

Abstract
The severity of West Nile virus (WNV) infection in immunocompetent animals is highly strain dependent, ranging from avirulent to highly neuropathogenic. Here, we investigate the nature of this strain-specific restriction by analyzing the replication of avirulent (WNV-MAD78) and highly virulent (WNV-NY) strains in neurons, astrocytes, and microvascular endothelial cells, which comprise the neurovascular unit within the central nervous system (CNS). We demonstrate that WNV-MAD78 replicated in and traversed brain microvascular endothelial cells as efficiently as WNV-NY. Likewise, similar levels of replication were detected in neurons. Thus, WNV-MAD78's nonneuropathogenic phenotype is not due to an intrinsic inability to replicate in key target cells within the CNS. In contrast, replication of WNV-MAD78 was delayed and reduced compared to that of WNV-NY in astrocytes. The reduced susceptibility of astrocytes to WNV-MAD78 was due to a delay in viral genome replication and an interferon-independent reduction in cell-to-cell spread. Together, our data suggest that astrocytes regulate WNV spread within the CNS and therefore are an attractive target for ameliorating WNV-induced neuropathology.
INTRODUCTION
The Flaviviridae include several globally important emerging arthropod-borne viruses, such as yellow fever virus, dengue virus, Japanese encephalitis virus, and West Nile virus (WNV). WNV has re-emerged as a pathogen that causes severe neurological disease. Prior to the 1990s, most WNV infections were asymptomatic or associated with a mild febrile illness known as West Nile fever. Since its introduction into the United States in 1999, annual outbreaks of WNV have resulted in ∼16,000 reported cases with neurological complications, including meningitis, encephalitis, and acute flaccid paralysis. These cases have resulted in greater than 1,500 deaths, making WNV the leading cause of mosquito-borne neuroinvasive disease in the United States (http://www.cdc.gov/ncidod/dvbid/westnile/index.htm). In addition, outbreaks have occurred in other parts of the world, including eastern and western Europe (1, 2). Nevertheless, the factors responsible for the increase in pathogenicity of WNV remain poorly understood.
Many neuroinvasive viruses, including WNV, preferentially enter the central nervous system (CNS) via the hematogenous route by crossing the blood-brain barrier (BBB). The BBB is comprised of specialized endothelial cells, which line the cerebral microvasculature, and the foot processes of astrocytes, which envelop >99% of the endothelium. Under normal physiological conditions, the BBB tightly regulates transport of molecules into and out of the CNS. The restrictive nature of the BBB is a consequence of the formation of complex cell-to-cell tight junctions and lower basal levels of pinocytosis and endocytosis (3, 4). Although astrocytes were once thought to serve a structural role in the BBB, it is now clear that they play an important role in maintaining its functional integrity. In vitro, cocultures of brain endothelial cells and astrocytes establish a tighter barrier than endothelial cells alone, and secretion and activation of matrix metalloproteases (MMPs) by astrocytes result in disruption of the BBB during disease (5, 6). Astrocytes also modulate neuronal health and activity through the uptake of excess neurotransmitters and secretion of nutrients (7–10). Thus, astrocytes serve as a structural and functional bridge between endothelial cells and neurons. Together, these three cell types form the neurovascular unit (NVU), which functions to regulate blood flow, the integrity of the BBB, and neuronal activity in response to environmental changes (11). Understanding how viruses replicate within the NVU may facilitate novel strategies for treating viral infection of the CNS.
The neuroinvasive potential of WNV is strain dependent. While most, if not all, strains of WNV are neurovirulent when mice are inoculated intracranially, only a subset of strains are neuroinvasive when inoculated via a peripheral route (12). The mechanistic basis for the increased neuroinvasiveness of some strains of WNV remains poorly understood. However, the observation that exogenous disruption of the BBB enables a nonneuropathogenic strain of WNV to enter the CNS (13) suggests that the capacity to traverse the BBB is a determining factor for neuropathogenicity. Here, we investigated the nature of the restriction at the BBB by comparing the ability of an avirulent lineage 2 African isolate, WNV-MAD78 (12), to a highly virulent lineage 1 North American strain isolated in 2000, West Nile virus New York (WNV-NY), (12, 14, 15) to replicate in various cell types comprising the NVU. While both strains replicated efficiently in brain microvascular endothelial cells and neurons, WNV-MAD78 replication was restricted within astrocytes compared to that of WNV-NY. WNV-MAD78 exhibited both a delay in viral genome synthesis and reduced cell-to-cell spread in astrocytes compared to WNV-NY. Moreover, the restriction of WNV-MAD78 replication and spread in astrocytes was independent of type I interferon (IFN). Together, our findings suggest that astrocytes play an important role in controlling WNV dissemination within the CNS.
MATERIALS AND METHODS
Cells and viruses.
Previously characterized human brain microvascular endothelial cells (HBMECs) were obtained from K. S. Kim (Baltimore, MD) (16). HBMECs were isolated from individuals with seizure disorders and transformed by stably transfecting cells with the simian virus 40 (SV40) large T antigen. HBMECs were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), antibiotic/antimycotic, nonessential amino acids, minimal essential medium (MEM) vitamins, 5 U/ml heparin, NuSerum (10%), 2 mM l-glutamine, 1 mM sodium pyruvate, and 150 μg/ml endothelial growth supplement. All HBMEC experiments were performed with cells passaged no more than 12 times. Donor-matched primary human brain cortical astrocytes (HBCAs; ABRI371) and human brain microvascular endothelial cells (HBMVECs; ABRI401) derived from normal human tissue were purchased from CellSystems (Kirkland, WA). Both cell types were maintained according to the manufacturer's instructions. All experiments with HBMVECs and HBCAs were performed on cells passaged no more than 14 times. Primary cortical neurons were prepared from day 15 C57BL/6 mouse embryos as previously described (17, 18). Cortical neuron experiments were performed using neurons that were cultured for 3 to 4 days in neurobasal medium containing B27 and l-glutamine (Invitrogen). Neuro2A and Vero cells were maintained in Dulbecco's modified essential medium (DMEM) supplemented with 10% FBS, 2 mM l-glutamine, 1 mM sodium pyruvate, antibiotic/antimycotic solution, and nonessential amino acids (complete DMEM). Neuro2A cells were differentiated 3 days prior to infection by reducing the FBS concentration to 0.5% and were maintained in differentiation media throughout the course of the experiment.
