

Abstract
Within target T lymphocytes, human immunodeficiency virus type I (HIV-1) encounters the retroviral restriction factor APOBEC3G (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G; A3G), which is counteracted by the HIV-1 accessory protein Vif. Vif is encoded by intron-containing viral RNAs that are generated by splicing at 3′ splice site (3′ss) A1 but lack splicing at 5′ss D2, which results in the retention of a large downstream intron. Hence, the extents of activation of 3′ss A1 and repression of D2, respectively, determine the levels of vif mRNA and thus the ability to evade A3G-mediated antiviral effects. The use of 3′ss A1 can be enhanced or repressed by splicing regulatory elements that control the recognition of downstream 5′ss D2. Here we show that an intronic G run (GI2-1) represses the use of a second 5′ss, termed D2b, that is embedded within intron 2 and, as determined by RNA deep-sequencing analysis, is normally inefficiently used. Mutations of GI2-1 and activation of D2b led to the generation of transcripts coding for Gp41 and Rev protein isoforms but primarily led to considerable upregulation of vif mRNA expression. We further demonstrate, however, that higher levels of Vif protein are actually detrimental to viral replication in A3G-expressing T cell lines but not in A3G-deficient cells. These observations suggest that an appropriate ratio of Vif-to-A3G protein levels is required for optimal virus replication and that part of Vif level regulation is effected by the novel G run identified here.
INTRODUCTION
Replication of human immunodeficiency virus type 1 (HIV-1) is counteracted by four major classes of host-encoded restriction factors: APOBEC3G (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G; A3G), TRIM5α (tripartite motif 5α), tetherin (BST-2, CD317, or HM1.24), and SAMHD1 (1–4). A3G (5) belongs to a family of cytidine deaminases that includes seven members (A3A to A3D and A3F to A3H) located in a gene cluster on chromosome 22 (6–8). It is encapsidated into newly assembled virions and introduces C-to-U substitutions during minus-strand synthesis, resulting in G-to-A hypermutations and aberrant DNA ends in the HIV-1 genome. Furthermore, A3G, independent of its deaminase activity, inhibits reverse transcriptase-mediated minus-strand elongation by direct binding to the viral RNA (9). This leads to massive impairment of viral replication (10). However, the HIV-1-encoded accessory protein Vif counteracts A3G by direct interaction, by inducing proteasomal degradation, and by repression of mRNA synthesis (10). Whereas HIV-1 is able to replicate efficiently in A3G-expressing cells, Vif-deficient virus strains are completely suppressed (5). Nevertheless, a narrowly restricted level of Vif is crucial for optimal HIV-1 replication since proteolytic processing of the Gag precursor at the p2/nucleocapsid processing site is inhibited by high levels of Vif (11).
During the course of infection, the HIV-1 9-kb single-sense pre-mRNA is processed into more than 40 alternatively spliced mRNA isoforms encoding 18 HIV-1 proteins, all of which interact with a wide variety of host cell proteins, complexes, and pathways (12). Furthermore, negative-sense mRNAs that lead to the expression of at least one antisense protein are also found in HIV-1-infected T cells (13).
The splicing process consists of a series of consecutive steps that are orchestrated by interactions of individual spliceosomal components (14). The initial binding of the U1 snRNP to the pre-mRNA is mediated by base pairing between the 5′ end of the U1 snRNA and the 5′ splice site (5′ss) initiating early E-complex formation (15). Subsequently, the 3′ss, consisting of the AG dinucleotide, the branch point sequence (BPS), and a polypyrimidine tract, is bound by the U2 snRNP at the BPS in an ATP-dependent manner, thus initiating A-complex formation. In a process named exon definition, U1 and U2 snRNPs bound to the exon-intron borders pair with each other (cross-exon interactions) and facilitate the removal of the flanking introns (16, 17).
Rev expression initiates the switch from the early to the late stage of viral replication (18). On binding to the Rev-responsive element (RRE), Rev oligomerizes cooperatively and, by interacting with the cellular Crm1 export pathway, facilitates the export of intron-containing viral mRNAs (19–21).
Since the vif AUG is localized within intron 2, this intron must be retained within the functional vif mRNA. Thus, the 5′ss D2 has to be rendered splicing incompetent, even though binding of U1 snRNP to this 5′ss is a prerequisite for efficient recognition of the upstream 3′ss A1 (22, 23). In general, 5′ss splice donor repression may be a common requirement for the generation of all spliced but intron-containing HIV-1 mRNAs, e.g., the env mRNAs (24). Indeed, recent studies demonstrated that the splicing regulatory element (SRE)-mediated binding of the U1 snRNP to a 5′ss does not necessarily result in its processing into the spliceosomal A-complex formation but can lead to a “dead-end” complex formation that prevents the splicing process (25–28).
Comparative analyses of the intrinsic strengths of all HIV-1 3′ss have shown that A1 is intrinsically inefficient (18) and, in the absence of other effects, would result in vif mRNA species representing only ∼1% of the total 4.0-kb mRNA class (29). However, in the presence of its downstream exonic sequence, A1 switches from being intrinsically very weak to being the most active HIV-1 3′ss (18). Indeed, two SREs within the noncoding leader exon 2 have been reported to enhance its splice site recognition, the SRSF1 (SF2/ASF)-dependent heptameric exonic splicing enhancers (ESEs) M1 and M2 (18) and the SRSF4 (SRp75)-dependent ESE Vif (22). Only the GGGG silencer element, which overlaps D2, is known to negatively act on exon 2 inclusion and vif mRNA production (22).
G runs (DGGGD, where D is U, G, or A) belong to those sequence motifs known to be bound by hnRNPs F and H (30) and often act as regulators within a variety of genes by modulating their splice site usage (30–38). Their influence is positive when they are localized immediately downstream of the 5′ss (39–43) or, alternatively, when multiple copies are present even further downstream (44–46).
It has been shown that overexpression of hnRNP F and H reduces the levels of HIV-1 transcripts by approximately 60% (hnRNP F) and 30% (hnRNP H) and affects the splicing pattern by increasing the relative amount of exon 3-containing mRNAs that splice to 3′ss A2 (Nef4, Env8, Vpr3, and Rev7/8/9). In line with this, knockdown of these splicing factors with small interfering RNA shows an inverse effect and results in mRNAs in which exon 3 skips, indicating that these proteins drive splicing toward 3′ss A2 (47, 48). Similarly, the ESS2p silencer downstream of 3′ss A3 (49) and the interaction of U1 snRNP with exon 6D 5′ss are regulated by hnRNPs F and H (50). Furthermore, in a subgenomic splicing reporter, the S3 G run in HIV-1 exon 1 was shown to modulate HIV-1 splicing by hnRNP H proteins (30). However, within a proviral context, the S3 G-run activity could not be confirmed (51).
Since there are many additional G runs present throughout the HIV-1 genome that might act as binding sites for hnRNP F/H family members, we sought to functionally analyze the impact of all of the G runs within the vif AUG-containing intron on the processing of the vif mRNA. We found that the G run localized directly upstream of the vif AUG was bound by members of the hnRNP F/H family and that binding was necessary for the maintenance of physiological vif mRNA levels.
MATERIALS AND METHODS
Plasmids.
The HIV-1 NL4-3 (GenBank accession no. M19921)-derived plasmid long terminal repeat (LTR) 4 exon was cloned by inserting the NdeI/EcoRI fragment from pNL4-3 (52) into the previously described LTR ex2 ex3 reporter (53). The minigene construct contains small noncoding leader exons 2 and 3, the 5′ part of tat exon 1, and the authentic NL4-3 full-length sequences for introns 2 and 3 in between. The coding regions for gag and pol within intron 1 (44 bp downstream of SD1 and 69 bp upstream of SA1) had been replaced with a short 13-bp linker fragment. LTR 4 exon GI2-1 mut was generated by PCR-directed mutagenesis with primer pair 1544/3307, subsequent digestion with BssHI/NdeI, and ligation into the LTR 4 exon. Cloning of plasmid LTR 4 exon GI2-2,3 mut was created by PCR-directed mutagenesis with primers 2339/3306 with LTR 4-exon as the template and subsequent digestion with SacI/PflMI and integration into the LTR 4 exon. LTR 4 exon GI2-4,5 was generated by amplifying 2710/3441 on the LTR 4 exon with subsequent digestion with NdeI/PflMI and ligation into the LTR 4 exon. LTR 4 exon GI2-1-5 mut was cloned by using the same primers, 2710/3441; LTR 4 exon GI2-4,5 as the PCR template; and integration into LTR 4 exon GI2-1 mut with SacI/PflMI. To generate HIV-1 molecular clone pNL4-3 GI2-1, the intron 1 sequence was extended up to the AgeI restriction site by cloning the PCR amplicon generated with primers 3375/3376 and digestion with EcoRI/NdeI, resulting in LTR 4 exon int1ext and LTR 4 exon int1ext GI2-1 mut, respectively. Cloning of pNL4-3 GI2-1 was then performed by using the AgeI/EcoRI fragment from LTR 4 exon int1ext GI2-1 mut, which was inserted into pNL4-3.
Rev expression plasmids SVcrev4 (4083/4084), SVcrev4b (4085/4086), SVcrev4 eff (4121/4122), and SVcrev4b eff (4123/42124) were cloned by replacement of the EcoRI/SacI fragment from SVcrev (54) with each indicated complementary oligonucleotide mixed at equimolar concentrations. Therefore, each oligonucleotide mixture was heated at 95°C for 5 min, cooled down to room temperature for 1 h, and subjected to DNA ligation. pXGH5 (55) was cotransfected to monitor transfection efficiency in quantitative and semiquantitative reverse transcription (RT)-PCR analyses. For minigene experiments, SVctat (54) was cotransfected to transactivate the HIV-1 LTR promoter. SVtat−rev−envRL was generated to replace the gene for chloramphenicol acetyltransferase (CAT) with a Renilla luciferase gene expression cassette by insertion of the XmaI/BbsI fragment from SVt−r−envCAT (simian virus 40 [SV40] early promoter, pNLA1 sequence nucleotides [nt] 5743 to 8887, tat− [ATG→AGG], rev− ATG→ACG; the sequence coding for Gp120 was replaced with the CAT-encoding gene sequence; SV40 polyadenylation signal) and the Esp3I/XmaI-restricted PCR amplicon (1271/1272) into pRLSV40 (Promega). pGL3-Control (Promega) was cotransfected to monitor transfection efficiency by firefly luciferase expression. All constructs were validated by DNA sequencing.
