

Abstract
There is a paradox between the remarkable genetic stability of measles virus (MV) in the field and the high mutation rates implied by the frequency of the appearance of monoclonal antibody escape mutants generated when the virus is pressured to revert in vitro (S. J. Schrag, P. A. Rota, and W. J. Bellini, J. Virol. 73:51–54, 1999). We established a highly sensitive assay to determine frequencies of various categories of mutations in large populations of wild-type and laboratory-adapted MVs using recombinant viruses containing an additional transcription unit (ATU) encoding enhanced green fluorescent protein (EGFP). Single and double mutations were made in the fluorophore of EGFP to ablate fluorescence. The frequencies of reversion mutants in the population were determined by measuring the appearance of fluorescence indicating a revertant virus. This allows mutation rates to be measured under nonselective conditions, as phenotypic reversion to fluorescence requires only either a single- or a double-nucleotide change and amino acid substitution, which does not affect the length of the nonessential reporter protein expressed from the ATU. Mutation rates in MV are the same for wild-type and laboratory-adapted viruses, and they are an order of magnitude lower than the previous measurement assessed under selective conditions. The actual mutation rate for MV is approximately 1.8 × 10−6 per base per replication event.
INTRODUCTION
Measles is still a leading cause of vaccine-preventable death among children. The virus exhibits extremely high levels of infectivity, witnessed by its ability to infect the rare susceptible individuals present in highly vaccinated populations successfully (1). Measles virus (MV) is a single-stranded RNA virus with a genome of negative polarity, 15,894 nucleotides in length (2). It is a typical member of the subfamily Paramyxovirinae, with a genome that encodes six transcription units expressing six structural and at least two nonstructural proteins. The program of gene expression and the functions of the genes were well reviewed previously (3) and in essence do not differ from those of the well-studied members of this group, such as Sendai virus and human parainfluenza virus 5 (hPIV5).
The MV substitution rate has been much debated (4–7) because of the relative stability of MV in the field, but only a single paper has made an estimate of the spontaneous mutation rate in vitro (8). The high spontaneous mutation rates for RNA viruses result in the generation of a viral quasispecies consisting of a swarm of different viruses (9). This has important consequences for the properties of these viruses, especially in relation to evolutionary adaptability and their potential to make cross-species jumps (9). It is thus important to assess the mutation rate of these RNA viruses. Several methods have been employed to measure the mutation rates, but it has generally not been recognized that the determination of the actual mutation rates is impossible unless a number of parameters that are difficult to establish have been evaluated (10). In the case of MV, the spontaneous mutation rate was reported to be 9 × 10−5 per nucleotide site per replication by analysis of the frequency of monoclonal antibody-resistant (MAR) mutants (8). This is at the higher end of the range of mutation rates for RNA viruses (11). However, as argued previously (9, 12), measurements of mutation rates made under selective conditions and using variations of the fluctuation test provide only an approximate average value for a mixture of transitions and transversions, as often, mutations at various nucleotide residues can contribute to the selection of the resistant phenotype. Furthermore, the measurements are also affected by phenotypic masking of mutant genomes in wild-type envelopes and by the lower fitness of the MAR mutants than the parental population from which they were isolated (9, 13).
We have developed a new method for the measurement of mutation rates in MV, but one that can be extrapolated to other viruses, under what are ostensibly nonselective conditions by using the following design. We generated viruses with mutations in the fluorophore of enhanced green fluorescent protein (EGFP), expressed as a supernumerary protein in the virus genome, such that the mutated viruses were nonfluorescent, and upon reversion, the active fluorophore was regenerated and fluorescence was restored. The required mutations were introduced into a wild-type MV, rMVIC323EGFP (14), that has been extensively used for studies of viral pathogenesis (15). This design has a further advantage in that rates can be measured for specific transitions and transversions required for the reconstitution of the fluorophore coding sequence and the conversion of a virus which is nonfluorescent (rMVIC323EGFPKO) to one that is fluorescent (revertant).
The mutation frequencies were determined by counting the number of revertants in rMVIC323EGFPKO populations grown from single plaques of viruses “rescued” from a cDNA clone. The reversion events were caused by single or double substitutions that occurred by errors of the viral polymerase during replication of the RNA genome at specific nucleotides in the mutated EGFP gene (EGFPKO), changing the nucleotides back to those in the original fluorophore of EGFP. The MV genome length (15,894 nucleotides) combined with the estimated mutation rate established previously by Schrag et al. (8) would give rise to the generation of 1.43 mutations per genome per replication. This rate is high but not unprecedented among RNA viruses. We find a mutation rate which is an order of magnitude lower than those from the previous estimate.
MATERIALS AND METHODS
Cell line and virus strains.
