

Abstract
Hepatitis B virus (HBV) replication requires reverse transcription of an RNA pregenome (pgRNA) by a multifunctional polymerase (HP). HP initiates viral DNA synthesis by using itself as a protein primer and an RNA signal on pgRNA, termed epsilon (Hε), as the obligatory template. We discovered a Mn2+-dependent transferase activity of HP in vitro that was independent of Hε but also used HP as a protein primer. This protein-primed transferase activity was completely dependent on the HP polymerase active site. The DNA products of the transferase reaction were linked to HP via a phosphotyrosyl bond, and replacement of the Y63 residue of HP, the priming site for templated DNA synthesis, almost completely eliminated DNA synthesis by the transferase activity, suggesting that Y63 also serves as the predominant priming site for the transferase reaction. For this transferase activity, HP could use all four deoxynucleotide substrates, but TTP was clearly favored for extensive polymerization. The transferase activity was highly distributive, leading to the synthesis of DNA homo- and hetero-oligomeric and -polymeric ladders ranging from 1 nucleotide (nt) to >100 nt in length, with single-nt increments. As with Hε-templated DNA synthesis, the protein-primed transferase reaction was characterized by an initial stage that was resistant to the pyrophosphate analog phosphonoformic acid (PFA) followed by PFA-sensitive DNA synthesis, suggestive of an HP conformational change upon the synthesis of a nascent DNA oligomer. These findings have important implications for HBV replication, pathogenesis, and therapy.
INTRODUCTION
Hepatitis B virus (HBV) is an important human pathogen and a member of the family Hepadnaviridae, which comprises a group of pararetroviruses replicating a double-stranded DNA genome through an RNA intermediate called pregenomic RNA (pgRNA) by a multifunctional viral polymerase protein (HP), a specialized reverse transcriptase (RT) (1–4). Structurally, HP is divided into four domains, including (from the N to the C terminus) the terminal protein (TP), spacer, RT, and RNase H domains (5–7). Although the RT and RNase H domains are conserved with those found in other RT enzymes, TP is unique to hepadnaviruses, and the spacer has no known function other than serving as a tether between the TP and RT domains (5, 7–11). As with other RTs, HP has RNA- and DNA-dependent DNA polymerase activity. Furthermore, HP also serves as a protein primer; specifically, a highly conserved Y residue in its TP domain (Y63) is used as a primer to initiate viral minus-strand DNA [(−)-DNA] synthesis (12, 13). This protein-primed reverse transcription requires a specific HBV RNA template called epsilon (Hε), located at the 5′ end of the pgRNA, which is specifically recognized by HP and is thought to be an integral component of the HP holoenzyme (14–21). HP-Hε interactions trigger conformational changes in both the protein and RNA of the resulting ribonucleoprotein (RNP) complex, which are thought to be required functionally for HP to gain catalytic activity and for Hε to serve its template function (22–25). The result of protein priming is a covalent HP-DNA complex in which a short DNA oligonucleotide consisting of the first 3 nucleotides (nt) of the viral (−)-DNA is attached to the Y63 primer of HP via a phosphotyrosyl linkage (16, 19, 26–28). In addition, HP-Hε interaction is required for packaging of HP and pgRNA into replication-competent nucleocapsids, in a process where Hε serves as the RNA packaging signal and HP and pgRNA packaging is mutually dependent (14–17, 19, 20, 29).
The availability of in vitro protein priming assays has facilitated detailed analysis of the hepadnavirus protein priming mechanism and the viral and host requirements for this complex reaction (21, 30–33). In particular, experiments using the duck hepatitis B virus (DHBV) polymerase (DP) protein have demonstrated that host chaperone proteins are required to establish a DP conformation competent for binding its cognate (DHBV) epsilon RNA signal (Dε) and that both the TP and RT domains, but not the spacer or the RNase H domain, are required for Dε binding and protein priming (34–41). Furthermore, DP undergoes a series of conformational changes during RNA binding and protein priming that can be manifested as changes in sensitivity to chemical inhibitors such as the pyrophosphate analog phosphonoformic acid (PFA) (31, 40). Interestingly, the choice of divalent metal ion cofactors, particularly Mn2+ versus Mg2+, can have profound effects on DP structures and functions during protein priming in vitro (42). While Mg2+ is generally assumed to be the polymerase cofactor in cells and also seems to preserve faithfully in vitro the requirements for protein priming as defined in cells, such as the strict dependence on Dε and the authentic priming site (Y96, equivalent to Y63 in HP) in the TP domain, Mn2+ is able to stimulate in vitro protein priming activity by an order of magnitude and allows for low levels of Dε-independent protein priming and priming from amino acid residues other than the authentic Y96 residue (i.e., cryptic site priming) (42–44).
