

Abstract
Subacute sclerosing panencephalitis (SSPE) is a fatal degenerative disease caused by persistent measles virus (MV) infection in the central nervous system (CNS). From the genetic study of MV isolates obtained from SSPE patients, it is thought that defects of the matrix (M) protein play a crucial role in MV pathogenicity in the CNS. In this study, we report several notable mutations in the extracellular domain of the MV fusion (F) protein, including those found in multiple SSPE strains. The F proteins with these mutations induced syncytium formation in cells lacking SLAM and nectin 4 (receptors used by wild-type MV), including human neuronal cell lines, when expressed together with the attachment protein hemagglutinin. Moreover, recombinant viruses with these mutations exhibited neurovirulence in suckling hamsters, unlike the parental wild-type MV, and the mortality correlated with their fusion activity. In contrast, the recombinant MV lacking the M protein did not induce syncytia in cells lacking SLAM and nectin 4, although it formed larger syncytia in cells with either of the receptors. Since human neuronal cells are mainly SLAM and nectin 4 negative, fusion-enhancing mutations in the extracellular domain of the F protein may greatly contribute to MV spread via cell-to-cell fusion in the CNS, regardless of defects of the M protein.
INTRODUCTION
Measles virus (MV) causes measles, a common acute infectious disease characterized by high fever and a maculopapular rash (1). Despite the availability of effective vaccines, measles still remains epidemic in developing countries. MV sometimes invades the central nervous system (CNS), causing a fatal degenerative disease, subacute sclerosing panencephalitis (SSPE), several years after acute measles (1–4). SSPE has been treated with broad-spectrum antiviral drugs, including ribavirin, interferons, and isoprinosine (4). Although these treatments may be beneficial, causing temporal stabilization of disease progression and prolonged survival, complete remission is not achieved. Therefore, establishment of effective therapy for SSPE is highly desirable, in addition to vaccination against measles to reduce SSPE occurrence.
MV, a member of the family Paramyxoviridae, is an enveloped virus with a nonsegmented, negative-strand RNA genome. The MV genome has six genes that encode the nucleocapsid (N), phospho- (P), matrix (M), fusion (F), hemagglutinin (H), and large (L) proteins. The genomic RNA is encapsidated with the N protein and, together with RNA-dependent RNA polymerase composed of the L and P proteins, forms a ribonucleoprotein (RNP) complex (3). There are two envelope glycoproteins, the H and F proteins, which are responsible for receptor binding and membrane fusion, respectively (3). The M protein plays a role in the assembly of virus particles by interacting with the RNP and the cytoplasmic tails of the H and F proteins. MV enters host cells by pH-independent membrane fusion at the cell surface. Binding of the H protein to a cellular receptor is thought to trigger the conformational changes of the F-protein trimer (5–7), thereby inducing the formation of the six-helix bundle (6-HB) structure involved in membrane fusion (8–11). The interaction between two heptad repeat (HR) domains, HR-A and HR-B, in individual F-protein monomers is responsible for the formation of 6-HB. While the F protein, upon activation, induces virus-to-cell fusion, it also causes cell-to-cell fusion in infected cells, producing syncytia (3).
Wild-type MV strains infect immune cells using signaling lymphocyte activation molecule (SLAM) (also called CD150) as a receptor (12), and the tissue distribution of SLAM is consistent with the lymphotropism and immunosuppressive nature of MV (13, 14). CD46, a complement-regulatory molecule expressed on all human cells except red blood cells, functions as a receptor for vaccine and laboratory-adapted strains of MV (15–17), but not for wild-type strains, as specific amino acid changes in the H protein are needed for MV to use CD46 (18, 19). Wild-type MV strains have also been shown to infect SLAM-negative cells, including polarized epithelial cells (20–22) and neuronal cells (23, 24). Recently, nectin 4 was identified as an epithelial cell receptor (25, 26).
A number of studies have reported successful virus isolation from brain tissues of SSPE cases (27–30). Sequencing analyses have revealed characteristic mutations in these viruses (SSPE strains) compared with wild-type MV isolates. In many SSPE strains, A-to-G-biased hypermutations occur in the genome, especially in the M gene (31–33). In some strains, a single deletion or mutation in the P gene causes a transcriptional error, leading to an increase in the level of dicistronic P-M mRNA compared with that of M mRNA. In yet other strains, mutations cause the elongation or shortening of the cytoplasmic tail of the F protein, affecting its interaction with the M protein (34–36). All these mutations in SSPE strains abrogate the expression or function of the M protein, thereby precluding the production of virus particles. Whereas standard MVs are efficiently eliminated by host defense mechanisms, assembly-defective MVs may persist in the body by evading immune responses (37). In addition, it was shown that the deletion of the M protein or the cytoplasmic domain of the F protein enhanced cell-cell fusion in SLAM- or CD46-dependent infection (38, 39). In short, defects of the M protein have been thought to play a crucial role in MV pathogenicity in the CNS.
In this study, we incidentally identified several substitutions in the extracellular domain of the F protein that enhance its fusion activity. We also noticed that most SSPE strains possess substitutions in the extracellular domain of the F protein that are likely to affect its fusion activity, judging from their positions within the protein. Therefore, we examined whether these substitutions in the F protein play a role in MV pathogenicity. Our results revealed that fusion-enhancing mutations in the extracellular domain of the F protein indeed facilitate MV spread in SLAM- and nectin 4-negative cells, as well as neuropathogenicity in suckling hamsters, independently of defects of the M protein.
MATERIALS AND METHODS
Cells.
Vero cells expressing human SLAM (Vero/hSLAM cells) (14), Vero cells, and IMR-32 cells were maintained in Dulbecco's minimum essential medium (DMEM) (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS). SYM-1 cells were maintained in 1:1 DMEM/RPMI medium supplemented with 10% FBS. Vero/hSLAM cells constitutively expressing the T7 RNA polymerase (Vero/hSLAM-T7 cells) were established using a retrovirus vector as follows. To generate the retrovirus expressing the T7 RNA polymerase, the cDNA encoding the T7 RNA polymerase was cloned into pMX-IRES-Puro (a gift from M. Shimojima and T. Kitamura) (40–42), producing pMX-T7pol-IRES-Puro. PLAT-gp cells (Invivogen, San Diego, CA) cultured in 6-well cluster plates were transfected with 7.5 μg of pMX-T7pol-IRES-Puro and 0.5 μg of pCVSV-G using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), and the retrovirus was recovered. Vero/hSLAM cells in 24-well plates were infected with this retrovirus, and at 72 h after transduction, the culture medium was replaced with complete DMEM containing 2 μg/ml puromycin (Invivogen). A single clone was isolated after several passages with puromycin and used for experiments. Vero/hSLAM-T7 cells were maintained in DMEM supplemented with 10% fetal bovine serum and 2 μg/ml puromycin.
Plasmid constructions.