Vero cells stably expressing canine CD150 (Vero-cCD150 cells) were used to propagate and titrate all viruses, as described previously (16). The recombinant virus used in this study is wild-type MV rMVIC323EGFP (14, 17). Nonfluorescent knockout (KO) viruses were generated by the introduction of one or two mutations into the fluorophore of the EGFP gene by mutagenesis (Fig. 1). Two additional viruses, rMVEdtagEGFPstop1 and MVEdtagEGFPstop2, carrying EGFP knockout mutations and based on the laboratory-adapted, Edmonston-derived Edtag virus (18) were also generated. In these EGFPKO viruses, the EGFP sequence was modified to introduce stop codons within the fluorophore (position 67 tyrosine) by changing TAC to TAA (stop1) or TAG (stop2).
Fig 1.

Design of EGFPKO viruses. (A) Schematic representation of the genomes of the recombinant wild-type viruses encoding EGFP in an additional transcription unit in position 1, i.e., before the gene encoding the nucleocapsid (N) protein. EGFPKO viruses for the laboratory-adapted strain expressed modified EGFP proteins from an ATU in the same position. The genome order from the 3′ end to the 5′ end is as follows: EGFP-nucleocapsid protein gene (N)-phosphoprotein gene (P/V/C)-matrix protein gene (M)-fusion protein gene (F)-hemagglutinin protein (H)-large protein (L). (B) Nucleotide (nt) and protein sequences of the fluorophore in the EGFPKO viruses. aa, amino acids. (C) False-color images of green fluorescence detection in cell monolayers infected with wild-type, EGFPKO, and revertant viruses. (D) Nucleotide and protein sequences of the fluorophore and genetic tag sequences of the wild type and a revertant virus.
Construction of KO viruses.
Plasmid p(+)MV323-EGFP (14) was digested with AscI and AatII to produce a vector into which were inserted appropriately mutagenized EGFP open reading frames (ORFs) in which the fluorophore was altered, as per design (Fig. 1B). The inserts were generated by overlap PCR employing appropriate mutagenic inner primers (sequences are available on request) and outer primers incorporating AscI and AatII restriction enzyme recognition sequences at the start and end of the product, respectively. The forward primer also introduced a SacI restriction enzyme recognition sequence by incorporating a synonymous mutation in codon 8 of the EGFP ORF. This single, silent mutation (G139C) acts as a genetic tag to distinguish revertant KO viruses from the wild type. The mutagenized PCR products were subcloned through the pGEM-T Easy vector system (Promega), and the resulting plasmids were sequenced to ensure that the appropriate restriction sites and mutations as well as the SacI genetic tag were present. The subcloned inserts were released by restriction with AscI and AatII, and the purified inserts were ligated with the prepared vector to generate plasmids that encoded the full-length rMVIC323EGFPKO genome with the EGFP fluorophore knocked out. Recombinant viruses were recovered from fowlpox virus T7-infected Vero-cCD150 cells transfected with the full-length template plasmids and plasmids expressing MV nucleoprotein (N), phosphoprotein (P), and large protein (L). Virus fitness of rMVIC323EGFP and rMVIC323EGFPa315c was compared in growth curves (16).
Determination of mutation frequency.
Each well in a 96-well tray was seeded with about 50,000 Vero-cCD150 cells and infected at a multiplicity of infection (MOI) of 0.2 with each of the EGFPKO viruses obtained by picking a plaque from a well in which the recombinant virus was freshly rescued from cDNA and amplifying this by maximally two passages on Vero-cCD150 cells to a population size of >1 million PFU. Maximum infection efficiency was attained by spinoculation of the viruses at 1,200 rpm for 10 min at 25°C. After infection, fusion-inhibitory peptide (FIP) (Z-d-Phe-Phe-Gly-OH) (19) was added (to a final concentration of 200 μC) to prevent fusion. The trays were incubated for 52 h at 37°C in an atmosphere of 5% (vol/vol) CO2, and the monolayers were then observed by using a UV microscope fitted with appropriate filter blocks (Leica Microsystems). Single and paired green cells were counted, and the numbers were used to establish the mutation frequency, i.e., the number of revertants divided by the total number of PFU in the population.
RESULTS
Rationale of the experiments and design of the KO mutants.
