

Abstract
Infection of mammalian cells by picornaviruses results in the nucleocytoplasmic redistribution of certain host cell proteins. These viruses interfere with import-export pathways, allowing for the cytoplasmic accumulation of nuclear proteins that are then available to function in viral processes. We recently described the cytoplasmic relocalization of cellular splicing factor SRp20 during poliovirus infection. SRp20 is an important internal ribosome entry site (IRES) trans-acting factor (ITAF) for poliovirus IRES-mediated translation; however, it is not known whether other picornaviruses utilize SRp20 as an ITAF and direct its cytoplasmic relocalization. Also, the mechanism by which poliovirus directs the accumulation of SRp20 in the cytoplasm of the infected cell is currently unknown. Work described in this report demonstrated that infection by another picornavirus (coxsackievirus B3) causes SRp20 to relocalize from the nucleus to the cytoplasm of HeLa cells, similar to poliovirus infection; however, SRp20 is relocalized to a somewhat lesser extent in the cytoplasm of HeLa cells during infection by yet another picornavirus (human rhinovirus 16). We show that expression of poliovirus 2A proteinase is sufficient to cause the nucleocytoplasmic redistribution of SRp20. Following expression of poliovirus 2A proteinase in HeLa cells, we detect cleavage of specific nuclear pore proteins known to be cleaved during poliovirus infection. We also find that expression of human rhinovirus 16 2A proteinase alone can cause efficient cytoplasmic relocalization of SRp20, despite the lower levels of SRp20 relocalization observed during rhinovirus infection compared to poliovirus. Taken together, these results further define the mechanism of SRp20 cellular redistribution during picornavirus infections, and they provide additional insight into some of the differences observed between human rhinovirus and other enterovirus infections.
INTRODUCTION
Members of the Picornaviridae family of viruses cause a range of diseases in humans, including poliomyelitis, myocarditis, and the common cold. Picornaviruses are small single-stranded, positive-sense RNA viruses that include the enteroviruses poliovirus and coxsackievirus, as well as human rhinoviruses (HRVs). They contain a ∼7.0-kb-to-8.5-kb genome that consists of a single open reading frame, which is translated to generate a polyprotein that is proteolytically processed by viral proteinases into structural and nonstructural proteins. Instead of a 7-methyl guanosine cap structure, picornaviruses contain a small viral protein, VPg, covalently linked to the 5′ end of the RNA. This unique protein-RNA linkage serves as a primer for viral RNA synthesis. Viral translation occurs via a cap-independent mechanism and is driven by an internal ribosome entry site (IRES) located in the long, highly structured 5′ noncoding region (5′ NCR).
In addition to proteolytic processing of the viral polyprotein, the viral proteinases 2A and 3C/3CD cleave several cellular proteins, including eukaryotic initiation factor 4G (eIF4G), to downregulate host cell translation during poliovirus, human rhinovirus, or coxsackievirus infection. Other work has shown that poliovirus and coxsackievirus proteinases cleave poly(A) binding protein (PABP), and this cleavage correlates with shutoff of host cell translation (1–6). Viral proteinases also play roles in the shutoff of cellular transcription during poliovirus infection (7–11). For enteroviruses and human rhinoviruses, the 2A proteinase catalyzes cleavage of viral and host proteins and may also play a role in protection against the cellular immune response (for a review, see reference 12). Incorporation of mutations in the 2A coding region of poliovirus has shown that this viral protein is required for replication of the virus, but it is not clear exactly what role 2A plays in viral RNA synthesis (13). Expression of poliovirus 2A proteinase in uninfected cells causes the cytoplasmic relocalization of some host nuclear factors, initially suggesting that 2A may be responsible for the degradation of nuclear pore complex proteins (or Nups) during infection (14). Specific Nups are cleaved during poliovirus or human rhinovirus infection, which results in the disruption of certain import/export pathways (15–20). More recent work has demonstrated that poliovirus and human rhinovirus 2A proteinases can directly cleave specific Nups (17, 18, 20, 21). Interestingly, human rhinovirus 2A proteinases from different species and serotypes show differential processing of nuclear pore complex proteins (20).
Viral protein 3CD is a precursor polypeptide containing the amino acid sequences of the 3C proteinase and the RNA-dependent RNA polymerase 3D. A major role of 3CD proteinase for enteroviruses and human rhinoviruses is the proteolytic processing of viral capsid precursors as well as the precursors leading to the production of mature nonstructural proteins (22, 23). In addition, 3C/3CD cleaves the cellular RNA binding protein poly(rC) binding protein 2 (PCBP2), which is thought to contribute to a switch from viral translation to RNA replication during poliovirus infection (24). Both the full-length and cleaved versions of PCBP2 can interact with stem-loop I RNA to form a ternary complex with 3CD. This complex is required for poliovirus RNA synthesis (24, 25). Protein 3CD interacts with other cellular proteins, including heterogeneous ribonucleoprotein C1/C2 (hnRNP C1/C2), which may be required for poliovirus positive-strand RNA synthesis (26, 27).
Picornaviruses carry out viral translation and RNA replication in the cytoplasm of the infected cell. Due to their limited coding capacity, these viruses have evolved to utilize host cell factors in concert with viral proteins and RNA secondary structures for viral translation and RNA replication. Several cellular proteins termed IRES trans-acting factors (or ITAFs) are known to bind to the 5′ NCRs of picornavirus RNAs, and some have been shown to stimulate viral translation (28–36). These include both canonical eukaryotic translation initiation factors (eIFs) as well as other noncanonical proteins. PCBP2 has been shown to bind to stem-loop IV of poliovirus and coxsackievirus in their respective IRESes, and the presence of this protein is important for efficient viral translation mediated by the poliovirus, coxsackievirus, and human rhinovirus IRESes (29, 30, 37, 38). In poliovirus-infected cells, it has been shown that PCBP2 interacts with cellular splicing factor SRp20, and these two proteins function synergistically in IRES-mediated translation (39, 40). A role for SRp20 in viral translation during other picornavirus infections has not yet been defined.
We have recently demonstrated the dramatic relocalization of SRp20 from the nucleus to the cytoplasm of cells during the course of poliovirus infection (40). In the present study, we examined the subcellular localization of SRp20 during infection with other picornaviruses and investigated the mechanism underlying SRp20 cytoplasmic accumulation following poliovirus infection. We determined that infection of HeLa cells with coxsackievirus B3 (CVB3) causes cytoplasmic redistribution of SRp20 similar to what was observed for poliovirus infection. However, infection of HeLa cells with human rhinovirus 16 (HRV16) resulted in a significantly lower level of SRp20 relocalization, highlighting differences between HRV and other enterovirus infections. In addition, we demonstrated that ectopic expression of poliovirus 2A proteinase, but not 3CD proteinase, results in the relocalization of SRp20 from the nucleus to the cytoplasm of cells. Using mutated poliovirus 2A proteins, we determined that the proteinase activity of 2A is required for SRp20 redistribution. With expression of wild-type poliovirus 2A, we observed cleavage of specific nuclear pore complex proteins that are known to be cleaved during poliovirus infection. Finally, we expressed HRV16 2A proteinase alone in cells and observed efficient relocalization of SRp20 from the nucleus to the cytoplasm, in contrast to what was seen during a HRV16 infection. Taken together, these results further define the mechanism of the cytoplasmic accumulation of SRp20 during poliovirus infection and provide additional insight into some of the differences observed in cells infected by HRV compared to those infected by other enteroviruses.
MATERIALS AND METHODS
Cell culture and DNA constructs.