The F genes of mutant viruses were amplified by PCR using the primer pair 5′-TTGAATTCGCCACCATGGGTCTCAAGGTGAACGT-3′ and 5′-TTGCGGCCGCTCAGAGCGACCTTACATAGG-3′. After digestion with EcoRI and NotI, the PCR products were cloned into pCA7, the eukaryotic expression vector (43), producing pCA7-ICF(I87T), pCA7-ICF(M94V), pCA7-ICF(S262R), pCA7-ICF(L354M), and pCA7-ICF(N462K). pCA7-ICH and pCA7-ICF, which encode the H and F proteins of the IC-B strain of MV, respectively, have been described previously (19). Six different sets of mutations were introduced independently into pCA7-ICF by site-directed mutagenesis using complementary primer pairs, producing pCA7-ICF(T461I), pCA7-ICF(S103I), pCA7-ICF(N462S), pCA7-ICF(N465S), pCA7-ICF(N462S/N465S), and pCA7-ICF(S103I/N462S/N465S). All full-length-genome plasmids were derived from pHHRz(+)MV323-EGFP (44, 45), which carries the antigenomic full-length cDNA of the wild-type IC-B strain together with enhanced green fluorescent protein (EGFP). The F gene of pHHRz(+)MV323-EGFP was replaced with those of pCA7-ICF-derived plasmids with different mutations, producing pHHRz(+)MV323-F(S262R)-EGFP, pHHRz(+)MV323-F(L354M)-EGFP, pHHRz(+)MV323-F(N462K)-EGFP, pHHRz(+)MV323-F(T461I)-EGFP, and pHHRz(+)MV323-F(S103I/N462S/N465S)-EGFP. To construct the plasmid pHHRz(+)MV323-ΔM-EGFP, the gene encoding the 5′ and 3′ untranslated regions (UTR) of the M protein but lacking the coding sequence of the M protein was amplified by the overlapping-PCR method with the primer set 5′-ATCCGCGGTCAGGGATCTGG-3′, 5′-CTGGGCACTGCGGTTGTGGAACTTAGGAGGCAATC-3′, 5′-CTCCTAAGTTCCACAACCGCAGTGCCCAGCAATACC-3′, and 5′-TGCCGCGGGCCGGGGTCTG-3′ and ligated to the plasmid pHHRz(+)MV323-EGFP after digestion with SacII. The plasmids pCITE-RL and pTKminusT7-FL used in the quantitative fusion assay were constructed as follows. To construct the plasmid pCITE-RL, the gene encoding Renilla luciferase was amplified by PCR from p18MGKLuc01 (46) and ligated to the plasmid pCITE-2a(+) (Novagen, Madison, WI). To construct the plasmid pTKminusT7-FL, the gene encoding firefly luciferase was amplified by PCR from p18MGFLuc01 (46) and ligated to the plasmid pTK-RL (Promega, Madison, WI), from which the T7 promoter and Renilla luciferase sequence had been removed.
Viruses.
Recombinant MVs were generated as reported previously (45). Briefly, BHK/T7-9 cells, which constitutively express T7 RNA polymerase, were transfected with full-length-genome plasmids carrying the antigenomes of MV and three support plasmids, pCITE-IC-N, pCITE-IC-PΔC, and pCITEko-9301B-L. At 2 days after transfection, the cells were cocultured with Vero/hSLAM cells. The recombinant viruses IC323-F(S262R)-EGFP, IC323-F(L354M)-EGFP, IC323-F(N462K)-EGFP, IC323-F(T461I)-EGFP, IC323-F(S103I/N462S/N465S)-EGFP, and IC323-ΔM-EGFP were generated from pHHRz(+)MV323-F(S262R)-EGFP, pHHRz(+)MV323-F(L354M)-EGFP, pHHRz(+)MV323-F(N462K)-EGFP, pHHRz(+)MV323-F(T461I)-EGFP, pHHRz(+)MV323-F(S103I/N462S/N465S)-EGFP, and pHHRz(+)MV323-ΔM-EGFP, respectively. The generated MVs were propagated in Vero/hSLAM cells. The number of PFU of each recombinant virus was determined in Vero/hSLAM cells.
Quantitative fusion assay.
293T cells cultured in a 12-well plate were transfected with pCA7-ICH (0.4 μg), an F-protein-encoding plasmid (pCA7-ICF or pCA7 encoding each mutant F protein; 0.1 μg) and pCITE-RL encoding Renilla luciferase driven by T7 polymerase (1.2 μg), using Polyethylenimine (PEI) “Max” high-potency linear PEI (Polysciences, Warrington, PA). These transfectants were used as effector cells. As an internal control, pTKminusT7-FL encoding firefly luciferase driven by the herpes simplex virus (HSV) thymidine kinase (TK) promoter was cotransfected into the effector cells. Vero/hSLAM-T7 cells were cultured in a 12-well plate and used as target cells. At 6 h posttransfection, effector cells were washed with PBS 3 times, detached with 0.05% EDTA in PBS, and centrifuged (400 × g; 5 min; 4°C). The cell pellet was resuspended in 500 μl of growth medium, and 100 μl of cell suspension was overlaid onto the target cells. After 18 h of incubation, cell-cell fusion was quantified by measuring the luciferase activity. Firefly and Renilla luciferase activities were independently assayed by using the dual-luciferase reporter assay system (Promega) with a Mithras LB940 plate reader (Berthold Technologies, Pforzheim, Germany). The relative fusion activity (Renilla luciferase activity divided by firefly luciferase activity) was calculated. The value obtained with pCA7-ICH and pCA7-ICF was set to 100%.
Plasmid-mediated fusion assay in Vero/hSLAM cells.
Vero/hSLAM cells cultured in a 12-well cluster plate were transfected with pCA7-ICH (0.2 μg) plus pCA7 encoding the wild-type or each mutant F protein (1.5 μg), using Lipofectamine 2000 (Invitrogen). One day after transfection, the cells were observed under a light microscope after Giemsa staining.
Plasmid-mediated fusion assay in Vero cells.
Vero cells cultured in a 24-well cluster plate were transfected with pCA7-ICH (0.75 μg) plus pCA7 encoding each mutant F protein (0.75 μg). Plasmid DNAs were incubated with 1.5 μl of Lipofectamine LTX Plus reagent (Invitrogen) for 5 min at room temperature and incubated with 4.5 μl of Polyethylenimine “Max” high-potency linear PEI for 30 min at room temperature, followed by dropping the mixture onto Vero cells. At 48 h posttransfection, the cells were observed under a light microscope after Giemsa staining. Then, the number of nuclei in syncytia per visual field was determined with a 10× objective lens.
Overlay fusion assay.
Recombinant viruses were rescued in BHK-T7/9 cells as described above. The transfected BHK-T7/9 cells were maintained in growth medium containing fusion block peptide (Z-D-Phe-Phe-Gly; Peptide Institute Inc., Osaka, Japan) to prevent cell-to-cell fusion. At 24 h posttransfection, the transfected BHK-T7/9 cells were washed 3 times with PBS and detached by trypsin digestion. The preparation, containing 200 EGFP-positive cells, was overlaid onto a confluent monolayer of Vero or IMR-32 cells cultured in a 10-cm dish. At 1 or 3 days after overlay, EGFP fluorescence was observed under a fluorescence microscope.
Surface biotinylation.
Vero/hSLAM cells were transfected with 4 μg of plasmid DNA encoding MV F variants as indicated. After washing with PBS three times, cells were incubated in PBS with 0.5 mg of NHS-SS-Biotin (Thermo Scientific, Rockford, IL)/ml for 20 min at 4°C, followed by washing and quenching for 5 min at 4°C in DMEM. The cells were scraped in 1 ml immunoprecipitation buffer (10 mM HEPES [pH 7.4], 50 mM sodium pyrophosphate, 50 mM sodium fluoride, 50 mM sodium chloride, 5 mM EDTA, 5 mM EGTA, 1% Triton X-100) containing protease inhibitor cocktail (Nacalai Tesque, Inc., Kyoto, Japan), and the lysates were cleared by centrifugation for 20 min at 20,000 × g and 4°C. The biotinylated proteins were adsorbed to Sepharose-coupled streptavidin (GE Healthcare, Waukesha, WI) for 90 min at 4°C, washed once in immunoprecipitation buffer, buffer 1 (100 mM Tris [pH 7.6], 500 mM lithium chloride, 0.1% Triton X-100) and buffer 2 (20 mM HEPES [pH 7.2], 2 mM EGTA, 10 mM magnesium chloride, 0.1% Triton X-100). Finally the proteins were incubated in urea buffer for 25 min at 50°C and subjected to Western blot analysis using antibodies specific for the MV-F tail. As a loading control, 10 μl of cell lysates was mixed with urea buffer and subjected to Western blot analysis using antibodies specific for beta-actin (Santa Cruz Biotechnology, Santa Cruz, CA). For densitometric quantification of F proteins, blots were analyzed using a VersaDoc digital imaging system (Bio-Rad, Richmond, CA), and the signals were quantified with QuantityOne software (Bio-Rad).
Virus challenge and histopathological examination.
