Dermatol. 128:917–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carretero M, Escamez MJ, Garcia M, Duarte B, Holguin A, Retamosa L, Jorcano JL, Rio MD, Larcher F. 2008. In vitro and in vivo wound healing-promoting activities of human cathelicidin LL-37. J. Invest. Dermatol. 128:223–236 [DOI] [PubMed] [Google Scholar]
- 7. Hong SP, Kim MJ, Jung MY, Jeon H, Goo J, Ahn SK, Lee SH, Elias PM, Choi EH. 2008. Biopositive effects of low-dose UVB on epidermis: coordinate upregulation of antimicrobial peptides and permeability barrier reinforcement. J. Invest. Dermatol. 128:2880–2887 [DOI] [PubMed] [Google Scholar]
- 8. Mendez-Samperio P. 2010. The human cathelicidin hCAP18/LL-37: a multifunctional peptide involved in mycobacterial infections. Peptides 31:1791–1798 [DOI] [PubMed] [Google Scholar]
- 9. Park K, Elias PM, Oda Y, Mackenzie D, Mauro T, Holleran WM, Uchida Y. 2011. Regulation of cathelicidin antimicrobial peptide expression by an endoplasmic reticulum (ER) stress signaling, vitamin D receptor-independent pathway. J. Biol. Chem. 286:34121–34130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Uchida Y, Houben E, Park K, Douangpanya S, Lee YM, Wu BX, Hannun YA, Radin NS, Elias PM, Holleran WM. 2010. Hydrolytic pathway protects against ceramide-induced apoptosis in keratinocytes exposed to UVB. J. Invest. Dermatol. 130:2472–2480 [DOI] [PubMed] [Google Scholar]
- 11. Uchida Y, Nardo AD, Collins V, Elias PM, Holleran WM. 2003. De novo ceramide synthesis participates in the ultraviolet B irradiation-induced apoptosis in undifferentiated cultured human keratinocytes. J. Invest. Dermatol. 120:662–669 [DOI] [PubMed] [Google Scholar]
- 12. Lei X, Zhang S, Bohrer A, Ramanadham S. 2008. Calcium-independent phospholipase A2 (iPLA2 beta)-mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. J. Biol. Chem. 283:34819–34832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hannun YA, Obeid LM. 2002. The ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J. Biol. Chem. 277:25847–25850 [DOI] [PubMed] [Google Scholar]
- 14. Zhang K, Kaufman RJ. 2008. From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang K, Kaufman RJ. 2008. Identification and characterization of endoplasmic reticulum stress-induced apoptosis in vivo. Methods Enzymol. 442:395–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Charruyer A, Bell SM, Kawano M, Douangpanya S, Yen TY, Macher BA, Kumagai K, Hanada K, Holleran WM, Uchida Y. 2008. Decreased ceramide transport protein, cert, function alters sphingomyelin production following UVB irradiation. J. Biol. Chem. 283:16682–16692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH, Jr, Milstien S, Spiegel S. 2005. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 280:37118–37129 [DOI] [PubMed] [Google Scholar]
- 18. Hagen N, Hans M, Hartmann D, Swandulla D, van Echten-Deckert G. 2011. Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell Death Differ. 18:1356–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanchez T, Hla T. 2004. Structural and functional characteristics of S1P receptors. J. Cell. Biochem. 92:913–922 [DOI] [PubMed] [Google Scholar]
- 20. Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, Spiegel S. 2009. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325:1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, Milstien S, Spiegel S. 2010. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465:1084–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee YS, Choi KM, Choi MH, Ji SY, Yoo JM, Lee YM, Hong JT, Yun YP, Yoo HS. 2009. Simultaneous HPLC analysis of ceramide and dihydroceramide in human hairs. Arch. Pharm. Res. 32:1795–1801 [DOI] [PubMed] [Google Scholar]