Working stocks of WNV-NY strain 3356 were generated from an infectious clone, pFL-WNV (19). Briefly, infectious particles were recovered as previously described (19), passaged once in 293 cells at a low multiplicity of infection (MOI), and subsequently passaged in Vero cells. WNV-MAD78 was obtained from the World Reference Center of Emerging Viruses and Arboviruses (Galveston, TX). Lyophilized virus was resuspended in complete DMEM supplemented with 20% FBS, amplified once in Vero cells, and plaque purified. Viral stocks were amplified once in 293 cells at a low MOI, and working stocks were generated by passaging once in Vero cells. All viral stocks were aliquoted and stored at −80°C.
Growth curves.
Cell cultures were infected with WNV-NY or WNV-MAD78 at the indicated MOI for 1 h at 37°C. Unless otherwise stated, the amount of virus added to cultures to achieve the indicated MOI was calculated using the titer of the viral stock on the respective cell line. The inoculum was removed and complete medium was added. Culture supernatants were recovered at the indicated times and clarified by low-speed centrifugation for 5 min. Supernatants were transferred to new tubes and stored at −80°C. Viral titers were determined by plaque assay on Vero cells.
Virus titration by plaque assay.
Monolayers of Vero cells in 6-well plates were washed once with phosphate-buffered saline (PBS) prior to the addition of serial dilutions of viral samples. The cells were incubated in a 5% CO2 incubator for 1 h at 37°C with rocking, the inocula were removed, and complete DMEM solution with 0.9% low-melting-point agarose (Fisher) was overlaid. Vesicular stomatitis virus (VSV) plaques were counted 24 h postinfection. For WNV titration, cell monolayers were incubated for 48 h, and a second overlay of agarose-containing complete DMEM supplemented with 0.003% neutral red (Sigma) was added. The plates were incubated for an additional 48 (WNV-NY) to 96 h (WNV-MAD78) prior to counting plaques. All titers were performed in duplicate.
Viral translocation assay.
HBMECs or HBMVECs (4 × 104 cells) were plated on the luminal side of a fibronectin-coated cell culture insert with a pore size of 3 μm (BD Biosciences) and incubated for 5 days at 37°C in a 5% CO2 incubator. The widely accepted methods of transendothelial electrical resistance (TEER) and translocation of fluorescein isothiocyanate (FITC)-labeled dextran were used to assess the integrity of the endothelial monolayers (16, 20). As previously reported, monolayers reaching TEER values between 250 and 300 Ω/cm2 were largely impermeable to FITC-labeled dextran (20). Therefore, monolayers were considered confluent once a minimum TEER value of 250 Ω/cm2 was achieved. Confluent cultures were infected at an MOI of 0.1 by adding virus to the upper, luminal chamber. Inoculum was removed after incubating for 1 h at 37°C, and 500 μl of appropriate complete medium was added to both chambers. Medium was collected from both chambers at the indicated times, and the level of infectious virus was determined by plaque assay.
In vitro BBB model.
HBMECs (4 × 104 cells) and HBCAs (7.5 × 103 cells) were plated on transwell inserts and bottom chambers, respectively. The TEER of the HBMEC monolayer was measured 5 days after plating using a Millicell ERS. Cultures were considered confluent when a resistance of 250 to 300 Ω/cm2 was recorded. The HBMEC monolayer was infected at an MOI of 0.1 by adding virus to the luminal chamber. Inoculum was removed after incubating for 1 h at 37°C, and 500 μl of appropriate complete medium was added to both chambers. Medium was collected from both chambers at the indicated times, and the level of infectious virus was determined by plaque assay.
Indirect immunofluorescence analysis (IFA).
At the indicated times postinfection, the HBCA monolayer was washed with PBS and fixed with 3% paraformaldehyde. Cell monolayers were permeabilized with a solution of PBS–0.2% Triton X-100, blocked with PBS containing 10% normal goat serum, and incubated with WNV hyperimmune ascitic fluid (1:1,000; World Reference Center of Emerging Viruses and Arboviruses) followed by goat anti-mouse IgG 549-nm Dylight-conjugated antibody (1:800; Jackson ImmunoLaboratories) and Hoescht stain (0.1 μg/ml). Cells were visualized with an Olympus IX51 microscope equipped with a digital camera.
Flow cytometry.
HBCA monolayers infected with WNV-NY or WNV-MAD78 (MOI of 0.01) were removed from plates by trypsinization, washed 2 times with PBS, and fixed in 3% paraformaldehyde. Cells were permeabilized with PBS–0.2% Triton X-100, blocked in PBS containing 0.5% heat-inactivated FBS, and probed with WNV hyperimmune ascitic fluid (1:1,000; World Reference Center of Emerging Viruses and Arboviruses) followed by goat anti-mouse IgG 633-nm Dylight-conjugated antibody. For flow cytometry analysis, 100,000 single cell events were collected using a FACS Canto (BD Biosciences).
Type I IFN bioassay.
A549s (7 × 104 cells) in 24-well plates were treated with 2-fold serial dilutions of human leukocyte IFN-α (BEI Biosciences) or cell-free, UV-inactivated supernatants recovered from WNV-infected HBCAs. Pretreated cells were infected with VSV (MOI of 1), and supernatants were collected at 24 h postinfection. Viral titers were determined by plaque assay on Vero cells as described above. IFN concentrations were determined based on a standard curve generated from the titers recovered from samples treated with IFN-α.
Neutralization of type I IFN.
The antibody concentration necessary to neutralize the IFN present in supernatants recovered from WNV-infected HBCAs was determined by pretreating A549 cells for 24 h with 160 IU/ml of IFN-α or -β in the presence of 10-fold dilutions of the antibodies to IFN-α (BEI) or IFN-β (BEI). Control wells consisted of cells treated with IFN only, no IFN, or isotype-matched antisera to IFN-α or IFN-β. Pretreated cells were infected with VSV (MOI of 1) and supernatants collected at 24 h postinfection. Viral titers were determined by plaque assay on Vero cells. For neutralization experiments, HBCAs were inoculated with WNV (MOI of 0.01) for 1 h at 37°C. The inoculum was replaced with complete DMEM containing 2 times the amount of antibody necessary to neutralize 160 IU/ml IFN-α and/or IFN-β or the appropriate control sera.
qRT-PCR.