In order to determine mutation frequencies under nonselective conditions, we generated a number of EGFPKO viruses, which express mutated forms of EGFP that do not fluoresce (Fig. 1A). In order to assess specific types of reversion frequencies, EGFPKO viruses were designed with both single- and double-nucleotide replacements. Only a reversion back to the original amino acid of the fluorophore will result in the restoration of the fluorescent phenotype. Hence, the mutations were designed in such a way that only the direct back mutation to the wild-type nucleotide would restore the amino acid and, hence, the fluorescence. The double replacements were either adjacent or close together in the fluorophore of the EGFP protein (Fig. 1B). For wild-type rMVIC323EGFP, four viruses containing single substitutions were generated, rMVIC323EGFPa315c and rMVIC323EGFPa315t, to examine revertants resulting from C→A and T→A transversions, and rMVIC323EGFPa315g and rMVIC323EGFPc312t, to examine revertants resulting from G→A and T→C transitions. Three double substitutions were generated to determine whether double mutations occurred with similar or reduced frequencies compared to single mutations and also to assess whether there were differences in reversion frequencies when the two nucleotides were adjacent, as in rMVIC323EGFPSii, or 2 to 3 nucleotides apart in the nucleotide sequence, as in rMVIC323EGFPSA and rMVIC323EGFPAS. MVEdtagEGFPstop1 and MVEdtagEGFPstop2 provided additional information for reversion frequencies of G→C and A→C transversions for a laboratory-adapted strain. None of the EGFPKO mutant viruses produced green fluorescence (Fig. 1C).
It was also important that we could demonstrate unambiguously that green fluorescent revertants were indeed true revertants and did not occur due to contamination. Therefore, we inserted a genetic tag, consisting of a silent mutation in codon 8 of the EGFP ORF, in the EGFPKO viruses, which generated a new SacI restriction enzyme recognition sequence. This allowed revertants to be distinguished from wild-type rMVIC323EGFP. A revertant virus of rMVIC323EGFPa315c which had reverted to express green fluorescence (Fig. 1C) was isolated. Data from sequencing showed that the mutation in the fluorophore (TCC) had reverted back to the wild-type fluorophore sequence (TAC), as expected, and also that the revertant fluorescent virus carried the genetic tag, demonstrating that it originated from the original EGFPKO virus (Fig. 1D).
Conditions for measurements of reversion frequencies are nonselective.
In order to assess whether an EGFPKO virus would have a selective advantage or disadvantage over the parental EGFP-expressing virus, we compared the growth characteristics of rMVIC323EGFP with those of an EGFPKO virus. We did not detect significant differences in the growth of rMVIC323EGFPa315c and wild-type rMVIC323EGFP in the released or cell-associated virus fractions (Fig. 2).
Fig 2.
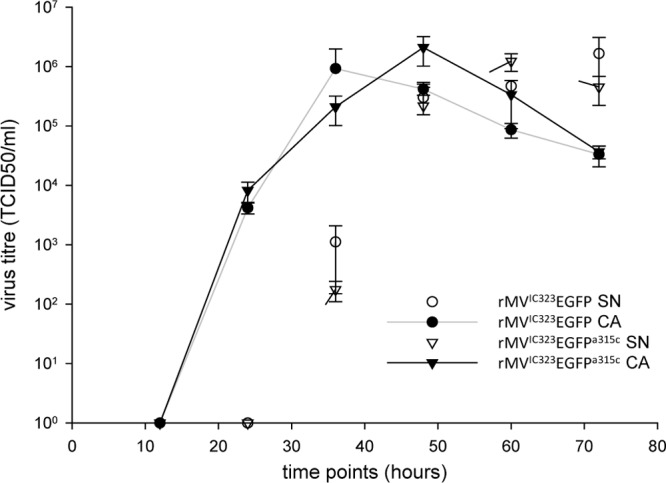
Replication kinetics of the wild type and a representative EGFPKO virus. Virus titers (50% tissue culture infective dose [TCID50]) were measured in the supernatant (SN) and cell-associated (CA) fractions of Vero-cCD150 cells infected with wild-type rMVIC323EGFP and the EFGPKO virus rMVIC323EGFPa315c.
Development of the reversion frequency detection assay.
Assessment of the frequency of revertant viruses in a specific viral population requires detection of a very small number of “green” viruses against the background of a large number of “nongreen” viruses. A single cell containing a fluorescent virus can readily be visualized (data not shown) against thousands of nonfluorescent EGFPKO viruses if syncytium formation is prevented by the use of FIP (19). Wild-type rMVIC323EGFP was used to demonstrate that FIP restricts virus infection to a single cell by preventing fusion (Fig. 3A). Pairs of green cells that were extremely close to each other were observed at a higher magnification (×200). Their spatial arrangements suggested that these were caused by the division of single infected cells rather than by cell-to-cell virus spread (17). These rare occurrences were therefore counted in the revertant frequency assay as single events (Fig. 3A).
Fig 3.