- 23. Bandhuvula P, Honbo N, Wang GY, Jin ZQ, Fyrst H, Zhang M, Borowsky AD, Dillard L, Karliner JS, Saba JD. 2011. S1P lyase: a novel therapeutic target for ischemia-reperfusion injury of the heart. Am. J. Physiol. Heart Circ. Physiol. 300:H1753–H1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bernard JJ, Gallo RL. 2010. Cyclooxygenase-2 enhances antimicrobial peptide expression and killing of Staphylococcus aureus. J. Immunol. 185:6535–6544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Siboo IR, Cheung AL, Bayer AS, Sullam PM. 2001. Clumping factor A mediates binding of Staphylococcus aureus to human platelets. Infect. Immun. 69:3120–3127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Inesi G, Wade R, Rogers T. 1998. The sarcoplasmic reticulum Ca2+ pump: inhibition by thapsigargin and enhancement by adenovirus-mediated gene transfer. Ann. N. Y. Acad. Sci. 853:195–206 [DOI] [PubMed] [Google Scholar]
- 27. Sultan I, Senkal CE, Ponnusamy S, Bielawski J, Szulc Z, Bielawska A, Hannun YA, Ogretmen B. 2006. Regulation of the sphingosine-recycling pathway for ceramide generation by oxidative stress, and its role in controlling c-Myc/Max function. Biochem. J. 393:513–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ren S, Xin C, Pfeilschifter J, Huwiler A. 2010. A novel mode of action of the putative sphingosine kinase inhibitor 2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole (SKI II): induction of lysosomal sphingosine kinase 1 degradation. Cell. Physiol. Biochem. 26:97–104 [DOI] [PubMed] [Google Scholar]
- 29. Yamasaki K, Schauber J, Coda A, Lin H, Dorschner RA, Schechter NM, Bonnart C, Descargues P, Hovnanian A, Gallo RL. 2006. Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J. 20:2068–2080 [DOI] [PubMed] [Google Scholar]
- 30. Schroder JM, Harder J. 1999. Human beta-defensin-2. Int. J. Biochem. Cell Biol. 31:645–651 [DOI] [PubMed] [Google Scholar]
- 31. Vogler R, Sauer B, Kim DS, Schafer-Korting M, Kleuser B. 2003. Sphingosine-1-phosphate and its potentially paradoxical effects on critical parameters of cutaneous wound healing. J. Invest. Dermatol. 120:693–700 [DOI] [PubMed] [Google Scholar]
- 32. Blom T, Bergelin N, Meinander A, Lof C, Slotte JP, Eriksson JE, Tornquist K. 2010. An autocrine sphingosine-1-phosphate signaling loop enhances NF-kappaB-activation and survival. BMC Cell Biol. 11:45 doi:10.1186/1471-2121-11-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kimura T, Tomura H, Mogi C, Kuwabara A, Ishiwara M, Shibasawa K, Sato K, Ohwada S, Im DS, Kurose H, Ishizuka T, Murakami M, Okajima F. 2006. Sphingosine 1-phosphate receptors mediate stimulatory and inhibitory signalings for expression of adhesion molecules in endothelial cells. Cell. Signal. 18:841–850 [DOI] [PubMed] [Google Scholar]
- 34. Okajima F, Tomura H, Sho K, Nochi H, Tamoto K, Kondo Y. 1996. Involvement of pertussis toxin-sensitive GTP-binding proteins in sphingosine 1-phosphate-induced activation of phospholipase C-Ca2+ system in HL60 leukemia cells. FEBS Lett. 379:260–264 [DOI] [PubMed] [Google Scholar]
- 35. Maceyka M, Milstien S, Spiegel S. 2009. Sphingosine-1-phosphate: the Swiss army knife of sphingolipid signaling. J. Lipid Res. 50(Suppl):S272–S276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee AS, Wells S, Kim KS, Scheffler IE. 1986. Enhanced synthesis of the glucose/calcium-regulated proteins in a hamster cell mutant deficient in transfer of oligosaccharide core to polypeptides. J. Cell. Physiol. 129:277–282 [DOI] [PubMed] [Google Scholar]