Ten-day-old Syrian golden hamsters (SLC-Japan, Shizuoka, Japan) were anesthetized with sevoflurane. Then, 25 μl of diluted viruses was inoculated into the right or left hemisphere of the brains of hamsters. After the inoculation, clinical symptoms were observed every day, and moribund hamsters were euthanized. The brains were collected from dead and moribund hamsters. Three hamsters inoculated with wild-type MV or IC323-F(L354M)-EGFP were euthanized 6 days postinoculation, and the brains were also collected. EGFP autofluorescence of the MV-infected cells in each brain was observed under a fluorescence stereomicroscope. All animal experiments were reviewed by the Institutional Committee of Ethics on Animal Experiments and carried out according to the Guidelines for Animal Experiments of the Faculty of Medicine, Kyushu University, Fukuoka, Japan. For histopathological analysis, the collected brain was fixed in 10% buffered formalin and processed into paraffin sections. Sections were stained with hematoxylin and eosin (HE). Immunohistochemical analysis was performed, using the following antibodies: mouse anti-NeuN (1:100; Chemicon, Temecula, CA), mouse anti-glial fibrillary acidic protein (GFAP) (1:1,000; Dako A/S, Glostrup, Denmark), rabbit anti-green fluorescent protein (GFP) (1:500; Invitrogen), and anti-MV-N (1:100; Novus Biological, Littleton, CO). Fluorescence images were captured with a confocal microscope (A1 Confocal Laser Microscope; Nikon Corporation, Tokyo, Japan).
RESULTS
Substitutions in the F protein that enhance its fusion activity.
In two different experiments, we identified substitutions in the F protein of IC323-EGFP (the EGFP-expressing recombinant MV based on the wild-type IC-B strain) (44) that enhanced its fusion activity. First, in our attempt to modify IC323-EGFP so that it possessed the H protein with an epitope tag at its C terminus, we found that the virus did not induce syncytia in Vero/hSLAM cells (14). This presumably occurred because the added tag adversely affected the interaction of the H protein with the receptor SLAM and/or the F protein. However, after a few passages in Vero/hSLAM cells, several mutant viruses emerged, which regained the ability to induce syncytia. These mutant viruses were plaque purified, and their H, F, and M genes, encoding the proteins involved in membrane fusion, were subjected to sequencing analysis. Three mutant viruses had single-amino-acid substitutions in the F protein but no substitutions in the H and M proteins (Table 1). Second, Vero cells (SLAM and nectin 4 negative) were infected with IC323-EGFP. As expected, no syncytia were detected at the beginning, but after a few passages, we found two syncytium-producing viruses that had single-amino-acid substitutions in the F protein but none in the H and M proteins (Table 1).
Table 1.
Single-amino-acid substitutions found in the F genes of mutant viruses with enhanced fusion activity
| Substitution | Virus used for identification | Cells used for identification |
|---|---|---|
| I87T | Epitope tagged | Vero/hSLAM |
| S262R | Epitope tagged | Vero/hSLAM |
| N462K | Epitope tagged | Vero/hSLAM |
| M94V | Wild type | Vero |
| L354M | Wild type | Vero |
To determine whether these single amino acid substitutions indeed endow the F protein with enhanced fusion activity, Vero/hSLAM cells were cotransfected with the expression plasmid encoding the F protein with each substitution and the plasmid encoding the H protein (without the epitope tag). Compared with the wild-type F protein, all mutant F proteins produced larger syncytia in Vero/hSLAM cells (Fig. 1A). To further evaluate the fusion activities of these F-protein mutants, a quantitative plasmid-mediated fusion assay was established. The S262R, L354M, and N462K substitutions allowed the F protein to exhibit more than 2-times-higher fusion activity than the wild-type F protein, while the M94V substitution slightly enhanced its fusion activity (Fig. 1B).
Fig 1.

Amino acid substitutions in the F protein enhance cell-cell fusion in Vero/hSLAM and Vero cells. (A) Vero/hSLAM cells were transfected with expression plasmids encoding wild-type (Wt) or mutant F proteins with the indicated substitutions, together with the plasmid encoding the wild-type H protein. Only the H-protein expression plasmid was transfected in H, F(-). At 24 h posttransfection, the cells were stained with a Giemsa solution and observed under a light microscope. A representative syncytium is shown. Scale bar, 200 μm. (B) Quantification of cell-cell fusion in Vero/hSLAM-T7 cells. 293T cells were transfected with expression plasmids encoding the H protein and the indicated mutant F protein, together with the T7 polymerase-driven expression vector encoding luciferase. At 6 h posttransfection, transfected 293T cells were cocultured with Vero/hSLAM-T7 cells. After 18 h of incubation, cell-cell fusion was quantified by measuring the luciferase activity. Luciferase activity with the wild-type H and F proteins was set to 100%. The bars indicate the means and standard deviations (SD) for triplicate samples. An asterisk indicates a statistically significant increase compared with the wild-type F protein (P < 0.05). A one-tailed Student's t test assuming unequal variance was used to analyze the data. (C) Vero cells were transfected and observed as in panel A. (D) Quantification of cell-cell fusion in Vero cells. Fusion activity is shown as the number of nuclei present in syncytia per visual field with a 10× objective lens by averaging the numbers in nine visual fields (+SD).
The fusion activities of these F-protein mutants were also examined in Vero cells by expressing them together with the wild-type H protein. While the wild-type F protein did not induce syncytium formation in Vero cells, all mutant proteins did so (Fig. 1C). Since the sizes of syncytia observed in Vero cells were much smaller than those in Vero/hSLAM cells, the quantitative fusion assay did not give large values so as to reliably evaluate fusion activity. Thus, fusion activity in Vero cells was evaluated by counting the nuclei in syncytia (Fig. 1D). The S262R and N462K substitutions conferred on the F protein more enhanced fusion activity than the other substitutions. When these mutant F proteins were expressed alone (without the H protein), they did not induce syncytia in Vero or Vero/hSLAM cells (data not shown).
Figure 2 shows the locations of these 5 substitutions within the full-length F-protein structure. The substitutions I87T, M94V, and S262R are located in the “microdomain,” the region near the HR-A domain that is involved in the initiation of fusion (47–49). The L354M substitution is located in the cysteine-rich region, which is thought to interact with the H protein (50). Mutant MVs resistant to a fusion-inhibitory peptide contain amino acid substitutions in this region (51). The N462K substitution is located in the HR-B domain. Mutant viruses resistant to fusion-inhibitory compounds contain substitutions at position 462 of the F protein, including the N462K substitution (47, 52). These three regions (the microdomain, cysteine-rich region, and HR-B domain) in the F protein have been implicated in controlling its fusion activity.
Fig 2.

Locations of substitutions within the full-length F-protein structure. A schematic representation of the MV F protein and the positions of identified substitutions enhancing fusion activity (solid lines) are shown. The positions of fusion-enhancing substitutions found in multiple SSPE strains are also shown (dashed lines). HR-A and HR-B, heptad repeat domains; TM, transmembrane domain.
Substitutions found in the F proteins of multiple SSPE strains confer increased fusion activity.
Although the amino acid sequences of the F protein are highly conserved among clinical isolates and vaccine strains (53), SSPE strains have many substitutions throughout the F protein. There are no apparently characteristic substitutions, but it is notable that most SSPE strains have substitutions in the microdomain, as well as the N-terminal region of the HR-B domain. We noted that the T461I and S103I/N462S/N465S substitutions are present in multiple SSPE strains and located in the fusion-controlling regions described above (Fig. 2). Using virus-like particles, Ayata et al. have reported that the neurovirulence of one SSPE strain (Osaka-2) is related to the ability of its F protein to induce syncytia in Vero cells, which is attributed to a single substitution (T461I) (54).
We examined whether the T461I and S103I/N462S/N465S substitutions confer enhanced fusion activity on the wild-type F protein. When expressed together with the wild-type H protein, the F protein containing the T461I or S103I/N462S/N465S substitutions induced larger syncytia in Vero/hSLAM cells than the wild-type F protein (Fig. 1A). The quantitative plasmid-mediated fusion assay also showed that these two mutant F proteins have 2.5- to 5-times-higher fusion activity in Vero/hSLAM cells than the wild-type F protein (Fig. 1B). The single substitution N462S or N465S slightly increased the fusion activity of the F protein, but the combined substitutions S103I/N462S/N465S additively enhanced fusion activity. The F protein containing the T461I or S103I/N462S/N465S substitutions also induced syncytia in receptor-negative (SLAM- and nectin 4-negative) Vero cells (Fig. 1C and D).
Surface expression of the mutant F proteins with enhanced fusion activity.