HeLa cells were grown as monolayers in Dulbecco's modified Eagle's medium (DMEM) supplemented with 8% newborn calf serum (NCS). HeLa T-REx cells (Invitrogen) stably express the tetracycline repressor protein, and were grown in DMEM supplemented with 10% fetal calf serum (FCS) and maintained in blasticidin (5 μg/ml). Enhanced green fluorescent protein (EGFP)-SRp20 plasmid DNA was kindly provided by Roz Sandri-Goldin. The FLAG-3CDμ10 construct was generated by cloning the FLAG and 3CDμ10 (41) coding sequences into the pcDNA4/TO tetracycline-inducible vector (Invitrogen). The FLAG-3CD wild-type (FLAG-3CDwt) construct was generated via site-directed mutagenesis of a FLAG-3CD-C147A construct (42), to change the alanine back to the wild-type cysteine (GCT → TGT). Poliovirus and HRV16 FLAG-2A constructs were generated via restriction digest of FLAG-3CDμ10 to remove the 3CDμ10 coding sequence, followed by PCR amplification and insertion of the 2A coding sequence derived from full-length cDNAs for either poliovirus or HRV16. cDNAs encoding full-length poliovirus containing previously characterized 2A mutations (2A-C109R and 2A-L98P) were generously provided by Karla Kirkegaard (43). The 2A region from each cDNA was PCR amplified and cloned into the same vector as for wild-type 2A.
DNA transfection and virus infection.
HeLa cells were seeded on coverslips and allowed to grow to approximately 60% confluence. Cells were transfected with pEGFP-SRp20 using Fugene 6 transfection reagent (Roche). Twenty-four hours posttransfection, cells were infected; for poliovirus and coxsackievirus infections, cells were infected with poliovirus type 1 or coxsackievirus B3 in 8% NCS–DMEM at a multiplicity of infection (MOI) of 25, following a 30-min adsorption at room temperature. For HRV infections, cells were infected with HRV16 (stock virus generated from a cDNA provided by Wai-Ming Lee [44]) in 8% NCS–DMEM additionally supplemented with 10 mM MgCl2 and 20 mM HEPES (pH 7.4) at an MOI of 10, following adsorption for 1 h at room temperature. Coxsackievirus infections were carried out at 37°C; poliovirus and HRV infections were individually carried out at both 34°C and 37°C. At specific times postinfection (pi), cells were washed once with 1× phosphate-buffered saline (1× PBS), and processed for imaging. For HRV16 infections, 100 cells at each time point were scored to determine the percentage of cells displaying any detectable SRp20 relocalization. In parallel experiments under the same conditions described above, mock- or HRV16-infected cells were harvested at each time point and cytoplasmic lysates were generated. Cells were lysed in NP-40 lysis buffer (50 mM Tris-HCl [pH 8], 5 mM EDTA [pH 8], 150 mM NaCl, 0.5% NP-40) on ice for 30 min, clarified, and subjected to SDS-PAGE and Western blot analysis.
DNA transfection and induction of viral proteinase expression.
HeLa T-REx cells were seeded on coverslips and allowed to grow to approximately 70% confluence. Cells were cotransfected with pEGFP-SRp20 and a plasmid encoding FLAG-tagged poliovirus 3CDμ10, 3CDwt, 2A, 2A-C109R, or 2A-L98P; HRV16 2A; or empty vector. Transfections were carried out using Lipofectamine transfection reagent (Invitrogen). Twenty-four hours posttransfection, the media were replaced with fresh media containing 1 μg/ml tetracycline (Sigma) to induce FLAG-tagged viral proteinase expression (GFP-SRp20 was constitutively expressed). Viral proteinase expression was induced for up to 6 h, and at specific times postinduction the cells were washed once with 1× PBS and processed for imaging. To investigate nuclear pore protein cleavage, HeLa cells were transfected with FLAG-2A for constitutive expression of the proteinase. Cells were harvested at 1, 6, 18, and 24 h posttransfection. Whole-cell lysates were subjected to SDS-PAGE and Western blot analysis.
Western blot analysis.
Rabbit polyclonal antibodies directed against HRV16 3D polymerase or 2A proteinase (generously provided by Ann Palmenberg) were used to detect expression of these viral proteins in cytoplasmic lysates from mock- or HRV16-infected cells. Membranes were incubated with anti-3D (1:5,000) or anti-2A (1:200) for 1 h, followed by incubation with goat anti-rabbit horseradish peroxidase (HRP) secondary antibody (Molecular Probes) for 45 min. Mouse monoclonal mAb414 (Covance) or rabbit polyclonal anti-Nup98 (Cell Signaling) antibody was used to detect nuclear pore complex proteins Nup153, Nup98, and Nup62 in whole-cell lysates. Rabbit polyclonal anti-tubulin (Abcam) antibody was used as a loading control. Rabbit monoclonal anti-eIF4G directed at an epitope surrounding Gly 188 of human eIF4G (Cell Signaling) was used to detect full-length eIF4G and its cleavage products. Mouse monoclonal anti-FLAG (Stratagene) antibody was used to detect expression of poliovirus FLAG-tagged 2A proteinase. Membranes were incubated with primary antibodies for 1 h, followed by goat anti-mouse HRP or goat anti-rabbit HRP secondary antibodies (Molecular Probes) for 45 min.
Laser scanning confocal microscopy.
Following virus infection or viral proteinase induction, cells were fixed with 3.7% paraformaldehyde at room temperature for 20 min. Cells were washed with 1× PBS twice, and cell membranes were permeabilized with 0.5% NP-40–PBS for 5 min. Cells from virus infections were washed with 1% NCS–PBS and incubated with 4′,6′diamidino-2-phenylindole (DAPI) to stain nuclei. Cells that were induced for viral proteinase expression were washed with 1% NCS–PBS and incubated with normal donkey serum (Jackson ImmunoResearch) for 1 h to block nonspecific interactions. Cells were then incubated with anti-FLAG antibody (Stratagene) for 1 h, washed, and incubated with Alexa Fluor 594 goat anti-mouse secondary antibody (Molecular Probes) for 45 min. Cells were washed with 1% NCS-PBS and incubated with DAPI to stain nuclei. All cells following DAPI incubation were washed with PBS, and coverslips were mounted on microscope slides with mounting media (Biomeda) and allowed to dry overnight at room temperature. Coverslips were sealed with nail polish, and cells were imaged using a Zeiss LSM 510 or LSM 700 laser scanning confocal microscope. Images were processed using the LSM 510 or Zen (Carl Ziess) confocal imaging software, respectively.
RESULTS
SRp20 relocalizes from the nucleus to the cytoplasm of the cell during coxsackievirus B3 infection.
We first investigated the subcellular localization of SRp20 during CVB3 infection. Because coxsackievirus and poliovirus are closely related picornaviruses, we predicted that we would observe a cytoplasmic redistribution of SRp20 during coxsackievirus infection, as has been seen during poliovirus infection (40). While the precise role of SRp20 during coxsackievirus infection has not yet been determined, we hypothesize that its function is equivalent to the role of this cellular factor during poliovirus infection, as an ITAF for viral IRES-mediated translation in the cytoplasm. To visualize SRp20 in the cell, we transfected a plasmid encoding a GFP-tagged version of the protein into HeLa cells prior to infection. We then utilized confocal microscopy to examine the localization of SRp20 in mock- and CVB3-infected cells during infection. The results are shown in Fig. 1. In the mock-infected cells (Fig. 1A), SRp20 is found predominantly in the nucleus, consistent with previously published data for this protein and its role as a cellular splicing factor (45). We determined that this nuclear localization in mock-infected cells did not change over the course of incubation (data not shown). At both 1 and 2 h postinfection (hpi) (Fig. 1B and C), SRp20 remained predominantly nuclear in localization. However, at 3 hpi, a low level of SRp20 was detected in the cytoplasm of the infected cells (Fig. 1D). This is somewhat different from what was observed during poliovirus infection, where low levels of SRp20 could be detected in the cytoplasm of infected cells at 2 hpi (40). The slight delay in the kinetics of SRp20 localization may be due to a slight growth delay observed for coxsackievirus compared to poliovirus, since poliovirus grows to peak titers at 5 to 6 h postinfection in HeLa cell monolayers at 37°C (46) compared to CVB3, which achieves peak titers under similar growth conditions at 7 to 8 h postinfection (37, 47, 48). Later in infection (4 and 5 hpi, Fig. 1E and F) a more dramatic relocalization of SRp20 could be observed, with much of the protein found in the cytoplasm. Therefore, coxsackievirus infection, like poliovirus infection, causes the nucleocytoplasmic redistribution of SRp20.