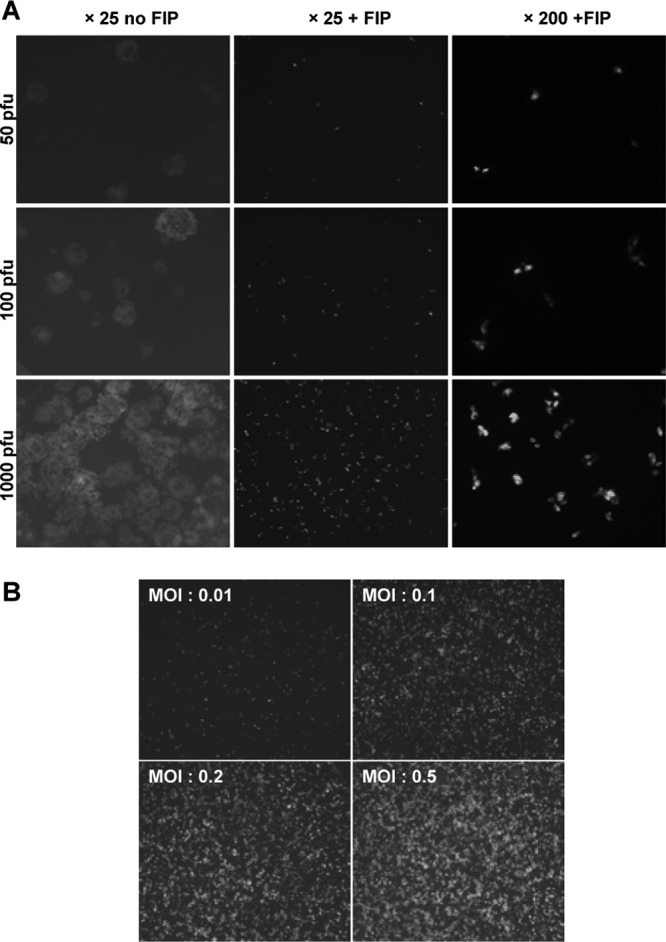
Development of the assay. (A) Effect of the fusion-inhibitory peptide (FIP). In the absence of FIP, syncytia form in Vero-cCD150 cell monolayers, which prevents enumeration of the revertant viruses. With FIP, the dynamic range for the measurement of revertant frequency is 1 to 1,000. At a higher magnification, paired green cells (the result of cell division) are detected. These are counted as single events in the measurement of mutation frequency in virus populations. (B) Establishment of the maximum MOI that results in discrete, countable infectious centers, reflecting the actual number of PFU used for the initial infection. Wells of a 96-well plate containing approximately 40,000 cells per well were infected at the indicated MOIs.
We mimicked the likely outcome of the reversion frequency detection assay by mixing small amounts of wild-type rMVIC323EGFP with a much larger amount of EGFPKO virus. The result showed that one fluorescent cell derived from 1 PFU of wild-type virus can be detected easily among 1,000 nonfluorescent viruses, proving that the use of FIP allowed the detection of very small numbers of revertants under the assay conditions. This experiment also established that it took 52 h before the number of “green” cells detected equated exactly to the number of wild-type PFU added (data not shown). Hence, this incubation time was used for the assay. This incubation time also allows only preexisting revertant virus in the starting population to be detected, while longer incubation times would potentially allow the detection of cells in which the reversion event occurred during replication of the EGFPKO virus in the cell during the assay. Cells in which a reversion event took place during the assay would have low levels of intensity of green fluorescence depending on when the reversion took place in the growth cycle of the virus. Green fluorescent cells observed in the assay were of fairly uniform intensity, indicating that the assay conditions allowed detection of preexisting mutants in the population and that reversion during the assay was below detection levels.
In order to obtain information on the upper limit of the amount of virus which could be assessed on one plate, it was paramount to determine the highest MOI that could be used in one well of a 96-well plate. Each well contains approximately 50,000 Vero-cCD150 cells when grown to confluence. Monolayers of Vero-cCD150 cells were infected at high MOIs with wild-type rMVIC323EGFP, and FIP was added to prevent fusion (Fig. 3B). It was determined that at an MOI of 0.2 the number of infected cells counted was directly proportional to the amount of input virus, indicating that there was a negligible chance of coinfection of cells. At an MOI of 0.5, we counted fewer infected cells than the input PFU would have predicted (presumably due to coinfections), and hence, we used an MOI of 0.2 for the assay. This allowed approximately 10,000 PFU of EGFPKO virus to be screened per well of a 96-well plate, thus allowing revertant frequencies to be measured in a population of almost 1 million PFU per plate.
Determination of the reversion frequency in viral populations.
Three independent populations were screened for each EGFPKO virus. Plaques were picked from the initial primary syncytia generated by recovery of the virus from a cDNA clone in Vero-cCD150 cells. Each of these plaques was expanded to a population of >1 million PFU by propagation in Vero-cCD150 cells at low MOIs. Depending on the titer of the virus stock, between 1 million and 8 million PFU of these populations were screened in the assay, as described above. For each well, the number of cells displaying green fluorescence was determined (data not shown). Replicate experiments with rMVIC323EGFPa315c demonstrated the reproducibility of the assay (data not shown), even though the generation of revertants is a chance event and therefore susceptible to stochastic variation.