Total RNA was extracted from HBCAs infected with WNV-MAD78 or WNV-NY (MOI of 0.01) using TRIzol (Invitrogen Life Technologies, Inc.) and treated with TurboDNase (Applied Biosystems). Total viral RNA levels were determined by quantitative real-time PCR (qRT-PCR) analysis on a Roche LC480 using Veriquest One-Step SYBR green MasterMix (Affymetrix Biosystems) with 50 ng of RNA. The following primers were used: WNV-NY (forward), 5′-GGA CCT TGT AAA GTT CCT ATC TCG-3′; WNV-NY (reverse), 5′-AGG GTT GAC AGT GAC CAA TC-3′; WNV-MAD78 (forward), 5′-CTG TAA GGT GCC CAT TTC C-3′; WNV-MAD78 (reverse), 5′-CCT CTT CCC ACC ACA ATG TAG-3′; human GAPDH (forward), 5′-CCA CTC CTC CAC CTT TGA C-3′; human GAPDH (reverse), 5′-ACC CTG TTG CTG TAG CCA-3′. Two-step quantitative PCR was used to determine strand-specific viral RNA levels. cDNA was generated from 500 ng of RNA using Moloney murine leukemia reverse transcriptase (NEB Biosciences) and gene-specific primers. The resulting cDNA was used as the template for qPCR with SYBR green 2× Veriquest MasterMix (Affymetrix). Primers used for two-step qPCR were the same as the primers described above.
Statistical analysis.
Graphpad Prism 5 was used to generate all statistical analyses. Standard errors and significance were determined using either one-way analysis of variance (ANOVA) with Bonferroni posttest correction or a two-tailed paired or unpaired Student t test.
RESULTS
WNV-NY and WNV-MAD78 replicate to similar levels in neuronal cells.
Because neurons constitute a primary target of WNV infection in vivo (21), we compared WNV-MAD78 to WNV-NY replication in primary cortical neurons derived from wild-type C57BL/6 mice and the mouse neuroblastoma cell line Neuro2A (Fig. 1A and B) at a multiplicity of infection (MOI) of 0.01 and 0.1, respectively. WNV-NY and WNV-MAD78 reached similar peak titers in both cell types, although the kinetics of WNV-MAD78 replication were slightly delayed at 24 h in cortical neurons (9.2-fold; P = 0.03). Despite the slight growth delay, neuronal cells were highly permissive for WNV-MAD78 replication, suggesting that the nonneuropathogenic phenotype documented in mice (12, 14) is not due to a reduced capacity to infect and replicate in neurons.
Fig 1.

WNV-NY and WNV-MAD78 replication in neuronal cells. Cortical neurons isolated from C57BL/6 mice (MOI of 0.01) (A) and differentiated Neuro2A cells (MOI of 0.1) (B) were infected with WNV-NY or WNV-MAD78. The MOI was calculated using the titer of the viral stocks on Vero cells. Culture supernatants were recovered at the indicated times, and titers were determined by plaque assay on Vero cells. Values represent the average numbers of PFU per ml (± standard errors) of supernatant and are from at least two separate experiments. A Student's unpaired t test was performed to determine statistical significance. Asterisks indicate differences that are statistically significant (*, P < 0.05).
WNV-MAD78 efficiently infects and traverses human brain endothelial cells.
Although many viruses achieve high levels of viremia, under normal conditions the BBB is highly effective at protecting the brain from circulating virus in the bloodstream. However, neuroinvasive viruses have evolved a variety of mechanisms to breach the BBB and gain access to the CNS (4). Because replication in brain endothelial cells is sufficient for transport of neuroinvasive strains of WNV across brain microvascular endothelial cells (20), we hypothesized that the decreased neuropathogenicity of WNV-MAD78 was due in part to a deficiency in replication in brain endothelial cells. Therefore, we monitored replication of WNV-NY and WNV-MAD78 in an established human brain microvascular endothelial cell line (HBMEC) that has been widely used as a model to study bacterial and parasitic neuroinvasion (16, 22–25) and primary brain microvascular endothelial cells (HBMVECs) derived from normal brain cortex tissue (20, 26). WNV-NY and WNV-MAD78 replicated at similar rates and to equivalent levels in both cell types, reaching peak titers between 32 and 48 h postinfection (Fig. 2A and B) without induction of cytopathic effect (CPE) (data not shown). However, we did observe a slight but statistically insignificant decrease in WNV-MAD78 titers very late in infection. These data suggest that replication in endothelial cells is not responsible for the decreased neuropathogenicity of WNV-MAD78.
Fig 2.

WNV-NY and WNV-MAD78 replication in human brain microvascular endothelial cells. Immortalized HBMECs (A) or primary HBMVECs (B) were infected with WNV-NY or WNV-MAD78 (MOI of 0.1). Culture supernatants were recovered at the indicated times and titers determined by plaque assay on Vero cells. Values represent the average numbers of PFU per ml (± standard errors) of supernatant from at least two separate experiments. (C) Traversal of HBMVEC by WNV-NY and WNV-MAD78. Confluent monolayers of HBMVECs were cultured on transwell inserts and infected with WNV-NY or WNV-MAD78 (MOI of 0.1). Culture supernatants were collected from the luminal and abluminal chambers at the indicated times, and titers were determined by plaque assay on Vero cells. Values represent the average numbers of PFU per ml (± standard errors) of supernatant from two separate experiments.
It was recently reported that nonreplicating virus-like particles (VLPs) generated from a lower virulence lineage 1 strain of WNV exhibited reduced transcellular migration across human umbilical vein endothelial cell monolayers compared to those generated from a highly virulent strain (27). Thus, nonneuropathogenic strains, such as WNV-MAD78, may have a reduced capacity to traverse the BBB despite replicating efficiently in brain microvascular endothelial cells. To test this, we infected HBMEC or HBMVEC monolayers grown on transwell supports and measured virus yields in both luminal (upper) and abluminal (lower) chambers. Equivalent levels of infectious particles were present in the luminal and abluminal chambers of WNV-NY- and WNV-MAD78-infected HBMECs (data not shown) and HBMVECs (Fig. 2C) at 24 and 48 h postinfection, demonstrating that both strains are capable of traversing the BBB at similar rates and levels. Therefore, the nonneuropathogenic phenotype of WNV-MAD78 does not correspond to a reduced capacity to replicate in or traverse brain microvascular endothelial cells.
WNV-MAD78 replication is restricted in cocultures of HBMECs and HBCAs.
We next tested whether astrocytes, which constitute the periparenchymal layer of the BBB, differentially limit WNV-MAD78 replication using an in vitro BBB model comprised of HBMECs cultured on transwell supports with primary human brain astrocytes derived from the cerebral cortex (HBCAs) in the bottom chamber. As previously observed, both strains of WNV were detected in medium recovered from the abluminal chamber 24 h after infection of the HBMEC monolayer (Fig. 3A). However, viral protein expression was not detected in the HBCAs until approximately 48 h after infection (Fig. 3B, panels iii and iv). At this time, equivalent numbers of infected cells were present in WNV-NY- and WNV-MAD78-infected cultures (144 ± 61 and 109 ± 63 WNV+ cells, respectively), suggesting that WNV-NY and WNV-MAD78 had similar capacities to establish an initial infection within the astrocyte monolayer. However, by 72 h postinfection, substantially more WNV-positive HBCAs were detected in WNV-NY-infected cultures than in WNV-MAD78-infected cultures (Fig. 3B, compare panel v to vi). Thus, WNV-MAD78 spread within the in vitro BBB model was constrained compared to that of WNV-NY.