Fig 1.

SRp20 relocalizes from the nucleus to the cytoplasm of HeLa cells during coxsackievirus B3 infection. Cells were transfected with GFP-SRp20 and either mock infected (A) or infected with CVB3 (B to F) at an MOI of 25. Infection was carried out at 37°C. Cells were fixed every hour postinfection (from 1 to 5 h [B to F]) and imaged. SRp20 localization was determined using confocal microscopy; nuclei were identified by DAPI staining.
SRp20 is relocalized to a lesser extent in the cytoplasm of cells infected with human rhinovirus 16.
We next determined the subcellular localization of SRp20 during infection of HeLa cells with HRV serotype 16. HRV16 is a picornavirus that has similarities to poliovirus and coxsackievirus, but also some significant differences; namely, the virus growth cycle is greatly protracted compared to that of poliovirus, with peak virus titers reached at later times (8 to 9 h postinfection) than poliovirus (49) (A. J. Chase and B. L. Semler, unpublished observations). Also, the peak titers reached for HRV infection are 1 to 2 log10 units lower than titers reached for poliovirus in HeLa cell monolayers (for example, refer to reference 50). Additionally, the optimal temperature for HRV16 infection is 34°C, compared to 37°C for poliovirus. Taking these differences into account, we investigated the localization of SRp20 in HeLa cells during HRV16 infection at 34°C from 0 to 12 hpi. The results of these experiments are shown in Fig. 2. SRp20 remained nuclear at 2 and 4 hpi (Fig. 2B and C). We observed relocalization of SRp20 to the cytoplasm of infected cells beginning about 6 h postinfection (Fig. 2D). The amount of SRp20 observed in the cytoplasm of infected cells continued to increase at later times postinfection (Fig. 2E to G). The cytoplasmic accumulation of SRp20 was somewhat asynchronous among the infected cells, despite a high multiplicity of infection (MOI, 10). A gradual increase in the percentage of cells displaying SRp20 relocalization during the course of infection was observed, as summarized in Table 1. It should be noted that under the conditions of these experiments (i.e., transfection of plasmid DNA expressing GFP-SRp20 followed by HRV16 infection), total cytopathic effects were not observed until after the final time point (12 hpi) used for confocal imaging. Additionally, the relocalization of SRp20 during HRV16 infection occurred to a lesser extent than relocalization during poliovirus infection. To verify efficient viral infection of our transfected HeLa cells, parallel experiments were carried out in which SRp20-transfected, HRV16-infected cells were harvested at the same times after infection to examine viral protein production. Cytoplasmic lysates were generated from mock- and HRV16-infected cells and subjected to SDS-PAGE and Western blot analysis (shown in Fig. 3). Viral polymerase 3D (top) was produced at high levels by 6 h postinfection. We also detected low levels of the proteinase 2A (bottom) at 6 h postinfection, increasing significantly by 8 h postinfection.
Fig 2.
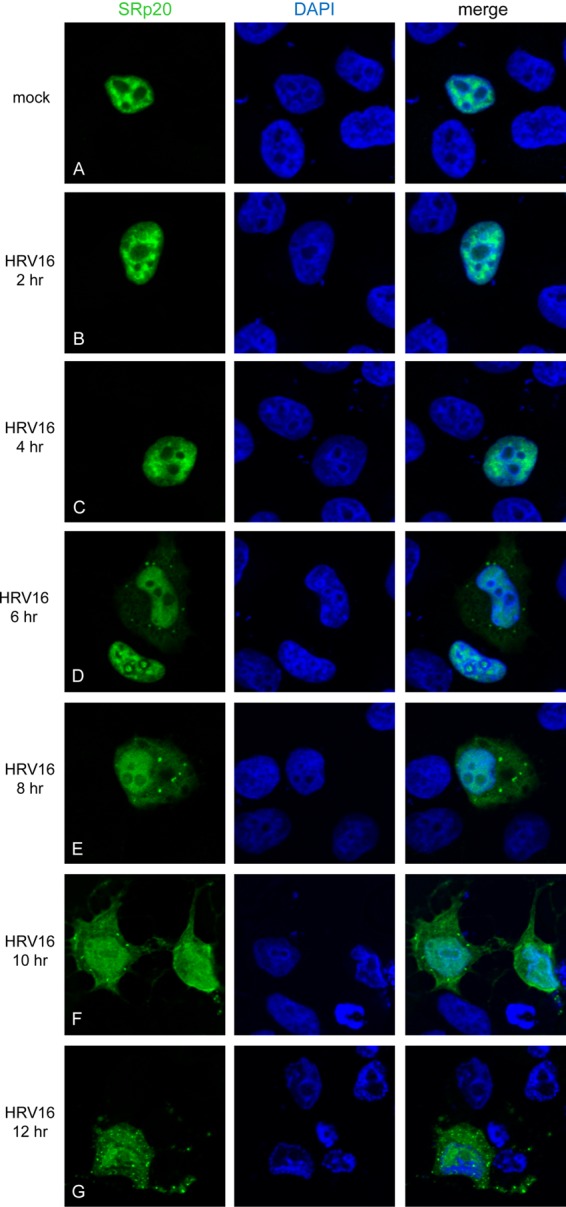
SRp20 is relocalized to a lesser extent in the cytoplasm of HeLa cells during human rhinovirus 16 infection. Cells were transfected with GFP-SRp20 and either mock infected (A) or infected with HRV16 (B to G) at an MOI of 10. Infection was carried out at 34°C; cells were fixed every 2 hpi (from 2 to 12 h [B to G]) and imaged. SRp20 localization was determined using confocal microscopy; DAPI staining indicated nuclei.
Table 1.
SRp20 relocalization during human rhinovirus 16 infection at 34°C and 37°Ca
| HRV16 infection time (hpi) | % cells displaying any detectable SRp20 nucleocytoplasmic relocalization |
|
|---|---|---|
| 34°C | 37°C | |
| Mock | 0 | 0 |
| 2 | 0 | 0 |
| 4 | 0 | 0 |
| 6 | 25 | 31 |
| 8 | 38 | 65 |
| 10 | 53 | 86 |
| 12 | 72 | 92 |
Cells fixed for imaging as described in the legend for Fig. 2 (and data not shown) were additionally scored for the percentage of cells displaying any detectable SRp20 nucleocytoplasmic relocalization. At each time point, 100 cells were counted and categorized as either displaying strictly nuclear SRp20 or displaying some detectable level of SRp20 cytoplasmic accumulation. At both temperatures of infection, there was no detectable cytoplasmic redistribution of SRp20 at 2 or 4 hpi. Relocalization could be visualized beginning at 6 hpi. The percentage of cells displaying SRp20 relocalization increased more rapidly during infection at 37°C than during infection at 34°C.
Fig 3.