Average frequencies of single-nucleotide substitutions in rMVIC323EGFP ranged from 1.3 × 10−6 substitutions per nucleotide (s/n) for the T→A transversion to 21.3 × 10−6 s/n for the G→A transition. The G→C and A→C transversions occurred at similar frequencies of 5.0 × 10−6 s/n and 8.7 × 10−6 s/n, respectively, in laboratory-adapted strain rMVEdtagEGFPstop (Fig. 4).
Fig 4.

Mutation frequencies. (A) Mutation frequencies of specific transitions and transversions. Closed circles refer to the rates found with the rMVIC323EGFPKO viruses. Open circles refer to mutation frequencies established for the rMVEdtagEGFPstop viruses. (B) Tabulated data for reversion of a range of EGFPKO mutant viruses.
The reversion frequencies for the EGFPKO viruses with double mutations, which required two substitutions for the restoration of the green phenotype, were below the lower detection limit, which was established by the population sizes that were entered into the assays, since, with one exception (discussed below), we did not observe any revertants in the populations of these EGFPKO viruses. This applied both to the situation of two substitutions adjacent to each other, which might potentially revert by a connected or dependent event, and when two substitutions within a 2- to 3-nucleotide distance from each other were required for reversion, possibly resulting from independent events.
DISCUSSION
On a theoretical basis, the conditions used in this assay are nonselective. The revertant mutations occur in an additional transcription unit (ATU) encoding the EGFP gene. Although there are some data which indicate that the presence and position of the ATU have an effect on the growth of recombinant viruses (16, 17), this is irrelevant, as the neutrality in the assay is determined by potential differences in growth parameters between the EGFPKO and revertant viruses. Both express the complete 239 amino acids of the EGFP protein, even though the mutations in the reporter lead to an observable phenotypic difference. Analysis of the growth kinetics demonstrated no differences in the growth of rMVIC323EGFPa315c and rMVIC323EGFP, thus providing experimental evidence that the single-nucleotide change in the EGFP gene did not affect replication ability and fitness. The two laboratory-adapted EGFPKO viruses rMVEdtagEGFPstop1 and rMVEdtagEGFPstop2 have a premature stop codon in the EGFP fluorophore sequence and therefore express only a truncated 66-amino-acid protein. This may theoretically provide a selective advantage over revertants that express the entire EGFP protein. Nevertheless, this difference did not lead to a change in the order or magnitude of the reversion frequencies. Thus, both theoretically and on the basis of the evidence supplied, it appears that the conditions of this assay are nonselective.
Population history is considered to be one of the important factors that affect mutation rate estimations but is difficult to control for. Therefore, independent plaques of virus rescued from cDNA were propagated to establish the virus stocks used in these experiments. Presumably, the resulting populations assayed in these experiments started as a homogeneous set of genomes with the same sequence. After two rounds of infection and replication, the population size of the stocks had been expanded to an adequate size for determination of the mutation frequency; we considered this to be 1 million to 10 million PFU based on the expected rates of mutation. We observed only single green cells per well of a 96-well plate in most assays, with the exception of the rMVIC323EGFPa315g, where a very large population size and a slightly higher rate of mutation led to the occasional observation of 2 to 3 green cells per well. An exceptional “jackpot” event occurred in only one of the three populations of rMVIC323EGFPSii that was assayed. This EGFPKO virus contains two adjacent mutations in the fluorophore, but since no revertant mutants were found in any of the populations of the other viruses that required two substitutions to restore the fluorescent phenotype, namely, rMVIC323EGFPAS and rMVIC323EGFPSA (data not shown), or in the other two rMVIC323EGFPSii populations, we have not counted the jackpot event in our calculations, and thus, it appears unlikely that the chance for the polymerase to make a second mistake immediately after a first one is not higher than it making two separate independent mistakes.
Two previous studies estimated MV mutation rates. Kalland et al. (20) previously looked at mutations in the P gene of MV after 100 passages of the virus and suggested that the mutation rate was 1.4 × 10−6 per base per replication. However, these data do not allow the number of genome replications to be determined or estimated. The second paper, by Schrag et al. (8), established the frequency of appearance of neutralizing monoclonal antibody escape (MAR) mutants on cloned populations of MV. The mutation frequency was estimated in what is called the Edmonston wild-type strain. However, this strain had already been passed in several nonhuman cells lines and is already adapted to growth on Vero cells. To address this issue, we used both CD150-using viruses and CD46-using laboratory-adapted viruses. rMVIC323EGFP uses CD150 as a receptor. We also applied our assay system to Edtag laboratory-adapted strain rMVEdtagEGFP (18) and found that there was no significant difference of the spontaneous mutation frequencies between the wild-type and laboratory-adapted strains. Therefore, it is assumed that the data for these two strains of MV probably reflect the fundamental situation for the species as a whole.
