

Abstract
Six novel insect-specific viruses, isolated from mosquitoes and phlebotomine sand flies collected in Brazil, Peru, the United States, Ivory Coast, Israel, and Indonesia, are described. Their genomes consist of single-stranded, positive-sense RNAs with poly(A) tails. By electron microscopy, the virions appear as spherical particles with diameters of ∼45 to 55 nm. Based on their genome organization and phylogenetic relationship, the six viruses, designated Negev, Ngewotan, Piura, Loreto, Dezidougou, and Santana, appear to form a new taxon, tentatively designated Negevirus. Their closest but still distant relatives are citrus leposis virus C (CiLV-C) and viruses in the genus Cilevirus, which are mite-transmitted plant viruses. The negeviruses replicate rapidly and to high titer (up to 1010 PFU/ml) in mosquito cells, producing extensive cytopathic effect and plaques, but they do not appear to replicate in mammalian cells or mice. A discussion follows on their possible biological significance and effect on mosquito vector competence for arboviruses.
INTRODUCTION
During the past decade, a growing number of novel insect-specific viruses have been detected in naturally infected mosquitoes. The term “insect-specific” was initially used to describe viruses in the genus Flavivirus (Flaviviridae) that replicate in mosquito cells but not in vertebrate cells. Although the insect-specific flaviviruses share the same genome organization and numerous amino acid motifs with the vertebrate flaviviruses, they do not infect vertebrates or participate in the classical arthropod-vertebrate transmission cycle of arboviruses (1). Culex flavivirus (CxFV) and cell fusing agent virus (CFAV) are probably the best-known members of the insect-specific flavivirus group (2–4).
Recently, an increasing number of nonflaviviral RNA viruses (rhabdoviruses, bunyaviruses, alphaviruses, nidoviruses, and reoviruses) have been isolated from pools of field-collected mosquitoes, suggesting that these types of agents are quite common in mosquitoes in nature (5–10). Here, we describe a novel group of insect-specific viruses occurring in mosquitoes and phlebotomine sandflies which appear to represent a new virus taxon that is distantly related phylogenetically to citrus leprosis virus C (CiLV-C), genus Cilevirus, a mite-transmitted virus causing disease in citrus plants (11–13).
MATERIALS AND METHODS
Viruses. All viruses used in this study were obtained from the World Reference Center for Emerging Viruses and Arboviruses (WRCEVA) at the University of Texas Medical Branch. Some were isolated by the authors (R. B. Tesh and H. Guzman) during arbovirus field studies; the remainder were isolated by other investigators and sent to the WRCEVA for identification and further characterization. The proposed names, GenBank accession numbers of the obtained sequences, original sources, and geographic origins of the 10 viruses included in this study are listed below and in Table 1.
Table 1.
Origin of the 10 viruses described in this study
| Proposed name | Strain designation | Host species | Collection locality | Collection date |
|---|---|---|---|---|
| Negev | EO239 | Anopheles coustani | Negev, Israel | 1983 |
| Negev | M33056 | Culex quinquefasciatus | Harris County, TX, USA | 2008 |
| Negev | M30957 | Culex coronator | Harris County, TX, USA | 2008 |
| Piura | P60 | Culex sp. | Piura, Peru | 1996 |
| Loreto | 3940-83 | Anopheles albimanus | Lima, Peru | 1983 |
| Loreto | Pe AR 2617/77 | Lutzomyia sp. | Iquitos, Loreto, Peru | 1977 |
| Loreto | Pe AR 2612/77 | Culex sp. | Iquitos, Loreto, Peru | 1977 |
| Dezidougou | ArA 20086 | Aedes aegypti | Dezidougou, Côte d'Ivoire | 1987 |
| Santana | BeAR 517449 | Culex sp. | Santana, Amapa, Brazil | 1992 |
| Ngewotan | JKT 9982 | Culex vishnui | Wotan, Java, Indonesia | 1981 |
Negev virus (NEGV) strain EO239, the prototype strain of Negev virus, was initially isolated by Joseph Peleg, Hebrew University, Jerusalem, from a pool of Anopheles coustani mosquitoes collected in the Negev Desert, Israel, in 1983.
Negev virus strains M30957 and M33056 were isolated at the WRCEVA from pools of Culex coronator and Cx. quinquefasciatus mosquitoes, respectively, collected in Houston, Harris County, TX, in 2008.
Piura virus (PIUV; strain P60) was isolated at the Naval Medical Research Unit 6, Lima, Peru, from a pool of Culex species mosquitoes collected in Piura, Peru, in 1996. The sample was provided by Douglas M. Watts.
Loreto virus (LORV) strain 3940-83 was isolated from a pool of Anopheles albimanus mosquitoes collected in Lima, Peru, in 1983.
Loreto virus strain PeAR 2612/77 was isolated from a pool of Culex sp. mosquitoes collected in Iquitos, Loreto, Peru, in 1977.
Loreto virus strain 2617/77 was isolated from a pool of phlebotomine sandflies (Lutzomyia spp.) collected in Iquitos, Peru, in 1977. James G. Olson, Centers for Disease Control and Prevention, Atlanta, Georgia, provided the three Loreto virus strains.
Dezidougou (DEZV; ArA 20086) was isolated at the Institute Pasteur, Dakar, Senegal, from a pool of Aedes aegypti collected in Dezidougou, Ivory Coast, in 1987. The sample was provided by Jean-Pierre Digoutte, Institute Pasteur, Dakar, Senegal.
Santana virus (SANV; BeAR 517449) was isolated at the WRCEVA from a pool of Culex species mosquitoes originally collected in Santana, Amapa, Brazil, in 1992. The mosquito pool was provided by Amelia P. A. Travassos da Rosa, Evandro Chagas Institute, Belem, Para, Brazil.
Ngewotan virus (NWTV; JKT 9982) was isolated by James D. Converse, Naval Medical Research 2, Jakarta, Indonesia, from a pool of Culex vishnui collected at Wotan, Central Java, Indonesia, in 1981.
All viruses were initially isolated from triturated pools of field-collected mosquitoes collected during arbovirus surveillance studies. The mosquito homogenates were inoculated into cultures of the C6/36 line of Aedes albopictus cells (14) or the AP-61 line of Aedes pseudoscutellaris (15). After inoculation, cultures were maintained in incubators at a constant temperature of 28°C and observed at regular intervals for evidence of viral cytopathic effect (CPE) (16).
Before sequencing, all virus stocks were grown in cultures of the C6/36 clone of Ae. albopictus cells (14) obtained from the American Type Culture Collection (ATCC), Manassas, VA. Infection was characterized by detachment of cells and cell lysis. Plaque assays were performed using the C7/10 (LTC-7) clone of Ae. albopictus cells (17).
Cell lines utilized for virus replication kinetics.
African green monkey kidney (Vero), baby hamster kidney (BHK-21), human embryonic kidney (HEK293), Drosophila melanogaster, and Ae. albopictus (C6/36 and C7/10) cell lines were obtained from the ATCC. Anopheles albimanus, An. gambiae, Culex tarsalis, and Phlebotomus papatasi cells were obtained from the WRCEVA (18–20). Monolayers of Vero, BHK-21, and HEK293 cells were grown at 37°C in Dulbecco's minimal essential medium (DMEM) (4.5 g/liter d-glucose) with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin-streptomycin. C6/36, C7/10, An. albimanus, An. gambiae, and Cx. tarsalis were grown at 28°C in Dulbecco's minimal essential medium (DMEM) (4.5 g/liter d-glucose) with 10% heat-inactivated FBS, 1% penicillin-streptomycin, 1% sodium pyruvate, and 1% tryptose phosphate broth (TPB). Phlebotomus papatasi cells were maintained in Schneider's medium (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum and penicillin (100 U/ml)-streptomycin (100 μg/ml). For replication kinetic studies, vertebrate and invertebrate cell lines were propagated in 6-well plates and infected at a multiplicity of infection (MOI) of 10 in duplicate. Plates with vertebrate cells were incubated for 1 h with periodic gentle rocking at 37°C, whereas plates containing invertebrate cells were incubated at 28°C. After three washes with phosphate-buffered saline (PBS) to remove unabsorbed virus, 2 ml of complete cell media was added to each well, and plates were incubated at 28 or 37°C for the invertebrate or vertebrate cell lines, respectively. Virus from individual wells was harvested at designated time points for 3 days postinfection (p.i.) and clarified by low-speed centrifugation, and the virus titer was determined by plaque assay in C7/10 cells. Virus yield at each time point was recorded as PFU/cell, represented as the ratio of the total amount of virus present in the sample to the number of cells originally infected.