Fig 3.
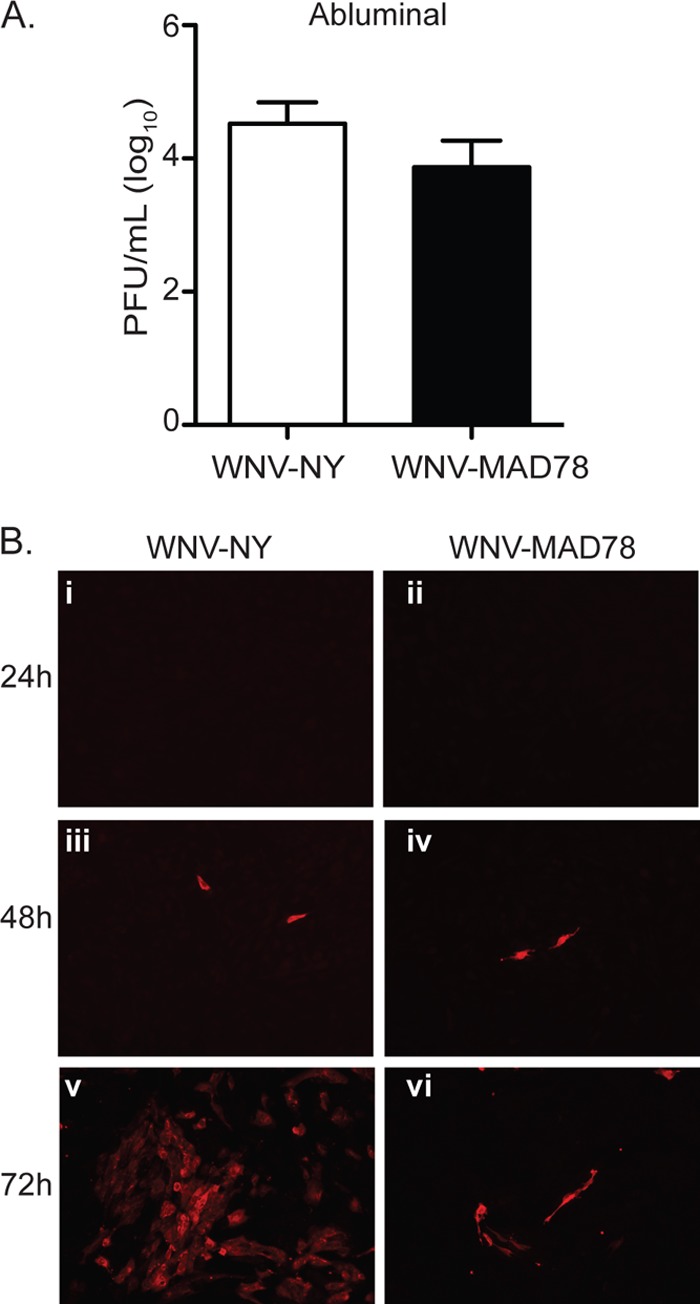
WNV replication in an in vitro model of the BBB. (A and B) HBMECs cultured on transwell inserts with HBCAs plated in the abluminal chamber were infected with WNV-NY or WNV-MAD78 (MOI of 0.1). (A) Culture supernatants were recovered from the abluminal chamber of the transwell at 24 h postinoculation, and viral titers were determined by plaque assays on Vero cells. Values represent the average numbers of PFU per ml (± standard errors) of supernatant from at least three separate experiments. (B) Viral replication in the HBCA layer of an in vitro BBB model. HBCAs were fixed with 3% PFA at the indicated times and probed with WNV hyperimmune ascitic fluid and goat anti-mouse IgG 549-nm Dylight-conjugated secondary antibody. Images are representative of at least three independent experiments.
WNV-MAD78 replication within astrocytes is restricted at multiple steps in the virus life cycle.
To further investigate the contribution of astrocytes in restricting WNV-MAD78 replication within the in vitro BBB, we examined WNV replication in HBCAs alone. In contrast to the donor-matched HBMVECs utilized in Fig. 2B and C, WNV-MAD78 infectious particle production in HBCAs was lower than that observed for WNV-NY throughout the course of infection (Fig. 4A). At the point of peak viral production, WNV-MAD78 titers were reduced approximately 45-fold (P < 0.005) compared to those of WNV-NY. Moreover, the latent period prior to the detection of extracellular infectious particles in WNV-MAD78-infected cultures was prolonged compared to that of WNV-NY-infected cultures, suggesting that WNV-MAD78 replication is delayed in HBCAs. A similar delay and reduction in WNV-MAD78 infectious particle production was observed when cultures were infected at an MOI of 2, which established a nearly synchronous infection of the monolayer (data not shown).
Fig 4.

WNV replication in HBCAs. HBCAs were infected with WNV-NY or WNV-MAD78 (MOI of 0.01). (A) WNV-NY and WNV-MAD78 infectious particle production in HBCAs. Culture supernatants were removed at the indicated times, and titers were determined by plaque assay on Vero cells. Values represent the average numbers of PFU per ml (± standard errors) of supernatant from at least three separate experiments. Asterisks indicate differences that are statistically significant (***, P < 0.005). (B to D) RNA synthesis of WNV-NY and WNV-MAD78 in HBCAs. Total RNA was recovered from HBCAs infected as described for panel A. Total (B) and strand-specific (C, positive; D, negative) viral RNA levels were determined by qRT-PCR. Values represent the averages (± standard errors) from at least three independent experiments. A Student's unpaired t test was performed to determine significance. Asterisks indicate differences that are statistically significant (*, P < 0.05; **, P < 0.01; ***, P < 0.005).
Accumulation of intracellular viral RNA also was delayed in WNV-MAD78-infected HBCAs compared to that in WNV-NY-infected HBCAs (Fig. 4B). Thus, the prolonged lag period prior to infectious particle production in the supernatants of WNV-MAD78-infected HBCAs may be due to a delay in viral RNA replication rather than assembly of virus particles. To better define the nature of the delay in WNV-MAD78 replication, we assessed the rate of accumulation of both positive- and negative-strand RNA at early times postinfection. Synthesis of both positive- and negative-strand WNV-MAD78 RNA was delayed compared to that of WNV-NY (Fig. 4C and D), suggesting that WNV-MAD78 replication in astrocytes is restricted at an early step in the virus life cycle.