In contrast to DP, HP expressed by in vitro translation in a rabbit reticulocyte lysate (RRL) containing all of the eukaryotic chaperones required for DP protein priming (31, 35, 36, 38) or expressed and purified using a bacterial system and reconstituted with eukaryotic chaperones that are sufficient to recapitulate DP priming (37, 39) does not show protein priming activity (17, 30). HP expressed in insect cells by use of the recombinant baculovirus system does show a low level of protein priming activity in vitro (12, 13, 45), leading to the covalent attachment of DNA strands to Y63 in its TP domain, the same primer residue used to initiate viral (−)-DNA synthesis in vivo (46, 47). However, it remains unresolved whether the protein priming activity of HP expressed using this heterologous system depends on Hε or not (13, 45). As with DP, the HP priming activity detected using the insect cell-derived HP was also shown to be influenced greatly by the divalent metal ion cofactor, i.e., Mn2+ versus Mg2+. In particular, Mg2+ appeared to stimulate mainly DNA strand elongation far beyond the synthesis of the nascent 3-nt (−)-DNA during protein priming in vitro by DP, whereas Mn2+ appeared to stimulate the synthesis in vitro of shorter DNA strands attached to HP (45). However, the DNA species synthesized by HP in these in vitro reactions were not characterized in detail.
Recently, we reported an authentic in vitro HP priming assay using HP expressed and purified using human cells (27). In a Mg2+-dependent reaction, the HP priming activity is completely dependent on the viral Hε template. Both initiation (the covalent attachment of the first nucleotide, dGMP, to Y63 in HP via a phosphotyrosyl linkage) and polymerization (the subsequent addition of two dAMP residues to dGMP), two distinct stages of protein priming defined initially using DP (40), can be demonstrated in this new in vitro HP priming system. While characterizing the in vitro HP priming activity using this system, we discovered a novel protein-primed transferase activity of HP that is independent of Hε but stimulated by Mn2+.
MATERIALS AND METHODS
Plasmids.
pcDNA-3FHP, used to express 3× FLAG-tagged full-length HP in human cells, and its mutant derivatives, pcDNA-3FHP-YMHD and pcDNA-3FHP-Y63D, defective in the HP polymerase and primer activities, respectively, have been described previously (16, 27). pCMV-HE, which expresses the Hε RNA derived from the 5′ end of HBV pgRNA, was also described recently (27). pSUMO-MiniRT2 was used to express in Escherichia coli a truncated DP protein termed SUMO-3FMiniRT2, which contains SUMO (small ubiquitin-like modifier) and 3× FLAG tags fused to the DHBV MiniRT2 protein (27, 34, 35, 44, 48).
Protein expression and purification.
HP, with or without bound Hε, was expressed and purified using HEK293T cells as previously described (27). Briefly, HEK293T cells were transfected with pCDNA-3FHP, pcDNA-3FHP-Y63D, or pcDNA-3FHP-YMHD, alone or together with pCMV-HE. Two days after transfection, cells were washed and then lysed at 4°C in FLAG lysis buffer (50 mM Tris [pH 7.0], 100 mM NaCl, 50 mM KCl, 10% glycerol, 1% NP-40, 1 mM EDTA) plus 1× Complete protease inhibitor (Roche), 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM β-mercaptoethanol (β-ME), 2 mM dithiothreitol (DTT), and 250 units RNasin Plus RNase inhibitor (Promega) per ml lysis buffer and scraped off the plate. HP was purified from the clarified lysate by using protein A/G beads (Pierce) bound to the M2 anti-FLAG antibody (Sigma). Unbound materials were removed by washing 5 times with FLAG lysis buffer at 4°C with individual protease inhibitors (28 μM E-64, 1 mM PMSF, and 5 μg/μl leupeptin), 2 mM DTT, 10 mM β-ME, and 10 units RNasin Plus RNase inhibitor per ml lysis buffer. Purified HP was stored in the same washing buffer while remaining bound to the M2 beads at −80°C until use.
In vitro HP priming assay.
The FLAG lysis buffer from HP purification was removed from the HP-bound M2 beads, which contained ca. 200 ng HP per aliquot. HP was then washed in TNK buffer (20 mM Tris-HCl, pH 7, 15 mM NaCl, 10 mM KCl) supplemented with individual protease inhibitors and 10 mM β-ME. TMnNK buffer (20 mM Tris-HCl, pH 7.0, 15 mM NaCl, 10 mM KCl, 1 mM MnCl2) (42) along with 1× EDTA-free protease inhibitor cocktail (Roche), 4 mM DTT, 1 mM PMSF, and 1 unit RNasin Plus RNase inhibitor (Promega) per μl buffer was added to the beads. One microliter of the indicated radiolabeled deoxynucleoside triphosphate ([α-32P]dNTP at 10 mCi/ml and 3,000 Ci/mmol) was then added, and the reaction mixtures were incubated at 25°C for 4 h with shaking. HP priming in the presence of Mg2+ (TMgNK; same as TMnNK, but containing 4 mM Mg2+ and no Mn2+) was performed as described previously (27). DP priming using SUMO-3FMiniRT2 in TMnNK buffer was carried out as previously described (27, 35, 42, 44). Modifications, if any, to this standard priming protocol are described further in the relevant figure legends. After priming, beads were washed in TNK buffer supplemented with individual protease inhibitors and 10 mM β-ME and were then boiled in 2× sodium dodecyl sulfate (SDS) sample buffer for 10 min. Radiolabeled HP as a result of protein priming was resolved by running the eluate in an SDS-12.5% polyacrylamide gel and detected by autoradiography.