Virus purification.
Virus purification was performed as previously described (21). Virus was amplified on C7/10 cells at an MOI of 0.5, harvested 48 h postinfection (hpi), and clarified by centrifugation at 2,000 × g for 10 min. Virus was precipitated overnight at 4°C by adding polyethylene glycol and NaCl to 7 and 2.3% (wt/vol) concentrations, respectively. Virus was pelleted by centrifugation at 4,000 × g for 30 min at 4°C, and the precipitate was then resuspended in TEN buffer (0.05 M Tris-HCl, pH 7.4, 0.1 M NaCl, 0.001 M EDTA) and loaded onto a 20 to 70% continuous sucrose (wt/vol) gradient in TEN buffer and centrifuged at 270,000 × g for 1 h. Following centrifugation, the visible virus band was harvested using a Pasteur pipette and centrifuged 4 times through an Amicon Ultra-4 100-kDa-cutoff filter (Millipore) and resuspended in 1 ml of TEN buffer.
Transmission electron microscopy.
For ultrastructural analysis, infected C6/36 cells were fixed for at least 1 h in a mixture of 2.5% formaldehyde prepared from paraformaldehyde powder and 0.1% glutaraldehyde in 0.05 M cacodylate buffer (pH 7.3), to which 0.03% picric acid and 0.03% CaCl2 were added. The monolayers were washed in 0.1 M cacodylate buffer, and cells were scraped off and processed further as a pellet. The pellets were postfixed in 1% OsO4 in 0.1 M cacodylate buffer (pH 7.3) for 1 h, washed with distilled water, and en bloc stained with 2% aqueous uranyl acetate for 20 min at 60°C. The pellets were dehydrated in ethanol, processed through propylene oxide, and embedded in Poly/Bed 812 (Polysciences, Warrington, PA). Ultrathin sections were cut on a Leica EM UC7 ultramicrotome (Leica Microsystems, Buffalo Grove, IL), stained with lead citrate, and examined in a Philips 201 transmission electron microscope at 60 kV.
Purified virus particles were also allowed to adhere to a Formvar carbon-coated copper grid for 10 min, negatively stained with either 2% aqueous uranyl acetate for 30 s or 2% phosphotungstic acid with pH adjusted to 6.8 with 1N KOH (30 s), and then examined in the electron microscope.
Plaque assay.
Virus titrations were performed on confluent C7/10 cell monolayers in 6-well plates. Duplicate wells were inoculated with 0.1-ml aliquots of serial 10-fold dilutions of virus in growth medium. An additional 0.4 ml of growth medium was added to each well to prevent cell desiccation, and virus was adsorbed for 2 h. Following incubation, the virus inoculum was removed by aspiration, and cell monolayers were overlaid with 3 ml of a medium consisting of a 1:1 mixture of 2% tragacanth suspension and 2× minimal essential medium (MEM) with 5% FBS, 2% tryptose phosphate broth solution, and 2% of a 100× solution of penicillin and streptomycin. Cells were incubated at 28°C in 5% CO2 for 2 days to allow plaque development, and then the overlay was removed and monolayers were fixed with 3 ml of 10% formaldehyde in PBS for 30 min. Cells were subsequently stained with 2% crystal violet in 30% methanol for 5 min at room temperature; excess stain was removed under running water, and plaques were counted and recorded as the number of plaques per ml of inoculum.
Virus stability in solvents.
To examine whether these viruses contain a glycoprotein-containing envelope, we assessed the virus sensitivity to ether. Negev virus strain EO239 was selected as our model strain.
Cold diethyl ether was added in a ratio of 1:2 to 2.0 ml of spent medium from a culture of C6/36 cells infected with Negev virus strain EO239. The mixture was shaken vigorously and placed overnight in a refrigerator at ∼4°C. A control sample of the infected cell culture medium without ether was held in the same manner. After 20 h, the ether was removed in a separatory funnel and by evaporation in a fume hood. The two samples of infected medium were then titrated by plaque assay in C7/10 cells. A loss of ≥1.5 log in virus titer in the ether-treated sample was considered evidence of solvent (ether) sensitivity (22).
Experimental infection of mosquitoes with Negev virus.
Laboratory colonies of Ae. aegypti and Ae. albopictus were used for experimental infections. The progenitors of both colonies were originally collected in Thailand and had been maintained in our insectary for about 10 generations. Six to 10 days after emergence, cohorts of 100 females of each species were allowed to feed on artificial blood meals containing three different concentrations (5, 7, and 9 log10 PFU/ml) of Negev virus strain EO239 made by serially diluting a virus stock of known titer in defibrinated sheep blood (Colorado Serum Company, Denver, CO) in MEM. Artificial blood meals were placed in vials covered with mouse skin and were warmed to 37°C using a Hemotek feeder (Discovery Workshops, Accrington, United Kingdom). Mosquitoes were allowed to feed for ∼1 h and then were cold anesthetized on ice for sorting. Engorged females were removed and placed in cages and maintained with 10% sucrose at 28°C with a relative humidity of ∼70%. Fourteen days after feeding, mosquitoes were cold anesthetized and the legs and wings were removed. Mosquito bodies and legs/wings were put in individual tubes containing 500 μl MEM with 10% FBS and a stainless steel bead for trituration. Each body and leg/wing sample was homogenized for 4 min using a Mixer Mill 300 (Retsch, Haan, Germany). Samples were centrifuged for 10 min at 5,000 rpm, and 100 μl of each sample supernatant was inoculated into individual 24-well plates containing C7/10 cells. Cultures were maintained with 2 ml of medium at 28°C and 5% CO2. CPE observed in C7/10 cultures was used as a surrogate indicator for the presence of virus.
Quantitative real-time reverse transcription (qRT)-PCR system.
To confirm the inability of NEGV to replicate in vertebrate cells, we developed a real-time RT-PCR assay. Primer Express 3.0 was used on the NEGV sequence (strain EO239; GenBank accession number JQ675605; Table 2) to design specific primers (NEGV_2+, 5′ TGTTCTCTGGTGATGACTCACTCC 3′ [nucleotide {nt} positions 6882 to 6905]; NEGV_2−, 5′ TGACGACGAGCAAGAACTTTGAG 3′ [nucleotide positions 7006 to 7028]) and TaqMan-MGB fluorescent probe (6-carboxyfluorescein-CAGCATTTCGGACTCAA-MGB-NFQ, nucleotide positions 6941 to 6957). Corresponding DNA standards ranging from 1 to 109 copies/μl were obtained to construct the standard curve. The real-time PCR assays were performed using the No AmpErase UNG kit (Applied Biosystem). First-strand cDNA was synthesized using random hexamers (Roche) and SuperScript III reverse transcriptase (Invitrogen). Triplicate reaction mixtures were set up for each sample, and 5 μl of cDNA, 0.2 μM probe, and 0.50 μl of each primer were used. The concentrations of primers and the probe were optimized. Real-time PCR was performed in a 96-well plate using the ABI 7900 HT sequence detection system (Applied Biosystems) under the following conditions: 95°C for 30 s, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The data were collected at the end of the elongation step.