The delay in synthesis of WNV-MAD78 viral RNA corresponded with a decrease in the number of extracellular infectious particles produced per infected cell compared to WNV-NY-infected cells at 24 h postinfection (Fig. 5A). However, by 48 h postinfection, WNV-NY- and WNV-MAD78-infected HBCAs produced similar levels of infectious particles per cell. In spite of this, the level of total infectious particles detected in WNV-MAD78-infected cultures remained substantially reduced compared to that of WNV-NY throughout the course of infection (Fig. 4A), suggesting that factors independent of viral RNA synthesis also are involved in restricting WNV-MAD78 replication within astrocytes. Indeed, visualization of WNV-infected astrocytes in the BBB coculture system indicated that WNV-MAD78 is also limited in cell-to-cell spread compared to WNV-NY (Fig. 3B, compare panel v to vi). To confirm these results, we quantitated the number of infected HBCAs over the course of infection using flow cytometry (Fig. 5B). Low levels of WNV-positive cells were detected at 12 h postinfection in WNV-NY-infected cultures. However, antigen-positive cells were not detected in WNV-MAD78-infected cultures until 16 h postinfection, which is consistent with the delay in viral replication. Importantly, the baseline number of WNV-NY-infected HBCAs at 12 h postinfection was comparable to the number of WNV-MAD78-infected HBCAs detected at 16 h postinfection, confirming that both strains are capable of establishing an initial infection within the astrocyte monolayer. While the number of WNV-NY-positive cells continued to increase throughout the course of infection, the number of WNV-MAD78-positive cells remained unchanged. By 48 h postinfection, there was a 15-fold (P < 0.01) increase in antigen-positive cells detected in WNV-NY-infected HBCAs compared to WNV-MAD78-infected HBCAs. Overall, our data suggest that WNV-MAD78 is both delayed in initiation of viral synthesis in HBCAs and impaired in its ability to spread to neighboring cells compared to WNV-NY.
Fig 5.

Infectious virions released per cell and cell-to-cell spread in HBCAs. HBCAs were infected with WNV-NY or WNV-MAD78 (MOI of 0.01). Culture medium was removed at the indicated times, and titers were determined by plaque assay on Vero cells. The number of infected cells within the monolayer was determined by flow cytometry. Cells were removed from the plate by trypsinization, fixed with 3% PFA, probed with WNV hyperimmune serum followed by goat anti-mouse IgG 633-nm Dylight-conjugated antibody, and subjected to flow cytometry. (A) Infectious particle production per infected cell. The number of infectious extracellular particles per cell was determined by dividing viral titers by the number of infected cells. Values represent the averages from three independent experiments. A Student's unpaired t test was performed to determine significance. Asterisks indicate differences that are statistically significant (*, P < 0.05) (B) WNV-positive cells per 105 cells from three independent experiments. Values represent the averages from three independent experiments. A Student's unpaired t test was performed to determine significance. Asterisks indicate differences that are statistically significant compared to WNV-NY at the 12-h time point (*, P < 0.05; ***, P < 0.005).
The reduced susceptibility of HBCAs to WNV-MAD78 is independent of type I IFN.
The restriction to WNV-MAD78 spread within astrocytes was suggestive of the paracrine protection of type I IFNs (28–30). Therefore, we examined supernatants recovered from WNV-infected HBCAs for the presence of type I IFNs using a bioassay. Measureable levels of IFN were first detected at 30 and 40 h postinfection in supernatants recovered from WNV-NY- and WNV-MAD78-infected HBCAs, respectively (Fig. 6A). Furthermore, infection of HBCAs with WNV-NY induced significantly higher levels of IFN than infection with WNV-MAD78 at all time points examined. Therefore, the kinetics and amplitude of induction of secreted IFN did not correlate with WNV-MAD78's diminished capacity to spread from cell to cell early in infection. However, WNV-MAD78 is more sensitive to IFN treatment than pathogenic strains of WNV in other cell types (14 and unpublished data). Thus, local levels of type I IFNs surrounding the WNV-MAD78-infected cells may be sufficient to inhibit viral spread. To assess this possibility, we examined viral spread and infectious particle production in HBCAs infected with WNV-NY or WNV-MAD78 in the presence or absence of neutralizing antibodies to IFN-α and -β (Fig. 6B to D). We consistently observed a significant increase in the number of WNV-NY-infected cells in the presence of neutralizing antibodies to IFN-α/β (Fig. 6B). Additionally, neutralizing antibodies to IFN-α/β or IFN-β enhanced infectious particle production in WNV-NY-infected HBCAs (Fig. 6C and D). In contrast, neutralizing antibodies to IFN-α and/or -β did not enhance viral spread or infectious particle production in WNV-MAD78-infected cells (Fig. 6B to D). These data suggest that IFN-β plays a role in restricting WNV-NY, but not WNV-MAD78, replication and spread in HBCAs.
Fig 6.

Role of type I IFN in limited WNV replication and spread in HBCAs. (A) Level of secreted type I IFN secretion in supernatants recovered from WNV-infected HBCAs. Culture supernatants were removed from HBCAs infected with WNV-NY or WNV-MAD78 (MOI of 0.01) at the indicated times postinfection. IFN levels were determined using a VSV-based bioassay. Values represent the level of type I IFN (IU/ml) (± standard errors) from a minimum of 2 independent experiments. A Student's unpaired t test was performed to determine significance. Asterisks indicate differences that are statistically significant (**, P < 0.01; ***, P < 0.005). (B to D) The effect of neutralization of type I IFN on WNV spread and replication in HBCAs. HBCAs were infected with WNV-NY or WNV-MAD78 (MOI of 0.01), and the inoculum was replaced with culture media containing control antisera or neutralizing antibodies to IFN-α and -β combined (B and C) or individually (D). (B) Cells were fixed at 48 h postinfection with 3% PFA, permeabilized, probed with WNV hyperimmune serum followed by goat anti-mouse IgG 633-nm Dylight-conjugated antibody, and subjected to flow cytometry. Values represent the average numbers of WNV-positive cells per 105 cells (± standard errors) from at least two independent experiments. A Student's unpaired t test was performed to determine significance. Asterisks indicate differences that are statistically significant (*, P < 0.05). (C and D) Culture supernatants were recovered at 48 h postinfection, and viral titers were determined by plaque assay on Vero cells. Values represent the average numbers of PFU per ml (± standard errors) of supernatant from at least two separate experiments. A Student's unpaired t test was performed to determine significance. Asterisks indicate differences that are statistically significant (*, P < 0.05; **, P < 0.01).