To determine why these mutant F proteins exhibit enhanced fusion activity, their cell surface expression levels were examined (Fig. 3). All mutant F proteins had surface expression levels comparable to or slightly lower than that of the wild-type F protein, unlike the F protein with C68S/C195S substitutions, which is known to exhibit markedly decreased surface expression (48). The results indicate that the enhanced fusion activity of the mutant F proteins is due to their intrinsic fusogenicity, not to an increase in their surface expression levels.
Fig 3.
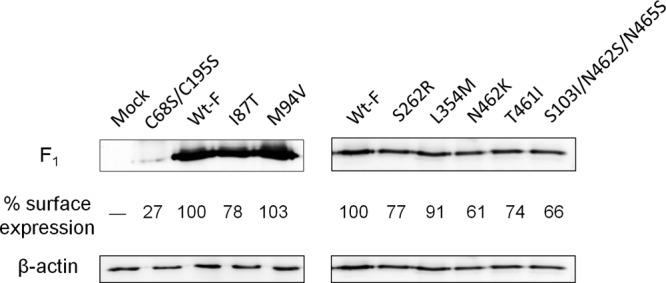
Expression levels of mutant F proteins at the cell surface. Biotinylated cell surface proteins separated by SDS-PAGE were reacted with antisera directed against the cytosolic domain of the F protein (detecting the F1 subunit of the F protein). The upper left gel was exposed longer to allow visualization of the F protein with C68S/C195S substitutions, which has been known to exhibit markedly decreased surface expression. As a loading control, cell lysates were also subjected to SDS-PAGE and reacted with antibodies against β-actin. The surface expression level of each F variant was quantified and normalized to that of β-actin. The relative expression levels of F proteins are indicated below the respective bands (the normalized value of the Wt-F protein was set to 100). The data shown are representative of three experiments.
Recombinant MVs with mutant F proteins induce syncytium formation in cells lacking both SLAM and nectin 4.
To examine the effect of enhanced fusion activity of the F protein on MV pathogenicity, we selected five different F-protein substitutions (S262R, L354M, N462K, T461I, and S103I/N462S/N465S) that can induce high levels of fusion in Vero cells (Fig. 1D). These substitutions were incorporated into IC323-EGFP by using reverse genetics, producing five recombinant viruses expressing the wild-type H protein (without the epitope tag) and the respective mutant F proteins. The parental IC323-EGFP induced syncytia in nectin 4-positive cells, as well as in SLAM-positive cells. However, IC323-EGFP never produced syncytia in Vero cells lacking effective receptors (SLAM and nectin 4) for wild-type MV, although it occasionally infected single cells (Fig. 4A). In contrast, all five recombinant viruses with the mutant F proteins induced syncytia even in Vero cells.
Fig 4.

Syncytium formation in SLAM- and nectin 4-negative cells infected with recombinant MVs. Vero (A), IMR-32 (B), and SYM-1 (C) cells were infected with the parental recombinant MV expressing EGFP (IC323-EGFP) or its mutants expressing the F protein with the indicated substitutions at an MOI of 0.1. At 72 h after infection, EGFP fluorescence in infected cell monolayers was observed under a fluorescence microscope. Representative images are shown.
These recombinant viruses were also examined for the ability to infect the human neuroblastoma cell lines IMR-32 and SYM-1, which do not express SLAM and nectin 4. The absence of nectin 4 in these neuroblastoma cell lines was confirmed by reverse transcription (RT)-PCR to detect its transcripts (data not shown). Infection of IMR-32 and SYM-1 cells with IC323-EGFP resulted in some infected cells, but syncytia were never produced (Fig. 4B and C). Anti-human nectin 4 monoclonal antibody, which completely blocked infection of H358 cells with IC323-EGFP, did not affect that of IMR-32 and SYM-1 cells (data not shown). The five viruses with the mutant F proteins again induced syncytia in IMR-32 and SYM-1 cells, although the virus with the L354M substitution [IC-F(L354M)-EGFP] did not spread as efficiently as the other mutant viruses (Fig. 4B and C). Moreover, these five viruses induced larger syncytia in H358 cells (nectin 4-positive cells) than IC323-EGFP (data not shown).
Neurovirulence of recombinant MVs with mutant F proteins.
To evaluate the neurovirulence of recombinant MVs with the mutant F proteins, we used a hamster model for MV infection with minor modifications (54). Six 10-day-old suckling hamsters were inoculated with IC323-EGFP or each mutant virus intracerebrally (10,000 PFU/brain). No hamsters inoculated with IC323-EGFP showed any symptoms for 4 weeks postinoculation (Fig. 5). In contrast, most of the hamsters inoculated with the recombinant viruses with the mutant F proteins, except IC-F(L354M)-EGFP, died or became moribund approximately 1 week postinoculation. Neurological signs (abnormal gait, convulsions, and continuous tremors) were observed 1 day before the hamsters became moribund. The L354M substitution has the weakest enhancing effect on the fusion activity of the F protein among the five substitutions (Fig. 1B and D and 4B and C). IC-F(L354M)-EGFP exhibited no neurovirulence in hamsters, while the other 4 mutant viruses showed high lethality (66 to 100%) (Fig. 5). Fusion activity in SLAM- and nectin 4-negative cells appeared to correlate well with the survival rate after virus challenge.
Fig 5.
Mortality of hamsters after intracerebral inoculation with the mutant viruses exhibiting enhanced fusion activity. Six 10-day-old hamsters were infected with 104 PFU of wild-type MV (IC323-EGFP) (Wt), MV-F(T461I) (T461I), MV-F(S103I/N462S/N465S) (S103I/N462S/N465S), MV-F(L354M) (L354M), MV-F(S262R) (S262R), or MV-F(N462K) (N462K). Mock-infected hamsters were inoculated with growth medium (Mock). The animals were monitored for 28 days.
The brains were collected from moribund and dead animals, and the spread of EGFP-expressing recombinant viruses was examined under a fluorescence stereomicroscope (Fig. 6). Three hamsters were additionally inoculated with IC323-EGFP or IC-F(L354M)-EGFP and euthanized 6 days postinoculation for microscopic observation. While EGFP was not detected in the brains of hamsters inoculated with IC323-EGFP, IC-F(L354M)-EGFP was found to spread locally in the cerebrum. In sharp contrast, hamsters inoculated with the other 4 mutant viruses showed strong EGFP expression widely in their brains. After the observation, the brains were cocultured with Vero/hSLAM cells to recover viruses from hamsters inoculated with the mutant viruses. No amino acid substitution was found in the H, F, and M proteins of any recovered mutant virus compared with those of the virus used for challenge (data not shown), excluding the possibility that some mutations newly acquired in vivo account for the observed phenotype.
Fig 6.

Spread of EGFP-expressing recombinant viruses in the brains of infected hamsters. The brains of euthanized or dead hamsters were observed under a fluorescence stereomicroscope, and the spread of EGFP-expressing recombinant viruses was examined. Light and EGFP images of the brains were photographed, and top and sagittal views (indicated in the inset by a rectangle and a dotted line, respectively) are shown. For IC323-EGFP and IC323-F(L354M)EGFP, high-sensitivity EGFP images (with a longer exposure time) are also shown.
After observation with a fluorescence stereomicroscope, some of the brains were also subjected to histopathological examination (Fig. 7). Inflammatory cell infiltration was observed widely in the cerebra of hamsters inoculated with recombinant viruses possessing the mutant F proteins, except IC-F(L354M)-EGFP. In some lesions, monocytic or histiocytic cells predominantly invaded the inflamed sites, whereas the infiltration of lymphoid cells was observed in others. Severe lesions were found, especially in the cerebral cortex (Fig. 7A, B, and E) and the pyramidal layer and dentate gyrus of the hippocampus (Fig. 7C, D, and F). In these lesions, nuclear fragmentation, indicative of apoptotic cell death, was also observed. Syncytial giant cells were not observed in any brain samples. In contrast, few changes were found in the brains of hamsters inoculated with IC323-EGFP or IC-F(L354M)-EGFP. Immunohistochemical analysis (data not shown) confirmed that the region where EGFP was detected also expressed MV proteins. Moreover, EGFP colocalized with an astrocyte marker, GFAP, or with a neuronal marker, NeuN, showing that the mutant viruses infected astrocytes, as well as neurons, although the number of EGFP-positive astrocytes was smaller than that of EGFP-positive neurons.