Expression of human rhinovirus 16 viral proteins 3D and 2A can be detected beginning at 6 h postinfection. HeLa cell cytoplasmic lysates generated from SRp20-transfected and subsequently mock- or HRV16-infected cells were analyzed for viral proteins by Western blotting, probing with polyclonal antibodies against 3D or 2A. When probed with anti-3D (top panel), the viral RNA polymerase could be detected at high levels by 6 hpi. When probed with anti-2A (bottom panel), a small amount of the viral proteinase could be detected at 6 hpi, with greater levels present by 8 hpi.
We next considered the possibility that SRp20 relocalization may somehow be affected and/or significantly delayed simply by the lower temperature of infection, and also carried out the infection at 37°C (data not shown). Notably, we have not observed significant differences in the growth kinetics or peak titers of HRV16 when HeLa cells are infected at either 34°C or 37°C (Chase and Semler, unpublished), and human rhinoviruses have been shown to replicate effectively at 37°C (51). As was observed at the lower temperature, SRp20 remained localized in the nucleus at 2 and 4 hpi. Interestingly, nucleocytoplasmic relocalization of SRp20 was also detected beginning around 6 hpi at 37°C. The asynchronous accumulation of cytoplasmic SRp20 among the infected cells was observed at the higher temperature as well (refer to Table 1), although the percentage of cells displaying cytoplasmic SRp20 increased more rapidly at 37°C than at 34°C.
SRp20 displays a dramatic nucleocytoplasmic relocalization during poliovirus infection at 34°C.
We also examined the relocalization of SRp20 during poliovirus infection at 34°C. While the kinetics of SRp20 relocalization have already been reported for poliovirus infection at 37°C (40), we wanted to compare these results with infection carried out at a lower than optimal temperature for poliovirus to determine whether the temperature of infection affects the relocalization of SRp20. The results of these experiments are shown in Fig. 4. We have previously demonstrated that SRp20 relocalizes to the cytoplasm of poliovirus-infected cells at 37°C beginning about 2 hpi, with a more dramatic relocalization observed over the course of infection (40). When poliovirus infection was carried out at 34°C, SRp20 was also observed to relocalize to the cytoplasm at about 2 hpi (Fig. 4B). This cytoplasmic redistribution became more apparent as infection progressed (Fig. 4C to E), although it did appear that slightly less total SRp20 was relocalized at earlier times postinfection than was observed during infection at 37°C (Fig. 4C and data not shown). Therefore, independent of whether poliovirus infection of HeLa cells is carried out at 34°C or 37°C, SRp20 relocalizes from the nucleus to the cytoplasm of the infected cells beginning about 2 hpi.
Fig 4.

SRp20 relocalizes from the nucleus to the cytoplasm of HeLa cells during poliovirus infection at 34°C. Cells were transfected with GFP-SRp20 and either mock-infected (A) or infected with poliovirus (PV; B to E) at an MOI of 25. Infection was carried out at 34°C. Cells were fixed every hour postinfection from 1 to 8 h and imaged; shown are cells at 2, 4, 6, and 8 hpi (B to E). SRp20 localization was determined using confocal microscopy; nuclei were identified with DAPI staining.
Poliovirus 3CD proteinase expression alone does not cause the cytoplasmic redistribution of SRp20.
The mechanism of SRp20 relocalization during poliovirus infection is still unknown. We hypothesized that a viral proteinase may cause SRp20 to redistribute to the cytoplasm of the cell, and we first investigated whether expression of poliovirus 3CD resulted in a change in SRp20 subcellular localization. The 3CD proteinase is found at higher concentrations in the cell during infection than 3C proteinase, and has self-cleavage properties to generate 3C proteinase and 3D polymerase. To prevent self-cleavage and assess only 3CD activity, we utilized a mutated version of 3CD, containing an amino acid insertion adjacent to the cleavage site to render the protein deficient in self-cleavage while retaining proteolytic activity (3CDμ10) (41). FLAG-3CDμ10 (or empty vector) and GFP-SRp20 plasmids were cotransfected into cells, and 3CDμ10 expression was controlled using an inducible promoter (described in Materials and Methods). 3CDμ10 expression was induced over a 6-h time course, and at specific times postinduction cells were fixed and processed for confocal imaging. The results of these experiments are shown in Fig. 5. Figure 5A and B show that SRp20 localization was unaffected by transfection of empty vector or by mock induction of 3CDμ10 expression (respectively), and no detectable 3CDμ10 was observed when cells were mock induced (Fig. 5B). At 1 h postinduction, none of the cells observed contained any detectable 3CDμ10 (see Fig. 5C); therefore, the cells imaged at this time point were expressing either no or very low levels of 3CDμ10, were untransfected, or were refractory to induction. At 3 and 6 h postinduction, however, 3CDμ10 could be detected in the cytoplasm of the transfected/induced cells (see Fig. 5D and E). Even with high levels of expression, 3CDμ10 did not cause any detectable relocalization of SRp20. We also carried out the time course to 12 h postinduction and did not observe any redistribution of SRp20 to the cytoplasm of the cells with 3CDμ10 expression (data not shown). Thus, 3CD expression alone is not sufficient to cause the nucleocytoplasmic relocalization of SRp20.
Fig 5.

Expression of poliovirus 3CDμ10 proteinase in HeLa T-REx cells does not cause SRp20 relocalization. Cells were cotransfected with GFP-SRp20 and FLAG-3CDμ10 (or empty vector). Cells were either mock induced (B) or induced with 1 μg/ml tetracycline (A and C to E) at 37°C for 3CDμ10 expression. At 1, 3, and 6 h postinduction (C to E), cells were fixed; cells that were mock induced (B) and cells transfected with empty vector (A) were fixed at 6 h postinduction. Cells were incubated with an anti-FLAG mouse monoclonal antibody and, subsequently, Alexa Fluor 594 goat anti-mouse secondary antibody. 3CDμ10 expression and SRp20 localization were determined using confocal microscopy; DAPI staining identified nuclei.
To address the issue of whether the μ10 mutation has any effect on the activity of 3CD to potentially cause a change in SRp20 localization, we also expressed wild-type 3CD in cells. In these experiments, we would expect 3CD as well as the 3C and 3D proteins to be expressed following induction, due to the self-cleavage activity of 3CD. However, because the FLAG tag is at the N terminus of the protein, only 3CD and 3C would be labeled for imaging. The results of this experiment are shown in Fig. 6. Transfection of the empty vector or mock induction of 3CD had no effect on SRp20 localization (shown in Fig. 6A and B). No detectable 3C/3CD was observed in any of the cells after 1 h of induction (Fig. 6C), although detectable levels of 3C/3CD could be observed at 3 and 6 h postinduction (Fig. 6D and E). Even with the self-cleavage activity of 3CD intact and the additional production of the 3C and 3D proteins, there was no effect on SRp20 localization. We conclude that expression of 3CD (or 3C/3CD) alone cannot cause the cytoplasmic relocalization of SRp20.
Fig 6.