DISCUSSION
The naturally occurring diversity in virulence among WNV strains provides an excellent model system to define the viral and host factors involved in pathogenesis. To better understand the mechanistic basis for the differential neuropathogenicity between WNV strains, we compared the replication of pathogenic and nonpathogenic strains of WNV within the various cell types of the NVU. Consistent with reports that WNV-MAD78 is neurovirulent when mice are infected via intracranial inoculation (15), we observed that WNV-NY and WNV-MAD78 replicate to equivalent levels in neuronal cells. This suggests that WNV-MAD78's nonneuropathogenic phenotype is due to an inability to access highly susceptible neurons under peripheral infection conditions. Indeed, the nonneuropathogenic phenotype of some strains of WNV has been attributed to an inability to invade the CNS (12, 13, 27). Increasing evidence suggests that WNV entry into the CNS is a multistep process that can occur through one of several routes (17, 20, 31–35). WNV entry into the CNS has been shown to precede disruption of the BBB and leukocyte infiltration (32, 33, 36, 37), suggesting that WNV utilizes a direct mechanism to initially invade the CNS, such as basolateral secretion of virus particles from infected brain endothelial cells or transcytosis. Thus, the brain endothelium likely constitutes a primary barrier to WNV neuroinvasion. Using an established endothelial cell line that exhibits the physiological characteristics of the brain endothelium (16, 22–25, 38) and primary brain endothelial cells, we have demonstrated that WNV-MAD78 can replicate in and traverse the brain endothelium as efficiently as WNV-NY. Our findings are consistent with reports that highly and mildly neuropathogenic strains of Semliki Forest virus replicate to equivalent levels in endothelial cells (39, 40), suggesting that the capacity to cross the BBB is not always the determining factor for neuropathogenicity.
The initial invasion of WNV into the CNS brings the virus into close proximity with the second component of the BBB, astrocytes. Indeed, infected astrocytes have been detected in some fatal human cases of WNV encephalitis, suggesting that these cells are also targeted by WNV in vivo (41). We observed both a delay in the kinetics of WNV-MAD78 replication and a reduction in peak infectious particle production in astrocytes compared to WNV-NY. Since the astrocyte and primary brain endothelial cell lines used in this study were recovered from the same donor, it is unlikely that the constrained replication of WNV-MAD78 in HBCAs was due to a donor-specific restriction. Further comparison of the growth characteristics of WNV-NY and WNV-MAD78 in HBCAs indicated that both strains were capable of establishing an initial infection within this cell type. However, accumulation of viral protein and RNA was delayed in WNV-MAD78-infected HBCAs compared to WNV-NY. Analysis of kinetics of accumulation of viral positive- and negative-strand RNA indicated that the delay in WNV-MAD78 replication was at, or prior to, the initiation of negative-strand synthesis. However, it is unlikely that a delay in viral replication alone would result in the significant reduction in peak viral titers observed in WNV-MAD78-infected HBCAs, suggesting that an additional factor(s) is involved in restricting WNV-MAD78 accumulation in astrocytes.
The host innate antiviral response plays a pivotal role in controlling WNV replication in many cell types (14, 42–46). This response consists of a direct IFN regulatory factor 3 (IRF-3)-dependent and an indirect IFN-dependent mechanism, which function to constrain viral replication within the infected cell and prevent viral spread to neighboring cells, respectively. Measureable levels of type I IFN were detected in supernatants recovered from both WNV-NY- and WNV-MAD78-infected HBCAs. However, significantly higher levels of IFN were detected in WNV-NY-infected HBCAs, which corresponded to the increased replication and spread of this strain within the monolayer. Moreover, the high levels of IFN induced by WNV-NY, specifically IFN-β, suppressed viral spread and replication in astrocytes. In contrast, the low levels of IFN induced by WNV-MAD78 late in infection did not substantially restrict replication or spread within astrocytes.
In addition, the observation that WNV-NY- and WNV-MAD78-infected HBCAs produced equivalent levels of infectious particles per cell at 48 h postinfection suggests that the IRF-3-dependent arm of the host antiviral response does not differentially restrict WNV-NY and WNV-MAD78 replication within infected cells. Furthermore, induction of the direct IRF-3 target proteins ISG15 and ISG56 in HBCAs corresponded with the rate of WNV-NY and WNV-MAD78 replication (data not shown), indicating that WNV-MAD78 does not preferentially induce antiviral programs within the infected cell. Combined, these data suggest that the restriction to WNV-MAD78 replication and spread in astrocytes occurs prior to the induction of the host antiviral response. Therefore, the host antiviral response appears to play a minimal role in the reduced susceptibility of astrocytes to WNV-MAD78.
Determining both the viral and host cell factors involved in constraining WNV-MAD78 replication in astrocytes will be necessary to fully elucidate the nature of the restriction of WNV-MAD78. One possible viral factor is the presence of specific N-linked glycans on the envelope protein, which correlates with the ability to invade the CNS (12, 47, 48). WNV-MAD78 lacks this glycosylation site; thus, our data suggest that glycosylation is not a determining factor in the initial infection of brain endothelial cells or subsequent replication in neurons. Consistent with this, WNV-MAD78 replicates efficiently in neurons when introduced directly into the brain by an intracranial route (15). Experiments are under way to determine whether glycosylation contributes to WNV replication and/or spread in astrocytes.
We hypothesize that the constrained replication of WNV-MAD78 in astrocytes has several effects on WNV-mediated neuropathology. First, the restricted replication of WNV-MAD78 in astrocytes may minimize the initial amplification of virus and reduce the rate of spread within the CNS, thus allowing more time for the host innate and adaptive immune responses to clear the virus prior to widespread infection of highly susceptible neurons. Second, as with other disease states, WNV infection induces astrocytes to release neurotoxic factors that exacerbate neuropathology (41). Therefore, suppression of viral replication within astrocytes may reduce the extent of bystander cell death of uninfected neurons. The combined effects of reduced replication in astrocytes and decreased production of neurotoxic molecules is consistent with reports that mortality is delayed and reduced in mice infected with WNV-MAD78 via intracranial inoculation (15).