Fig 7.

Histopathological examination of the brains of infected hamsters. The sections from the brains of infected hamsters were stained with HE. (A and B) Lesions in the cerebral cortexes of hamsters inoculated with IC-F(S262R)-EGFP. Invasion of monocytic or histiocytic cells into the parenchyma (A) or perivascular space (B) is shown. (C and D) Lesions in the hippocampuses of hamsters inoculated with IC-F(T461I)-EGFP. Infiltration of monocytic or histiocytic cells into the pyramidal layer (C) or dentate gyrus (D) of the hippocampus is shown. The arrows indicate nuclear fragmentation. (E and F) Lesions in the brains of hamsters inoculated with IC-F(N462K)-EGFP. Invasion of lymphoid cells into the cerebral cortex (E) or the pyramidal layer of the hippocampus (F) is shown.
Taken together, the results indicate that neurovirulence is correlated with the severity of lesions induced by viruses [IC323-EGFP and IC-F(L354M)-EGFP versus recombinant viruses with the other mutant F proteins].
Infection of SLAM- and nectin 4-negative cells with the recombinant MV lacking the M protein or the cytoplasmic domain of the F protein.
To compare the contributions of fusion-enhancing mutations in the extracellular domain of the F protein to SLAM- and nectin 4-independent MV infection with that of defects of the M protein, Vero and IMR-32 cells were infected with the recombinant virus lacking the M protein or the cytoplasmic domain of the F protein. First, the recombinant MV lacking the M protein (M-less MV) was generated by using reverse genetics, and the absence of the M protein was confirmed in virus-infected cells. Consistent with a previous report (37), M-less MV induced larger syncytia in Vero/hSLAM cells than wild-type MV (data not shown). As M-less MV could reach only low titers, it was not possible to perform infection of SLAM- and nectin 4-negative cells at sufficiently high multiplicities of infection (MOIs). Instead, BHK-T7/9 cells used for rescue of the recombinant M-less MV were detached by trypsin digestion and overlaid onto Vero or IMR-32 cells. Unexpectedly, apparent syncytium formation was not observed in either cell line (Fig. 8). Although small syncytium-like cells were occasionally observed in the cells cocultured with the rescued M-less MV, they were much smaller than syncytia formed by recombinant MVs possessing the mutant F proteins that were similarly rescued and processed (Fig. 8) (syncytia were observed 3 days, but not 1 day, after overlay). Moreover, expansion of these small syncytium-like cells was not found, even when the cells were observed 7 days after overlay.
Fig 8.

Limited fusogenic activity of the recombinant virus lacking the M protein in SLAM- and nectin 4-negative cells. IC323-EGFP, mutant viruses expressing the F protein with the indicated substitutions, and M-less MV (IC323-ΔM-EGFP) were rescued by using reverse genetics. BHK-T7/9 cells used for virus rescue were detached by trypsin digestion at 24 h after transfection with full-length-genome plasmids, and the preparation containing 200 EGFP-positive cells was overlaid onto a confluent monolayer of Vero (A) or IMR-32 (B) cells. At 1 or 3 days after overlay, EGFP fluorescence was observed under a fluorescence microscope. Representative images are shown.
Second, we examined the recombinant MV carrying the F protein lacking the cytoplasmic tail (IC323-EGFP-FΔ30) (39). Although we found that IC323-EGFP-FΔ30 induced larger syncytia in SLAM-positive cells than wild-type MV (39), the virus did not induce syncytia in Vero and IMR-32 cells (Fig. 9A and B). In addition, 8 hamsters were inoculated with IC323-EGFP-FΔ30, and 3 of them were euthanized 6 days postinoculation for microscopic observation (Fig. 9C). EGFP was detected in the brains of hamsters inoculated with IC323-EGFP-FΔ30 or IC323-EGFP only when observed in a high-sensitivity mode (with a long exposure time). The spread of IC323-EGFP-FΔ30 was locally restricted in the cerebrum, although it was more widespread than that of IC323-EGFP. The remaining 5 hamsters were examined daily for clinical symptoms of infection for 4 weeks postinoculation. No hamsters showed any symptoms, and EGFP was not detected in the brains of the hamsters at 4 weeks postinoculation (data not shown). The recombinant M-less MV could not be tested for in vivo infection experiments because of its low titers.
Fig 9.

Infection with the recombinant MV with the F protein lacking the cytoplasmic tail (IC323-EGFP-FΔ30). SLAM- and nectin 4-negative Vero (A) and IMR-32 (B) cells were infected with IC323-EGFP or IC323-EGFP-FΔ30 at an MOI of 0.1. At 72 h after infection, EGFP fluorescence in infected cell monolayers was observed under a fluorescence microscope. Representative images are shown. (C) Spread of IC323-EGFP or IC323-EGFP-FΔ30 in the brains of infected hamsters. The brains of euthanized hamsters were observed as described in the legend to Fig. 6. High-sensitivity EGFP images with longer exposure times are also shown.
DISCUSSION
In this study, we have demonstrated that recombinant MVs bearing the mutant F proteins with enhanced fusion activity induce cell fusion in SLAM- and nectin 4-negative cells and exhibit neurovirulence in hamsters, unlike the parental wild-type MV. The mutations introduced into the F proteins of these recombinant viruses are (i) those found in multiple SSPE strains (T461I and S103I/N462S/N465S) and (ii) those not found in SSPE strains (S262R, L354M, and N462K). IC-F(L354M)-EGFP, possessing the least enhanced fusion activity among these viruses, spread locally in the brains of inoculated hamsters but did not cause lethality. These results suggest that enhanced fusion activity is one of the major determinants for MV neurovirulence and that MV must have a certain level of fusion activity in cells lacking both SLAM and nectin 4 in order to exhibit neurovirulence.
From the genetic study of SSPE strains, it has been thought that defects of the M protein play a crucial role in MV neuropathogenicity (55, 56). While cumulative mutations in the M protein may lead to the lack of virus particle formation and escape from host immune responses, the deletion of the M protein can also enhance membrane fusion (37). In addition, the cytoplasmic domain of the F protein, which interacts with the M protein, is elongated or shortened in some SSPE strains (34–36), and its deletion has been shown to enhance cell-cell fusion (38, 39). These fusion-enhancing effects were thought to be important for MV spread in the CNS. However, the effects of the deletion of the M protein or the F-protein cytoplasmic domain on MV fusion activity have been studied in SLAM- or CD46-dependent infection, but not in SLAM- and nectin 4-independent infection. Since SLAM and nectin 4 (receptors used by wild-type MV) were found to be scarcely expressed in the human brain (26, 57, 58), the relevance of the above-mentioned fusion-enhancing mutations in the M and F proteins should be reexamined in the context of SLAM- and nectin 4-independent infection. In fact, we could not detect the expression of nectin 4 in human neuroblastoma cells used in the present study, and anti-nectin 4 antibody did not block inefficient infection of these neuroblastoma cells with wild-type MV. In addition, Zhang et al. reported that other human neuroblastoma cell lines hardly express nectin 4 (59). Importantly, several whole-transcriptome analyses showed that the expression level of nectin 4 mRNA in the human brain is extremely low. These transcriptome data are accessible at the databases Gene Expression Atlas (http://www.ebi.ac.uk/gxa/) and RefExA (http://157.82.78.238/refexa/main_search.jsp). In contrast, it was reported that nectin 4 is expressed in the dog brain and is involved in the neurovirulence of canine distemper virus, which is closely related to MV and belongs to the genus Morbillivirus (60). The difference in neuropathogenicity between MV and canine distemper virus may be explained by the species difference in the tissue distribution of nectin 4.