RNA and protein isolation from transiently transfected and infected cells.
HeLa-T4+ and HEK 293T cell cultures were prepared with Dulbecco's high-glucose modified Eagle's medium (Invitrogen) supplemented with 10% fetal calf serum and penicillin and streptomycin each at 50 μg/ml (Invitrogen). Jurkat, CEM-A, CEM-T4, and CEM-SS cells were maintained in RPMI 1640 medium (Invitrogen) under the same conditions. Transient-transfection experiments were performed with six-well plates at 2.5 × 105 cells per plate by using TransIT-LT1 transfection reagent (Mirus Bio LLC) according to the manufacturer's instructions. Total RNA and proteins were isolated by using the AllPrep DNA/RNA/Protein minikit (Qiagen) according to the manufacturer's instructions. Alternatively, total RNA was isolated by using acid guanidinium thiocyanate-phenol-chloroform as described previously (56).
Quantitative and semiquantitative RT-PCRs.
For reverse transcription (RT), 5 μg of RNA was digested with 10 U of DNase I (Roche). The DNase was subsequently heat inactivated at 70°C for 5 min, and cDNA synthesis was performed for 1 h at 50°C and 15 min at 72°C by using 200 U Superscript III RNase H reverse transcriptase (Invitrogen), 7.5 pmol oligo(dT)12-18 (Invitrogen), 20 U of RNasin (Promega), and 10 mM each deoxynucleoside triphosphate (Qiagen). For semiquantitative analysis of minigene and viral mRNAs, cDNA was used as the template for a PCR with the indicated primers. For transfection controls, PCRs were performed with primers 2258 and 2263 to detect spliced GH1 mRNA and with primers 3153 and 3154 for glyceraldehyde 3-phosphate dehydrogenase (GAPDH), respectively. PCR products were separated on 10% nondenaturing polyacrylamide gels, stained with ethidium bromide, and visualized with an F1 Lumi-Imager (Roche). Quantitative RT-PCR analysis was performed by using the LightCycler DNA Master SYBR green I kit (Roche) and LightCycler 1.5 (Roche). For normalization, primers 3387 and 3388 were used, detecting all HIV-1 mRNAs. Detection of vif mRNA was performed by using primer pair 3395/3396.
Oligonucleotides.
All of the DNA and RNA oligonucleotides used in this study were obtained from Metabion (Table 1).
Table 1.
DNA oligonucleotides used in this work
| Primer | Sequence |
|---|---|
| 1271 | 5′-AGCAGTCGTCTCGTGGCCAAGAAATGGCTTCGAAAGTTTATGAT-3′ |
| 1272 | 5′-TAGCCCGGGCTACTATTATTGTTCATTTTTGAGA-3′ |
| 1544 | 5′-CTTGAAAGCGAAAGTAAAGC-3′ |
| 2258 | 5′-TCTTCCAGCCTCCCATCAGCGTTTGG-3′ |
| 2263 | 5′-CAACAGAAATCCAACCTAGAGCTGCT-3′ |
| 2339 | 5′-TGGGAGCTCTCTGGCTAACTAGGGAACCCACTGCTTAAGC-3′ |
| 2588 | 5′-CTTTACGATGCCATTGGGA-3′ |
| 2710 | 5′-GGGGGGATCGATAATTAAGGAGTTTATATGGAAACCCTTAAAGGTAAAGGGGCAGTAGTAATACAA-3′ |
| 3153 | 5′-ACCACAGTCCATGCCATCAC-3′ |
| 3154 | 5′-TCCACCACCCTGTTGCTGTA-3′ |
| 3306 | 5′-TTCCTCCATTCTATGGAGACGCCTTGACCCAAATGCCAGTCTCTTTCTCCTGTATGCAGACCCCAATATGTTGTTATTACTAATTTAGCATCGCCTAGTGGGATGTGTACTTCTG-3′ |
| 3307 | 5′-TATACATATGGTGTTTTACTAATCTTTTCCATGTGTTAATCCTCATCCTGTCTACTTGCCACACAATCATCACCTGCCATCTGTTTTCCATAATCGCGGATGATCTTTGCTTTTCTTCTTGGC-3′ |
| 3375 | 5′-GGCCTGAATTCAGGGAGATTCTAAAAGAACCGGTACAT-3′ |
| 3376 | 5′-TATACATATGGTGTTTTACTAATCTTTTCC-3′ |
| 3387 | 5′-TTGCTCAATGCCACAGCCAT-3′ |
| 3388 | 5′-TTTGACCACTTGCCACCCAT-3′ |
| 3392 | 5′-CGTCCCAGATAAGTGCTAAGG-3′ |
| 3395 | 5′-GGCGACTGGGACAGCA-3′ |
| 3396 | 5′-CCTGTCTACTTGCCACAC-3′ |
| 3441 | 5′-TTTCCTCCATTCTATGGAGACGCCTTGGCCGAGATGCCAGTCTCTTTCTCCTGTATGCAGGCCGAAGTATGTTGTTATTACTAATTTAGCATCGCCT-3′ |
| 4083 | 5′-AATTCCTCCTATGGCAGGAAGAAGCGGAGACAGCGACGAAGAGCT-3′ |
| 4084 | 5′-CTTCGTCGCTGTCTCCGCTTCTTCCTGCCATAGGAGG-3′ |
| 4085 | 5′-AATTCGGATTATGGAAAACAGATGGCAGGCATCTCCTATGGCAGGAAGAAGCGGAGACAGCGACGAAGAGCT-3′ |
| 4086 | 5′-CTTCGTCGCTGTCTCCGCTTCTTCCTGCCATAGGAGATGCCTGCCATCTGTTTTCCATAATCCG-3′ |
| 4121 | 5′-AATTCCAGCCATGGCAGGAAGAAGCGGAGACAGCGACGAAGAGCT-3′ |
| 4122 | 5′-CTTCGTCGCTGTCTCCGCTTCTTCCTGCCATGGCTGG-3′ |
| 4123 | 5′-AATTCCAGCCATGGAAAACAGATGGCAGGCATCTCCTATGGCAGGAAGAAGCGGAGACAGCGACGAAGAGCT-3′ |
| 4124 | 5′-CTTCGTCGCTGTCTCCGCTTCTTCCTGCCATAGGAGATGCCTGCCATCTGTTTTCCATGGCTGG-3′ |
| 4355 | 5′-TTCATCGAATTCAGTGCCAAGAAGAAAAGCAAAGATCA-3′ |
RNA pulldown.
Three thousand picomoles of high-performance liquid chromatography-purified RNA oligonucleotides 3600 (GATCATCAGGGATTATGGA [underlining indicates the wild-type 3600 and the mutant 3601 GI2-1 sequences]) and 3601 (GATCATCCGCGATTATGGA) was adjusted to a total volume of 340 μl. Twenty microliters of a saturated sodium metaperiodate solution and 40 μl of sodium acetate (1 M, pH 5) were added, and after incubation for 1 h at room temperature in the dark, the RNAs were precipitated by the addition of 80 μl of sodium acetate (1 M, pH 5) and 1 ml of ethanol (96%) and incubation at −80°C for 5 min and subsequently pelleted at full speed for 30 min at 4°C. The RNAs were covalently linked to adipic acid dihydrazide-agarose beads (Sigma) at 4°C overnight and washed twice with sodium chloride (2 M) and three times with Dignam buffer D (20 mM HEPES-KOH [pH 7.6], 6.5% [vol/vol] glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM dithiothreitol [DTT]). Two hundred microliters of HeLa cell nuclear extract (∼6 mg/ml; CilBiotech) was added to the immobilized RNA, and the mixture was incubated for 20 min at 30°C and washed five times with Dignam buffer D plus MgCl2 (20 mM HEPES-KOH [pH 7.6], 6.5% [vol/vol] glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM DTT, 1 M MgCl2). Agarose beads were resuspended in 2× protein sample buffer (0.75 M Tris-HCl [pH 6.8], 20% [vol/vol] glycerol, 10% [vol/vol] β-mercaptoethanol, 4% [wt/vol] SDS), and the proteins were eluted by heating at 95°C for 10 min and subjected to SDS-PAGE and Coomassie staining.
Immunoblot analysis.
Proteins were subjected to SDS-PAGE under denaturing conditions (57) in 12% polyacrylamide gels (Rotiphorese Gel 30; Roth) using Bio-Rad Protean II Mini electrophoresis systems (Bio-Rad). Gels were run in Laemmli running buffer (1% SDS, 0.25 M Tris base, 1.9 M glycine) for 45 min at 25 mA and 4°C. The proteins were transferred to a polyvinylidene difluoride membrane (pore size, 0.45 mm; Protran) by using the Bio-Rad Protean II Mini tank blotting system (Bio-Rad) in transfer buffer (0.1% SDS, 192 mM glycine, 25 mM Tris [pH 8.8], 20% methanol). The membrane was washed twice in TBS-T (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% [vol/vol] Tween 20), blocked in TBS-T with 10% nonfat dry milk for 1 h at room temperature, and then incubated overnight at 4°C with the primary antibody in TBS-T with 5% dry milk. Sheep antibody against HIV-1 p24 CA was obtained from Aalto Bioreagents Ltd. (Dublin, Ireland), and rabbit antiserum against Vif was obtained through the NIH AIDS Research and Reference Reagent Program from Dana Gabuzda and Jeffrey Kopp (58). Anti-A3G immunoblot assays were performed with anti-ApoC17 antibody from Klaus Strebel (59, 60). The membrane was washed three times with TBS-T for 10 min each time and incubated with anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (A6154) from Sigma-Aldrich and HRP-conjugated anti-sheep antibody from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA) for 1 h, respectively. The blot was washed four times, rinsed with water, visualized by a ECL chemiluminescence detection system (Amersham), and exposed to photosensitive film (GE) or Lumi-Imager F1 (Roche). Immunoblotting employing the Gp41 monoclonal antibody Chessie 8 (61) was performed as previously described (62). The p24-CA protein in the cell-free supernatant was concentrated by using sucrose centrifugation at 50,000 × g for 4 h and subsequently subjected to immunoblot analysis as described above.