DNase, pronase, and RNase treatment.
DNase or pronase treatment of the in vitro protein priming products was carried out as recently described (27). For RNase treatment, purified HP bound to M2 beads was pretreated in FLAG lysis buffer with 2 μg DNase-free RNase (Sigma) for 2 h at 4°C with shaking. The supernatant was then removed, the beads were washed in TNK buffer plus individual protease inhibitors and 10 mM β-ME, and priming reactions were conducted as described above, using [α-32P]TTP (27). After priming, RNase-treated samples were washed to remove unincorporated nt. To monitor the HP protein levels before and after RNase treatment, HP was resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and detected by Western blot analysis using the anti-FLAG M2 antibody (27).
Tdp2-mediated release of DNA covalently linked to HP.
To detect the DNA products attached to HP following in vitro priming, the 32P-labeled HP after protein priming was incubated with Tdp2 to cleave the tyrosyl-DNA linkages between HP and the DNA (or the single nt) as previously described (27, 49, 50), except with 200 to 400 ng Tdp2. The released DNA was collected and resolved in an 8 M urea-20% polyacrylamide sequencing gel as described previously, and the residual HP bound to beads was resolved by SDS-PAGE (27).
Apyrase treatment.
α-32P-labeled dGTP, dCTP, dATP, or TTP was mock treated (with 1 μl double-distilled water [ddH2O]) or treated with 1 μg apyrase (Sigma) for 1 min at 25°C in 150 mM Tris, pH 6.0, 5 mM CaCl2, and 1× EDTA-free protease inhibitor cocktail (Roche). The treated samples were then resolved in an 8 M urea-20% polyacrylamide gel as described previously (27).
Limited Exo I digestion of protein priming products.
HP bound to M2 beads was primed with [α-32P]TTP and Tdp2 treated as outlined above. Purified DP was bound via the M2 anti-FLAG antibody to protein A/G beads (27, 44). Priming reactions for DP bound to protein A/G beads followed the same protocol as that for HP, but with 0.1% NP-40 and 1 μg in vitro-transcribed Dε and with a combination of 1 μl each of [α-32P]dATP, [α-32P]dGTP, [α-32P]TTP, and [α-32P]dCTP to create uniformly labeled 32P-labeled DNA species (27, 41, 42). DP priming reaction mixtures were washed and treated with Tdp2 as described for HP. To produce a 3′-end-labeled 10-bp DNA ladder, 250 ng of a 10-bp ladder (Invitrogen) was incubated with 5 units Taq polymerase (Gene Choice) in 20 mM Tris, pH 8.4, 50 mM KCl, 1.5 mM MgCl2, and 5 μl [α-32P]dATP in a total volume of 50 μl. The labeling reaction was carried out at 72°C for 15 min. Prior to digestion, the 3′-end-labeled 10-bp ladder was denatured by boiling for 5 min followed by cooling on ice for 30 s. The Tdp2-released DNA products from protein priming as well as the 3′-end-labeled and denatured DNA ladder were then treated with exonuclease I (Exo I) under limited digestion conditions. All Exo I digestion reactions were done in 1× Tdp2 buffer with 10 units Exo I (NEB) for 10 min at 25°C and then boiled for 5 min to terminate the digestion. Samples were resolved in an 8 M urea-20% polyacrylamide gel (27).
Calf thymus TdT reaction.
Terminal transferase reactions were conducted with 20 units of E. coli-produced calf thymus terminal deoxynucleotidyltransferase (TdT; New England BioLabs) or ddH2O (mock reaction) in 1× terminal transferase reaction buffer [20 mM Tris-OAc, 50 mM KOAc, 10 mM Mg(OAc)2, pH 7.9; New England BioLabs] with 0.25 mM CoCl2, 1 μl [α-32P]TTP, and 500 ng of a 29-nt DNA oligonucleotide. Reaction mixtures were incubated at 37°C for 1 h, cleared of unincorporated [α-32P]TTP by passing through a Quick Spin Sephadex G-25 RNA column (Roche), and resolved by urea-PAGE analysis as described previously (27).
RESULTS
In vitro Hε-independent priming activity of purified HP in the presence of Mn2+.