Interestingly, neither M-less MV nor the recombinant MV carrying the F protein lacking the cytoplasmic tail (IC323-EGFP-FΔ30) induced apparent syncytia in Vero and IMR-32 cells (Fig. 8 and 9), although their fusion-enhancing effects were evident in SLAM-positive cells (37–39). In addition, IC323-EGFP-FΔ30 spread only locally in the brains of inoculated hamsters, unlike recombinant viruses with mutations in the extracellular domain of the F protein (Fig. 9). It is known that the M protein interacts with the cytoplasmic domains of viral envelope glycoproteins, as well as the viral RNP complex. Defects of the M protein, including those caused by deletion of the M protein or the cytoplasmic domain of the F protein may affect the structure and/or stability of viral envelope glycoproteins, leading to fusion enhancement in SLAM-, nectin 4-, or CD46-dependent infection. Moreover, the M protein inhibits MV RNA synthesis through its interaction with the viral RNP complex (via direct interaction with the N protein) (61). Thus, defects of the M protein may also increase the production of envelope glycoproteins. However, these effects caused by defects of the M protein are not enough to induce cell-cell fusion in cells lacking SLAM and nectin 4. In contrast, recombinant MVs with mutations in the extracellular domain of the F protein caused cell-cell fusion even in SLAM- and nectin 4-negative cells. Since the surface expression levels of these mutant F proteins were not affected significantly (Fig. 3), the intrinsic fusogenicity of these F proteins must be increased to induce cell-to-cell fusion in cells lacking SLAM and nectin 4.
In MV entry and virus-mediated cell-cell fusion, binding of the H protein to a cellular receptor triggers a series of conformational changes of the F protein, leading to membrane fusion. Thus, wild-type MV usually does not infect SLAM- and nectin 4-negative cells, nor does it induce syncytia in them. However, it is known that MV can infect various cultured cells (including neuronal cells and Vero cells) independent of known receptors, albeit at low efficiencies (100- to 1,000-fold lower than that of SLAM-dependent infection) (44). This inefficient infection produces solitary infected cells but does not induce syncytia. This presumably occurs because the F protein may sometimes be activated when the H protein interacts with an unidentified “inefficient receptor(s).” A previous study reported that the affinity of the H protein for a receptor had little impact on virus attachment, but it is nevertheless a key determinant of infectivity and cell-to-cell fusion (62). The expected low affinity of the H protein for the inefficient receptor might lead to inefficient infection but fail to induce cell-to-cell fusion (62). Certain substitutions in the extracellular domain of the F protein, especially the microdomain and HR-B domain, may decrease threshold levels for fusion triggering by the H protein, resulting in syncytium formation even in SLAM- and nectin 4-negative cells (via the inefficient receptor).
Notably, enhanced fusion activity of the F protein may be detrimental to the virus, as it can result in stronger cytopathogenicity and decreased virus production in SLAM-positive cells (43). This may be a reason why the F protein is highly conserved among clinical isolates and vaccine strains. However, the situation may be different in the CNS, where there are few efficient receptors available for wild-type MV and enhanced fusion activity allows the virus to spread via cell-to-cell fusion. Indeed, we demonstrated that MVs possessing enhanced fusion activity spread widely in the cerebra of hamsters lacking effective MV receptors, unlike the wild-type MV. Syncytial giant cells were not present in the brain samples as examined histopathologically, although viral proteins and GFP were detected widely in the cerebrum, including the cerebral cortex and hippocampus, by immunohistochemistry. This is consistent with the clinical observation that syncytia are not detected in the brains of SSPE patients (63). Recombinant MVs having the mutant F proteins were found to infect not only neurons, but also astrocytes (data not shown). The cell-cell contacts between these cells may be limited to small areas, such as synapses, and may be mostly hindered by other supporting cells and myelinated nerve fibers. This spatial arrangement may be a reason why neuronal cells do not form syncytia in MV-infected brains.
Recently, Seki et al. reported that an SSPE-derived strain (SI) uses CD46 and exhibits reduced fusion activity (45). The ability to use CD46 as a receptor, rather than enhanced fusion activity, may be a critical determinant for the strain's neuropathogenicity. It is possible that the ability to use the CD46 receptor has been acquired during virus isolation. In fact, many SSPE strains, including the SI strain, were isolated with Vero cells (27, 28, 64). Shingai et al. have reported that SSPE strains isolated with SLAM-positive cells do not utilize CD46 as a receptor (65). At any rate, the use of the CD46 receptor may be another strategy for MV to spread in the CNS, although its occurrence in vivo should be established by isolating more SSPE strains by using SLAM-positive (or nectin 4-positive) cells.
The present study indicates that even single-amino-acid substitutions (including those thus far unreported among SSPE strains) in its extracellular domain can confer enhanced fusion activity on the F protein, thereby allowing the virus to exhibit neurovirulence. Indeed, many SSPE strains have substitutions at different positions of the extracellular domain of the F protein, especially in the regions critical for controlling fusion activity, the microdomain and HR-B domain. Since nucleotide sequences of the F gene have been determined from only a limited number of SSPE strains, analysis of more SSPE strains, especially those isolated in SLAM-positive cells, may reveal unidentified mutations of the F gene, including those found in our study.
In the present study, we used 10-day-old suckling hamsters, as 3-week-old weanling hamsters were not susceptible to the viruses, unlike the prior report using virus-like particles (54). This suggests that the maturity of the host immune system is also important for MV neuropathogenicity in hamsters. At present, we do not know how MVs with mutant F proteins infect hamster neurons and astrocytes. It should be noted that receptors in hamsters may not be the same as the molecules used by MV in human patients with SSPE. Furthermore, in humans, acquisition of enhanced fusion activity due to mutations in the extracellular domain of the F protein may be a late event following several years of persistent MV infection in the CNS. Once acquisition of enhanced fusion activity occurs, viruses may spread effectively in the brain and cause SSPE. On the other hand, by using recombinant MVs with enhanced fusion activity, we can observe neurovirulence in hamsters within ∼7 days.
Fusion-inhibitory reagents that efficiently block paramyxovirus-mediated fusion in vitro are available (66–69), and some of them are effective even for in vivo infection (70). Thus, blocking MV-mediated fusion with these reagents has the potential to be a good therapeutic approach for SSPE. To test these fusion-inhibitory reagents, the hamster model used in this study would be useful, and the development of effective therapy targeting MV-mediated fusion is the next step of the current study.
In conclusion, our data indicate that fusion-enhancing mutations in the extracellular domain of the F protein can cause SLAM- and nectin 4-independent cell fusion. Since human neuronal cells are mainly SLAM and nectin 4 negative, enhanced fusion activity may be one of the major determinants of MV spread and pathogenicity in the CNS, independently of defects of the M protein.
ACKNOWLEDGMENTS
We thank M. Nakashima and S. Yamamoto for helping establish the quantitative fusion assay. We also thank R. Cattaneo for providing the antibodies against MV-F and M. Tahara, T. Hashiguchi, M. Ayata, and J. Arii for providing technical information.
This study was supported by grants from the Ministry of Health, Labor and Welfare (the Research Committee of Prion Disease and Slow Virus Infection) of Japan and by JSPS KAKENHI grant numbers 21249032, 24115005, and 24790444.