Studies with Icam1−/−, Mmp9−/−, or Drak2−/− mice suggest that WNV undergoes a second round of entry and dissemination within the CNS as a result of recruitment of infected leukocytes and/or perturbation of the BBB (49–51). There is circumstantial evidence that astrocytes contribute to the second wave of WNV neuroinvasion through the upregulation of MMPs, which disrupt the BBB, and proinflammatory cytokines, which recruit infected leukocytes (5, 41, 52, 53). Indeed, propagation within astrocytes, neurons, and glial cells prior to the breakdown of the BBB is believed to be a common strategy of neuroinvasive viruses, including tick-borne encephalitis virus and HIV, to enhance dissemination within the CNS (54, 55). Therefore, the inability of WNV-MAD78 to replicate in astrocytes may limit the capacity of the virus to undergo a second round of entry into the CNS (32, 33). Thus, astrocytes may play a central role in both the initial dissemination of WNV to the CNS as well as secondary waves of spread, which are likely to exacerbate WNV-mediated neuropathology.
Overall, our data suggest that WNV-MAD78 is capable of invading the CNS. However, the inability to amplify and spread within astrocytes may block WNV-MAD78 dissemination to neurons. Furthermore, our findings suggest that astrocytes play an essential role in initiating and regulating WNV infection in the CNS and may act as a critical determinant of differential WNV neuropathology. Determining which astrocyte factors limit WNV infection may ultimately promote the development of therapies that regulate neuronal injury after the onset of WNV infection.
ACKNOWLEDGMENTS
We thank Jonathan Dinman for critical evaluation of the manuscript and Kenneth Class (flow cytometry facility at the University of Maryland) for assistance with the acquisition and analysis of flow cytometry data.
This work was supported by NIH grant AI083397 (B.L.F.). Katherine L. Hussmann is supported by graduate research fellowship support from the NSF (NSF GFRP).
Footnotes
Published ahead of print 26 December 2012
REFERENCES
- 1. Papa A, Bakonyi T, Xanthopoulou K, Vazquez A, Tenorio A, Nowotny N. 2011. Genetic characterization of West Nile virus lineage 2, Greece, 2010. Emerg. Infect. Dis. 17:920–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papa A, Danis K, Baka A, Bakas A, Dougas G, Lytras T, Theocharopoulos G, Chrysagis D, Vassiliadou E, Kamaria F, Liona A, Mellou K, Saroglou G, Panagiotopoulos T. 2010. Ongoing outbreak of West Nile virus infections in humans in Greece, July-August 2010. Euro Surveill. 15:19644 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19644 [DOI] [PubMed] [Google Scholar]
- 3. Abbott NJ. 2002. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 200:629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim KS. 2008. Mechanisms of microbial traversal of the blood-brain barrier. Nat. Rev. Microbiol. 6:625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Verma S, Kumar M, Gurjav U, Lum S, Nerurkar VR. 2010. Reversal of West Nile virus-induced blood-brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology 397:130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gralinski LE, Ashley SL, Dixon SD, Spindler KR. 2009. Mouse adenovirus type 1-induced breakdown of the blood-brain barrier. J. Virol. 83:9398–9410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldstein GW. 1988. Endothelial cell-astrocyte interactions. A cellular model of the blood-brain barrier. Ann. N. Y. Acad. Sci. 529:31–39 [DOI] [PubMed] [Google Scholar]
- 8. Risau W, Wolburg H. 1990. Development of the blood-brain barrier. Trends Neurosci. 13:174–178 [DOI] [PubMed] [Google Scholar]
- 9. Risau W. 1991. Induction of blood-brain barrier endothelial cell differentiation. Ann. N. Y. Acad. Sci. 633:405–419 [DOI] [PubMed] [Google Scholar]
- 10. Hayashi Y, Nomura M, Yamagishi S, Harada S, Yamashita J, Yamamoto H. 1997. Induction of various blood-brain barrier properties in non-neural endothelial cells by close apposition to cocultured astrocytes. Glia 19:13–26 [PubMed] [Google Scholar]
- 11. Iadecola C. 2004. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat. Rev. Neurosci. 5:347–360 [DOI] [PubMed] [Google Scholar]
- 12. Beasley DW, Li L, Suderman MT, Barrett AD. 2002. Mouse neuroinvasive phenotype of West Nile virus strains varies depending upon virus genotype. Virology 296:17–23 [DOI] [PubMed] [Google Scholar]
- 13. Lustig S, Danenberg HD, Kafri Y, Kobiler D, Ben-Nathan D. 1992. Viral neuroinvasion and encephalitis induced by lipopolysaccharide and its mediators. J. Exp. Med. 176:707–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keller BC, Fredericksen BL, Samuel MA, Mock RE, Mason PW, Diamond MS, Gale M., Jr 2006. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J. Virol. 80:9424–9434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shrestha B, Zhang B, Purtha WE, Klein RS, Diamond MS. 2008. Tumor necrosis factor alpha protects against lethal West Nile virus infection by promoting trafficking of mononuclear leukocytes into the central nervous system. J. Virol. 82:8956–8964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stins MF, Badger J, Sik Kim K. 2001. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb. Pathog. 30:19–28 [DOI] [PubMed] [Google Scholar]
- 17. Samuel MA, Wang H, Siddharthan V, Morrey JD, Diamond MS. 2007. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl. Acad. Sci. U. S. A. 104:17140–17145 doi:10.1073/pnas.0705837104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. 2005. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 79:11457–11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi PY, Tilgner M, Lo MK, Kent KA, Bernard KA. 2002. Infectious cDNA clone of the epidemic West Nile virus from New York City. J. Virol. 76:5847–5856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, Luo H, Nakatsuka A, Nerurkar VR. 2009. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: transmigration across the in vitro blood-brain barrier. Virology 385:425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Diamond MS, Shrestha B, Mehlhop E, Sitati E, Engle M. 2003. Innate and adaptive immune responses determine protection against disseminated infection by West Nile encephalitis virus. Viral Immunol. 16:259–278 [DOI] [PubMed] [Google Scholar]
- 22. Xie Y, Kim KJ, Kim KS. 2004. Current concepts on Escherichia coli K1 translocation of the blood-brain barrier. FEMS Immunol. Med. Microbiol. 42:271–279 [DOI] [PubMed] [Google Scholar]
- 23. Stins MF, Gilles F, Kim KS. 1997. Selective expression of adhesion molecules on human brain microvascular endothelial cells. J. Neuroimmunol. 76:81–90 [DOI] [PubMed] [Google Scholar]
- 24. Huang SH, Jong AY. 2001. Cellular mechanisms of microbial proteins contributing to invasion of the blood-brain barrier. Cell Microbiol. 3:277–287 [DOI] [PubMed] [Google Scholar]