Table 2.
Summary of genome organization
| Virus and strain | GenBank assession no. | Genome length (bp) | Size (nt) of: |
Poly(A) tail | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 5′-UTR | ORF1 | Intergenic region | ORF2 | Intergenic region | ORF3 | 3′-UTR | ||||
| Negev | ||||||||||
| EO329 | JQ675605 | 9536 | 232 | 7107 | 33 | 1203 | 50 | 627 | 288 | p(A)33 |
| M30957 | JQ675608 | 9538 | 234 | 7107 | 33 | 1203 | 50 | 627 | 288 | p(A)34 |
| M33056 | JQ675609 | 9532 | 232 | 7107 | 33 | 1203 | 50 | 627 | 284 | p(A)30 |
| Ngewotan | ||||||||||
| JKT9982 | JQ686833 | 9240 | 227 | 6849 | 33 | 1197 | 51 | 624 | 263 | p(A)13 |
| Piura | ||||||||||
| P60 | JQ675607 | 10059 | 730 | 7011 | 44 | 1203 | 142 | 618 | 315 | p(A)31 |
| Loreto | ||||||||||
| 3940-83 | JQ675610 | 9207 | 285 | 7014 | 35 | 1206 | 21 | 642 | 142 | p(A)31 |
| PeAr2612/77 | JQ675611 | 9011 | 151 | 6705 | 35 | 1200 | 26 | 642 | 122 | p(A)32 |
| PeAr2617/77 | JQ675612 | 9011 | 285 | 6705 | 35 | 1206 | 21 | 642 | 121 | p(A)52 |
| Dezidougou | ||||||||||
| ArA20086 | JQ675604 | 9290 | 72 | 6741 | 30 | 1284 | 110 | 615 | 442 | p(A)32 |
| Santana | ||||||||||
| JQ675606 | 9266 | 224 | 6774 | 14 | 1209 | 175 | 699 | 174 | p(A)27 | |
Genome sequencing.
All virus sequences were obtained using 454 pyrosequencing (Roche Life Sciences, Branford, CT), except for the Dezidougou genomic sequences, which were obtained by Illumina sequencing (Illumina, San Diego, CA).
Pyrosequencing.
RNA was extracted from virus stocks using TRIzol LS (Invitrogen, Carlsbad, CA) and treated with DNase I (DNA-Free; Ambion, Austin, TX). cDNA was generated using the Superscript II system (Invitrogen) employing random hexamers linked to an arbitrary 17-mer primer sequence (23). Resulting cDNA was treated with RNase H and then randomly amplified by PCR with a 9:1 mixture of primer corresponding to the 17-mer sequence and the random hexamer-linked 17-mer primer (23). Products greater than 70 bp were selected by column chromatography (MinElute; Qiagen, Hilden, Germany) and ligated to specific adapters for sequencing on the 454 Genome Sequencer FLX (454 Life Sciences, Branford, CT) without fragmentation (24–26). Software programs accessible through the analysis applications at the GreenePortal website (http://tako.cpmc.columbia.edu/Tools/) were used for removal of primer sequences, redundancy filtering, and sequence assembly. No more than 20% of any of the genomes identified here were identified from 454 data by using the BLAST algorithm suite. Although after reconstruction of the genome we recognized additional reads that were part of the virus genomes, they did not form part of the initial scaffold, since they were not recognized as viral by the algorithms utilized. Sanger sequencing was used to fill in gaps as large as 3 kb between next-generation sequencing (NGS) contigs. These sequence gaps were completed by RT-PCR amplification using primers based on pyrosequencing data. Amplification products were size fractionated on 1% agarose gels, purified (MiniElute; Qiagen, Hilden, Germany), and directly sequenced in both directions with ABI PRISM BigDye Terminator 1.1 cycle sequencing kits on ABI PRISM 3700 DNA analyzers (Perkin-Elmer Applied Biosystems, Foster City, CA). The terminal sequences for each virus were amplified using the Clontech SMARTer RACE kit (Clontech, Mountain View, CA). Genome sequences were verified by Sanger dideoxy sequencing using primers designed from the draft sequence to create products of 1,000 bp with 500-bp overlaps.
Illumina sequencing.
Viral RNA (0.1 to 0.2 μg) was fragmented by incubation at 94°C for 8 min in 19.5 μl of fragmentation buffer (Illumina 15016648). First- and second-strand synthesis, adapter ligation, and amplification of the library were performed using the Illumina TruSeq RNA sample preparation kit under conditions prescribed by the manufacturer (Illumina, San Diego, CA). Samples were tracked using the index tags incorporated into the adapters as defined by the manufacturer. Cluster formation of the library DNA templates was performed using the TruSeq PE Cluster kit (v3; Illumina, San Diego, CA) and the Illumina cBot workstation using conditions recommended by the manufacturer. Paired-end 50-base sequencing by synthesis was performed using a TruSeq SBS kit (v3; Illumina, San Diego, CA) on an Illumina HiSeq 1000 using protocols defined by the manufacturer. Cluster density per lane was 645 to 980 k/mm2, and postfilter reads ranged from 148 to 178 million per lane. Base call conversion to sequence reads was performed using CASAVA-1.8.2. Virus sequences were edited and assembled using the SeqMan and NextGen modules of the DNAStar Lasergene 7 program (Bioinformatics Pioneer DNAStar, Inc., Madison, WI). In certain cases, prefiltering of reads to remove host sequence enhanced the assembly process.
RNA analysis.
C7/10 cell monolayers were infected with Negev and Piura viruses at an MOI of 10. [3H]uridine (20 μCi/ml) was added 1 or 24 hpi, respectively, and incubated for an additional 24 h. Supernatants were harvested and virus was purified via rate-zonal centrifugation (see below). Viral RNA was isolated by TRIzol LS (Invitrogen, Grand Island, NY), denatured with glyoxal in dimethyl sulfoxide, and analyzed by agarose gel electrophoresis using previously described conditions (27).
Genomic analysis.
The genome of Negev virus strain EO329 was used to determine protein domains. The genome was translated into proteins and then submitted to the NCBI conserved domain prediction tool http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi. The nucleotide and protein identities for the open reading frames (ORFs) of all of the 10 viruses generated were determined in EnzymeX (EnzymeX, Aalsmeer, Netherlands).
Phylogenetic analysis.
Completed genomes of the 10 sequences were first aligned using translated protein sequences before being toggled back to nucleotides while maintaining the alignment. To determine areas of alignment that had sufficient confidence to determine phylogenetic relationships, the alignment was run using the GUIDANCE software (28, 29). Areas with sufficient confidence were selected for further phylogenetic analysis. The phylogenetic analyses were undertaken using PAUP* version 4.0, 10b (30). The optimal evolutionary model for each data set was estimated from 56 models implemented using Modeltest version 3.06 (31). An optimal maximum-likelihood (ML) tree was then estimated using the appropriate model and a heuristic search with tree-bisection-reconstruction branch swapping and 10 replicates, estimating variable parameters from the data where necessary. Bootstrap replicates were calculated for each data set under the same models mentioned above. Bayesian analysis was undertaken using MrBayes v3.1 (32, 33), and data sets were run for 500,000 generations until they reached congruence. The models used were HKY+G and HKY+I+G.
Nucleotide sequence accession numbers.
Virus genome sequences obtained in this study are included in Table 1. The genomic sequences of CiLV-C, which are available in GenBank (NC_008169, DQ388512, and DQ157466), were included in the phylogenetic analyses.
RESULTS
Genome organization and analysis.