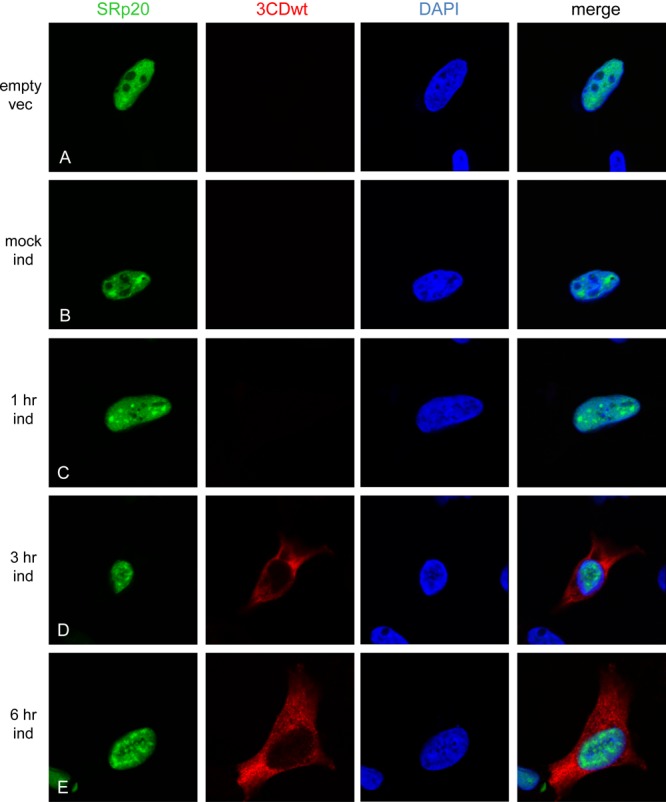
Poliovirus 3CDwt proteinase expression does not result in SRp20 relocalization. Cells were cotransfected with GFP-SRp20 and FLAG-3CDwt (or empty vector). Cells were either mock induced (B) or induced with 1 μg/ml tetracycline (A and C to E) at 37°C for 3CDwt expression and processed as described in the legend for Fig. 5.
Poliovirus 2A proteinase expression alone is sufficient to cause SRp20 relocalization from the nucleus to the cytoplasm of cells.
We next addressed the ability of poliovirus 2A proteinase to cause the redistribution of SRp20 to the cytoplasm of cells. Poliovirus 2A is known to cleave a number of cellular proteins during infection, including several nuclear pore proteins, which is thought to interfere with nucleocytoplasmic transport (14–17). We investigated whether these properties of 2A could contribute to the cytoplasmic relocalization of SRp20 by expression of the proteinase alone. The results of these experiments are shown in Fig. 7. Similar to the experiments described in the previous section, FLAG-2A (or empty vector) and GFP-SRp20 plasmids were cotransfected into cells, and 2A expression was controlled by induction. Transfection of empty vector or mock induction of 2A did not affect SRp20 localization, and it remained predominantly in the nucleus (shown in Fig. 7A and B). None of the cells expressed any detectable levels of 2A at 1 h postinduction (Fig. 7C), but 2A expression was readily detectable at 3 and 6 h postinduction (Fig. 7D and E). Correlating with 2A proteinase expression, SRp20 was observed to dramatically relocalize to the cytoplasm of cells. Therefore, poliovirus 2A expression alone is sufficient to cause the nucleocytoplasmic redistribution of SRp20.
Fig 7.

Expression of poliovirus 2A proteinase in HeLa T-REx cells results in the cytoplasmic accumulation of SRp20. Cells were cotransfected with GFP-SRp20 and poliovirus FLAG-2A (or empty vector). Cells were either mock induced (B) or induced with 1 μg/ml tetracycline (A and C to E) at 37°C for 2A expression. At 1, 3, and 6 h postinduction (C to E), cells were fixed; cells that were mock induced (B) and cells transfected with empty vector (A) were fixed at 6 h postinduction. Cells were incubated with an anti-FLAG mouse monoclonal antibody and, subsequently, Alexa Fluor 594 goat anti-mouse secondary antibody. 2A expression and SRp20 localization were determined using confocal microscopy; nuclei were stained with DAPI.
Expression of mutated poliovirus 2A proteins that lack proteinase activity no longer causes the nucleocytoplasmic redistribution of SRp20.
To determine if the proteinase activity of poliovirus 2A protein is required for SRp20 cytoplasmic accumulation, we examined the effect of expressing two different mutated versions of 2A lacking proteinase activity on SRp20 localization. The two mutated 2A proteins (2A-C109R and 2A-L98P) have been previously characterized (43, 52), and the sequences encoding these proteins were each inserted into the inducible vector. Figure 8 shows the results of expression of 2A-C109R with respect to SRp20 localization. While detectable levels of the mutated 2A protein were observed at 3 and 6 h postinduction, no effect on SRp20 localization was seen (see Fig. 8D and E). Expression of the other 2A mutated protein, 2A-L98P, resulted in very similar findings (data not shown). We conclude that the proteinase activity of poliovirus 2A is required for the relocalization of SRp20 to the cytoplasm of cells.
Fig 8.

SRp20 remains nuclear in localization following expression of poliovirus 2A-C109R. Cells were cotransfected with GFP-SRp20 and poliovirus FLAG-2A-C109R (or empty vector). Cells were either mock induced (B) or induced with 1 μg/ml tetracycline (A and C to E) at 37°C for 2A-C109R expression and processed as described in the legend for Fig. 7.
Expression of poliovirus wild-type 2A proteinase results in the cleavage of nuclear pore complex proteins Nup153, Nup98, and Nup62.
It has been previously demonstrated that specific nuclear pore complex proteins (Nups) are cleaved during poliovirus infection, resulting in the disruption of certain nuclear import/export pathways (15–17). More recent work has demonstrated that poliovirus 2A proteinase can directly cleave specific Nups (17, 21). We determined whether expression of poliovirus wild-type 2A (using the expression vectors and transfection protocols employed in this study) resulted in the cleavage of Nup153, Nup98, and Nup62, possibly contributing to the cytoplasmic accumulation of SRp20. HeLa cells were transfected with a plasmid encoding FLAG-tagged 2A (or transfected with empty vector), and 2A was constitutively expressed. Whole-cell lysates were generated from cells harvested at specific times posttransfection. The results are shown in Fig. 9. Nup98 was the most dramatically affected by 2A expression (Fig. 9A); its levels were reduced at 18 h posttransfection compared to the vector control, and Nup98 was virtually undetectable at 24 h posttransfection. Nup153 levels were also reduced at 18 h and 24 h posttransfection, and a smaller reduction in Nup62 levels was observed at the same time points. There was a slight decrease in the loading control (tubulin) as a result of some cell death due to the toxicity of 2A proteinase. We were also able to detect cleavage of eIF4G with 2A expression as early as 6 h posttransfection (Fig. 9B). These results verify that following expression in our assays, 2A has catalytic activity and recognizes specific cellular targets (e.g., eIF4G), in agreement with known functions of this proteinase in mammalian cells. Our findings are consistent with previously published data indicating that expression of poliovirus 2A proteinase results in the cleavage of Nup153, Nup98, and Nup62, albeit to differing extents.
Fig 9.

Wild-type poliovirus 2A proteinase expression in HeLa cells results in the cleavage of nuclear pore proteins Nup153, Nup98, and Nup62. HeLa cells were transfected with poliovirus FLAG-2A (or empty vector) for constitutive expression. Whole-cell lysates were generated from cells harvested at 1, 6, 18, or 24 h posttransfection. Lysates were subjected to SDS-PAGE and Western blot analysis. (A) mAb414 monoclonal antibody detected Nup153 and Nup62; anti-Nup98 polyclonal antibody detected Nup98. Cleavage of Nups following 2A proteinase expression resulted in loss of the full-length proteins. Tubulin was used as a loading control. (B) Utilizing a monoclonal anti-eIF4G antibody, cleavage of eIF4G could be observed beginning as early as 6 h posttransfection with FLAG-2A. A monoclonal anti-FLAG antibody determined expression of 2A proteinase, which could also be detected beginning about 6 h posttransfection. Tubulin was used as a loading control. Twice the volume of lysate was loaded as described for panel A, in order to detect FLAG-2A.