The spontaneous mutation rate measured previously by Schrag et al. (8) was estimated under selective conditions, which, as described in the introduction, affects the estimated rates (9, 13). MAR mutants that arose were sequenced, and four different nucleotide changes and amino acid changes that led to the obliteration of the ability of the antibody to neutralize the MAR mutants were found, and therefore, Schrag et al. (8) divided the rate estimated from the fluctuation tests by 4 after multiplying the raw rates by a factor of 3 to take account of the fact that there are 3 unique changes per site. The determination of mutation rates was critically reassessed in a paper by Sanjuán et al. (10). Taking account of the fact that the mutation rates determined by measuring the rate of appearance of neutralizing monoclonal antibody-resistant mutants in a population are neither neutral nor lethal, Sanjuán et al. (10) applied a correction to the mutation rate determined by Schrag et al. (8) to be 9 × 10−5 substitutions per nucleotide site per replication. This correction for selection bias was based on empirical information about the distribution of mutational fitness effects previously obtained for several viruses (10), and when applied, the mutation rate of MV was converted to 4.4 × 10−5 substitutions per nucleotide per strand copying, indicating that the use of selective conditions increased the estimation of the mutation rate by a factor of 2 (10). This reduces the reported genome mutation rate for MV from 1.43 to 0.7 nucleotide substitutions per replication.
The present study used mutations in an ATU containing a reporter gene in which revertant point mutations lead to an observable phenotype. This provides a novel and, in our opinion, optimal assay system, as it is nonselective, making the conversion from mutation frequencies to mutation rates straightforward. By definition, the number of copying cycles is maximal under binary replication. Under binary replication, progeny strands immediately serve as templates for further replication, and the number of molecules doubles in each cycle of strand copying, increasing geometrically (10). In contrast, under linear replication, multiple copies are made sequentially from the same template, and the resulting progeny strands do not become templates until the progeny virions infect another cell. In the replication cycle of MV, the negative-sense genome in the ribonucleoprotein complex (−RNP) is replicated by the viral RNA polymerase into positive-sense RNA, which is also encapsidated into RNPs (+RNP). Both −RNP and +RNP serve as templates for further replication events, and transcription of the −RNPs, after expansion of their numbers, is required for virus growth. We are not aware of any evidence in the literature on MV which would contradict the assumption of binary replication in this system (3). Assuming binary replication, the formulae developed by Sanjuán et al. (10) give the lower limit estimate of the mutation rate as min[μ] = 3f/lnN, where μ refers to the mutation rate (substitutions per nucleotide site per strand copying [s/n/r]), f is the mutation frequency (per nucleotide site), and N represents the population size (number of PFU). The multiplication factor 3 is introduced because there are three possible unique substitutions at any given site, assuming that all three possible base substitutions occur at equal frequencies. The upper limit estimate is max[μ] = 3f (10). The unit here is substitutions per nucleotide per cell infection (s/n/c) and is equivalent to the rate per nucleotide per strand copying if replication is linear. However, as single-stranded RNA viruses produce an intermediate strand of opposite polarity, full linear replication is certainly not the case in MV. Thus, the actual mutation rates should be lower than the maximum derived from a linear replication model and is probably closer or equal to the minimum rate estimated from the formula μ = 3f/lnN (Table 1). As all possible reversion mutations for the nucleotide at position 315 in the EGFP ORF were determined, the mutation rate could be obtained by averaging the three values, giving an interval with a maximum limit of 2.78 × 10−5 substitutions per nucleotide per cell infection and a minimum limit of 1.84 × 10−6 substitutions per nucleotide site per strand copying. If one assumes that the mutation rates determined for this nucleotide apply to all nucleotides in the 15,894-nucleotide-long MV genome, the genome mutation rate is <0.44 nucleotides per cell infection and could be as low as 0.03 nucleotides per strand copying if the replication mode is completely binary. These figures are substantially lower than those measured previously (10) for vesicular stomatitis virus (genome size of 11.2 kb and mutation rate of 3.5 × 10−5 s/n/c) and influenza A virus (total segmented genome size of 13.6 kb and mutation rate of 2.3 × 10−5 s/n/c) and more in line with data reported for influenza B virus (genome size of 14.5 kb and mutation rate of 1.7 × 10−6 s/n/c). These three negative-stranded RNA viruses have genome sizes similar to that of MV.
Table 1.