As both HP and DP protein priming activities are influenced by the metal ion Mn2+ versus Mg2+ (see the introduction), we were interested in testing the effects of Mn2+ on the in vitro priming activity carried out by HP purified from human cells, which we recently showed to display authentic, Hε-dependent protein priming activity in vitro in the presence of Mg2+ (27). HP purified with or without coexpression of Hε (Fig. 1A, lanes 2 and 3) was thus incubated with [α-32P]dGTP in a Mn2+ priming buffer (see Materials and Methods) similar to that used for in vitro priming reactions with DP (42, 44). In sharp contrast to HP priming carried out in the presence of Mg2+, which is completely dependent on Hε (27), HP priming in the presence of Mn2+ was not dependent on Hε at all (Fig. 1B, lanes 3 and 4). The small amount (200 to 400 pg) of Hε bound to the HP (200 ng) (27) did not stimulate the Hε-independent priming under these conditions. As with Mg2+, the Y63D mutation eliminated almost all of the priming signal (Fig. 1B, lanes 6 and 8), suggesting that HP priming with Mn2+ also occurred predominantly on Y63 of HP, as with Mg2+. Very low levels of cryptic site priming, i.e., independent of Y63, also appeared to occur with HP and Mn2+ (Fig. 1B, lanes 6 and 8), as recently reported for DP (43, 44). On the other hand, priming with Mn2+, as with Mg2+, was completely dependent on the HP polymerase activity, as no priming signal was detected with the YMHD catalytic mutant (Fig. 1B, lanes 5 and 7). Although it is formally possible that instead of serving as a primer Y63 may somehow be required for the HP enzymatic activity with Mn2+, we consider this to be very unlikely given the known role of Y63 as a primer with Mg2+, the known role of the YMDD active site in catalysis with both metal ions, and the phosphotyrosyl nature of the covalent DNA-HP linkage formed by protein priming with Mn2+ (see Fig. 2), as with Mg2+. It is also unlikely that the Y63D mutation causes global misfolding of HP, as it shows no effect on HP-Hε interaction (27) or HP function in pgRNA packaging (47). As expected from protein priming, the labeled HP signal, with or without Hε, was sensitive to pronase but not DNase digestion (Fig. 1C).
Fig 1.
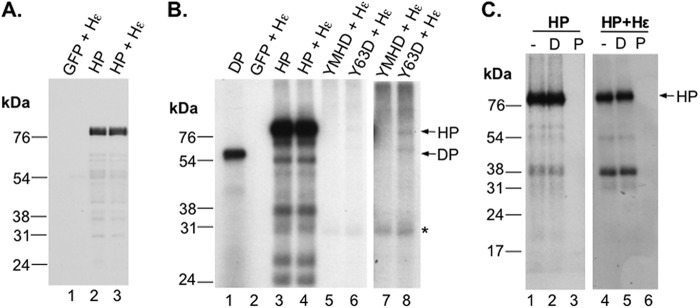
Hε-independent priming activity in vitro by purified HP in the presence of Mn2+. HP, YMHD, or Y63D polymerase was purified using anti-FLAG-bound protein A/G beads as outlined in Materials and Methods. HP + Hε, YMHD + Hε, and Y63D + Hε refer to purified WT or mutant HP from cotransfections with Hε. Samples labeled “GFP” were from a GFP control transfection that was put through a parallel purification with HP. (A) After purification, HP-bound beads were boiled in 2× SDS sample buffer, and HP was resolved in a 12.5% polyacrylamide-SDS gel and detected by Western blotting using the anti-FLAG antibody. (B) In vitro priming reactions were performed by incubating immunoaffinity-purified HP with or without Hε coexpression (lanes 3 and 4) or the indicated polymerase mutants with Hε coexpression (lanes 5 to 8) (ca. 200 ng HP per reaction mix) in TMnNK buffer with [α-32P]dGTP. After priming, the beads were washed in TNK buffer with individual protease inhibitors and 10 mM β-ME to remove unincorporated labeled nucleotides. The priming products were then resolved by SDS-PAGE and detected by autoradiography. Lanes 7 and 8 show a longer exposure of lanes 5 and 6. A GFP + Hε cotransfection product was included in a priming reaction mix as a negative control (lane 2). A priming reaction was also performed with DP (10 ng in total) in TMnNK buffer and resolved in the same gel for comparison (lane 1). The labeled products below full-length HP represent likely HP degradation fragments, which were found at various levels for different priming reactions (see also Fig. 2 and 6 to 8). *, weakly labeled unknown species unrelated to HP activity (also present in the YMHD mutant priming reaction mix [lane 7]). It could represent a cellular protein or an HP degradation product that was tightly bound to very low levels of the radiolabeled nucleotide or was somehow labeled at a very low level by a contaminating activity in the immunoaffinity purified HP. (C) After protein priming as outlined for panel B, primed HP was left untreated (−) (lanes 1 and 4) or treated with DNase I (D) (lanes 2 and 5) or pronase (P) (lanes 3 and 6) before analysis by SDS-PAGE. The positions of the protein molecular mass markers (in kDa) are indicated.
Fig 2.