Expression of human rhinovirus 16 2A proteinase is sufficient to cause relocalization of SRp20 from the nucleus to the cytoplasm of cells.
While poliovirus and coxsackievirus infections were found to cause the efficient nucleocytoplasmic redistribution of SRp20, this relocalization was not observed to the same extent when cells were infected with HRV16. Because expression of poliovirus 2A proteinase alone could cause efficient SRp20 relocalization, we examined whether the same effect was observed with expression of HRV16 2A proteinase. The results are shown in Fig. 10. HRV16 2A expression could be detected at 3 and 6 h postinduction (see Fig. 10D and E). Interestingly, expression of the HRV16 2A proteinase alone caused the efficient cytoplasmic accumulation of SRp20, despite the fact that the viral infection resulted in lower detectable levels of SRp20 relocalization than poliovirus infection. Thus, the 2A proteinase of HRV16 can function in the nucleocytoplasmic relocalization of SRp20. When expressed alone the proteinase is sufficient for directing the redistribution of SRp20, although the extent of this effect is different from what is observed during the course of a HRV16 infection.
Fig 10.

Expression of human rhinovirus 16 2A proteinase in HeLa T-REx cells results in the efficient relocalization of SRp20. Cells were cotransfected with GFP-SRp20 and HRV16 FLAG-2A (or empty vector). Cells were either mock induced (B) or induced with 1 μg/ml tetracycline (A and C to E) at 37°C for 2A expression. At 1, 3, and 6 h postinduction (C to E), cells were fixed; cells that were mock induced (B) and cells transfected with empty vector (A) were fixed at 6 h postinduction. Cells were incubated with an anti-FLAG mouse monoclonal antibody and, subsequently, Alexa Fluor 594 goat anti-mouse secondary antibody. 2A expression and SRp20 localization were determined using confocal microscopy; nuclei were stained with DAPI.
DISCUSSION
The results obtained in this study provide important new insights into some of the similarities and differences among picornaviruses and the functions of their encoded proteins. Due to their small genome size, picornaviruses take advantage of host cell factors in addition to their viral proteins for the processes of viral translation and RNA replication. Poliovirus directs the relocalization of different nuclear proteins, including splicing factor SRp20, for utilization during viral infection. SRp20 has been shown to be an important ITAF for poliovirus IRES-mediated translation (39), and we predict that the related picornavirus CVB3 also requires the function of this host factor. Indeed, our experiments show that CVB3 infection causes nucleocytoplasmic relocalization of SRp20 similar to what is seen for poliovirus infection. Additional studies are needed to define the predicted role of SRp20 in CVB3 translation, as well as to examine any significance in the slight delay in the kinetics of SRp20 relocalization compared to poliovirus infection.
We further investigated the molecular mechanism(s) responsible for SRp20 relocalization by separately expressing the poliovirus proteinases 3CD and 2A and found that poliovirus 2A alone is sufficient to cause SRp20 cytoplasmic accumulation. We also determined that the proteinase activity of 2A was required for the relocalization of SRp20. It was not surprising that expression of poliovirus 3CD (wild type or μ10) proteinase alone resulted in a purely cytoplasmic distribution of labeled proteins, as it has been shown that poliovirus 2A is responsible for inducing the nuclear translocation of 3CD and 3C′ (53). Poliovirus 2A proteinase is known to cleave several nuclear pore complex proteins (Nups) and disrupt some, but not all, nucleocytoplasmic trafficking during infection (17, 21, 54). We found that expression of poliovirus 2A proteinase alone results in the cleavage of nuclear pore complex proteins Nup153, Nup98, and Nup62, which is consistent with other published work (21). We predict that the cleavage of certain Nups likely contributes to the accumulation of SRp20 in the cytoplasm of poliovirus-infected cells, where it can function in viral translation. Additional studies will be required to examine a direct link between specific Nup cleavage and SRp20 cytoplasmic relocalization.
Human rhinoviruses are picornaviruses with similarities to the enteroviruses poliovirus and coxsackievirus, but there are also some significant differences. Ongoing studies are aimed at elucidating the cause of some of these differences; namely, HRV RNA replication is less efficient than poliovirus RNA replication in vitro and in infected cells, although levels of viral translation in vitro are similar (50). Our work has revealed that while poliovirus and coxsackievirus direct the efficient cytoplasmic relocalization of SRp20, HRV16 infection causes SRp20 relocalization to a lesser extent. We considered the possibility that the temperature of infection may contribute to SRp20 localization in the cell during infection, and carried out poliovirus and HRV16 infections in parallel, at both 34°C and 37°C for each virus. HRV16 has been shown to replicate efficiently at both temperatures in vivo, with slightly lower titers reached at 37°C (51). In addition, a minor delay in poliovirus growth kinetics is observed when infection is carried out at 34°C compared to 37°C, although the virus grows to similar titers at both temperatures (A. J. Chase, J. H. Nguyen, and B. L. Semler, unpublished data). Surprisingly, poliovirus directed the relocalization of SRp20 at both temperatures of infection with similar kinetics. It was somewhat unexpected that the cytoplasmic redistribution of SRp20 was observed at the same time postinfection at a lower, suboptimal temperature of infection. We predicted that we would detect a delay in relocalization, due to the general delay in virus growth observed at 34°C and/or a reduction in the activities of the viral proteins produced. Thus, the slight delay in poliovirus growth seen at the lower temperature does not correlate with the relocalization of SRp20. HRV16 infection of HeLa cells resulted in small amounts of SRp20 relocalization beginning at 6 h postinfection at both 34°C and 37°C, with a gradual increase in the percentage of cells showing detectable cytoplasmic SRp20. Taken together, these results lead us to conclude that the temperature of infection alone does not account for differences observed in SRp20 localization between the viruses. It is possible that HRV16 does not require high levels of SRp20 for viral translation at early times of infection, or it can utilize other cellular proteins to provide a similar function. Future experiments will be required to investigate whether SRp20 plays a role in HRV16 translation and to examine the localization of other nuclear proteins during HRV16 infection that are known to redistribute to the cytoplasm of poliovirus-infected cells and function in poliovirus translation.
Finally, we expressed the HRV16 2A proteinase in the same inducible system as the poliovirus 2A proteinase and found that expression of HRV16 2A alone could indeed cause the efficient cytoplasmic redistribution of SRp20 in HeLa T-REx cells. These data conflict somewhat with what we observed during the course of an authentic HRV16 infection, where SRp20 is relocalized to the cytoplasm of infected cells to a lesser extent than the relocalization seen during poliovirus infection. The question remaining is why the HRV16 infection itself does not result in efficient SRp20 relocalization. We hypothesize that the levels of 2A proteinase produced during HRV16 infection are significantly lower than the levels of 2A produced during poliovirus infection. With lower levels of 2A present during infection, it is possible that the mechanism by which 2A directs the relocalization of SRp20 is dramatically impaired. In agreement with this prediction is the observation that the overexpression of HRV16 2A from an inducible plasmid resulted in SRp20 relocalization, possibly because the levels of 2A achieved via cytomegalovirus (CMV) promoter-driven expression are sufficient to direct the efficient redistribution of SRp20. Importantly, our data imply that the HRV16 2A proteinase is not deficient in its ability to cause SRp20 redistribution, and its activity does not necessarily differ from that of poliovirus 2A.