Luciferase measurement.
HeLa-T4+ cells (2.5 × 105) were transiently transfected with 2 μg SVtat−rev−envRL reporter plasmid, 0.5 μg pGL3-Control (Promega) for normalization and expression plasmids as indicated and pcDNA3.1(+) to adjust the amount of transfected DNA to 3.5 μg per sample. For luciferase measurement, cultured cells were rinsed in phosphate-buffered saline (PBS), dispensed into 500 μl passive lysis buffer (Promega), and shaken for 15 min at ambient temperature, and the cell lysates were cleared by centrifugation for 10 s at 20,000 × g. Firefly and Renilla luciferase activities were measured by a dual-luciferase program by adding 100 μl Beetle-Juice and Renilla-Juice (p · j · k) with Mithras LB 940 (Berthold), respectively.
Northern blot analysis.
Total RNA (2 μg) was electrophoresed on a denaturing 1% agarose gel and capillary blotted onto a positively charged nylon membrane by using 20× SSC (3 M NaCl, 300 mM sodium citrate). The RNA was UV cross-linked to the membrane, and the large and small rRNAs were marked. Subsequently, the membrane was prehybridized with 10 ml 1× DIG Easy Hyb hybridization solution (Roche) for 1 h at 65°C. The membrane was then hybridized with probes for 5 h at 55°C. The probe for HIV-1 mRNAs was based on a 153-bp digoxigenin (DIG)-labeled PCR product using the primer pair 3387/3388 (HIV-1 exon 7). The PCR product was purified by using phenol and chloroform-isoamyl alcohol (24:1 ratio), and a second PCR was performed by using DIG-labeled dUTPs (alkali-labile DIG-11-dUTP; Roche), the same primer pair, and the first PCR product as the template. The probe was heated in 1× DIG Easy Hyb hybridization solution (Roche) at 95°C for 5 min and chilled on ice. Following overnight hybridization at 55°C, the membranes were washed twice with wash buffer 2× SSC (300 mM NaCl, 30 mM sodium citrate) with 0.1% SDS at room temperature, followed by two 20-min washes in 0.2× SSC (30 mM NaCl, 3 mM sodium citrate) with 0.1% SDS at 68°C. The membrane was then washed twice with double-distilled H2O and maleic acid buffer (0.1 M maleic acid, 0.15 M NaCl) and blocked with blocking solution (Roche) for 1 h. Alkaline phosphatase (AP)-conjugated anti-DIG-antibody (Fab fragments from sheep antibody; Roche) was diluted 1:20,000 in 1× blocking solution (Roche) and incubated for 1 h. The membrane was washed three times with maleic acid buffer, and the RNA bands were visualized by using CDP-Star for chemiluminescent reactions (1:100 in AP buffer [0.1 M Tris HCl, 0.1 M NaCl, pH 9.5]; Roche).
Measurement of HIV-1 replication kinetics.
CEM-A, CEM-T4, and CEM-SS cells (4 × 105) were infected with 1.6 ng p24 CA of wild-type and mutant viruses in serum-free RPMI medium at 37°C for 6 h. Infected cells were then centrifuged, washed in PBS (Invitrogen), and resuspended in RPMI medium (Invitrogen) containing 10% fetal calf serum (FCS; Invitrogen). Aliquots of cell-free medium were harvested at regular intervals, and virus production was measured by p24 enzyme-linked immunosorbent assay (ELISA). Peripheral blood mononuclear cells (PBMCs) were isolated from 15-ml whole-blood samples from two healthy donors by Ficoll gradient centrifugation. PBMCs were cultured in RPMI 1640 GlutaMax medium containing 10% FCS and 1% penicillin-streptomycin, activated with phytohemagglutinin (5 μg/ml), and treated with interleukin-2 (30 mg/ml; Roche) after 48 h. Cells (8 × 105) were infected with 16 ng of p24 CA of wild-type and mutant viruses and treated as described above.
Next-generation sequencing (NGS) and read mapping.
Total RNA was extracted from 5 × 106 Jurkat cells per sample by using the AllPrep DNA/RNA/Protein minikit (Qiagen), and the preparations were checked for RNA integrity with an Agilent 2100 Bioanalyzer. All of the samples in this study showed common high-quality RNA integrity numbers (9.1 to 10; mean, 9.9). RNA was quantified by photometric measurement (NanoDrop 1000 Spectrophotometer, ND-1000 version 3.7.0; Thermo Scientific). Syntheses of cDNA libraries were performed with the TruSeq RNA Sample Prep kit (Illumina) according to the manufacturer's protocol. One microgram of total RNA was used for poly(A) RNA enrichment, followed by cDNA synthesis, adapter ligation, and PCR amplification. The resulting cDNA libraries were validated by Agilent DNA 1000 chip, quantified by fluorometric measurement (Qubit dsDNA HS Assay kit; Invitrogen), and adjusted to 10 nM.
Clonal amplification of cDNA on two Illumina flow cells (v1.5) was done by using the appropriate cBot recipe (version 7) at a final library concentration of 10 pM. Sequencing was carried out on a HiSeq 2000 according to the manufacturer's protocol (HiSeq 2000 User Guide, part 15011190, revision H; Illumina, Inc.) using TruSeq SBS kits v1. The resulting 101-nt sequence reads were converted to fastq by CASAVA 1.8.2. The 101-nt reads were mapped to an exon junction database by using the reference mapping algorithm within the CLC Genomics Workbench software (v4.9; CLC bio). On the basis of those reads mapping to HIV-1 exon junctions, we then estimated relative splice site usage. Read sequences that mapped to more than one exon junction were excluded from quantification. In order to identity unannotated splice events, Reads were mapped with TopHat2.0.0 to HIVNL43 sequence (downloaded from http://www.ncbi.nlm.nih.gov/nuccore/M19921).
RESULTS
A guanosine (G) run element (GI2-1) embedded within HIV-1 intron 2 controls splicing at 5′ss D2b.
To analyze whether one of the five G runs identified within HIV-1 intron 2 (GI2-1-5) is involved in splice site regulation and processing of vif mRNA, we performed a mutational analyses of these motifs (Fig. 1). Using an HIV-1 subgenomic splicing reporter (Fig. 1C), we disrupted the G runs individually or in combination (GI2-1-5 mut) and determined individual splicing outcomes. For the analysis in the context of infectious provirus, mutations were chosen that did not change the coding sequence of the overlapping IN (integrase) and Vif open reading frames (ORFs). Only mutation GI2-4 resulted in an amino acid substitution (W70F) within the Vif protein (Fig. 1D).
Fig 1.

HIV-1 intron 2 contains several G runs and an alternative 5′ss. The diagram shows the locations of splice sites, exons, introns, and SREs in the HIV-1 NL4-3 genome. (A) Schematic overview of the HIV-1 NL4-3 genome, including the locations of 5′ss and 3′ss, respectively. The RRE is indicated by an open box. The positions of the PCR primers used in this analysis are indicated by black triangles. (B) vif mRNA is formed primarily by splicing of 5′ss D1 to 3′ss A1 where noncoding exon 2 (50 nt) is included and vif AUG-containing intron 2 is retained. Rev4 is formed by splicing of 5′ss D1 to 3′ss A4b and spliced downstream from 5′ss D4 to 3′ss A7. In contrast, rev4b is formed by splicing from alternative 5′ss D2b to 3′ss A4b up, taking the vif AUG into the rev ORF. (C) Vif-coding intron 2 sequence (427 nt) including the locations of the vif ATG and G run GI2-1. The intrinsic strengths of 5′ss D2 and D2b are indicated (HBS, MAXENT). (D) Representation of the mutant constructs used in this work. The mutant constructs were based on the reference sequence from infectious proviral clone NL4-3. With the exception of GI2-4, silent mutations were introduced to maintain the coding sequence for the amino acids of integrase and Vif. GI2-4 led to a tryptophan-to-phenylalanine amino acid substitution in the vif ORF.
Total RNA was isolated 24 h following the transient transfection of HeLa-T4+ cells, and the splicing pattern was analyzed by RT-PCR and quantitative RT-PCR (Fig. 2). Mutating G run GI2-1, which is highly conserved among most HIV-1 strains and subtypes, although not among HIV-2 nor simian immunodeficiency virus strains (Los Alamos HIV database 2011, data not shown), led to an increased amount of both the exon 2-containing transcript tat2 (Fig. 2B, 1544/2588, cf. lanes 2 and 3) and the intron 2-retaining vif2 mRNA (Fig. 2B, 1544/3396, cf. lanes 2 and 3). Additionally, we detected an unknown RT-PCR product migrating slower than tat3 (Fig. 2B, line 3, [1.2b.4]). Sequencing analysis showed this to be a splicing isoform resulting from splicing at an alternative 5′ss downstream of D2 (Fig. 1D and 2A). This 5′ss, referred to here as D2b, has been described previously as being a rarely detectable cryptic 5′ss at position 5059 (29). Of note, 5′ss D2b has an even slightly stronger intrinsic strength (CAGGTgAtgAT, HBS 12,4/MAXENT 5,99) than D2 (aAGGTgAaggg, HBS 10,7/MAXENT 5,79) (45, 63) and, like GI2-1, is remarkably conserved among most HIV-1 strains and subtypes (Los Alamos HIV database 2011, data not shown). Therefore, the efficiency of D2b use would have been expected to be similar to that of D2. In order to confirm splicing at 5′ss D2b in the context of nonmutated pre-mRNAs, HeLa-T4+ or HEK 293T cells were transfected with the splicing reporter and splicing patterns analyzed. Indeed, using a 5′ primer positioned between D2 and D2b, we could confirm detectable, but weak, D2b usage even in the absence of the GI2-1 mutation (data not shown). Remarkably, disruption of GI2-1 also led to the increased activation of 3′ss A1 and vif mRNA formation in semiquantitative (Fig. 2B, 1544/3396) and quantitative RT-PCR analyses (Fig. 2C, 3395/3396).