The size of the positive-sense, single-strand genomes of the 10 viruses identified ranged in size from approximately 9 to 10 kb (Table 2). Three open reading frames (ORFs) are flanked by untranslated regions (UTRs) at the 5′ and 3′ ends, while each ORF is separated by short intergenic regions. Using Negev virus strain EO329 as the prototype, we determined that ORF1 and ORF2 encode the nonstructural proteins and structural proteins, respectively. No function was found for ORF3 by utilizing ORF prediction programs (EnzymeX, Aalsmeer, Netherlands).
5′- and 3′-UTRs.
The sequences of the 5′- and 3′-UTRs vary in length among these viruses, ranging in length from 72 to 730 and 121 to 442 nt, respectively. For all viruses a polyadenylate tail is present in the distal sites of each genome, from 13 to 52 nt in length (Table 2).
ORFs.
A large ORF was found at nt 233 to 7339. Two small ORFs were identified at the 3′ end of the genome (Fig. 1A). The large ORF contains putative protein domains that correspond to nonstructural proteins (Fig. 1B). Using the protein domain prediction software in the BLAST suite of programs, we determined four functional domains: (i) a methyltransferase domain at nt 522 to 1386; (ii) an RNA ribosomal methyltransferase domain at nt 2511 to 3072; (iii) a helicase domain at nt 4182 to 4908; and (iv) an RNA-dependent RNA polymerase domain (RdRp) at nt 5802 to 6927 (Fig. 1B). We could not identify regions similar to known proteins in the GenBank database in the two smaller ORFs. However, by using the PsiPRED server (http://bioinf.cs.ucl.ac.uk/psipred//?program=psipred) (34), ORF2 was predicted to contain three transmembrane regions according to the algorithm prediction software MEMSAT-SVM (35–37), as depicted in Fig. 2A. The presence of these transmembrane regions suggests that this protein is contained within a viral envelope and is a glycoprotein. To confirm that ORF2 is a glycoprotein, Negev virus EO239 was subjected to treatment with ether, which led to a 3-log10 decrease in titer (untreated control at 3.7 × 108 50% tissue culture infectious doses [TCID50]/ml versus 2 × 105 TCID50/ml for the ether-treated virus).
Fig 1.
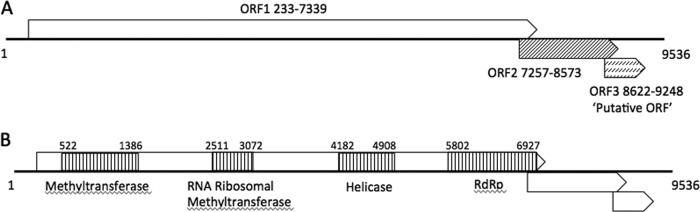
Genome organization and position of the open reading frames (A) and the conserved protein domains (B) for Negev virus strain EO239. All 5 identified viruses showed similar genome organization and protein domains.
Fig 2.

Predicted transmembrane domains and the orientation of ORF2 by MAMSAT-SVM for Negev virus (A), Piura virus (B), Loreto virus (C), Dezidougou virus (D), Santana virus (E), and Ngewotan virus (F).
Analysis of all ORF2s from Negev, Piura, Loreto, Dezidougou, Santana, and Ngewotan viruses showed that all contained transmembrane helices (Fig. 2A to F). However, the number and arrangement of these helices differed for all 6 viruses, suggesting a significant difference in the arrangement of the viral surface. For ORF3, no putative functions or domain homologies were identified. Short intergenic regions ranging from 14 to 44 nt and 21 to 175 nt long intersect the junctions of ORF1/ORF2 and ORF2/ORF3, respectively.
To confirm monosegmentation, we infected monolayers of C7/10 cells with Negev virus EO239 and Piura P60 viruses at an MOI of 10 in the presence of [3H]uridine (20 μCi/ml). Viral RNA was isolated from purified virus and analyzed by agarose gel electrophoresis. These results showed an abundant large genomic RNA species as well as the presence of several less abundant smaller RNA species (Fig. 3). Similar observations were obtained with Northern blot analysis (data not shown). There are three possible explanations for these results: first, that the bands represent nonspecific host RNA packaged into virions; second, that the genomes of these viruses are segmented; and third, that viral mRNA species may be packaged in the virions of these viruses, a possibility that has been shown for other arboviruses (38, 39). To investigate these possibilities further, a reverse genetic system was generated using EO239 as a model virus; the resulting data suggest that these viruses are nonsegmented (R. V. Gorchakov, F. Nasar, R. B. Tesh, and S. C. Weaver, unpublished data).
Fig 3.

Analysis of genomic RNA of NEGV and PIUV labeled with [3H]uridine in the presence of dactinomycin (ActD) for 12 h. Both viruses were purified via rate-zonal centrifugation. Viral RNA was analyzed by agarose gel electrophoresis. Lane 1, mock treatment; 2, NEGV; 3, PIUV.
Phylogenetic analysis.
Alignments of ORF1 were created for all of the virus sequences. As initial BLAST results had indicated that the nearest viral relative was the citrus leprosis C viruses (CiLV-C) (12), we aligned the ORF1 sequences of the 10 newly identified sequences to the three full-length RNA sequences of CiLV-C present in GenBank. The sequences were first aligned as proteins and then toggled back to nucleotides. These alignments were run through the GUIDANCE algorithm, which shows the level of confidence in the alignment, to ensure that regions of the genome exhibited no more evidence of homology than random assembly of protein codes would show. Two regions of the ORF1, nt 626 to 2908 and nt 4316 to 7309, exhibited confidence levels sufficient to perform further phylogenetic analysis. Phylogenetic trees generated under ML and Bayesian algorithms exhibited the same topology, thus only the ML trees are shown (Fig. 4 and 5, respectively). Six distinct viruses can be identified in these analyses: Negev, Loreto, Ngewotan, Piura, Dezidougou, and Santana viruses. As the regions used to generate the phylogeny were highly conserved, the relationships presented in the trees do not necessarily reflect the diversity among the viruses. Therefore, a full-length alignment of the ORF1 polyprotein was used to determine similarities in nucleotide and protein sequences of these viruses (Fig. 6). The three Negev viruses and the three Loreto viruses exhibited nucleotide and protein identities between 95.6 to 100% and 98.6 to 100%, respectively. However, among the viruses identified in this study (excluding CiLV-C), the nucleotide identity ranged from 33.2 to 70.6% and the protein identity ranged from 20 to 79.2%. The 3 strains of the CiLV-C viruses were nearly identical but showed little similarity to the newly identified viruses, with nucleotide identity ranging from 30.7 to 34.7% and protein identity ranging from 16.9 to 20.6%.
Fig 4.

Phylogenetic trees produced using maximum-likelihood methods of the 10 genomes determined in the study plus three genomes of CiCLV. The trees were rooted using the CiCLV viruses as an outgroup. The region of the genome corresponds to nt 626 to 2908 (Negev EO239), which corresponds to the helicase region of the genome. The model used was the TrN+G model with 1,000 bootstrap replications. Bootstrap replications are presented on the major branches.
Fig 5.

Phylogenetic trees produced using maximum-likelihood methods of the 10 genomes determined in the study plus three genomes of CiCLV. The trees were midpoint rooted. The region of the genome corresponds to nt 4316 to 7309 (Negev virus EO239), which corresponds with the RNA-dependent RNA polymerase of the genome. The model used was the GTR+G model with 1,000 bootstrap replications. Bootstrap replications are presented on the major branches.
Fig 6.

Cladistic tree showing the relationships between the viruses along with the nucleotide identity and the protein identity for ORF1 of the nine viruses and the RNA species one of CiCLV. Alignments were performed as proteins and then toggled back to nucleotide forms. The branch lengths of the tree do not reflect genetic distance but have the same topology as the trees shown in Fig. 3 and 4.