Characterization of protein-primed DNA synthesis by purified HP in the presence of Mn2+. The purified HP protein was assayed for protein priming with each of the indicated 32P-labeled dNTPs. Primed HP was extensively washed to remove unincorporated nucleotides and then treated with Tdp2 or mock treated. (A and B) The beads, which contained the primed HP, were processed for SDS-PAGE to visualize the labeled HP. (C and D) The supernatant, which contained the released nucleotides/DNA, was collected and resolved in an 8 M urea-20% polyacrylamide gel. Radiolabeled proteins and nucleotides/DNA were detected by autoradiography. (A) HP purified either with (lanes 1 to 8) or without (lanes 9 to 16) coexpressed Hε was assayed for priming activity in the presence of [α-32P]TTP (T) (lanes 1, 2, 9, and 10), [α-32P]dGTP (G) (lanes 3, 4, 11, and 12), [α-32P]dCTP (C) (lanes 5, 6, 13, and 14), or [α-32P]dATP (A) (lanes 7, 8, 15, and 16). Primed HP was mock treated (odd-numbered lanes) or treated with Tdp2 (even-numbered lanes). (B) Priming assays were performed with [α-32P]TTP alone (T) (lanes 1 and 2) or [α-32P]TTP plus 10 μM unlabeled dGTP, dCTP, and dATP [T+(N)] (lanes 3 and 4). Priming products were then mock treated (odd-numbered lanes) or treated with Tdp2 (even-numbered lanes). (C) HP purified either without (lanes 2, 3, 6, 7, 10, 11, 14, and 15) or with (lanes 4, 5, 8, 9, 12, 13, 16, and 17) coexpressed Hε was assayed for priming activity in the presence of [α-32P]TTP (T) (lanes 2 to 5), [α-32P]dGTP (G) (lanes 6 to 9), [α-32P]dCTP (C) (lanes 10 to 13), or [α-32P]dATP (A) (lanes 14 to 17). Primed HP was mock treated (even-numbered lanes) or treated with Tdp2 (odd-numbered lanes, except for lane 1). Lane 1 shows the positions of a 5′-end-labeled 10-nt marker (Invitrogen) and DNA oligonucleotides (dTG, dTGA, and dTGAA). The positions of the various dNMPs and dNTPs are also indicated, save dCTP, which migrated off the gel (see Fig. 3 for further verification of the migration of the dNTPs and dNMPs). (D) Priming assays were performed with [α-32P]TTP alone (T) (lanes 2 to 5) or [α-32P]TTP plus unlabeled dGTP, dCTP, and dATP [T+(N)] (lanes 6 and 7). After priming, labeled products were either mock treated (lanes 2, 4, and 6) or treated with Tdp2 (lanes 3, 5, and 7). 32P-labeled dTMP homopolymers (T2 to T10) (lane 1) were run in parallel to confirm the mobility of the Mn2+ TTP priming reaction products. *, unknown, Tdp2-independent species that was detected only in reaction mixtures with HP purified after coexpression with Hε and in the presence of [α-32P]dGTP. It could represent a degraded Hε fragment that was somehow labeled by dGTP.
Nucleotide selectivity of HP priming with Mn2+ was different from that with Mg2+.
As HP priming in vitro with Mg2+, like the case in vivo, is templated by the Hε internal bulge sequence, it displays strict nucleotide selectivity; in particular, priming initiation shows a strong selectivity for dGTP, as dictated by the 3′-most Hε template sequence (rC) (27). In contrast, the nucleotide selectivity of HP priming in Mn2+ was very different. Without Hε, the clearly preferred nucleotide of HP was TTP (Fig. 2A, lane 1), followed by dGTP (Fig. 2A, lane 3). Both dCTP and dATP gave only weak signals (Fig. 2A, lanes 5 and 7). In the presence of Hε, HP showed a similarly strong preference for TTP, and the reactions with dGTP and dCTP were not significantly affected (Fig. 2A, lanes 9, 11, and 13). However, in contrast to HP alone, the small amounts of Hε associated with purified HP had a strong stimulatory effect on in vitro DNA synthesis in the presence of dATP (Fig. 2A, lane 15). As we have shown that a population of HP bound to Hε is able to carry out protein priming in vivo prior to HP purification (27), the increased dATP priming signal obtained with HP with Hε versus HP alone was likely a result, in part, of in vitro extension of the in vivo-primed HP as templated by the associated Hε RNA, which has the potential to code for five consecutive dAMP residues in the DNA product following the initiating dGMP residue (see also the next section and Discussion).
Analysis of DNA synthesis products covalently attached to HP.
To analyze the DNA products covalently linked to HP as a result of in vitro protein priming, the labeled HP priming products were treated with Tdp2, an enzyme that is able to specifically break the phosphotyrosyl linkage between Y63 of HP and the 5′ phosphate of covalently attached (−)-DNA (27, 49, 50). As with DNA synthesized in vitro during HP priming with Mg2+ (27), DNA products synthesized by HP with Mn2+ were susceptible to Tdp2 cleavage, which decreased the labeled HP protein signal detected by SDS-PAGE (Fig. 2A, even-numbered lanes) and released labeled DNA species as detected by urea-PAGE (Fig. 2C, odd-numbered lanes other than lane 1). The efficiency of Tdp2 release was less than complete and somewhat variable depending on the batch of Tdp2, as we noted previously (27). These results thus confirmed that the DNA products synthesized in the Mn2+ priming reactions, as in the Mg2+ priming reaction, were attached to HP via a phosphotyrosyl linkage, consistent with the result above showing that elimination of the Y63 priming site eliminated the vast majority of the Mn2+ priming signal (Fig. 1B).