It is possible that the HRV IRES may not interact as productively with the cellular translation machinery as the poliovirus IRES, or that the HRV RNA itself may be inherently less stable. Based upon in vitro translation experiments, HRV RNA is translated slightly less efficiently than that of poliovirus (50). If recapitulated in vivo, this 2-to-3-fold reduction in translation efficiency compared to poliovirus may result in the production of slightly lower levels of viral proteins required for RNA synthesis during infection. A minor reduction in the levels of viral proteins could have a cumulative effect over the course of infection, potentially contributing to the significant reduction in HRV RNA replication that is observed and/or lowering the levels of nucleocytoplasmic redistribution of host factors, thereby generating or amplifying other deficiencies for the virus in the infected cell. The accumulation of these events would lead to the overall reduction in viral titers for HRV via a mass-action effect. In addition, slightly lower levels of viral translation may require lower levels of SRp20, and HRV may be able to utilize the SRp20 that is already present in the cytoplasm of the cell without requiring any additional SRp20 from the nucleus. Alternatively, HRV infection might result in a low level of SRp20 relocalization during the early stages of infection that we are unable to detect by confocal microscopy due to the abundance of the protein in the nucleus. Finally, we also consider the possibility that the cell type utilized for infection may contribute to differences in the subcellular environment generated by human rhinoviruses versus other enteroviruses. Additional experiments are necessary to address the localization of SRp20 in cell types other than HeLa cells, for example, in bronchial cell lines, which could be more relevant for rhinovirus infection.
While there is known overlap in the host cell factors that are cleaved by 2A between poliovirus and HRV during infection, it is still unclear what contributes to the overall protracted time course of infection for HRV compared to poliovirus, and what specific replication events may be affected. Questions remain regarding the specific cellular targets and enzymatic activities for poliovirus versus HRV16 2A proteinases. Indeed, recent work has shown that the 2A proteinases from several different serotypes of HRV differentially cleave nuclear complex proteins (20). While the catalytic core of 2A is conserved among poliovirus and human rhinoviruses, there may be other differences between the proteinases (such as targets of cleavage, or other protein-protein interactions) that play a role in the subsequent differences observed between the two virus infections. In addition, there may be certain pathways or signaling cascades that are activated, or conversely downregulated, differentially by these two picornaviruses. Taken together, there are likely many yet undiscovered factors contributing to the overall differences observed between HRV and other enterovirus infections.
ACKNOWLEDGMENTS
We thank Rozanne Sandri-Goldin for providing the GFP-SRp20 expression plasmid and Karla Kirkegaard for her gift of the poliovirus cDNA expression plasmids for the generation of mutated versions of 2A proteinase. We also thank Wai-Ming Lee for providing the cDNA encoding the full-length HRV16 genome. We are indebted to Ann Palmenberg for kindly providing the polyclonal antibodies to detect HRV16 3D and 2A proteins. Confocal images were generated with assistance from Adeela Syed at the UCI Optical Biology Core, which is supported by Comprehensive Cancer Center award P30CA062203 from the National Cancer Institute, and at the UCI Stem Cell Research Center, with assistance from Marek Mandau.
K.D.F. was a predoctoral trainee of the Public Health Service (training grant AI 07319). This work was supported by Public Health Service grant AI 26765 from the National Institutes of Health and by the California Center for Antiviral Drug Discovery (a Multicampus Research Program Initiative from the University of California).
Footnotes
Published ahead of print 19 December 2012
REFERENCES
- 1. Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JW. 1982. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J. Biol. Chem. 257:14806–14810 [PubMed] [Google Scholar]
- 2. Joachims M, Van Breugel PC, Lloyd RE. 1999. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 73:718–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kerekatte V, Keiper BD, Badorff C, Cai A, Knowlton KU, Rhoads RE. 1999. Cleavage of poly(A)-binding protein by coxsackievirus 2A protease in vitro and in vivo: another mechanism for host protein synthesis shutoff? J. Virol. 73:709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kräusslich HG, Nicklin MJ, Toyoda H, Etchison D, Wimmer E. 1987. Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J. Virol. 61:2711–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuyumcu-Martinez NM, Joachims M, Lloyd RE. 2002. Efficient cleavage of ribosome-associated poly(A)-binding protein by enterovirus 3C protease. J. Virol. 76:2062–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lloyd RE, Jense HG, Ehrenfeld E. 1987. Restriction of translation of capped mRNA in vitro as a model for poliovirus-induced inhibition of host cell protein synthesis: relationship to p220 cleavage. J. Virol. 61:2480–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yalamanchili P, Datta U, Dasgupta A. 1997. Inhibition of host cell transcription by poliovirus: cleavage of transcription factor CREB by poliovirus-encoded protease 3Cpro. J. Virol. 71:1220–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clark ME, Hammerle T, Wimmer E, Dasgupta A. 1991. Poliovirus proteinase 3C converts an active form of transcription factor IIIC to an inactive form: a mechanism for inhibition of host cell polymerase III transcription by poliovirus. EMBO J. 10:2941–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clark ME, Lieberman PM, Berk AJ, Dasgupta A. 1993. Direct cleavage of human TATA-binding protein by poliovirus protease 3C in vivo and in vitro. Mol. Cell. Biol. 13:1232–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Das S, Dasgupta A. 1993. Identification of the cleavage site and determinants required for poliovirus 3CPro-catalyzed cleavage of human TATA-binding transcription factor TBP. J. Virol. 67:3326–3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yalamanchili P, Banerjee R, Dasgupta A. 1997. Poliovirus-encoded protease 2APro cleaves the TATA-binding protein but does not inhibit host cell RNA polymerase II transcription in vitro. J. Virol. 71:6881–6886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Castelló A, Alvarez E, Carrasco L. 2011. The multifaceted poliovirus 2A protease: regulation of gene expression by picornavirus proteases. J. Biomed. Biotechnol. 2011:369648 doi:10.1155/2011/369648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li X, Lu HH, Mueller S, Wimmer E. 2001. The C-terminal residues of poliovirus proteinase 2A(pro) are critical for viral RNA replication but not for cis- or trans-proteolytic cleavage. J. Gen. Virol. 82:397–408 [DOI] [PubMed] [Google Scholar]
- 14. Belov GA, Lidsky PV, Mikitas OV, Egger D, Lukyanov KA, Bienz K, Agol VI. 2004. Bidirectional increase in permeability of nuclear envelope upon poliovirus infection and accompanying alterations of nuclear pores. J. Virol. 78:10166–10177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Belov GA, Evstafieva AG, Rubtsov YP, Mikitas OV, Vartapetian AB, Agol VI. 2000. Early alteration of nucleocytoplasmic traffic induced by some RNA viruses. Virology 275:244–248 [DOI] [PubMed] [Google Scholar]