Ultrastructural characteristics.
The most prominent ultrastructural characteristic of infected C6/36 mosquito cells was expansion of perinuclear spaces, which became filled with vesicles or microtubules (Fig. 7A). These had a universal diameter of 20 nm. In some cells, the expansions were filled only with vesicles, while in others they were also filled with microtubules, so the vesicles appeared as cross sections of the microtubules. These microtubules were up to 160 nm long and in rare instances were even longer. In some cells, they formed paracrystalline arrays (Fig. 7B). Expanded perinuclear space filled with vesicles or tubules could occupy most of the cell volume, pushing the cytoplasm to the cell periphery as a thin rim.
Fig 7.

Transmission electron microscopy analysis of infected cells and purified suspensions. (A) Expanded perinuclear space (the arrow indicates its membrane) of an Ae. albopictus C6/36 cell infected with Negev (EO239) virus is filled with microtubules 20 nm in diameter and up to 160 nm long. Bar, 0.5 μm. (B) Portion of a tremendous perinuclear space-granular endoplasmic reticulum extension loaded with microtubules forming paracrystalline arrays in cross sections in a C6/36 cell infected with ArA 20086 virus. The arrow indicates a limiting membrane with ribosomes at the outer surface. Bar, 0.5 μm. (C) Cytopathic vacuole with spherules at its periphery (arrow) surrounded by microtubules in a perinuclear space of a C6/36 cell infected with ArA 20086 virus. Bar, 100 nm. (D) Negatively stained (2% uranyl acetate) suspension of purified suspension of P60 virus contains particles mostly ∼50 nm in diameter. Bar, 100 nm. (E) Expanded perinuclear space (arrow) filled with microtubules and a cytoplasmic vacuole with spherules (arrowhead) in cytoplasm of a C6/36 cell infected with JKT-9982 virus. N, host cell nucleus. Bar, 0.5 μm.
The second peculiar feature of many of these viruses was the formation of cytoplasmic cytopathic vacuoles (CPVs), similar to those seen with alphaviruses, containing spherules ∼50 nm in diameter at the inner surface of their limiting membrane. In alphavirus-infected cells, CPVs are modified endosomes and lysosomes in which translation, transcription, and assembly of viral nucleocapsids occurs (40). Some CPVs reached a diameter of 1.4 μm and could be found in almost all viruses studied (Table 3) and sometimes were found among the microtubules of the expanded perinuclear space (Fig. 7C and E). In a negatively stained purified suspension of the Piura virus (P60), spherical particles with diameters of ∼45 and ∼55 nm were found (Fig. 7D).
Table 3.
Summary of some ultrastructural characteristics of the 10 negeviruses, as observed in infected mosquito (C6/36) cells
| Strain designation | Host (mosquito) species | Presence of CPVs | Expansion of perinuclear space |
|---|---|---|---|
| M30957 | Culex coronator | Yes | Yes |
| M33056 | Culex quinquefaciatus | Yes | Yes |
| EO239 | Anopheles coustani | Yes | Yes |
| P60 | Culex sp. | Yes | Yes |
| 3940-83 | Anopheles albimanus | Yes | Not seen |
| Pe AR 2617/77 | Lutzomyia sp. | Yes | Not seen |
| Pe AR 2612/77 | Culex sp. | Yes | Not seen |
| ArA 20086 | Aedes aegypti | Yes | Yes |
| BeAR 517449 | Culex sp. | Yes | Not seen |
| JKT 9982 | Culex vishnui | Yes | Yes |
Phenotypic characterization and host range.
Negev virus strain EO239-infected C6/36 and C7/10 cells and produced extensive CPE 12 hpi (Fig. 8A and B); however, no overt cytopathic effects were observed in the three vertebrate cell lines at either 37 or 28°C up to 6 days postinfection (data not shown). Negev virus EO239 formed 3- to 4-mm-size plaques on C7/10 cells at 36 hpi (Fig. 8C).
Fig 8.

Cytopathic effects of Negev virus (EO239) infection in C7/10 cells. (A) Mock-infected C7/10 monolayers observed with bright-field microscopy at 12 hpi; (B) NEGV-infected C7/10 cells at an MOI of 10 at 12 hpi observed with bright-field microscopy; and (C) representative plaques of Negev virus-infected C7/10 cells 36 hpi. Cells were fixed with 10% formalin and stained with crystal violet dye.
Representative vertebrate (African green monkey kidney [Vero], hamster kidney [BHK-21], and human embryonic kidney [HEK293]) as well as invertebrate (Ae. albopictus [C6/36 and C7/10], An. albimanus, An. gambiae, Cx. tarsalis, P. papatasi, and D. melanogaster) cell lines were used to determine the in vitro host range of Negev virus EO239. That stated, we acknowledge that cultured cells, particularly Vero cells and C6/36 cells, which are deficient in the interferon (41) and RNA interference responses (42, 43), respectively, are imperfect models of host range. The cell-free supernatants of the infected cell lines were collected at 12, 24, 48, and 72 hpi, and viral output was evaluated by plaque-forming assay (measured in PFU) on C7/10 cells. Negev virus EO239 failed to replicate in vertebrate cells at 37°C (Fig. 9A) or 28°C (data not shown), as mean replication titers remained steady or declined. To further investigate whether vertebrate cells could support virus replication in the absence of CPE, we generated a real-time RT-PCR assay. The cell-free supernatants of the infected vertebrate cell lines (Vero and BHK-21) with Negev virus EO239 were collected at 1, 4, 6, and 8 days p.i., and viral output was evaluated by the real-time RT-PCR assay. As outlined in Table 4, Negev virus EO239 failed to replicate in either vertebrate cell line tested. However, these cell lines are permissive for replication with a wide range of other arthropod-transmitted viruses (44). Mean replication titers of Negev virus showed significant differences in levels of replication in invertebrate cell lines (Fig. 9B). Mean replication titers peaked consistently at 24 hpi (Fig. 9B) and plateaued thereafter in all cell lines, except in P. papatasi, where maximum titers were reached at 48 hpi, and An. gambiae and D. melanogaster cells, where mean replication titers remained steady or declined (Fig. 8B). Cell lysis (as CPE) was readily evident in Aedes (Fig. 8B, depicting CPE in C7/10 cells) and Culex (data not shown) cell lines at 12 hpi, whereas no overt CPE was observed in An. albimanus or P. papatasi cells at any time point (data not shown). The other 9 viruses included in this study demonstrated a similar phenotype, namely, rapid growth and CPE in C6/36 cells but no CPE in Vero or BHK-21 cells. Likewise, none of the viruses produced illness in newborn mice after intracerebral inoculation.
Fig 9.

Comparative replication curves of prototype Negev virus (EO239). (A) Virus outputs from 12 to 72 h following infection at an MOI of 10 by Negev virus EO239 in the vertebrate cell lines Vero (African green monkey kidney), BHK-21 (baby hamster kidney), and HEK293 (human embryonic kidney). (B) Virus outputs from 12 to 72 h following infection at an MOI of 10 by Negev virus in the insect cell lines Ae. albopictus (C6/36 and C7/10), An. albimanus, An. gambiae, Cx. tarsalis, P. papatasi, and D. melanogaster. The limit of detection of the assay is 1.0 log10 PFU/ml.
Table 4.