When priming was carried out with [α-32P]TTP, [α-32P]dGTP (see Fig. 1B), or [α-32P]dCTP as the substrate, the primed (labeled) HP signals were similar with and without Hε coexpression (Fig. 2A, lanes 1 to 6 versus 9 to 14), and the amounts and patterns of DNA released by Tdp2 were also identical in the presence and absence of coexpressed Hε RNA (Fig. 2C, lanes 3, 5, 7, 9, 11, and 13). Upon Tdp2 release of protein priming products obtained with [α-32P]TTP, the TMP residue was clearly detected (Fig. 2C, lanes 3 and 5, and 3A), indicating that the initiation of priming (i.e., the covalent attachment of TMP to HP) occurred under these conditions. Single dCMP or dGMP residues released by Tdp2 could also be visualized when [α-32P]dCTP or [α-32P]dGTP, respectively, was used for priming (Fig. 2C, lanes 6 to 13, and 3B and C), although detection of these released single nucleotides was made more difficult by the presence of background signals in the mock (no Tdp2)-treated samples, which apparently resulted from nucleotide binding to the immunoprecipitation resin, as previously noted (27). These results thus indicated that HP priming with Mn2+ could be initiated with multiple dNTPs.
Fig 3.

Verification of nucleotide mobility on urea-PAGE. HP (expressed alone, without Hε) was primed in the presence of [α-32P]TTP (A), [α-32P]dGTP (B), [α-32P]dCTP (C), or [α-32P]dATP (D). The priming products were then mock treated (lanes 1) or treated with Tdp2 (lanes 2) to release the phosphotyrosyl-linked nucleotides/DNA, which were resolved by urea-PAGE. To verify the mobility of the single nucleotide species, fresh [α-32P]TTP (A), [α-32P]dGTP (B), [α-32P]dCTP (C), or [α-32P]dATP (D) was mock treated (−) (lanes 3) or treated with apyrase (apy +) (lanes 4) and resolved in parallel. Apyrase converted the faster-migrating species (dNTP) to the slower-migrating species (dNMP), which comigrated with the corresponding dNMP released from the primed HP by Tdp2.
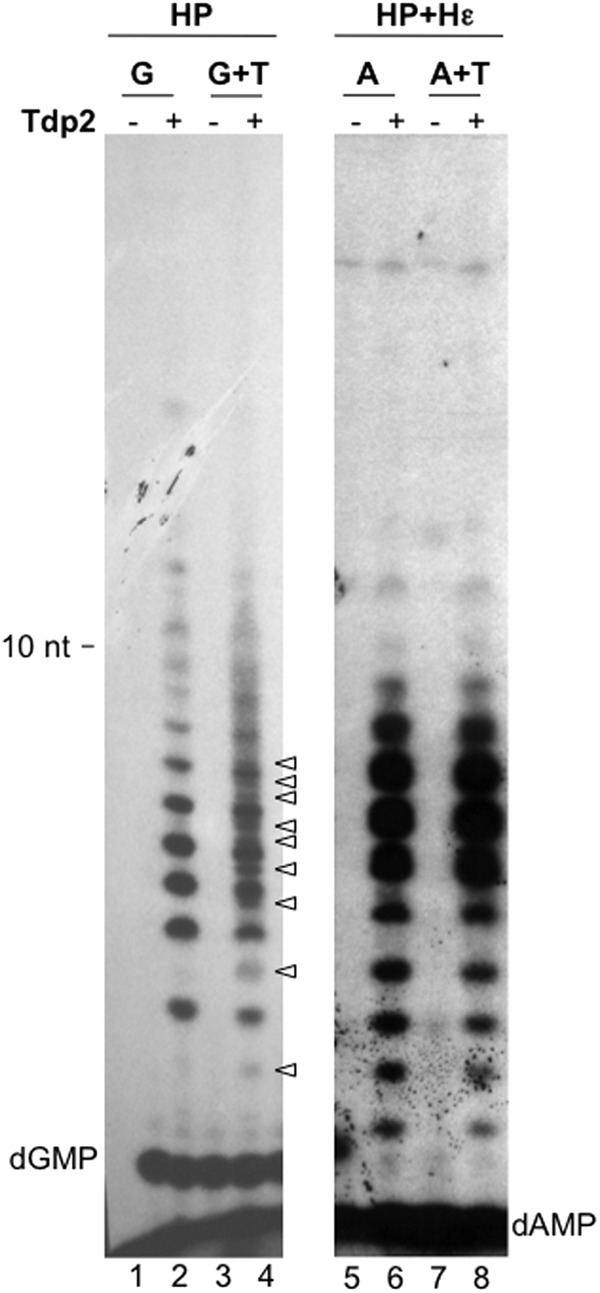
DNA polymerization with purified HP and Mg2+ is Hε (template) dependent and is efficient only when both dGTP and dATP are present, resulting in the production of 3- to 6-nt species (dGA2–5; predominantly the dGAA oligomer) attached to HP (27) (see Fig. 8D, lane 2). In sharp contrast, when priming was carried out with [α-32P]TTP and Mn2+, an apparent DNA ladder of up to and more than 100 nt in length was also released by Tdp2 (Fig. 2C, lanes 3 and 5). The lower-molecular-size species of this DNA ladder comigrated with the DNA oligonucleotide standards T2 to T10 (with 2 to 10 consecutive TMPs) in single-nt increments (Fig. 2D, lanes 1 and 3), indicating that the released DNA species from HP represented TMP homo-oligomers and -polymers synthesized in a highly distributive manner. Priming with dGTP also produced a ladder of labeled DNA oligomers ranging from 1 to 9 nt in length, while priming with dCTP produced a ladder of approximately 1 to 8 nt in length (Fig. 2C, lanes 7, 9, 11, and 13), both also with single-nt increments. Again, it was evident that the DNA level and pattern synthesized by HP priming with TTP, dGTP, or dCTP were not affected by Hε coexpression. In contrast, the HP priming signal on SDS-PAGE with [α-32P]dATP was much stronger when Hε was coexpressed with HP (Fig. 2A, lane 15 versus lane 7), as described above. Furthermore, Tdp2 released a DNA ladder of 1 to 8 nt without Hε copurification, whereas HP with Hε produced much stronger DNA species of 5 to 10 nt in length but approximately the same levels of DNA species of up to 4 nt (Fig. 2C, lanes 15 and 17) (see Fig. 4 and 8 and Discussion).