- 37. Ross SE, Radomska HS, Wu B, Zhang P, Winnay JN, Bajnok L, Wright WS, Schaufele F, Tenen DG, MacDougald OA. 2004. Phosphorylation of C/EBPalpha inhibits granulopoiesis. Mol. Cell. Biol. 24:675–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang P, Liu B, Jenkins GM, Hannun YA, Obeid LM. 1997. Expression of neutral sphingomyelinase identifies a distinct pool of sphingomyelin involved in apoptosis. J. Biol. Chem. 272:9609–9612 [DOI] [PubMed] [Google Scholar]
- 39. Jeschke MG, Finnerty CC, Herndon DN, Song J, Boehning D, Tompkins RG, Baker HV, Gauglitz GG. 2012. Severe injury is associated with insulin resistance, endoplasmic reticulum stress response, and unfolded protein response. Ann. Surg. 255:370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Richardson CE, Kinkel S, Kim DH. 2011. Physiological IRE-1-XBP-1 and PEK-1 signaling in Caenorhabditis elegans larval development and immunity. PLoS Genet. 7:e1002391 doi:10.1371/journal.pgen.1002391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Richardson CE, Kooistra T, Kim DH. 2010. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature 463:1092–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gombart AF, Borregaard N, Koeffler HP. 2005. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 19:1067–1077 [DOI] [PubMed] [Google Scholar]
- 43. Lepine S, Allegood JC, Park M, Dent P, Milstien S, Spiegel S. 2011. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 18:350–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van der Does AM, Beekhuizen H, Ravensbergen B, Vos T, Ottenhoff TH, van Dissel JT, Drijfhout JW, Hiemstra PS, Nibbering PH. 2010. LL-37 directs macrophage differentiation toward macrophages with a proinflammatory signature. J. Immunol. 185:1442–1449 [DOI] [PubMed] [Google Scholar]
- 45. Davidson DJ, Currie AJ, Reid GS, Bowdish DM, MacDonald KL, Ma RC, Hancock RE, Speert DP. 2004. The cationic antimicrobial peptide LL-37 modulates dendritic cell differentiation and dendritic cell-induced T cell polarization. J. Immunol. 172:1146–1156 [DOI] [PubMed] [Google Scholar]
- 46. Spiegel S, Milstien S. 2011. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 11:403–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arikawa K, Takuwa N, Yamaguchi H, Sugimoto N, Kitayama J, Nagawa H, Takehara K, Takuwa Y. 2003. Ligand-dependent inhibition of B16 melanoma cell migration and invasion via endogenous S1P2 G protein-coupled receptor. Requirement of inhibition of cellular RAC activity. J. Biol. Chem. 278:32841–32851 [DOI] [PubMed] [Google Scholar]
- 48. Gonzalez-Diez M, Rodriguez C, Badimon L, Martinez-Gonzalez J. 2008. Prostacyclin induction by high-density lipoprotein (HDL) in vascular smooth muscle cells depends on sphingosine 1-phosphate receptors: effect of simvastatin. Thromb. Haemost. 100:119–126 [DOI] [PubMed] [Google Scholar]
- 49. Ryu Y, Takuwa N, Sugimoto N, Sakurada S, Usui S, Okamoto H, Matsui O, Takuwa Y. 2002. Sphingosine-1-phosphate, a platelet-derived lysophospholipid mediator, negatively regulates cellular Rac activity and cell migration in vascular smooth muscle cells. Circ. Res. 90:325–332 [DOI] [PubMed] [Google Scholar]
- 50. Maceyka M, Harikumar KB, Milstien S, Spiegel S. 2011. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 22:50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sugiura K, Muro Y, Futamura K, Matsumoto K, Hashimoto N, Nishizawa Y, Nagasaka T, Saito H, Tomita Y, Usukura J. 2009. The unfolded protein response is activated in differentiating epidermal keratinocytes. J. Invest. Dermatol. 129:2126–2135 [DOI] [PubMed] [Google Scholar]