- 16. Gustin KE, Sarnow P. 2001. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J. 20:240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park N, Katikaneni P, Skern T, Gustin KE. 2008. Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J. Virol. 82:1647–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park N, Skern T, Gustin KE. 2010. Specific cleavage of the nuclear pore complex protein Nup62 by a viral protease. J. Biol. Chem. 285:28796–28805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gustin KE, Sarnow P. 2002. Inhibition of nuclear import and alteration of nuclear pore complex composition by rhinovirus. J. Virol. 76:8787–8796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Watters K, Palmenberg AC. 2011. Differential processing of nuclear pore complex proteins by rhinovirus 2A proteases from different species and serotypes. J. Virol. 85:10874–10883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Castelló A, Izquierdo JM, Welnowska E, Carrasco L. 2009. RNA nuclear export is blocked by poliovirus 2A protease and is concomitant with nucleoporin cleavage. J. Cell Sci. 122:3799–3809 [DOI] [PubMed] [Google Scholar]
- 22. Ypma-Wong MF, Dewalt PG, Johnson VH, Lamb JG, Semler BL. 1988. Protein 3CD is the major poliovirus proteinase responsible for cleavage of the P1 capsid precursor. Virology 166:265–270 [DOI] [PubMed] [Google Scholar]
- 23. Parsley TB, Cornell CT, Semler BL. 1999. Modulation of the RNA binding and protein processing activities of poliovirus polypeptide 3CD by the viral RNA polymerase domain. J. Biol. Chem. 274:12867–12876 [DOI] [PubMed] [Google Scholar]
- 24. Perera R, Daijogo S, Walter BL, Nguyen JH, Semler BL. 2007. Cellular protein modification by poliovirus: the two faces of poly(rC)-binding protein. J. Virol. 81:8919–8932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parsley TB, Towner JS, Blyn LB, Ehrenfeld E, Semler BL. 1997. Poly (rC) binding protein 2 forms a ternary complex with the 5′-terminal sequences of poliovirus RNA and the viral 3CD proteinase. RNA 3:1124–1134 [PMC free article] [PubMed] [Google Scholar]
- 26. Brunner JE, Nguyen JHC, Roehl HH, Ho TV, Swiderek KM, Semler BL. 2005. Functional interaction of heterogeneous nuclear ribonucleoprotein C with poliovirus RNA synthesis initiation complexes. J. Virol. 79:3254–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Belov GA, Habbersett C, Franco D, Ehrenfeld E. 2007. Activation of cellular Arf GTPases by poliovirus protein 3CD correlates with virus replication. J. Virol. 81:9259–9267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meerovitch K, Svitkin YV, Lee HS, Lejbkowicz F, Kenan DJ, Chan EK, Agol VI, Keene JD, Sonenberg N. 1993. La autoantigen enhances and corrects aberrant translation of poliovirus RNA in reticulocyte lysate. J. Virol. 67:3798–3807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blyn LB, Towner JS, Semler BL, Ehrenfeld E. 1997. Requirement of poly(rC) binding protein 2 for translation of poliovirus RNA. J. Virol. 71:6243–6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walter BL, Nguyen JH, Ehrenfeld E, Semler BL. 1999. Differential utilization of poly(rC) binding protein 2 in translation directed by picornavirus IRES elements. RNA 5:1570–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Borman A, Howell MT, Patton JG, Jackson RJ. 1993. The involvement of a spliceosome component in internal initiation of human rhinovirus RNA translation. J. Gen. Virol. 74:1775–1788 [DOI] [PubMed] [Google Scholar]
- 32. Costa-Mattioli M, Svitkin Y, Sonenberg N. 2004. La autoantigen is necessary for optimal function of the poliovirus and hepatitis C virus internal ribosome entry site in vivo and in vitro. Mol. Cell. Biol. 24:6861–6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hellen CU, Witherell GW, Schmid M, Shin SH, Pestova TV, Gil A, Wimmer E. 1993. A cytoplasmic 57-kDa protein that is required for translation of picornavirus RNA by internal ribosomal entry is identical to the nuclear pyrimidine tract-binding protein. Proc. Natl. Acad. Sci. U. S. A. 90:7642–7646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaminski A, Hunt SL, Patton JG, Jackson RJ. 1995. Direct evidence that polypyrimidine tract binding protein (PTB) is essential for internal initiation of translation of encephalomyocarditis virus RNA. RNA 1:924–938 [PMC free article] [PubMed] [Google Scholar]
- 35. Hunt SL, Jackson RJ. 1999. Polypyrimidine-tract binding protein (PTB) is necessary, but not sufficient, for efficient internal initiation of translation of human rhinovirus-2 RNA. RNA 5:344–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hunt SL, Hsuan JJ, Totty N, Jackson RJ. 1999. unr, a cellular cytoplasmic RNA-binding protein with five cold-shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev. 13:437–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sean P, Nguyen JH, Semler BL. 2009. Altered interactions between stem-loop IV within the 5′ noncoding region of coxsackievirus RNA and poly(rC) binding protein 2: effects on IRES-mediated translation and viral infectivity. Virology 389:45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Blyn LB, Swiderek KM, Richards O, Stahl DC, Semler BL, Ehrenfeld E. 1996. Poly(rC) binding protein 2 binds to stem-loop IV of the poliovirus RNA 5′ noncoding region: identification by automated liquid chromatography-tandem mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 93:11115–11120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bedard KM, Daijogo S, Semler BL. 2007. A nucleo-cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J. 26:459–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fitzgerald KD, Semler BL. 2011. Re-localization of cellular protein SRp20 during poliovirus infection: bridging a viral IRES to the host cell translation apparatus. PLoS Pathog. 7:e1002127 doi:10.1371/journal.ppat.1002127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Semler BL, Johnson VH, Dewalt PG, Ypma-Wong MF. 1987. Site-specific mutagenesis of cDNA clones expressing a poliovirus proteinase. J. Cell. Biochem. 33:39–51 [DOI] [PubMed] [Google Scholar]
- 42. Lawson MA, Semler BL. 1991. Poliovirus thiol proteinase 3C can utilize a serine nucleophile within the putative catalytic triad. Proc. Natl. Acad. Sci. U. S. A. 88:9919–9923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Crowder S, Kirkegaard K. 2005. Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nat. Genet. 37:701–709 [DOI] [PubMed] [Google Scholar]
- 44. Lee WM, Wang W, Rueckert RR. 1995. Complete sequence of the RNA genome of human rhinovirus 16, a clinically useful common cold virus belonging to the ICAM-1 receptor group. Virus Genes 9:177–181 [DOI] [PubMed] [Google Scholar]
- 45. Neugebauer KM, Roth MB. 1997. Distribution of pre-mRNA splicing factors at sites of RNA polymerase II transcription. Genes Dev. 11:1148–1159 [DOI] [PubMed] [Google Scholar]
- 46. Dewalt PG, Blair WS, Semler BL. 1990. A genetic locus in mutant poliovirus genomes involved in overproduction of RNA polymerase and 3C proteinase. Virology 174:504–514 [DOI] [PubMed] [Google Scholar]
- 47. Harkins S, Cornell CT, Whitton JL. 2005. Analysis of translational initiation in coxsackievirus B3 suggests an alternative explanation for the high frequency of R+4 in the eukaryotic consensus motif. J. Virol. 79:987–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chapman NM, Ragland A, Leser JS, Hofling K, Willian S, Semler BL, Tracy S. 2000. A group B coxsackievirus/poliovirus 5′ nontranslated region chimera can act as an attenuated vaccine strain in mice. J. Virol. 74:4047–4056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee WM, Wang W. 2003. Human rhinovirus type 16: mutant V1210A requires capsid-binding drug for assembly of pentamers to form virions during morphogenesis. J. Virol. 77:6235–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Todd S, Towner JS, Semler BL. 1997. Translation and replication properties of the human rhinovirus genome in vivo and in vitro. Virology 229:90–97 [DOI] [PubMed] [Google Scholar]
- 51. Papadopoulos NG, Sanderson G, Hunter J, Johnston SL. 1999. Rhinoviruses replicate effectively at lower airway temperatures. J. Med. Virol. 58:100–104 [DOI] [PubMed] [Google Scholar]
- 52. Yu SF, Lloyd RE. 1991. Identification of essential amino acid residues in the functional activity of poliovirus 2A protease. Virology 182:615–625 [DOI] [PubMed] [Google Scholar]
- 53. Tian W, Cui Z, Zhang Z, Wei H, Zhang X. 2011. Poliovirus 2A(pro) induces the nucleic translocation of poliovirus 3CD and 3C' proteins. Acta Biochim. Biophys. Sin. (Shanghai) 43:38–44 [DOI] [PubMed] [Google Scholar]
- 54. Gustin KE. 2003. Inhibition of nucleo-cytoplasmic trafficking by RNA viruses: targeting the nuclear pore complex. Virus Res. 95:35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]