Fig 8.

Selective inhibition of late but not early DNA polymerization by PFA during priming. (A and C) HP was primed in TMnNK buffer with [α-32P]TTP in the presence (lanes 3 and 4) or absence (lanes 1 and 2) of 1 mM PFA, and after extensive washes, it was then mock treated (lanes 1 and 3) or treated with Tdp2 (lanes 2 and 4). Priming products were then resolved by SDS-PAGE (A), and Tdp2-released DNA products were resolved by urea-PAGE (C). The positions of the dTGAA, dTGA, dTG, and 10-nt DNA markers are indicated on the left in panel C. (B) Priming reactions were performed using HP purified with Hε coexpression and [α-32P]dGTP (initiation reaction) (lanes 1 and 2) in TMgNK buffer without (lane 1) or with (lane 2) PFA. Additionally, priming reaction mixtures were also set up in TMgNK buffer with 1.5 μM unlabeled dGTP for 2 h to initiate priming and were then incubated with [α-32P]dATP for two more hours to extend the HP-dGMP complex (polymerization reaction) (lanes 3 to 6), in the presence (lanes 5 and 6) or absence (lanes 3 and 4) of PFA. The polymerization products were then extensively washed before mock (lanes 3 and 5) or Tdp2 (lanes 4 and 6) treatment. Priming products were then resolved by SDS-PAGE. (D) Polymerization reaction mixtures from panel B, in the absence (lanes 1 and 2) or presence (lanes 3 and 4) of PFA, were mock treated (odd-numbered lanes) or treated with Tdp2 (even-numbered lanes). The Tdp2-released DNA products were resolved by urea-PAGE. (E and F) HP copurified with Hε was primed in TMnNK buffer with [α-32P]dATP in the presence (E, lanes 3 and 4, and F, lanes 5 and 6) or absence (E, lanes 1 and 2, and F, lanes 3 and 4) of PFA. Washed priming products were then mock treated (E, lanes 1 and 3, and F, lanes 3 and 5) or treated with Tdp2 (E, lanes 2 and 4, and F, lanes 4 and 6). The Tdp2-released DNA products (in the supernatant) were resolved by urea-PAGE (F), while the pellets containing the primed HP were resolved by SDS-PAGE (E). The dAn (E, lane 2) and dGAn (E, lane 1) oligonucleotide markers were also loaded into the urea-PAGE gel to help identify the DNA synthesis products. (G) A calf thymus TdT (lanes 3 and 4) or mock (no TdT; lane 2) reaction mixture was incubated with a 29-nt ssDNA oligonucleotide and [α-32P]TTP as substrates, in the presence (lane 4) or absence (lanes 2 and 3) of PFA (1 mM). A 10-nt marker (Invitrogen) was loaded in lane 1. The weak DNA signals in panel F, lane 3, are presumably contaminating DNA species and were not observed in other experiments.
Fig 4.
Chasing of HP priming products with unlabeled TTP. HP purified alone (lanes 1 to 4) or after coexpression with Hε (lanes 5 to 8) was first primed for 2 h in TMnNK buffer with [α-32P]dGTP (lanes 1 to 4) or [α-32P]dATP (lanes 5 to 8). Priming products were washed twice in TNK buffer with individual protease inhibitors and 10 mM β-ME to remove labeled nucleotides, and then 10 μM unlabeled TTP (lanes 3, 4, 7, and 8) or water (lanes 1, 2, 5, and 6) was added along with fresh TMnNK buffer and incubated for 2 more hours at room temperature with shaking to allow for extension of previously labeled priming products. After extensive washes to remove unincorporated nucleotides, priming products were then mock treated (odd-numbered lanes) or treated with Tdp2 (even-numbered lanes). The Tdp2-released DNA products were resolved by urea-PAGE. The arrowheads denote DNA products containing both labeled dGMP and unlabeled TMP, with distinct mobilities compared to those containing dGMP alone. Additional DNA species containing both dGMP and TMP (in various combinations) are likely represented by the smearing of the DNA signals approaching the 10-nt marker in lane 4. dGMP and dAMP species are denoted.
When the labeled TTP and unlabeled dGTP, dCTP, and dATP were incubated together with HP in a Mn2+ priming reaction, unlabeled dNTPs stimulated overall DNA synthesis, and a smear of extended HP-linked DNA species above the major HP band was visualized by SDS-PAGE (Fig. 2B, lane 3). Furthermore, the DNA products released by Tdp2 in this case showed a migration pattern on urea-PAGE that was more complex than, and distinct from, those produced in the presence of labeled TTP alone, with new DNA species migrating between the TMP homo-oligomers (Fig. 2D, lane 7), consistent with the incorporation of the unlabeled deoxynucleoside monophosphates (dNMPs) in addition to the labeled TMP into the DNA strands.