- 25. Grab DJ, Perides G, Dumler JS, Kim KJ, Park J, Kim YV, Nikolskaia O, Choi KS, Stins MF, Kim KS. 2005. Borrelia burgdorferi, host-derived proteases, and the blood-brain barrier. Infect. Immun. 73:1014–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chapagain ML, Verma S, Mercier F, Yanagihara R, Nerurkar VR. 2007. Polyomavirus JC infects human brain microvascular endothelial cells independent of serotonin receptor 2A. Virology 364:55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hasebe R, Suzuki T, Makino Y, Igarashi M, Yamanouchi S, Maeda A, Horiuchi M, Sawa H, Kimura T. 2010. Transcellular transport of West Nile virus-like particles across human endothelial cells depends on residues 156 and 159 of envelope protein. BMC Microbiol. 10:165 doi:10.1186/1471-2180-10-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Isaacs A, Lindenmann J. 1957. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 147:258–267 [PubMed] [Google Scholar]
- 29. Isaacs A, Lindenmann J, Valentine RC. 1957. Virus interference. II. Some properties of interferon. Proc. R. Soc. Lond. B Biol. Sci. 147:268–273 [PubMed] [Google Scholar]
- 30. Perry AK, Chen G, Zheng D, Tang H, Cheng G. 2005. The host type I interferon response to viral and bacterial infections. Cell Res. 15:407–422 [DOI] [PubMed] [Google Scholar]
- 31. Diamond MS. 2009. Virus and host determinants of West Nile virus pathogenesis. PLoS Pathog. 5:e1000452 doi:10.1371/journal.ppat.1000452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bai F, Kong KF, Dai J, Qian F, Zhang L, Brown CR, Fikrig E, Montgomery RR. 2010. A paradoxical role for neutrophils in the pathogenesis of West Nile virus. J. Infect. Dis. 202:1804–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang P, Bai F, Zenewicz LA, Dai J, Gate D, Cheng G, Yang L, Qian F, Yuan X, Montgomery RR, Flavell RA, Town T, Fikrig E. 2012. IL-22 signaling contributes to West Nile encephalitis pathogenesis. PLoS One 7:e44153 doi:10.1371/journal.pone.0044153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. 2004. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 10:1366–1373 [DOI] [PubMed] [Google Scholar]
- 35. Samuel MA, Morrey JD, Diamond MS. 2007. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J. Virol. 81:2614–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morrey JD, Olsen AL, Siddharthan V, Motter NE, Wang H, Taro BS, Chen D, Ruffner D, Hall JO. 2008. Increased blood-brain barrier permeability is not a primary determinant for lethality of West Nile virus infection in rodents. J. Gen. Virol. 89:467–473 [DOI] [PubMed] [Google Scholar]
- 37. Getts DR, Terry RL, Getts MT, Muller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. 2008. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J. Exp. Med. 205:2319–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van Sorge NM, Quach D, Gurney MA, Sullam PM, Nizet V, Doran KS. 2009. The group B streptococcal serine-rich repeat 1 glycoprotein mediates penetration of the blood-brain barrier. J. Infect. Dis. 199:1479–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Soilu-Hanninen M, Eralinna JP, Hukkanen V, Roytta M, Salmi AA, Salonen R. 1994. Semliki Forest virus infects mouse brain endothelial cells and causes blood-brain barrier damage. J. Virol. 68:6291–6298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fazakerley JK. 2002. Pathogenesis of Semliki Forest virus encephalitis. J. Neurovirol. 8(Suppl. 2):66–74 [DOI] [PubMed] [Google Scholar]
- 41. van Marle G, Antony J, Ostermann H, Dunham C, Hunt T, Halliday W, Maingat F, Urbanowski MD, Hobman T, Peeling J, Power C. 2007. West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J. Virol. 81:10933–10949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Daffis S, Suthar MS, Szretter KJ, Gale M, Jr, Diamond MS. 2009. Induction of IFN-beta and the innate antiviral response in myeloid cells occurs through an IPS-1-dependent signal that does not require IRF-3 and IRF-7. PLoS Pathog. 5:e1000607 doi:10.1371/journal.ppat.1000607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M., Jr 2008. Establishment and maintenance of the innate antiviral response to West Nile virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 82:609–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lazear HM, Pinto AK, Vogt MR, Gale M, Jr, Diamond MS. 2011. Beta interferon controls West Nile virus infection and pathogenesis in mice. J. Virol. 85:7186–7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Samuel MA, Diamond MS. 2006. Pathogenesis of West Nile virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J. Virol. 80:9349–9360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, Diamond MS, Gale M., Jr 2010. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 6:e1000757 doi:10.1371/journal.ppat.1000757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Botha EM, Markotter W, Wolfaardt M, Paweska JT, Swanepoel R, Palacios G, Nel LH, Venter M. 2008. Genetic determinants of virulence in pathogenic lineage 2 West Nile virus strains. Emerg. Infect. Dis. 14:222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Beasley DW, Whiteman MC, Zhang S, Huang CY, Schneider BS, Smith DR, Gromowski GD, Higgs S, Kinney RM, Barrett AD. 2005. Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 West Nile virus strains. J. Virol. 79:8339–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dai J, Wang P, Bai F, Town T, Fikrig E. 2008. Icam-1 participates in the entry of West Nile virus into the central nervous system. J. Virol. 82:4164–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang S, Welte T, McGargill M, Town T, Thompson J, Anderson JF, Flavell RA, Fikrig E, Hedrick SM, Wang T. 2008. Drak2 contributes to West Nile virus entry into the brain and lethal encephalitis. J. Immunol. 181:2084–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang P, Dai J, Bai F, Kong KF, Wong SJ, Montgomery RR, Madri JA, Fikrig E. 2008. Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain. J. Virol. 82:8978–8985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Verma S, Kumar M, Nerurkar VR. 2011. Cyclooxygenase-2 inhibitor blocks the production of West Nile virus-induced neuroinflammatory markers in astrocytes. J. Gen. Virol. 92:507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kumar M, Verma S, Nerurkar VR. 2010. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J. Neuroinflammation 7:73 doi:10.1186/1742-2094-7-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Eugenin EA, Clements JE, Zink MC, Berman JW. 2011. Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J. Neurosci. 31:9456–9465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ruzek D, Salat J, Singh SK, Kopecky J. 2011. Breakdown of the blood-brain barrier during tick-borne encephalitis in mice is not dependent on CD8+ T-cells. PLoS One 6:e20472 doi:10.1371/journal.pone.0020472 [DOI] [PMC free article] [PubMed] [Google Scholar]