Fig 2.
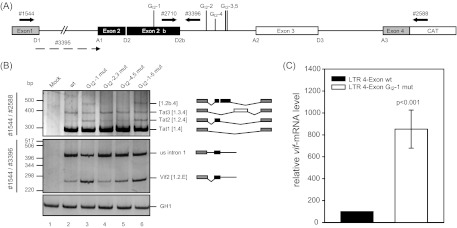
Alternative 5′ss within HIV-1 NL4-3 intron 2 leads to alternative transcript isoforms. (A) Schematic drawing of the HIV-1 pNL4-3-derived subgenomic splicing reporter (LTR 4 exon). The reporter contains noncoding leader exons 2 and 3 flanked by authentic NL4-3 sequences. Coding regions for gag and pol in intron 1 have been deleted and replaced with a shorter linker sequence (see Materials and Methods). The 5′ss and 3′ss, including D2b, are indicated. The positions of the RT-PCR primers used are depicted by arrows. (B) RT-PCR analysis of total RNA isolated from HeLa-T4+ cells transiently transfected with the subgenomic splicing reporter shown in panel A or its mutant derivate at 24 h posttransfection. Cells were cotransfected with SVctat and pXGH5 (GH1). The primer pairs used are indicated on the left (see panel A). Every band was isolated from the gel and confirmed by sequencing analysis. Transcript isoforms are depicted on the right. To compare the total RNA amount and transfection efficiency, a separate RT-PCR was performed by using primer pair 2263/2858 to amplify a spliced GH1 sequence. PCR amplicons were separated on a nondenaturing 10% polyacrylamide gel and stained with ethidium bromide. (C) Quantitative RT-PCR of total RNA from panel B using exon junction primers specific for vif mRNA (3395/3396). The authentic splice pattern (wt) was set to 100%. The relative splice site usage was normalized to cotransfected GH1.
In support of a functionally exclusive role for GI2-1 in Vif splice site regulation, the mutation of any of the other G runs (GI2-2, 3 and GI2-4, 5) did not affect the splicing pattern of the splicing reporter.
Members of the hnRNP F/H protein family bind to the intronic G run (GI2-1).
Since G-run motifs have been shown to bind members of the hnRNP F/H protein family, RNA affinity purification assays were performed to determine if this were also the case with the viral GI2-1 sequence. RNA oligonucleotides containing the wild-type or mutant GI2-1 sequence were immobilized on agarose beads and incubated with HeLa cell nuclear extract. Bound proteins were eluted and separated by SDS-PAGE (Fig. 3). Protein bands bound to the wild-type GI2-1 oligonucleotide but exhibiting less efficient binding to the mutant GI2-1 RNA oligonucleotide were analyzed by mass spectrometry. These analyses confirmed the binding of hnRNP F/H proteins to the wild-type sequence but not to the mutated sequence (Table 2). This, in turn, suggests that these proteins play a role in GI2-1-mediated splicing regulation.
Fig 3.

Proteins hnRNP F, hnRNP H, and hnRNP A2/B1 bind to the intronic G run (GI2-1) but not to the mutated sequence. Shown is a Coomassie-stained protein gel of a GI2-1 RNA pulldown experiment. Short (19-nt) RNA oligonucleotides (wt, GATCATCAGGGATTATGGA; GI2-1 mut, GATCATCCGCGATTATGGA) of authentic or mutated GI2-1 sequence were linked to adipic acid dihydrazide-agarose beads and incubated with HeLa cell nuclear protein extract. The precipitated proteins were resolved by SDS-PAGE (12%) and stained with Coomassie brilliant blue. Bands were eluted and analyzed by mass spectrometry. The identified proteins are indicated by arrows.
Table 2.
Results of mass spectrometric analysis of proteins of the GI2-1 RNA pulldown experiment shown in Fig. 3
GI2-1 controls the levels of vif mRNA and Vif protein expression from replication-competent virus.
Next we analyzed the impact of the silent but inactivating mutation of GI2-1 on gene expression from proviral plasmids. HEK 293T cells were transiently transfected with pNL4-3 or the GI2-1 mutant proviral plasmid pNL4-3 GI2-1 mut, and total RNA and proteins were harvested at 48 h posttransfection. The RNAs were subjected to Northern blotting and probed with an exon 7 fragment detecting all viral mRNAs. As shown in Fig. 4A, the GI2-1 mutation led to an increased amount of vif mRNA (cf. lanes wt and GI2-1 mut). In order to quantify the GI2-1 mutation-mediated change in vif mRNA levels, a quantitative real-time PCR was performed using vif mRNA-specific exon junction primers. As a control, we determined the amounts of all viral mRNAs by using a primer pair amplifying exon 7 sequences that are present in all viral mRNA species. In agreement with the result obtained from the Northern blot analysis, vif mRNA levels were upregulated 7-fold following the disruption of GI2-1 (Fig. 4B). Furthermore, immunoblot analysis showed an increase in the amount of Vif protein expressed within transfected cells (Fig. 4C). Taken together, these results demonstrate that GI2-1 is important for splicing regulation of 3′ss A1 and the generation of functional vif mRNA and confirm previous observations that an increase in 5′ss D2 usage leads to an increase in exon 2 inclusion and levels of vif mRNA (22). However, our results additionally show that the increase in vif mRNA can also be mediated by activation of D2b, which is located further downstream. These data are comparable to the concomitantly observed increase in 5′ss D3 usage and vpr mRNA levels (see the accompanying paper by Erkelenz et al. [81]).
Fig 4.

The silent mutation of GI2-1 increases both vif mRNA and protein levels. (A) Northern blot analysis of total RNA from pNL4-3-transfected HEK 293T cells isolated at 48 h posttransfection. RNA was separated in a 1% RNA agarose gel, capillary blotted, and cross-linked on a positively charged nylon membrane and UV cross-linked. The membrane was treated with a DIG-labeled DNA fragment binding to exon 7. (B) RNA from panel A was subjected to quantitative RT-PCR analysis using an exon junction primer pair specific for vif mRNA (3395/3396) and for exon 7 (3387/3388) for normalization of the viral load. (C) Immunoblot analysis of proteins from transiently transfected HEK 293T cells with proviral plasmids pNL4-3 and pNL4-3 GI2-1 mut. Proteins were separated by SDS-PAGE, blotted, and analyzed with antibodies specific to Vif and p24 as a loading control for the viral load. (D) RT-PCR analysis of total RNA isolated from panel A with primer pairs specific for the 2-kb mRNA class. The primer pairs used are indicated on the left (see Fig. 1). PCR amplicons were separated on a nondenaturing 10% polyacrylamide gel and stained with ethidium bromide. Transcript isoforms are depicted on the right. To compare the total RNA amounts, a separate RT-PCR was performed with the primer pair 3502/3503 to amplify the GAPDH sequence.
To quantify the frequency of D2b usage and to identify its 3′ss targets, we performed RT-PCR analysis of total RNA isolated from HEK 293T cells transfected with pNL4-3. To also monitor the impact of the GI2-1 inactivation on D2b usage, we included the pNL4-3 GI2-1 mut proviral DNA and focused on transcripts that were spliced not at D2 but at D2b. In addition to the published transcript isoform, 1.2b.5.7 (29), we identified three novel D2b spliced transcript isoforms (1.2b.3.5.7, 1.2b.4.7, and 1.2b.3.4.7) covering either the Tat or the Nef ORF (Fig. 4D, 2710/3392). In addition, we identified an alternatively spliced vif1 mRNA, 1.2E.4.7, not previously found among the Rev-independent 1.8-kb species (29). Inactivation of GI2-1 led to the increased inclusion of exon 2b in an mRNA, 1.2b.5.7, encoding a Vif-Tat-Gp41 fusion protein. Concomitantly, there was then a reduction in the amount of the exon 3-containing transcript 1.2b.3.4.7 encoding a putative 9 amino-acid (aa)-long Vif peptide (Fig. 4D, cf. lanes wt and GI2-1 mut). The use of a primer pair covering the 5′ half of the HIV-1 genome allowed the detection of a significant amount of an mRNA 1.2b.4.7 coding for a putative 12-aa-long Vif peptide when GI2-1 was inactivated. The levels of this transcript were below the analytical limit of detection in the wild-type context (Fig. 4D, 1544/3632). Furthermore, we observed an increased amount of tat2 mRNA 1.2.4.7, indicating more-efficient recognition of 3′ss A1. These data are in agreement with those obtained from the subgenomic minigene constructs demonstrating that GI2-1 acts to repress 3′ss A1 recognition. In conclusion, these data support the hypothesis that 5′ss D2b, which is negatively regulated by GI2-1, might safeguard D2 with respect to its exon-bridging function, thus supporting 3′ss A1 activation and maintaining Vif protein levels.
GI2-1 in intron 2 is necessary for efficient virus replication in nonpermissive cells.
Since the Vif protein level was considerably increased upon the inactivation of GI2-1 (Fig. 4C), we wanted to analyze the impact of this SRE on multiround HIV-1 replication in the presence and absence of A3G. Therefore, we produced virus by transient transfection of HEK 293T cells with the proviral plasmid pNL4-3 or its mutant derivate NL4-3 GI2-1 mut and harvested virus-containing supernatants at 48 h posttransfection. pNL4-3 Δvif proviral DNA (64) served as a control for a vif-deficient virus. To monitor virus replication, the permissive host cell line CEM-SS lacking A3G (5) and the nonpermissive A3G-expressing CEM-A cell line (65) were infected with equal amounts of p24 CA (multiplicity of infection [MOI], 0.01). The heterogeneous A3G-expressing cell line CEM-T4 was also infected to analyze the replication ability of GI2-1 mutant virus under semipermissive host cell restriction pressure conditions (66). The A3G expression levels of all three cell lines were determined by immunoblot analysis (Fig. 5B). Cell culture supernatants were harvested at different time points after infection, and p24 capsid protein (CA) was monitored by immunoblotting (Fig. 5C) and quantified by capture ELISA (Fig. 5A) to record virus replication.