Quantitative real-time RT-PCR of Negev virus strain EO239 on vertebrate cell linesa
| Standard EO239 curve (copy/tube) and cell line | CT | Detection on day p.i.: |
|||
|---|---|---|---|---|---|
| 1 | 4 | 6 | 8 | ||
| BHK-21 | |||||
| 108 | 6 | <LD | <LD | <LD | <LD |
| Vero | |||||
| 107 | 10 | <LD | <LD | <LD | <LD |
| 106 | 14 | ||||
| 105 | 17 | ||||
| 104 | 21 | ||||
| 103 | 24 | ||||
| 102 | 28 | ||||
| 101 | 31 | ||||
| 1 | <LD | ||||
| Negative control | <LD | ||||
Each cell line was exposed to strain EO239 at a dose of infection equal to a CT value of 25. <LD, below the level of detection. CT, threshold cycle.
Mosquito susceptibility studies.
We also investigated whether the prototype Negev virus, EO239, could infect and disseminate after ingestion by two common anthropophilic mosquito vectors, Ae. aegypti and Ae. albopictus. As shown in Table 5, when Ae. aegypti ingested various concentrations of Negev virus, the level of infection varied in a dose-dependent manner. At the highest dose of 109 PFU/ml, 91% of midguts were infected, decreasing to 57 and 8% for the 107 and 105 PFU/ml doses, respectively. Table 5 also shows that the percentage of mosquitoes with virus dissemination (total number of disseminated infections divided by total number of engorged mosquitoes) also decreased from 73 to 50% for the higher doses to 0% for the lowest dose. Furthermore, dissemination rates from the infected midguts (total number of disseminated infections divided by total number of infected mosquitoes) ranged from 80 to 87.5% for the highest two doses to 0% for the lowest dose. In contrast to Ae. aegypti, Ae. albopictus mosquitoes were relatively refractory to oral infection with Negev virus midgut and disseminated infection rates in the 5 to 6% range for all doses (Table 5). No mortality other than regularly observed attrition was observed in any of the mosquitoes during the 14-day incubation. Thus, infection with Negev virus did not appear to have a deleterious effect on the insects.
Table 5.
Infection and dissemination of NEGV in the domestic and peridomestic vectors Ae. aegypti aegypti and Ae. albopictus
| Species | Bloodmeal titer (log10 PFU/ml) | No. infected/no. engorged | % Infected | No. disseminated/no. engorged | % Absolute dissemination | No. disseminated/no. infected | % Disseminated from infected midgut |
|---|---|---|---|---|---|---|---|
| Ae. aegypti aegypti | 109 | 20/22 | 91 | 16/22 | 73 | 16/20 | 80 |
| 107 | 8/14 | 57 | 7/14 | 50 | 7/8 | 87.5 | |
| 105 | 2/25 | 8 | 0/25 | 0 | 0/2 | 0 | |
| Ae. albopictus | 109 | 1/20 | 5 | 1/20 | 5 | 1/1 | 100 |
| 107 | 6/35 | 6 | 2/35 | 6 | 2/6 | 33.3 |
DISCUSSION
We report the isolation and characterization of 10 novel viruses from mosquitoes and phlebotomine sand flies collected in Brazil, Peru, the United States, Ivory Coast, Israel, and Indonesia. Their genomes are single-stranded, positive-sense RNAs with poly(A) tails. Based on their genome organization and phylogenetic relationships, the 10 viruses appear to fall into six distinct species, which we have designated Negev, Ngewotan, Piura, Loreto, Dezidougou, and Santana viruses. We propose the genus name Negevirus for this new group (taxon) of viruses, since Negev virus was the first virus that we characterized and appears to have the widest geographic distribution. It is noteworthy that the original Negev virus isolate EO239, from Anopheles mosquitoes collected in Israel in 1983, showed nucleotide and protein sequence identities between 95.6 to 100% and 98.6 to 100%, respectively, to two Negev isolates from Culex mosquitoes collected in Texas in 2008. During arbovirus surveillance studies in Houston between 2005 and 2010, other isolates of Negev virus were also made, but only two were included in this study (R. B. Tesh and H. Guzman, unpublished data).
The biological and potential public health importance of the negeviruses has yet to be determined, but some possible scenarios and areas of future research are outlined below.
In addition to their broad geographic distribution, the negeviruses appear to infect a wide range of hematophagous insects (mosquitoes of the genera Culex, Aedes, and Anopheles as well as sand flies of the genus Lutzomyia). The three isolates of Negev and of Loreto viruses were each made from pools of 3 different insect genera and/or species from two different localities. This suggests that these viruses are not species specific and have a broad host range among dipteran species. All of these viruses were obtained from hematophagous insects collected during arbovirus surveillance studies; however, this may reflect sampling bias given that arbovirologists generally only sample biting or blood-sucking arthropods. There are many other nonbiting dipteran species that could be infected with such viruses but that are not routinely cultured for viruses. Furthermore, these 10 negeviruses were only recognized because they produced CPE in cultures of mosquito cells, a phenotype that may not be found among all members of this new virus group. It seems probable that there are other negeviruses that do not have this phenotypic characteristic and thus would not be detected by culture. Based on these observations, we predict that other novel viruses in this group will be found.
A second consideration is how these viruses are transmitted and maintained among their insect hosts in nature. Our attempts to orally infect adult Ae. aegypti and Ae. albopictus with Negev virus indicated a high threshold for oral infection. Perhaps the wrong mosquito species or life stage was used; however, it seems more likely that oral infection is not the natural route of infection. Vertical or transovarial transmission seem to be more likely modes of transmission for such viruses among their insect hosts.
The rapid and high levels of Negev virus replication (up to 1010 PFU/ml) in some mosquito and sand fly cell lines suggest that high viral loads would also be obtained in susceptible naturally infected mosquitoes unless the insect's innate immune system could somehow downregulate virus replication. Nonetheless, the potential impact of negevirus infection on the insect's behavior, fertility, fecundity, and survival could also be important and should be investigated. Therefore, these aspects could be further exploited in the future to develop some of these viral agents as biological control agents.
The results of our studies on the growth of the six negeviruses in vertebrate and insect cells and in newborn mice indicate that they are mosquito-specific viruses; however, we did not have the facilities to test their growth in plant cells. Because of their distant genetic relationship with the cileviruses, we cannot eliminate the possibility that the negeviruses are also plant viruses. However, a scenario where mosquitoes acquire virus from plants seems unlikely in view of the relative refractoriness of adult mosquitoes to oral infection with Negev virus. Both adult mosquitoes and sand flies feed on plant sugars (floral and extrafloral nectars, damaged fruit, etc.) as an energy source (45, 46). However, if our studies of oral infection of Ae. aegypti and Ae. albopictus with Negev virus are indicative of mosquitoes' susceptibility by this route, then the insects would need to ingest plant juices containing 108 to 1010 PFU/ml of virus. It seems unlikely that floral nectars or fruit juices would contain such high viruses titers. Instead, the genetic relationship between the cileviruses and the negeviruses indicates that they are members of a larger virus family.
All of these viruses originally were isolated from naturally infected mosquito and sand fly genera that also serve as arbovirus vectors. Consequently, another consideration is the potential effect of negevirus infection on the susceptibility and vector competence of a mosquito or sandfly for viral pathogens of vertebrates. Recent experimental studies with Ae. aegypti infected with certain strains of the bacterial endosymbiont Wolbachia indicate that the presence of Wolbachia infection upregulates or primes the mosquito's innate immune system, which in turn interferes with dengue virus replication and decreases vector competence (47, 48). Similar results have been reported for Wolbachia-infected Ae. aegypti with chikungunya virus (47) and with Wolbachia-infected Cx. quinquefaciatus and West Nile virus (49). If a bacterial endosymbiont can alter a mosquito's vector competence for arboviruses, it seems plausible that a viral symbiont could have a similar effect. This is another potentially important area of investigation.