Maximum and minimum mutation rates
| Nucleotide | Type of mutation | Max[μ] limit (s/n/c) | Min[μ] (s/n/r) |
|---|---|---|---|
| 315 | C→A | 1.56 × 10−5 | 1.08 × 10−6 |
| G→A | 6.38 × 10−5 | 4.18 × 10−6 | |
| T→A | 0.39 × 10−5 | 0.27 × 10−6 | |
| 312 | T→C | 1.38 × 10−5 | 0.90 × 10−6 |
Misincorporation rates have not been measured directly for MV as for poliovirus and vesicular stomatitis virus (21, 22), as appropriate in vitro replication systems have not yet been developed. Studies using deep sequencing of populations of viral RNA molecules extracted from human tissues have not yet been reported, and hence, in vivo mutation fixation rates and the direct observation of quasispecies compositions have not been made. Interestingly, a recent study reported the in vivo mutation rate estimates for hepatitis C virus to be approximately 100× lower than what would be expected from mutation rates measured in vitro reflecting the pressure of selection (23). A similar argument can be applied to MV. The mutation rate for MV measured here is still high enough to provide a paradox with the low replacement rate for the H gene of MV from isolates in the field (24, 25). The number of replications involved in the transmissions of virus between at least 26 infected patients in an outbreak sustained over 1 year must be in the order of trillions, and multiplying this with the lowest rate determined here of 0.03 per genome replication still indicates the tremendous selection pressure that is required to explain the low replacement rate for MV in the field. Replacement rates for other RNA virus genomes, such as those of influenza A virus, foot-and-mouth disease virus, and poliovirus, are between 10−2 and 10−3 per site per annum (9). In comparison, MV is much more stable in the field, and a substitution rate of approximately 4 × 10−4 per annum per nucleotide position was estimated for a single outbreak in Spain (24). These observations would place MV in a class of viruses with a relatively low mutation rate on a relatively flat fitness surface (26) and could explain the stability of the over 23 different genotypes that have been described (27), in addition to other factors contributing to this stability, including a lack of recombination in morbilliviruses, strict constraints on insertions and deletions due to the need to comply with the rule of six, the limited host range of MV, and functional constraints due to the use of protein receptors by the virus (8). In the context of MV elimination efforts, a relatively low mutation rate reduces the possibility of strains that may escape neutralization by vaccine. It is also consistent with the fact that no vaccine escape mutants have been seen to date.
ACKNOWLEDGMENTS
We gratefully acknowledge grant support by the United Kingdom Medical Research Council (grant number G0501427) and Queen's University Belfast for a postgraduate studentship that supported X.Z.
Footnotes
Published ahead of print 19 December 2012
REFERENCES
- 1. Anonymous 2009. Global reductions in measles mortality 2000-2008 and the risk of measles resurgence. Wkly. Epidemiol. Rec. 84:509–516 [PubMed] [Google Scholar]
- 2. Griffin DE. 2007. Measles virus, p 1551–1585 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3. Rima BK, Duprex WP. 2009. The measles virus replication cycle. Curr. Top. Microbiol. Immunol. 329:77–102 [DOI] [PubMed] [Google Scholar]
- 4. Furuse Y, Suzuki A, Oshitani H. 2010. Origin of measles virus: divergence from rinderpest virus between the 11th and 12th centuries. Virol. J. 7:52 doi:10.1186/1743-422X-7-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Holmes EC. 2009. RNA virus genomics: a world of possibilities. J. Clin. Invest. 119:2488–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Woelk CH, Jin L, Holmes EC, Brown DW. 2001. Immune and artificial selection in the haemagglutinin (H) glycoprotein of measles virus. J. Gen. Virol. 82:2463–2474 [DOI] [PubMed] [Google Scholar]
- 7. Woelk CH, Pybus OG, Jin L, Brown DW, Holmes EC. 2002. Increased positive selection pressure in persistent (SSPE) versus acute measles virus infections. J. Gen. Virol. 83:1419–1430 [DOI] [PubMed] [Google Scholar]
- 8. Schrag SJ, Rota PA, Bellini WJ. 1999. Spontaneous mutation rate of measles virus: direct estimation based on mutations conferring monoclonal antibody resistance. J. Virol. 73:51–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Domingo E, Holland JJ. 1994. Mutation rates and rapid evolution of RNA viruses, p 161–184 In Morse SS. (ed), The evolutionary biology of viruses. Raven Press Ltd, New York, NY [Google Scholar]