Fig 5.
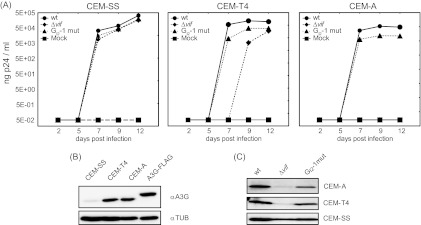
The G run in intron 2 is necessary for efficient virus replication in nonpermissive cells. (A) CEM-SS, CEM-T4, and CEM-A cells were infected with the NL4-3 virus or mutant derivatives (MOI, 0.01). Virus production was determined by p24 CA capture ELISA of cell-free supernatant collected at the indicated time points. (B) Immunoblot analysis showing A3G expression in CEM-SS, CEM-T4, and CEM-A cells. As a positive control, HEK 293T cells were transiently transfected with a FLAG-tagged A3G expression plasmid. (C) Immunoblot analysis of p24-CA protein in the cell-free supernatant concentrated by sucrose centrifugation at 12 dpi.
As expected, in A3G-deficient CEM-SS cells, the presence of Vif was not required for efficient virus particle production, as neither inactivation of the GI2-1 SRE nor vif deficiency (NL4-3 Δvif) impaired viral replication (Fig. 5A and C). On the other hand, NL4-3 Δvif was not capable to replicate in A3G-positive CEM-A cells and newly synthesized virus was not detectable, even after 12 days postinfection (dpi) (Fig. 5A). Unexpectedly, the p24 CA production of GI2-1 mutant virus was decreased almost 5-fold despite increased Vif protein expression and a comparable reduction in p24 CA levels was observed in CEM-T4 cells. According to the semipermissiveness of the mixed cell population, Δvif mutant virus was able to replicate, but with a delay of 2 days, presumably due to viral replication in only a subset of the CEM-T4 cells. These results are in agreement with previous observations demonstrating that the amount of Vif required for optimal viral replication is in a narrow range and that higher levels of Vif decrease viral infectivity, perhaps by modulating proteolytic processing of the Gag precursor at the p2/nucleocapsid processing site (11). To further characterize the impact of GI2-1 on viral replication, we assayed HIV infection of human peripheral blood mononuclear cells (PBMCs) from two healthy donors with an MOI of 0.5. In cells from both donors inactivation of GI2-1 caused a moderate increase in virus replication (Fig. 6A). As expected, HIV-1 Δvif did not replicate in the PBMC cultures (Fig. 6A and C). Interestingly, the activated PBMCs showed higher expression of A3G than the CEM-A and CEM-T4 cells did (Fig. 6B).
Fig 6.

Inactivation of GI2-1 enhances virus replication in PBMCs. (A) PBMCs were infected with the NL4-3 virus or mutant derivatives (MOI, 0.5). Virus production was determined by p24 CA capture ELISA of cell-free supernatant collected at the indicated time points. (B) Immunoblot analysis showing A3G expression in CEM-SS, CEM-T4, and CEM-A cells and PBMCs of two human donors. αTUB, antitubulin antibody. (C) Immunoblot analysis of p24-CA protein in cell-free supernatant at 12 dpi.
In summary, these results demonstrate the impact of GI2-1 as a critical SRE to maintain the balanced vif mRNA levels necessary for optimal HIV-1 replication in nonpermissive cells and that an appropriate ratio of Vif to A3G protein levels is required for optimal virus replication under different physiological environments.
NGS analysis of NL4-3-infected Jurkat T cells reveals that 5′ss D2b is used, albeit with low efficiency.
Since we observed 5′ss D2b usage in pNL4-3-transfected HEK 293T cells, we were interested in the frequency of D2b-containing exon junctions during the course of T cell infection. Therefore, we analyzed splice site selection by RNA deep sequencing of NL4-3-infected Jurkat T cells. Total RNAs from uninfected and HIV-1-infected Jurkat T cells were isolated at 24 (MOI, 0.1) and 144 (MOI, 0.01) h postinfection (hpi), and cDNA libraries were created from poly(A)-selected mRNAs and subjected to NGS analysis. Using the Illumina sequencing protocol, we obtained ∼170 million 101-nt reads per sample and mapped them to an HIV-1 exon junction database. Whereas, D2b usage was not detectable at 24 hpi, most likely due to a low number of overall exon junction reads, at 144 hpi, D2b was used in 0.2% of the reads that were mapped to this genomic region (Fig. 7 and Table 3). Since we could not map any reads to an exon junction using D1a or D5 (both of which were not expected to be used [67, 68]), D2b usage should be classified as an additional alternative HIV-1 5′ss. Thus, one or several of the alternative D2b spliced transcripts may encode an additional, as yet unidentified, HIV-1 protein or protein isoform.
Fig 7.

NGS analysis reveals usage of alternative 5′ss D2b. Total RNA of HIV-1 NL4-3-infected Jurkat cells was isolated and subjected to NGS analysis. The resulting 101-nt sequence reads were mapped to the HIV-1 exon junction database, and the relative usage of every splice site was estimated. Read sequences that mapped to more than one exon junction were excluded from quantification. Shown is the relative splice site usage in samples obtained at 24 and 144 hpi.
Table 3.
Percentages of spliced and unspliced reads obtained from HIV-1 infected T cells mapped to exon junction database
| 5′ss | 24 hpia |
144 hpib |
||||
|---|---|---|---|---|---|---|
| Total no. of reads | % Spliced | % Unspliced | Total no. of reads | % Spliced | % Unspliced | |
| D1 | 28 | 60.7 | 39.3 | 1,852 | 61.1 | 38.9 |
| D1a | 0 | 0.0 | 0.0 | 0 | 0.0 | 0.0 |
| D2 | 31 | 12.9 | 87.1 | 3,401 | 5.3 | 94.7 |
| D2b | 38 | 0.0 | 100 | 3,919 | 0.2 | 99.8 |
| D3 | 33 | 6.1 | 93.9 | 2,669 | 6.7 | 93.3 |
| D4 | 78 | 53.8 | 46.2 | 4,501 | 27.2 | 72.8 |
| D5 | 0 | 0.0 | 0.0 | 0 | 0.0 | 0.0 |
MOI, 0.1.
MOI, 0.01.
Newly identified D2b-derived transcripts coding for Rev and Gp41 isoforms.
Since we identified D2b-A2, D2b-A3, D2b-A4b, and D2b-A5 exon junction reads (Table 4), we aimed at identifying those transcript isoforms covering these junctions. Therefore, total RNA of NL4-3-infected Jurkat cells (144 hpi) was isolated and reverse transcribed and the resulting cDNAs were subsequently amplified with primers located immediately upstream of the vif ATG codon (4355) and downstream of SA7 (3392). By cloning and sequencing, we identified three cDNAs coding for new proteins, Rev4b, Gp41a, and Gp41b, all carrying 6 aa derived from Vif (MENRWQ) at their N termini (Fig. 1E).
Table 4.
5′ss D2b-derived exon junctions, transcripts, and ORFs identified in this work
| Junction | Exon | No. of reads | cDNA | Transcript | Length (bp) | Protein (molecular mass [kDa]) | Length (aa) |
|---|---|---|---|---|---|---|---|
| D2b_A2 | 1.2b.3.7 | 9 | Identified | 27 | Short ORF | 9 | |
| D2b_A3 | 1.2b.4.7 | 9 | Identified | 33 | Short ORF | 11 | |
| D2b_A4c | 1.2b.4c.7 | NDa | ND | 24 | Short ORF | 8 | |
| D2b_A4a | 1.2b.4a.7 | ND | Identified | 27 | Short ORF | 9 | |
| D2b_A4b | 1.2b.4b.7 | 2 | Identified | rev4b | 376 | Rev4b (14.21) | 125 |
| D2b_A7 | 1.2b.7 | ND | Identified | gp41 1 | 432 | Gp41a (16.93) | 144 |
| D2b_A5 | 1.2b.5.7 | 4 | Identified | gp41 2 | 501 | Gp41b (19.32) | 167 |
ND, none detected.
To answer the question of whether the Gp41 isoforms could be detected in infected cells, C8166 T cells were infected with wild-type NL4-3 and NL4-3 Env-Tr712 with a stop codon at position 713 in Env employing vesicular stomatitis virus G-protein-pseudotyped virions as previously described (62, 69). HEK 293T cells were transfected with an expression vector coding for the larger Gp41 isoform, i.e., Gp41b, and the protein lysates of the infected and transfected cells were subjected to immunoblotting employing Chessie 8 antibodies directed against the C-terminal domain of Gp41 (61, 62). As expected, Gp160 and Gp41 were detected in the lysate of pNL-4-3-infected cells (Fig. 8B) but absent from the lysates of cells infected with pNL-Env-Tr712 virions encoding truncated Env lacking the Chessie 8 epitope. However, in both cases, additional reactive protein entities, with sizes ranging from approximately 13 to 23 kDa, were detected. These were absent from lysates of mock-transfected cells or when unrelated antibodies were used (not shown), meaning that they represent novel, currently undefined, proteins containing the HIV Chessie 8 epitope. Expression of the larger Gp41 isoform led to the generation of a band comparable in size to one of the Env-CT variants detectable in infected T cells (Fig. 8B). This could mean that this comigrating protein band in the infected T cells corresponds to the cloned Gp41 isoform but, in the absence of mass spectrometric identification, this remains only a postulate.
Fig 8.