Experimental studies are also needed to determine how and where these viruses replicate in mosquitoes. Our preliminary studies indicate that Negev virus is disseminated in some of the insects, since it could be detected in their legs and/or wings 14 days after ingestion of an infectious blood meal. If a virus infects and disseminates in a mosquito, then it may infect the insect's salivary glands and be transmitted to a vertebrate host during blood feeding (50). In this scenario, humans and other vertebrate hosts of infected hematophagous insects would have intimate contact with negeviruses, raising the possibility that some of these viruses can adapt to vertebrates and eventually emerge as vertebrate pathogens. Some eminent virologists (51, 52) have previously suggested that many arthropod-borne viruses of vertebrates and of plants originally were arthropod viruses. As arthropods evolved and developed blood-feeding or sap-sucking habits, some of their viruses developed the ability to infect the new vertebrate or plant host and eventually become vertebrate or plant pathogens (51, 52). If true, then such a scenario might be possible with some negeviruses.
ACKNOWLEDGMENTS
We are grateful to Joseph Peleg for providing Negev virus strain EO239, James G. Olson for providing the three Loreto virus strains, Vincent Deubel and Jean-Pierre Digoutte for sending Dezidougou virus, and James Converse for providing Ngewotan virus. Ae. aegypti and Ae. albopictus, used to initiate our laboratory colonies, were kindly provided by the Armed Forces Research Institute of Medical Sciences (AFRIMS), Bangkok, Thailand. We also thank Frederick A. Murphy for help in interpreting the electron micrographs.
This work was supported in part by the Department of Pathology startup funds to N.V., NIH contract HHSN272201000040I/HHSN27200004/D04 to R.B.T., an NIH T-32 training grant to A.D.H. and S.L.R., AI157158 (Northeast Biodefense Center-Lipkin), and the Defense Threat Reduction Agency. A.V.G. received a grant from Sociedad Española de Enfermedades Infecciosas y Microbiologia Clínica (SEIMC) to support her sabbatical to UTMB.
We have no conflicting financial interests.
Footnotes
Published ahead of print 19 December 2012
REFERENCES
- 1. Kuno G. 2004. A survey of the relationships among the viruses not considered arboviruses, vertebrates, and arthropods. Acta Virol. 48:135–143 [PubMed] [Google Scholar]
- 2. Hoshino K, Isawa H, Tsuda Y, Yano K, Sasaki T, Yuda M, Takasaki T, Kobayashi M, Sawabe K. 2007. Genetic characterization of a new insect flavivirus isolated from Culex pipiens mosquito in Japan. Virology 359:405–414 [DOI] [PubMed] [Google Scholar]
- 3. Igarashi A, Harrap KA, Casals J, Stollar V. 1976. Morphological, biochemical, and serological studies on a viral agent (CFA) which replicates in and causes fusion of Aedes albopictus (Singh) cells. Virology 74:174–187 [DOI] [PubMed] [Google Scholar]
- 4. Stollar V, Thomas VL. 1975. An agent in the Aedes aegypti cell line (Peleg) which causes fusion of Aedes albopictus cells. Virology 64:367–377 [DOI] [PubMed] [Google Scholar]
- 5. Marklewitz M, Handrick S, Grasse W, Kurth A, Lukashev A, Drosten C, Ellerbrok H, Leendertz FH, Pauli G, Junglen S. 2011. Gouleako virus isolated from West African mosquitoes constitutes a proposed novel genus in the family Bunyaviridae. J. Virol. 85:9227–9234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nasar F, Palacios G, Gorchakov R, Guzman H, Travassos Da Rosa AP, Popov VL, Sherman MB, Lipkin WI, Tesh RB, Weaver SC. 2012. Eilat virus, a newly identified host restricted alphavirus. Proc. Natl. Acad. Sci. U. S. A. 109:14622–14627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nga PT, Parquet Mdel C, Lauber C, Parida M, Nabeshima T, Yu F, Thuy NT, Inoue S, Ito T, Okamoto K, Ichinose A, Snijder EJ, Morita K, Gorbalenya AE. 2011. Discovery of the first insect nidovirus, a missing evolutionary link in the emergence of the largest RNA virus genomes. PLoS Pathog. 7:e1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quan PL, Junglen S, Tashmukhamedova A, Conlan S, Hutchison SK, Kurth A, Ellerbrok H, Egholm M, Briese T, Leendertz FH, Lipkin WI. 2010. Moussa virus: a new member of the Rhabdoviridae family isolated from Culex decens mosquitoes in Cote d'Ivoire. Virus Res. 147:17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamao T, Eshita Y, Kihara Y, Satho T, Kuroda M, Sekizuka T, Nishimura M, Sakai K, Watanabe S, Akashi H, Rongsriyam Y, Komalamisra N, Srisawat R, Miyata T, Sakata A, Hosokawa M, Nakashima M, Kashige N, Miake F, Fukushi S, Nakauchi M, Saijo M, Kurane I, Morikawa S, Mizutani T. 2009. Novel virus discovery in field-collected mosquito larvae using an improved system for rapid determination of viral RNA sequences (RDV ver4.0). Arch. Virol. 154:153–158 [DOI] [PubMed] [Google Scholar]
- 10. Zirkel F, Kurth A, Quan PL, Briese T, Ellerbrok H, Pauli G, Leendertz FH, Lipkin WI, Ziebuhr J, Drosten C, Junglen S. 2011. An insect nidovirus emerging from a primary tropical rainforest. mBio 2:e00077–11 doi:10.1128/mBio.00077-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Locali-Fabris EC, Freitas-Astua J, Machado MA. 2012. Genus Cilevirus, p 1169–1172 In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed), Virus taxonomy. Ninth report of the International Committee on Taxonomy of Viruses. Elsevier, San Diego, CA [Google Scholar]
- 12. Locali-Fabris EC, Freitas-Astua J, Souza AA, Takita MA, Astua-Monge G, Antonioli-Luizon R, Rodrigues V, Targon ML, Machado MA. 2006. Complete nucleotide sequence, genomic organization and phylogenetic analysis of citrus leprosis virus cytoplasmic type. J. Gen. Virol. 87:2721–2729 [DOI] [PubMed] [Google Scholar]
- 13. Pascon RC, Kitajima JP, Breton MC, Assumpcao L, Greggio C, Zanca AS, Okura VK, Alegria MC, Camargo ME, Silva GG, Cardozo JC, Vallim MA, Franco SF, Silva VH, Jordao H, Jr, Oliveira F, Giachetto PF, Ferrari F, Aguilar-Vildoso CI, Franchiscini FJ, Silva JM, Arruda P, Ferro JA, Reinach F, da Silva AC. 2006. The complete nucleotide sequence and genomic organization of citrus leprosis associated virus, cytoplasmatic type (CiLV-C). Virus Genes 32:289–298 [DOI] [PubMed] [Google Scholar]
- 14. Igarashi A. 1978. Isolation of a Singh's Aedes albopictus cell clone sensitive to dengue and chikungunya viruses. J. Gen. Virol. 40:531–544 [DOI] [PubMed] [Google Scholar]
- 15. Varma MG, Pudney M, Leake CJ. 1974. Cell lines from larvae of Aedes (Stegomyia) malayensis Colless and Aedes (S) pseudoscutellaris (Theobald) and their infection with some arboviruses. Trans. R. Soc. Trop. Med. Hyg. 68:374–382 [DOI] [PubMed] [Google Scholar]
- 16. Kim DY, Guzman H, Bueno R, Jr, Dennett JA, Auguste AJ, Carrington CV, Popov VL, Weaver SC, Beasley DW, Tesh RB. 2009. Characterization of Culex flavivirus (Flaviviridae) strains isolated from mosquitoes in the United States and Trinidad. Virology 386:154–159 [DOI] [PubMed] [Google Scholar]
- 17. Sarver N, Stollar V. 1977. Sindbis virus-induced cytopathic effect in clones of Aedes albopictus (Singh) cells. Virology 80:390–400 [DOI] [PubMed] [Google Scholar]