- 10. Sanjuán R, Nebot MR, Chirico N, Mansky LM, Belshaw R. 2010. Viral mutation rates. J. Virol. 84:9733–9748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gago S, Elena SF, Flores R, Sanjuán R. 2009. Extremely high mutation rate of a hammerhead viroid. Science 323:1308 doi:10.1126/science.1169202 [DOI] [PubMed] [Google Scholar]
- 12. Ojosnegros S, Perales C, Mas A, Domingo E. 2011. Quasispecies as a matter of fact: viruses and beyond. Virus Res. 162:203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martinez A, Carrillo C, Gonzalez-Candelas F, Moya A, Domingo E, Sobrino F. 1991. Fitness alteration of foot-and-mouth disease virus mutants: measurement of adaptability of viral quasispecies. J. Virol. 65:3954–3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takeda M, Takeuchi K, Miyajima N, Kobune F, Ami Y, Nagata N, Suzaki Y, Nagai Y, Tashiro M. 2000. Recovery of pathogenic measles virus from cloned cDNA. J. Virol. 74:6643–6647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Swart RL, Ludlow M, de Witte L, Yanagi Y, van Amerongen G, McQuaid S, Yüksel S, Geijtenbeek TBH, Duprex WP, Osterhaus ADME. 2007. Predominant infection of CD150+ lymphocytes and dendritic cells during measles virus infection of macaques. PLoS Pathog. 3:e178 doi:10.1371/journal.ppat.0030178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lemon K, de Vries RD, Mesman AW, McQuaid S, van Amerongen G, Yuksel S, Ludlow M, Rennick LJ, Kuiken T, Rima BK, Geijtenbeek TB, Osterhaus AD, Duprex WP, de Swart RL. 2011. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog. 7:e1001263 doi:10.1371/journal.ppat.1001263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hashimoto K, Ono N, Tatsuo H, Minagawa H, Takeda M, Takeuchi K, Yanagi Y. 2002. SLAM (CD150)-independent measles virus entry as revealed by recombinant virus expressing green fluorescent protein. J. Virol. 76:6743–6749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dotsch C, Christiansen G, Billeter MA. 1995. Rescue of measles viruses from cloned DNA. EMBO J. 14:5773–5784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richardson CD, Choppin PW. 1983. Oligopeptides that specifically inhibit membrane fusion by paramyxoviruses: studies on the site of action. Virology 131:518–532 [DOI] [PubMed] [Google Scholar]
- 20. Kalland KH, Havarstein LS, Endresen C, Haukenes G. 1990. Stability of the nucleotide sequence of the phosphoprotein gene of measles virus during lytic infections. APMIS 98:327–335 [DOI] [PubMed] [Google Scholar]
- 21. Steinhauer DA, Holland JJ. 1986. Direct method for quantitation of extreme polymerase error frequencies at selected single base sites in viral RNA. J. Virol. 57:219–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ward CD, Stokes MAM, Flanegan JB. 1988. Direct measurement of the poliovirus RNA polymerase error frequency in vitro. J. Virol. 62:558–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ribeiro RM, Li H, Wang S, Stoddard MB, Learn GH, Korber BT, Bhattacharya T, Guedj J, Parrish EH, Hahn BH, Shaw GM, Perelson AS. 2012. Quantifying the diversification of hepatitis C virus (HCV) during primary infection: estimates of the in vivo mutation rate. PLoS Pathog. 8:e1002881 doi:10.1371/journal.ppat.1002881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rima BK, Earle JA, Baczko K, ter Meulen V, Liebert UG, Carstens C, Carabana J, Caballero M, Celma ML, Fernandez-Munoz R. 1997. Sequence divergence of measles virus haemagglutinin during natural evolution and adaptation to cell culture. J. Gen. Virol. 78:97–106 [DOI] [PubMed] [Google Scholar]
- 25. Rota JS, Rota PA, Redd SB, Redd SC, Pattamadilok S, Bellini WJ. 1998. Genetic analysis of measles viruses isolated in the United States, 1995-1996. J. Infect. Dis. 177:204–208 [DOI] [PubMed] [Google Scholar]
- 26. Lauring AS, Andino R. 2010. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 6:e1001005 doi:10.1371/journal.ppat.1001005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rota PA, Brown K, Mankertz A, Santibanez S, Shulga S, Muller CP, Hübschen JM, Siqueira M, Beirnes J, Ahmed H, Triki H, Al-Busaidy S, Dosseh A, Byabamazima C, Smit S, Akoua-Koffi C, Bwogi J, Bukenya H, Wairagkar N, Ramamurty N, Incomserb P, Pattamadilok S, Jee Y, Lim W, Xu W, Komase K, Takeda M, Tran T, Castillo-Solorzano C, Chenoweth P, Brown D, Mulders MN, Bellini WJ, Featherstone D. 2011. Global distribution of measles genotypes and measles molecular epidemiology. J. Infect. Dis. 204(Suppl 1):S514–S523 doi:10.1093/infdis/jir118 [DOI] [PubMed] [Google Scholar]