Sequence alignment of authentic Gp41 and D2b-derived isoforms Gp41a and Gp41b. From position 208, all of the protein isoforms show sequence identity. (B) Western blot analysis with Gp41-CT antibodies employing lysates of cells transfected or infected with the indicated constructs. The positions of molecular mass markers are shown on the right, and those of the provirally expressed Env-CT-containing proteins are shown on the left. The Env-CT bands are a group of specifically detected low-molecular-mass protein entities.
Rev4b mediates Rev-dependent luciferase expression.
In order to compare the activities of Rev4b and Rev, we measured their abilities to mediate luciferase expression. HeLa-T4+ cells were cotransfected with the Rev-dependent luciferase reporter SVtat−rev−envRL and increasing amounts of the expression plasmids coding for Rev4b or Rev (Fig. 9). When HeLa-T4+ cells were transfected with the same amount of expression plasmid Rev or Rev4b, the latter yielded 2-fold higher luciferase activity (Fig. 9C), suggesting that the N-terminal amino acids of Rev4b resulted in higher protein activity. Alternatively, the better match of the AUG nucleotide surrounding of vif with the Kozak consensus sequence may be more efficiently recognized than that of rev, thus leading to a greater amount of Rev4b protein. We thus repeated the luciferase assay with Rev4b and Rev expression plasmids with identical AUG nucleotide surroundings. As shown in Fig. 8B and C, equalization of the AUG nucleotide surroundings of both the rev4b and rev ORFs resulted in equally strong luciferase expression (Fig. 9B and C), demonstrating functional equivalence of Rev4b and Rev protein isoforms in this assay.
Fig 9.
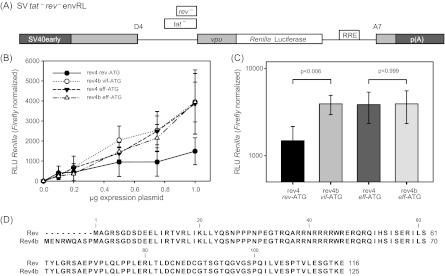
The usage of alternative 5′ss D2b allows the translation of a novel functional Rev protein variant by using the vif AUG. (A) Schematic drawing of the Rev-dependent luciferase reporter SVtat−rev−envRL. The 5′ss and 3′ss D4 and A7 are indicated. The RRE is represented by an open box. SV40early, SV40 early promoter; p(A), SV40 polyadenylation signal. (B) HeLa-T4+ cells were transiently transfected with pGL3-Control and cDNA expression vectors as indicated. Rev activity was quantified by Renilla luciferase activity and normalized to firefly luciferase activity. (C) The same as in panel B but with 1 μg expression plasmid. (D) Alignment of Rev and Rev4b sequences.
DISCUSSION
In this study, we have performed a mutational analysis of the G runs located in the HIV-1 NL4-3 intron 2 sequence and demonstrated that vif mRNA splicing is tightly regulated by the intronic G run GI2-1 that is localized at nt 73 to 76. So far, only the GGGG motif overlapping 5′ss D2 from its intronic position has been shown to have splicing regulatory activity by repressing exon 2 splicing (22). Here we have discovered that the hnRNP family members F/H bound to GI2-1 and acted on vif mRNA expression levels through repression of the alternative 5′ss, termed D2b. Whereas the importance of the functional strength of 5′ss D2 for vif expression has already been demonstrated by up and down mutations of its intrinsic strength (22, 70), as well as by the identification of SRE elements in exon 2 (18, 22), the inactivation of GI2-1 now confirms that it is the functional strength of the exon 2 or/and 2b 5′ss that regulates vif mRNA expression levels.
Mutation of GI2-1 abrogated hnRNP F/H binding and relieved silencing of the downstream alternative 5′ss D2b that led to the emergence of several viral mRNA isoforms, including an exon formed by 3′ss A1 and 5′ss D2b. However, mutation of GI2-1 also increased the levels of vif mRNA, suggesting that an increase in 5′ss D2b recognition might also parallel an enhanced formation of exon definition complexes and thus enhanced 3′ss A1 usage. Indeed, endowing viral 5′ss D2 or D4 with a higher complementarity to the cellular U1 snRNA facilitates activation of the upstream 3′ss A1 and A5 (22, 24). Furthermore, coexpression of fully complementary U1 snRNAs directed against either 5′ss D2 or D3 (71) strongly enhances the usage of the respective upstream 3′ss A1 or A2, which is evident by high levels of vif and vpr mRNAs. Therefore, it appears likely that inactivation of GI2-1 promotes the recruitment of the U1 snRNP to 5′ss D2b and enhances the formation of exon definition complexes leading to both increased inclusion of exon 2b in the different viral mRNA species and higher levels of vif mRNA.
G runs mediate splicing regulation throughout evolution by multiple mechanisms, depending on their position relative to the splice site, distance, sequence context, and complex interactions with other splicing factors (72, 73). Therefore, the proper position of the G run seems to be decisive for its acting either as a silencer or as an enhancer. Thus, the position of GI2-1, i.e., downstream of 5′ss D2 but upstream of D2b, seems to be a key factor rendering it a negative regulator of vif mRNA by repressing 5′ss D2b.
Analyses of the hydrogen bonding patterns of U1 snRNA binding (63; http://www.uni-duesseldorf.de/rna/html/hbond_score.php) and maximum-entropy estimation (45; http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) revealed that the alternative 5′ss D2b was, in fact, intrinsically stronger (HBond score, 12.4; MaxEnt score, 5.99) than the upstream 5′ss D2 (HBond score, 10.7; MaxEnt score, 5.73). Thus, according to their intrinsic strength, the usage of D2b would be expected to be more frequent. However, quite on the contrary, by RNA deep sequencing of HIV-1-infected T cells, we found 26.5-fold less D2b- than D2-containing exon junction reads (a relative D2b usage of 0.2% versus a relative D2 usage of 5.3%, i.e., a 26.5-fold difference). Even though D2b usage was underrepresented in the pool of exon junction reads (Table 4) and not found in a recent transcriptome analysis by the Katze laboratory (74), it appears from our results unlikely that it is the result of mere “biological noise.” We rather favor the view that it is a bona fide NL4-3 alternative 5′ss. This assumption is based on the finding that, unlike 5′ss D2b-containing exon junction reads, D1a-containing exon junction reads could not be detected in our analysis, which is compatible with the observation that 5′ss D1a appears to be tightly repressed and only infrequently used in vivo (68). Moreover, the usage of 5′ss D2b could be confirmed by RT-PCR, which revealed the existence of several D2b-derived transcripts when a forward primer positioned between D2 and D2b was used. Thus, the repression of D2b appears to result from the silencing activity mediated by G run GI2-1, which concomitantly has an indirect enhancing activity in favor of the intrinsically weaker D2 5′ss. These results also demonstrated that the functional strength of a 5′ss D2b is not only determined by its intrinsic strength, i.e., its complementarity to the 5′ end of U1 snRNA (63, 75), but also is controlled by specific SREs.
The data demonstrated in this and previous work (18, 22) suggested that splicing at 3′ss A1 and processing of vif mRNA are modulated by the formation of bridging interactions across exons 2 and 2b, respectively (Fig. 10). On the one hand, two SREs within the noncoding leader exon 2 enhance its splice site recognition (the SRSF1 [SF2/ASF]-dependent heptameric ESEs M1 and M2 [18] and the SRSF4 [SRp75]-dependent ESE Vif [22]). On the other hand, the intronic G run GGGG, which overlaps 5′ss D2 [22), and GI2-1 negatively regulate exon 2 and 2b inclusion and levels of vif mRNA. Mechanistically, the repressing capacity of such G runs, which overlap the 11 nt of a 5′ss, is likely based on the restricted accessibility of the U1 snRNA due to competing binding of hnRNP F/H (76). From its position upstream of 5′ss D2b, GI2-1 could negatively regulate the bridging interaction across exon 2b by inhibiting the usage of 5′ss D2b. In line with this, inactivation of GI2-1 increased the usage of 5′ss D2b and, as a result, disproportionally facilitated 3′ss A1 recognition. The inhibition could be a result of the interaction of hnRNP F/H proteins with components of the spliceosome at various steps. The influence of G runs on the progression on the spliceosomal cycle has been observed for the major myelin proteolipid protein and its isoform DM20 lacking the alternatively spliced exon 3B. Here, G run G1M2 promoted the ATP-independent formation of the E complex, which initiates the spliceosomal cycle (73). In contrast, the S3 G run of HIV-1 tat exon 1 promotes ATP-dependent spliceosomal A-complex formation but had no effect on E-complex formation (30). Regarding this, recent studies demonstrated that the SRE-mediated binding of the U1 snRNP to a 5′ss does not necessarily imply processing into the spliceosomal A-complex formation, but can lead to a “dead-end” complex formation that prevents the splicing process (25–28). One can imagine that such a mechanism could be responsible for the formation of vif mRNA when 3′ss A1 is recognized by cross-exon interactions, which seems to be facilitated by the binding of U1 snRNP to the 5′ss without splicing at this position. For further information, see the accompanying paper by Erkelenz et al. (81).
Fig 10.
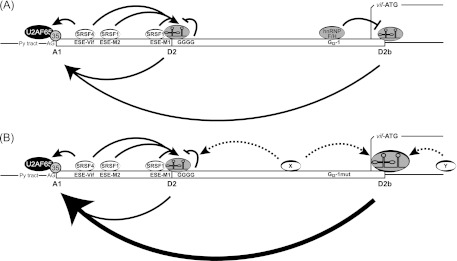
Splicing regulatory model for GI2-1. (A) Splicing at 5′ss D2 and D2b and 3′ss A1 and processing of vif mRNA are modulated by the formation of bridging interactions across exons 2 and 2b. SRSF1-dependent heptameric ESEs M1 and M2 (ESEM1/2) and SRSF4-dependent ESE Vif (ESE-Vif) facilitate the recognition of exon 2. The intronic G runs GGGG and GI2-1 negatively regulate exon 2 and 2b inclusion and levels of vif mRNA. (B) Inactivation of GI2-1 increased the usage of 5′ss D2b and facilitated the recognition of 3′ss A1. The increased usage of the intrinsic weak 5′ss D2b in the absence of GI2-1 could be due to the wide-range effect of SR proteins located upstream (X) and/or downstream (Y) of 5′ss D2b. Py tract, polypyrimidine tract.