- 18. Bello Garcia FJ, Boshell J, Rey G, Morales A, Olano VA. 1995. Initiation of primary cell cultures from embryos of the mosquitoes Anopheles albimanus and Aedes taeniorhynchous (Diptera: Culicidae). Mem. Inst. Oswaldo Cruz 90:547–551 [Google Scholar]
- 19. Chao J, Ball GH. 1976. A comparison of amino acid utilization by cell lines of Culex tarsalis and Culex pipiens, p 263–266 In Kurstak E, Maramorosch K. (ed), Invertebrate tissue culture applications in medicine, biology and agriculture. Academic Press, New York, NY [Google Scholar]
- 20. Marhoul Z, Pudney M. 1972. A mosquito cell line (MOS.55) from Anopheles gambiae larva. Trans. R. Soc. Trop. Med. Hyg. 66:183–184 [DOI] [PubMed] [Google Scholar]
- 21. Sherman MB, Weaver SC. 2010. Structure of the recombinant alphavirus Western equine encephalitis virus revealed by cryoelectron microscopy. J. Virol. 84:9775–9782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shope RE, Sather GE. 1979. Arboviruses. In Lennette EH, Schmidt NJ. (ed), Diagnostic procedures for viral, rickettsial and chlamydial infections, 5th ed American Public Health Association, Washington, DC [Google Scholar]
- 23. Palacios G, Quan PL, Jabado OJ, Conlan S, Hirschberg DL, Liu Y, Zhai J, Renwick N, Hui J, Hegyi H, Grolla A, Strong JE, Towner JS, Geisbert TW, Jahrling PB, Buchen-Osmond C, Ellerbrok H, Sanchez-Seco MP, Lussier Y, Formenty P, Nichol MS, Feldmann H, Briese T, Lipkin WI. 2007. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerg. Infect. Dis. 13:73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cox-Foster DL, Conlan S, Holmes EC, Palacios G, Evans JD, Moran NA, Quan PL, Briese T, Hornig M, Geiser DM, Martinson V, van Engelsdorp D, Kalkstein AL, Drysdale A, Hui J, Zhai J, Cui L, Hutchison SK, Simons JF, Egholm M, Pettis JS, Lipkin WI. 2007. A metagenomic survey of microbes in honey bee colony collapse disorder. Science 318:283–287 [DOI] [PubMed] [Google Scholar]
- 25. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, Conlan S, Quan PL, Hui J, Marshall J, Simons JF, Egholm M, Paddock CD, Shieh WJ, Goldsmith CS, Zaki SR, Catton M, Lipkin WI. 2008. A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med. 358:991–998 [DOI] [PubMed] [Google Scholar]
- 27. Gorchakov R, Hardy R, Rice CM, Frolov I. 2004. Selection of functional 5′ cis-acting elements promoting efficient Sindbis virus genome replication. J. Virol. 78:61–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Penn O, Privman E, Ashkenazy H, Landan G, Graur D, Pupko T. 2010. GUIDANCE: a web server for assessing alignment confidence scores. Nucleic Acids Res. 38:W23–W28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Penn O, Privman E, Landan G, Graur D, Pupko T. 2010. An alignment confidence score capturing robustness to guide tree uncertainty. Mol. Biol. Evol. 27:1759–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Swofford D. 2000. PAUP*. Phylogenetic analysis using parsimony (* and other methods). Version 4 Sinauer Associates, Sunderland, MA [Google Scholar]
- 31. Posada DC, Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818 [DOI] [PubMed] [Google Scholar]
- 32. Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogeny. Bioinformatics 17:754–755 [DOI] [PubMed] [Google Scholar]
- 33. Ronquist F, Huelsenbeck JP. 2003. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574 [DOI] [PubMed] [Google Scholar]
- 34. Buchan DW, Ward SM, Lobley AE, Nugent TC, Bryson K, Jones DT. 2010. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 38:W563–W568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jones DT. 2007. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23:538–544 [DOI] [PubMed] [Google Scholar]
- 36. Jones DT, Taylor WR, Thornton JM. 1994. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 33:3038–3049 [DOI] [PubMed] [Google Scholar]
- 37. Nugent T, Jones DT. 2009. Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics 10:159 doi:10.1186/1471-2105-10-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rumenapf T, Brown DT, Strauss EG, Konig M, Rameriz-Mitchel R, Strauss JH. 1995. Aura alphavirus subgenomic RNA is packaged into virions of two sizes. J. Virol. 69:1741–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rumenapf T, Strauss EG, Strauss JH. 1994. Subgenomic mRNA of Aura alphavirus is packaged into virions. J. Virol. 68:56–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Froshauer S, Kartenbeck J, Helenius A. 1988. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell Biol. 107:2075–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mosca JD, Pitha PM. 1986. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol. Cell. Biol. 6:2279–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brackney DE, Scott JC, Sagawa F, Woodward JE, Miller NA, Schilkey FD, Mudge J, Wilusz J, Olson KE, Blair CD, Ebel GD. 2010. C6/36 Aedes albopictus cells have a dysfunctional antiviral RNA interference response. PLoS Negl. Trop. Dis. 4:e856 doi:10.1371/journal.pntd.0000856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scott JC, Brackney DE, Campbell CL, Bondu-Hawkins V, Hjelle B, Ebel GD, Olson KE, Blair CD. 2010. Comparison of dengue virus type 2-specific small RNAs from RNA interference-competent and -incompetent mosquito cells. PLoS Negl. Trop. Dis. 4:e848 doi:10.1371/journal.pntd.0000848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Karabatsos N. 1985. International catalogue of arboviruses, including certain other viruses of vertebrates, 3rd ed American Society of Tropical Medicine and Hygiene, San Antonio, TX: [DOI] [PubMed] [Google Scholar]
- 45. Clements AN. 1992. The biology of mosquitoes: development, nutrition and reproduction, vol 1 Chapman and Hall, London, United Kingdom [Google Scholar]
- 46. Lane RP. 1993. Sandflies (Phlebotominae). In La Crosskey RW. (ed), Medical insects and arachnids. Chapman and Hall, London, United Kingdom [Google Scholar]
- 47. Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, Lu G, Pyke AT, Hedges LM, Rocha BC, Hall-Mendelin S, Day A, Riegler M, Hugo LE, Johnson KN, Kay BH, McGraw EA, van den Hurk AF, Ryan PA, O'Neill SL. 2009. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, chikungunya, and Plasmodium. Cell 139:1268–1278 [DOI] [PubMed] [Google Scholar]
- 48. Rances E, Ye YH, Woolfit M, McGraw EA, O'Neill SL. 2012. The relative importance of innate immune priming in Wolbachia-mediated dengue interference. PLoS Pathog. 8:e1002548 doi:10.1371/journal.ppat.1002548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Glaser RL, Meola MA. 2010. The native Wolbachia endosymbionts of Drosophila melanogaster and Culex quinquefasciatus increase host resistance to West Nile virus infection. PLoS One 5:e11977 doi:10.1371/journal.pone.0011977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Higgs S, Beaty BJ. 2005. Natural cycles of vector-borne pathogens, p 167–185 In Marquardt WC. (ed), Biology of disease vectors. Elsevier Academic Press, San Diego, CA [Google Scholar]
- 51. Maramorosch K. 1955. Multiplication of plant viruses in insect vectors. Adv. Virus Res. 3:221–249 [DOI] [PubMed] [Google Scholar]
- 52. Schlesinger RW. 1980. Evolutionary aspects of Togaviridae, p 40–46 In Schlesinger RW. (ed), The Togaviridae: biology, structure, replication. Academic Press, New York, NY [Google Scholar]