Footnotes
Published ahead of print 19 December 2012
REFERENCES
- 1. Rima BK, Duprex WP. 2006. Morbilliviruses and human disease. J. Pathol. 208:199–214 [DOI] [PubMed] [Google Scholar]
- 2. Bellini WJ, Rota JS, Lowe LE, Katz RS, Dyken PR, Zaki SR, Shieh WJ, Rota PA. 2005. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than was previously recognized. J. Infect. Dis. 192:1686–1693 [DOI] [PubMed] [Google Scholar]
- 3. Griffin DE. 2007. Measles virus, p1551–1585 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4. Gutierrez J, Issacson RS, Koppel BS. 2010. Subacute sclerosing panencephalitis: an update. Dev. Med. Child Neurol. 52:901–907 [DOI] [PubMed] [Google Scholar]
- 5. Hashiguchi T, Ose T, Kubota M, Maita N, Kamishikiryo J, Maenaka K, Yanagi Y. 2011. Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat. Struct. Mol. Biol. 18:135–141 [DOI] [PubMed] [Google Scholar]
- 6. Plemper RK, Brindley MA, Iorio RM. 2011. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog. 7:e1002058 doi:10.1371/journal.ppat.1002058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yanagi Y, Takeda M, Ohno S. 2006. Measles virus: cellular receptors, tropism and pathogenesis. J. Gen. Virol. 87:2767–2779 [DOI] [PubMed] [Google Scholar]
- 8. Eckert DM, Kim PS. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777–810 [DOI] [PubMed] [Google Scholar]
- 9. Prussia AJ, Plemper RK, Snyder JP. 2008. Measles virus entry inhibitors: a structural proposal for mechanism of action and the development of resistance. Biochemistry 47:13573–13583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Russell CJ, Jardetzky TS, Lamb RA. 2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20:4024–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Samuel O, Shai Y. 2001. Participation of two fusion peptides in measles virus-induced membrane fusion: emerging similarity with other paramyxoviruses. Biochemistry 40:1340–1349 [DOI] [PubMed] [Google Scholar]
- 12. Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897 [DOI] [PubMed] [Google Scholar]
- 13. Erlenhofer C, Duprex WP, Rima BK, ter Meulen V, Schneider-Schaulies J. 2002. Analysis of receptor (CD46, CD150) usage by measles virus. J. Gen. Virol. 83:1431–1436 [DOI] [PubMed] [Google Scholar]
- 14. Ono N, Tatsuo H, Hidaka Y, Aoki T, Minagawa H, Yanagi Y. 2001. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J. Virol. 75:4399–4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dorig RE, Marcil A, Chopra A, Richardson CD. 1993. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 75:295–305 [DOI] [PubMed] [Google Scholar]
- 16. Manchester M, Liszewski MK, Atkinson JP, Oldstone MB. 1994. Multiple isoforms of CD46 (membrane cofactor protein) serve as receptors for measles virus. Proc. Natl. Acad. Sci. U. S. A. 91:2161–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdin-Combe C, Gerlier D. 1993. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J. Virol. 67:6025–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hsu EC, Sarangi F, Iorio C, Sidhu MS, Udem SA, Dillehay DL, Xu W, Rota PA, Bellini WJ, Richardson CD. 1998. A single amino acid change in the hemagglutinin protein of measles virus determines its ability to bind CD46 and reveals another receptor on marmoset B cells. J. Virol. 72:2905–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tahara M, Takeda M, Seki F, Hashiguchi T, Yanagi Y. 2007. Multiple amino acid substitutions in hemagglutinin are necessary for wild-type measles virus to acquire the ability to use receptor CD46 efficiently. J. Virol. 81:2564–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leonard VH, Sinn PL, Hodge G, Miest T, Devaux P, Oezguen N, Braun W, McCray PB, Jr, McChesney MB, Cattaneo R. 2008. Measles virus blind to its epithelial cell receptor remains virulent in rhesus monkeys but cannot cross the airway epithelium and is not shed. J. Clin. Invest. 118:2448–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takeda M, Tahara M, Hashiguchi T, Sato TA, Jinnouchi F, Ueki S, Ohno S, Yanagi Y. 2007. A human lung carcinoma cell line supports efficient measles virus growth and syncytium formation via a SLAM- and CD46-independent mechanism. J. Virol. 81:12091–12096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takeuchi K, Miyajima N, Nagata N, Takeda M, Tashiro M. 2003. Wild-type measles virus induces large syncytium formation in primary human small airway epithelial cells by a SLAM(CD150)-independent mechanism. Virus Res. 94:11–16 [DOI] [PubMed] [Google Scholar]
- 23. Herndon RM, Rubinstein LJ. 1968. Light and electron microscopy observations on the development of viral particles in the inclusions of Dawson's encephalitis (subacute sclerosing panencephalitis). Neurology 18:8–20 [DOI] [PubMed] [Google Scholar]
- 24. Ishida H, Ayata M, Shingai M, Matsunaga I, Seto Y, Katayama Y, Iritani N, Seya T, Yanagi Y, Matsuoka O, Yamano T, Ogura H. 2004. Infection of different cell lines of neural origin with subacute sclerosing panencephalitis (SSPE) virus. Microbiol. Immunol. 48:277–287 [DOI] [PubMed] [Google Scholar]
- 25. Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 7:e1002240 doi:10.1371/journal.ppat.1002240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cattaneo R, Schmid A, Billeter MA, Sheppard RD, Udem SA. 1988. Multiple viral mutations rather than host factors cause defective measles virus gene expression in a subacute sclerosing panencephalitis cell line. J. Virol. 62:1388–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Homma M, Tashiro M, Konno H, Ohara Y, Hino M, Takase S. 1982. Isolation and characterization of subacute sclerosing panencephalitis virus (Yamagata-1 strain) from a brain autopsy. Microbiol. Immunol. 26:1195–1202 [DOI] [PubMed] [Google Scholar]
- 29. Ito N, Ayata M, Shingai M, Furukawa K, Seto T, Matsunaga I, Muraoka M, Ogura H. 2002. Comparison of the neuropathogenicity of two SSPE sibling viruses of the Osaka-2 strain isolated with Vero and B95a cells. J. Neurovirol. 8:6–13 [DOI] [PubMed] [Google Scholar]
- 30. Makino S, Sasaki K, Nakagawa M, Saito M, Shinohara Y. 1977. Isolation and biological characterization of a measles virus-like agent from the brain of an autopsied case of subacute sclerosing panencephalitis (SSPE). Microbiol. Immunol. 21:193–205 [DOI] [PubMed] [Google Scholar]
- 31. Ayata M, Komase K, Shingai M, Matsunaga I, Katayama Y, Ogura H. 2002. Mutations affecting transcriptional termination in the p gene end of subacute sclerosing panencephalitis viruses. J. Virol. 76:13062–13068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cattaneo R, Rebmann G, Schmid A, Baczko K, ter Meulen V, Billeter MA. 1987. Altered transcription of a defective measles virus genome derived from a diseased human brain. EMBO J. 6:681–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cattaneo R, Schmid A, Rebmann G, Baczko K, Ter Meulen V, Bellini WJ, Rozenblatt S, Billeter MA. 1986. Accumulated measles virus mutations in a case of subacute sclerosing panencephalitis: interrupted matrix protein reading frame and transcription alteration. Virology 154:97–107 [DOI] [PubMed] [Google Scholar]
- 34. Billeter MA, Cattaneo R, Spielhofer P, Kaelin K, Huber M, Schmid A, Baczko K, ter Meulen V. 1994. Generation and properties of measles virus mutations typically associated with subacute sclerosing panencephalitis. Ann. N. Y. Acad. Sci. 724:367–377 [DOI] [PubMed] [Google Scholar]
- 35. Ning X, Ayata M, Kimura M, Komase K, Furukawa K, Seto T, Ito N, Shingai M, Matsunaga I, Yamano T, Ogura H. 2002. Alterations and diversity in the cytoplasmic tail of the fusion protein of subacute sclerosing panencephalitis virus strains isolated in Osaka, Japan. Virus Res. 86:123–131 [DOI] [PubMed] [Google Scholar]
- 36. Schmid A, Spielhofer P, Cattaneo R, Baczko K, ter Meulen V, Billeter MA. 1992. Subacute sclerosing panencephalitis is typically characterized by alterations in the fusion protein cytoplasmic domain of the persisting measles virus. Virology 188:910–915 [DOI] [PubMed] [Google Scholar]