The importance of GI2-1 in the regulation of appropriate vif expression levels maintaining optimal viral replication in A3G-deficient, as well as A3G-expressing, cells was shown by multiround infection experiments in cell lines, as well as PBMCs from healthy donors. An appropriate ratio of Vif-to-A3G protein levels was required for optimal virus replication, and this ratio was highly dependent on the physiological environment. In accordance with the observations by Strebel and coworkers (11), Vif may function at very low levels when restriction pressure, i.e., A3G expression, is also low, whereas higher Vif levels negatively affect viral replication (Fig. 5). However, as seen in PBMCs, much higher Vif levels were required for efficient viral replication, when the host cell restriction pressure was high (Fig. 6). Thus, the higher levels of Vif due to the inactivated GI2-1 facilitated viral replication in the presence of large amounts of A3G. Since the expression of splicing regulatory proteins changes during the course of the infection (77), the presence of multiple SREs may be required to optimize the amount of Vif protein in different cell types or at various phases of the infection.
ACKNOWLEDGMENTS
We are grateful to Björn Wefers and Thorsten Wachtmeister for excellent technical assistance. T.W. prepared the DNA libraries and performed the quality control for NGS DNA analysis. We thank Nathaniel R. Landau for providing pNL4-3Δvif (64).
These studies were funded by the DFG (SCHA 909/3-1), the Heinz Ansmann Foundation for AIDS Research, Düsseldorf, Germany (H.S.), and the Jürgen Manchot Stiftung (M.W., A.K., C.M., H.S.). C.M. is supported by the Heinz Ansmann Foundation for AIDS Research. The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: CEM-T4 (catalog no. 117) from J. P. Jacobs (78), CEM-SS (catalog no. 776) from Peter L. Nara (78–80), CEM-A cells from Mark Wainberg and James McMahon, HIV-1HXB2 Vif antiserum from Dana Gabuzda (58), HIV-1NL4-3 Vpr antiserum (1-46) from Jeffrey Kopp, and anti-ApoC17 antibody from Klaus Strebel.
Footnotes
Published ahead of print 19 December 2012
REFERENCES
- 1. Berger A, Sommer AF, Zwarg J, Hamdorf M, Welzel K, Esly N, Panitz S, Reuter A, Ramos I, Jatiani A, Mulder LC, Fernandez-Sesma A, Rutsch F, Simon V, Konig R, Flory E. 2011. SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutieres syndrome are highly susceptible to HIV-1 infection. PLoS Pathog. 7:e1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, de Carvalho LP, Stoye JP, Crow YJ, Taylor IA, Webb M. 2011. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480:379–382 [DOI] [PubMed] [Google Scholar]
- 3. Kirchhoff F. 2010. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8:55–67 [DOI] [PubMed] [Google Scholar]
- 4. Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650 [DOI] [PubMed] [Google Scholar]
- 6. Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS. 2005. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol. Biol. Evol. 22:367–377 [DOI] [PubMed] [Google Scholar]
- 7. Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. 2002. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 79:285–296 [DOI] [PubMed] [Google Scholar]
- 8. Münk C, Willemsen A, Bravo IG. 2012. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol. Biol. 12:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Holmes RK, Malim MH, Bishop KN. 2007. APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci. 32:118–128 [DOI] [PubMed] [Google Scholar]
- 10. Wissing S, Galloway NL, Greene WC. 2010. HIV-1 Vif versus the APOBEC3 cytidine deaminases: an intracellular duel between pathogen and host restriction factors. Mol. Aspects Med. 31:383–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akari H, Fujita M, Kao S, Khan MA, Shehu-Xhilaga M, Adachi A, Strebel K. 2004. High level expression of human immunodeficiency virus type-1 Vif inhibits viral infectivity by modulating proteolytic processing of the Gag precursor at the p2/nucleocapsid processing site. J. Biol. Chem. 279:12355–12362 [DOI] [PubMed] [Google Scholar]
- 12. Jäger S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, Hernandez H, Jang GM, Roth SL, Akiva E, Marlett J, Stephens M, D'Orso I, Fernandes J, Fahey M, Mahon C, O'Donoghue AJ, Todorovic A, Morris JH, Maltby DA, Alber T, Cagney G, Bushman FD, Young JA, Chanda SK, Sundquist WI, Kortemme T, Hernandez RD, Craik CS, Burlingame A, Sali A, Frankel AD, Krogan NJ. 2012. Global landscape of HIV-human protein complexes. Nature 481:365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clerc I, Laverdure S, Torresilla C, Landry S, Borel S, Vargas A, Arpin-Andre C, Gay B, Briant L, Gross A, Barbeau B, Mesnard JM. 2011. Polarized expression of the membrane ASP protein derived from HIV-1 antisense transcription in T cells. Retrovirology 8:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wahl MC, Will CL, Luhrmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136:701–718 [DOI] [PubMed] [Google Scholar]
- 15. Michaud S, Reed R. 1991. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes Dev. 5:2534–2546 [DOI] [PubMed] [Google Scholar]
- 16. Hoffman BE, Grabowski PJ. 1992. U1 snRNP targets an essential splicing factor, U2AF65, to the 3′ splice site by a network of interactions spanning the exon. Genes Dev. 6:2554–2568 [DOI] [PubMed] [Google Scholar]
- 17. Robberson BL, Cote GJ, Berget SM. 1990. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell. Biol. 10:84–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kammler S, Otte M, Hauber I, Kjems J, Hauber J, Schaal H. 2006. The strength of the HIV-1 3′ splice sites affects Rev function. Retrovirology 3:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cullen BR. 2003. Nuclear mRNA export: insights from virology. Trends Biochem Sci. 28:419–424 [DOI] [PubMed] [Google Scholar]
- 20. Hoffmann D, Schwarck D, Banning C, Brenner M, Mariyanna L, Krepstakies M, Schindler M, Millar DP, Hauber J. 2012. Formation of trans-activation competent HIV-1 Rev:RRE complexes requires the recruitment of multiple protein activation domains. PLoS One 7:e38305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pollard VW, Malim MH. 1998. The HIV-1 Rev protein. Annu. Rev. Microbiol. 52:491–532 [DOI] [PubMed] [Google Scholar]
- 22. Exline CM, Feng Z, Stoltzfus CM. 2008. Negative and positive mRNA splicing elements act competitively to regulate human immunodeficiency virus type 1 vif gene expression. J. Virol. 82:3921–3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mandal D, Exline CM, Feng Z, Stoltzfus CM. 2009. Regulation of Vif mRNA splicing by human immunodeficiency virus type 1 requires 5′ splice site D2 and an exonic splicing enhancer to counteract cellular restriction factor APOBEC3G. J. Virol. 83:6067–6078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Asang C, Hauber I, Schaal H. 2008. Insights into the selective activation of alternatively used splice acceptors by the human immunodeficiency virus type-1 bidirectional splicing enhancer. Nucleic Acids Res. 36:1450–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Domsic JK, Wang Y, Mayeda A, Krainer AR, Stoltzfus CM. 2003. Human immunodeficiency virus type 1 hnRNP A/B-dependent exonic splicing silencer ESSV antagonizes binding of U2AF65 to viral polypyrimidine tracts. Mol. Cell. Biol. 23:8762–8772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Motta-Mena LB, Smith SA, Mallory MJ, Jackson J, Wang J, Lynch KW. 2011. A disease-associated polymorphism alters splicing of the human CD45 phosphatase gene by disrupting combinatorial repression by heterogeneous nuclear ribonucleoproteins (hnRNPs). J. Biol. Chem. 286:20043–20053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sharma S, Kohlstaedt LA, Damianov A, Rio DC, Black DL. 2008. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat. Struct. Mol. Biol. 15:183–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sharma S, Maris C, Allain FH, Black DL. 2011. U1 snRNA directly interacts with polypyrimidine tract-binding protein during splicing repression. Mol. Cell 41:579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Purcell DF, Martin MA. 1993. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J. Virol. 67:6365–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schaub MC, Lopez SR, Caputi M. 2007. Members of the heterogeneous nuclear ribonucleoprotein H family activate splicing of an HIV-1 splicing substrate by promoting formation of ATP-dependent spliceosomal complexes. J. Biol. Chem. 282:13617–13626 [DOI] [PubMed] [Google Scholar]
- 31. Caputi M, Zahler AM. 2001. Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H'/F/2H9 family. J. Biol. Chem. 276:43850–43859 [DOI] [PubMed] [Google Scholar]
- 32. Carlo T, Sterner DA, Berget SM. 1996. An intron splicing enhancer containing a G-rich repeat facilitates inclusion of a vertebrate micro-exon. RNA 2:342–353 [PMC free article] [PubMed] [Google Scholar]
- 33. Chen CD, Kobayashi R, Helfman DM. 1999. Binding of hnRNP H to an exonic splicing silencer is involved in the regulation of alternative splicing of the rat beta-tropomyosin gene. Genes Dev. 13:593–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Del Gatto F, Breathnach R. 1995. Exon and intron sequences, respectively, repress and activate splicing of a fibroblast growth factor receptor 2 alternative exon. Mol. Cell. Biol. 15:4825–4834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marcucci R, Baralle FE, Romano M. 2007. Complex splicing control of the human thrombopoietin gene by intronic G runs. Nucleic Acids Res. 35:132–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCarthy EM, Phillips JA., III 1998. Characterization of an intron splice enhancer that regulates alternative splicing of human GH pre-mRNA. Hum. Mol. Genet. 7:1491–1496 [DOI] [PubMed] [Google Scholar]
- 37. McCullough AJ, Berget SM. 1997. G triplets located throughout a class of small vertebrate introns enforce intron borders and regulate splice site selection. Mol. Cell. Biol. 17:4562–4571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCullough AJ, Berget SM. 2000. An intronic splicing enhancer binds U1 snRNPs to enhance splicing and select 5′ splice sites. Mol. Cell. Biol. 20:9225–9235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chou MY, Rooke N, Turck CW, Black DL. 1999. hnRNP H is a component of a splicing enhancer complex that activates a c-src alternative exon in neuronal cells. Mol. Cell. Biol. 19:69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garneau D, Revil T, Fisette JF, Chabot B. 2005. Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator Bcl-x. J. Biol. Chem. 280:22641–22650 [DOI] [PubMed] [Google Scholar]
- 41. Han K, Yeo G, An P, Burge CB, Grabowski PJ. 2005. A combinatorial code for splicing silencing: UAGG and GGGG motifs. PLoS Biol. 3:e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hastings ML, Wilson CM, Munroe SH. 2001. A purine-rich intronic element enhances alternative splicing of thyroid hormone receptor mRNA. RNA 7:859–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Min H, Chan RC, Black DL. 1995. The generally expressed hnRNP F is involved in a neural-specific pre-mRNA splicing event. Genes Dev. 9:2659–2671 [DOI] [PubMed] [Google Scholar]
- 44. Xiao X, Wang Z, Jang M, Nutiu R, Wang ET, Burge CB. 2009. Splice site strength-dependent activity and genetic buffering by poly-G runs. Nat. Struct. Mol. Biol. 16:1094–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yeo G, Burge CB. 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11:377–394 [DOI] [PubMed] [Google Scholar]
- 46. Zhang XH, Leslie CS, Chasin LA. 2005. Computational searches for splicing signals. Methods 37:292–305 [DOI] [PubMed] [Google Scholar]
- 47. Jablonski JA, Caputi M. 2009. Role of cellular RNA processing factors in human immunodeficiency virus type 1 mRNA metabolism, replication, and infectivity. J. Virol. 83:981–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lund N, Milev MP, Wong R, Sanmuganantham T, Woolaway K, Chabot B, Abou Elela S, Mouland AJ, Cochrane A. 2012. Differential effects of hnRNP D/AUF1 isoforms on HIV-1 gene expression. Nucleic Acids Res. 40:3663–3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jacquenet S, Mereau A, Bilodeau PS, Damier L, Stoltzfus CM, Branlant C. 2001. A second exon splicing silencer within human immunodeficiency virus type 1 tat exon 2 represses splicing of Tat mRNA and binds protein hnRNP H. J. Biol. Chem. 276:40464–40475 [DOI] [PubMed] [Google Scholar]
- 50. Caputi M, Zahler AM. 2002. SR proteins and hnRNP H regulate the splicing of the HIV-1 tev-specific exon 6D. EMBO J. 21:845–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Asang C, Erkelenz S, Schaal H. 2012. The HIV-1 major splice donor D1 is activated by splicing enhancer elements within the leader region and the p17-inhibitory sequence. Virology 432:133–145 [DOI] [PubMed] [Google Scholar]
- 52. Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Singh KK, Erkelenz S, Rattay S, Dehof AK, Hildebrandt A, Schulze-Osthoff K, Schaal H, Schwerk C. 2010. Human SAP18 mediates assembly of a splicing regulatory multiprotein complex via its ubiquitin-like fold. RNA 16:2442–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krummheuer J, Lenz C, Kammler S, Scheid A, Schaal H. 2001. Influence of the small leader exons 2 and 3 on human immunodeficiency virus type 1 gene expression. Virology 286:276–289 [DOI] [PubMed] [Google Scholar]
- 55. Selden RF, Howie KB, Rowe ME, Goodman HM, Moore DD. 1986. Human growth hormone as a reporter gene in regulation studies employing transient gene expression. Mol. Cell. Biol. 6:3173–3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chomczynski P, Sacchi N. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162:156–159 [DOI] [PubMed] [Google Scholar]
- 57. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 58. Goncalves J, Jallepalli P, Gabuzda DH. 1994. Subcellular localization of the Vif protein of human immunodeficiency virus type 1. J. Virol. 68:704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kao S, Miyagi E, Khan MA, Takeuchi H, Opi S, Goila-Gaur R, Strebel K. 2004. Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology 1:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Khan MA, Kao S, Miyagi E, Takeuchi H, Goila-Gaur R, Opi S, Gipson CL, Parslow TG, Ly H, Strebel K. 2005. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J. Virol. 79:5870–5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Abacioglu YH, Fouts TR, Laman JD, Claassen E, Pincus SH, Moore JP, Roby CA, Kamin-Lewis R, Lewis GK. 1994. Epitope mapping and topology of baculovirus-expressed HIV-1 gp160 determined with a panel of murine monoclonal antibodies. AIDS Res. Hum. Retrovir. 10:371–381 [DOI] [PubMed] [Google Scholar]
- 62. Holtkotte D, Pfeiffer T, Bosch V. 2007. Cell-free infectivity of HIV type 1 produced in nonpermissive cells is only moderately impacted by C-terminal Env truncation despite abrogation of viral spread. AIDS Res. Hum. Retrovir. 23:729–740 [DOI] [PubMed] [Google Scholar]
- 63. Freund M, Asang C, Kammler S, Konermann C, Krummheuer J, Hipp M, Meyer I, Gierling W, Theiss S, Preuss T, Schindler D, Kjems J, Schaal H. 2003. A novel approach to describe a U1 snRNA binding site. Nucleic Acids Res. 31:6963–6975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Münk C, Nymark-McMahon H, Landau NR. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21–31 [DOI] [PubMed] [Google Scholar]
- 65. Tremblay M, Sullivan AK, Rooke R, Geleziunas R, Tsoukas C, Shematek G, Gilmore N, Wainberg MA. 1989. New CD4(+) cell line susceptible to infection by HIV-1. J. Med. Virol. 28:243–249 [DOI] [PubMed] [Google Scholar]
- 66. Haché G, Harris RS. 2009. CEM-T4 cells do not lack an APOBEC3G cofactor. PLoS Pathog. 5:e1000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Benko DM, Schwartz S, Pavlakis GN, Felber BK. 1990. A novel human immunodeficiency virus type 1 protein, tev, shares sequences with tat, env, and rev proteins. J. Virol. 64:2505–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lützelberger M, Reinert LS, Das AT, Berkhout B, Kjems J. 2006. A novel splice donor site in the gag-pol gene is required for HIV-1 RNA stability. J. Biol. Chem. 281:18644–18651 [DOI] [PubMed] [Google Scholar]
- 69. Wilk T, Pfeiffer T, Bosch V. 1992. Retained in vitro infectivity and cytopathogenicity of HIV-1 despite truncation of the C-terminal tail of the env gene product. Virology 189:167–177 [DOI] [PubMed] [Google Scholar]
- 70. Madsen JM, Stoltzfus CM. 2006. A suboptimal 5′ splice site downstream of HIV-1 splice site A1 is required for unspliced viral mRNA accumulation and efficient virus replication. Retrovirology 3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mandal D, Feng Z, Stoltzfus CM. 2010. Excessive RNA splicing and inhibition of HIV-1 replication induced by modified U1 small nuclear RNAs. J. Virol. 84:12790–12800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Voelker RB, Erkelenz S, Reynoso V, Schaal H, Berglund JA. 2012. Frequent gain and loss of intronic splicing regulatory elements during the evolution of vertebrates. Genome Biol Evol. 4:659–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang E, Mueller WF, Hertel KJ, Cambi F. 2011. G run-mediated recognition of proteolipid protein and DM20 5′ splice sites by U1 small nuclear RNA is regulated by context and proximity to the splice site. J. Biol. Chem. 286:4059–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chang ST, Sova P, Peng X, Weiss J, Law GL, Palermo RE, Katze MG. 2011. Next-generation sequencing reveals HIV-1-mediated suppression of T cell activation and RNA processing and regulation of noncoding RNA expression in a CD4+ T cell line. mBio 2(5):e00134–11 doi:10.1128/mBio.00134-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kammler S, Leurs C, Freund M, Krummheuer J, Seidel K, Tange TO, Lund MK, Kjems J, Scheid A, Schaal H. 2001. The sequence complementarity between HIV-1 5′ splice site SD4 and U1 snRNA determines the steady-state level of an unstable env pre-mRNA. RNA 7:421–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Buratti E, Baralle M, De Conti L, Baralle D, Romano M, Ayala YM, Baralle FE. 2004. hnRNP H binding at the 5′ splice site correlates with the pathological effect of two intronic mutations in the NF-1 and TSHbeta genes. Nucleic Acids Res. 32:4224–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dowling D, Nasr-Esfahani S, Tan CH, O'Brien K, Howard JL, Jans DA, Purcell DF, Stoltzfus CM, Sonza S. 2008. HIV-1 infection induces changes in expression of cellular splicing factors that regulate alternative viral splicing and virus production in macrophages. Retrovirology 5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Foley GE, Lazarus H, Farber S, Uzman BG, Boone BA, McCarthy RE. 1965. Continuous culture of human lymphoblasts from peripheral blood of a child with acute leukemia. Cancer 18:522–529 [DOI] [PubMed] [Google Scholar]
- 79. Nara PL, Hatch WC, Dunlop NM, Robey WG, Fischinger PJ. 1987. Simple, rapid quantitative, syncytium-forming microassay for the detection of human immunodeficiency virus neutralizing antibody. AIDS Res. Hum. Retroviruses 3:283–302 [DOI] [PubMed] [Google Scholar]
- 80. Nara PL, Fischinger PJ. 1988. Quantitative infectivity assay for HIV-1 and -2. Nature 332:469–470 [DOI] [PubMed] [Google Scholar]
- 81. Erkelenz S, Poschmann G, Theiss S, Stefanski A, Hillebrand F, Otte M, Stühler K, Schaal H. 2013. Tra2-mediated recognition of HIV-1 5′ splice site D3 as a key factor in the processing of vpr mRNA. J. Virol. 87:2721–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]