- 37. Cathomen T, Mrkic B, Spehner D, Drillien R, Naef R, Pavlovic J, Aguzzi A, Billeter MA, Cattaneo R. 1998. A matrix-less measles virus is infectious and elicits extensive cell fusion: consequences for propagation in the brain. EMBO J. 17:3899–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cathomen T, Naim HY, Cattaneo R. 1998. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J. Virol. 72:1224–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tahara M, Takeda M, Yanagi Y. 2007. Altered interaction of the matrix protein with the cytoplasmic tail of hemagglutinin modulates measles virus growth by affecting virus assembly and cell-cell fusion. J. Virol. 81:6827–6836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kitamura T, Morikawa Y. 2000. Isolation of T-cell antigens by retrovirus-mediated expression cloning. Methods Mol. Biol. 134:143–152 [DOI] [PubMed] [Google Scholar]
- 41. Onishi M, Kinoshita S, Morikawa Y, Shibuya A, Phillips J, Lanier LL, Gorman DM, Nolan GP, Miyajima A, Kitamura T. 1996. Applications of retrovirus-mediated expression cloning. Exp. Hematol. 24:324–329 [PubMed] [Google Scholar]
- 42. Shirogane Y, Takeda M, Tahara M, Ikegame S, Nakamura T, Yanagi Y. 2010. Epithelial-mesenchymal transition abolishes the susceptibility of polarized epithelial cell lines to measles virus. J. Biol. Chem. 285:20882–20890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takeda M, Ohno S, Seki F, Nakatsu Y, Tahara M, Yanagi Y. 2005. Long untranslated regions of the measles virus M and F genes control virus replication and cytopathogenicity. J. Virol. 79:14346–14354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hashimoto K, Ono N, Tatsuo H, Minagawa H, Takeda M, Takeuchi K, Yanagi Y. 2002. SLAM (CD150)-independent measles virus entry as revealed by recombinant virus expressing green fluorescent protein. J. Virol. 76:6743–6749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seki F, Yamada K, Nakatsu Y, Okamura K, Yanagi Y, Nakayama T, Komase K, Takeda M. 2011. The si strain of measles virus derived from a patient with subacute sclerosing panencephalitis possesses typical genome alterations and unique amino Acid changes that modulate receptor specificity and reduce membrane fusion activity. J. Virol. 85:11871–11882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Komase K, Nakayama T, Iijima M, Miki K, Kawanishi R, Uejima H. 2006. The phosphoprotein of attenuated measles AIK-C vaccine strain contributes to its temperature-sensitive phenotype. Vaccine 24:826–834 [DOI] [PubMed] [Google Scholar]
- 47. Doyle J, Prussia A, White LK, Sun A, Liotta DC, Snyder JP, Compans RW, Plemper RK. 2006. Two domains that control prefusion stability and transport competence of the measles virus fusion protein. J. Virol. 80:1524–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Plemper RK, Compans RW. 2003. Mutations in the putative HR-C region of the measles virus F2 glycoprotein modulate syncytium formation. J. Virol. 77:4181–4190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Plemper RK, Lakdawala AS, Gernert KM, Snyder JP, Compans RW. 2003. Structural features of paramyxovirus F protein required for fusion initiation. Biochemistry 42:6645–6655 [DOI] [PubMed] [Google Scholar]
- 50. Wild TF, Fayolle J, Beauverger P, Buckland R. 1994. Measles virus fusion: role of the cysteine-rich region of the fusion glycoprotein. J. Virol. 68:7546–7548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hull JD, Krah DL, Choppin PW. 1987. Resistance of a measles virus mutant to fusion inhibitory oligopeptides is not associated with mutations in the fusion peptide. Virology 159:368–372 [DOI] [PubMed] [Google Scholar]
- 52. Plemper RK, Erlandson KJ, Lakdawala AS, Sun A, Prussia A, Boonsombat J, Aki-Sener E, Yalcin I, Yildiz I, Temiz-Arpaci O, Tekiner B, Liotta DC, Snyder JP, Compans RW. 2004. A target site for template-based design of measles virus entry inhibitors. Proc. Natl. Acad. Sci. U. S. A. 101:5628–5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rota JS, Hummel KB, Rota PA, Bellini WJ. 1992. Genetic variability of the glycoprotein genes of current wild-type measles isolates. Virology 188:135–142 [DOI] [PubMed] [Google Scholar]
- 54. Ayata M, Takeuchi K, Takeda M, Ohgimoto S, Kato S, Sharma LB, Tanaka M, Kuwamura M, Ishida H, Ogura H. 2010. The F gene of the Osaka-2 strain of measles virus derived from a case of subacute sclerosing panencephalitis is a major determinant of neurovirulence. J. Virol. 84:11189–11199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reuter D, Schneider-Schaulies J. 2010. Measles virus infection of the CNS: human disease, animal models, and approaches to therapy. Med. Microbiol. Immunol. 199:261–271 [DOI] [PubMed] [Google Scholar]
- 56. Rima BK, Duprex WP. 2005. Molecular mechanisms of measles virus persistence. Virus Res. 111:132–147 [DOI] [PubMed] [Google Scholar]
- 57. McQuaid S, Cosby SL. 2002. An immunohistochemical study of the distribution of the measles virus receptors, CD46 and SLAM, in normal human tissues and subacute sclerosing panencephalitis. Lab. Invest. 82:403–409 [DOI] [PubMed] [Google Scholar]
- 58. Reymond N, Fabre S, Lecocq E, Adelaide J, Dubreuil P, Lopez M. 2001. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with nectin1/PRR1 through V domain interaction. J. Biol. Chem. 276:43205–43215 [DOI] [PubMed] [Google Scholar]
- 59. Zhang SC, Cai WS, Zhang Y, Jiang KL, Zhang KR, Wang WL. 2012. Engineered measles virus Edmonston strain used as a novel oncolytic viral system against human neuroblastoma through a CD46 and nectin 4-independent pathway. Cancer Lett. 325:227–237 [DOI] [PubMed] [Google Scholar]
- 60. Pratakpiriya W, Seki F, Otsuki N, Sakai K, Fukuhara H, Katamoto H, Hirai T, Maenaka K, Techangamsuwan S, Lan NT, Takeda M, Yamaguchi R. 2012. Nectin4 is an epithelial cell receptor for canine distemper virus and involved in neurovirulence. J. Virol. 86:10207–10210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Iwasaki M, Takeda M, Shirogane Y, Nakatsu Y, Nakamura T, Yanagi Y. 2009. The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J. Virol. 83:10374–10383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hasegawa K, Hu C, Nakamura T, Marks JD, Russell SJ, Peng KW. 2007. Affinity thresholds for membrane fusion triggering by viral glycoproteins. J. Virol. 81:13149–13157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schneider-Schaulies J, Meulen V, Schneider-Schaulies S. 2003. Measles infection of the central nervous system. J. Neurovirol. 9:247–252 [DOI] [PubMed] [Google Scholar]
- 64. Mirchamsy H, Bahrami S, Shafyi A, Shahrabady MS, Kamaly M, Ahourai P, Razavi J, Nazari P, Derakhshan I, Lotfi J, Abassioun K. 1978. Isolation and characterization of a defective measles virus from brain biopsies of three patients in Iran with subacute sclerosing panencephalitis. Intervirology 9:106–118 [DOI] [PubMed] [Google Scholar]
- 65. Shingai M, Ayata M, Ishida H, Matsunaga I, Katayama Y, Seya T, Tatsuo H, Yanagi Y, Ogura H. 2003. Receptor use by vesicular stomatitis virus pseudotypes with glycoproteins of defective variants of measles virus isolated from brains of patients with subacute sclerosing panencephalitis. J. Gen. Virol. 84:2133–2143 [DOI] [PubMed] [Google Scholar]
- 66. Lambert DM, Barney S, Lambert AL, Guthrie K, Medinas R, Davis DE, Bucy T, Erickson J, Merutka G, Petteway SR., Jr 1996. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. U. S. A. 93:2186–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Richardson CD, Scheid A, Choppin PW. 1980. Specific inhibition of paramyxovirus and myxovirus replication by oligopeptides with amino acid sequences similar to those at the N-termini of the F1 or HA2 viral polypeptides. Virology 105:205–222 [DOI] [PubMed] [Google Scholar]
- 68. Singethan K, Hiltensperger G, Kendl S, Wohlfahrt J, Plattet P, Holzgrabe U, Schneider-Schaulies J. 2010. N-(3-Cyanophenyl)-2-phenylacetamide, an effective inhibitor of morbillivirus-induced membrane fusion with low cytotoxicity. J. Gen. Virol. 91:2762–2772 [DOI] [PubMed] [Google Scholar]
- 69. Wild TF, Buckland R. 1997. Inhibition of measles virus infection and fusion with peptides corresponding to the leucine zipper region of the fusion protein. J. Gen. Virol. 78:107–111 [DOI] [PubMed] [Google Scholar]
- 70. Porotto M, Rockx B, Yokoyama CC, Talekar A, Devito I, Palermo LM, Liu J, Cortese R, Lu M, Feldmann H, Pessi A, Moscona A. 2010. Inhibition of Nipah virus infection in vivo: targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog. 6:e1001168 doi:10.1371/journal.ppat.1001168 [DOI] [PMC free article] [PubMed] [Google Scholar]