HP-dGn complex could be extended further with the addition of TTP.
As fairly extensive DNA polymerization in vitro appeared to occur in the presence of any one of the four dNTPs or their combinations, and the DNA products synthesized in the presence of TTP alone comigrated with the TMP homo-oligomers, the most likely explanation for these results was that HP synthesized, in a template-independent fashion, in vitro homo-oligomeric and -polymeric DNA species in the presence of a single dNTP and DNA hetero-oligomers and -polymers in the presence of two or more dNTPs. That is, in the presence of Mn2+, HP displayed a novel deoxynucleotidyltransferase activity that was independent of the viral Hε RNA or any other template. To further determine the template independence of protein-primed DNA synthesis by HP with Mn2+, we tested the ability of the HP-attached oligo(dG) synthesized in a Mn2+ buffer to be extended further with a different nucleotide. If there was a homo-C (or -dC) template coding for a dG homo-oligomer, then dTTP would not be incorporated. In contrast, if DNA synthesis under these conditions was indeed template independent, we would expect to see further elongation of the HP-dGn products upon the addition of TTP.
Purified HP was incubated with [α-32P]dGTP to produce labeled HP with one or more dGMP residues attached. Following the removal of the labeled dGTP, the labeled HP-dGn products were “chased” with unlabeled TTP. As anticipated, new DNA species could indeed be detected after release by Tdp2 following the TTP chase, which migrated between the dG homo-oligomers, representing the production of dG-T hetero-oligomers from the addition of one or more TMP residues to the preexisting dGMP homo-oligomers (Fig. 4, lanes 1 to 4). As in vitro DNA synthesis by the HP-Hε complex in the presence of [α-32P]dATP and Mn2+ was likely templated by Hε (Fig. 2C, lane 17), we predicted that the HP priming products labeled with dA could not be chased by the addition of TTP, due to the lack of rA residues immediately following rU in the template (see Fig. 9 for a diagram of the Hε template sequence). Indeed, the DNA products synthesized by the HP-Hε complex were not changed by the TTP chase (Fig. 4, lanes 5 to 8). Thus, these Hε-templated priming reaction products served as excellent controls in the chasing experiment for template-independent DNA synthesis products (lanes 1 to 4).
Fig 9.
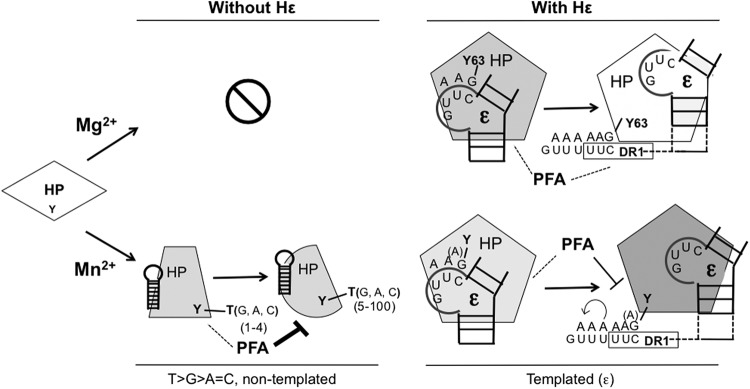
Hε-dependent and -independent protein-primed DNA synthesis by HP. In the presence of Mg2+ (upper section), HP in vitro priming can occur only when Hε (ε; only the internal bulge template region and part of the upper and lower stems are shown, for clarity) is bound to HP and serves as the obligatory viral template, which dictates the nucleotide selectivity of DNA synthesis primed by Y63 of HP. Protein priming (either the initiation or polymerization stage) as templated by Hε is insensitive to PFA inhibition. Furthermore, the addition of the first few dAMPs to the nascent HP-DNA complex following its translocation to DR1 appears to remain resistant to PFA. In the presence of Mn2+ (lower section), HP in vitro priming can occur independently of Hε. In the absence of Hε, a cellular or viral RNA(s) (indicated by the hairpin structure, different from that of Hε) is presumably bound to HP and serves as a scaffold or allosteric activator of HP but not as a template for DNA synthesis by HP. Protein-primed and template-independent DNA synthesis (i.e., terminal transferase) activity of HP in the presence of Mn2+ results in the distributive synthesis of homopolymeric and heteropolymeric DNA products of 1 to more than 100 nt, which is also primed predominantly by (and thus attached to) a Y residue(s) of HP (most likely Y63). As indicated by PFA sensitivity, the HP undergoes a conformational transition from the initial addition of nt 1 to 4 (PFA insensitive) to the subsequent DNA synthesis (PFA sensitive). In the presence of Mn2+, HP can also use its cognate Hε to carry out Hε (and DR1)-templated, protein-primed DNA synthesis with slippage and reannealing (stuttering, as denoted by the curved arrow), facilitated by the five consecutive U residues at DR1, leading to the synthesis of DNA oligomers containing more than five dA residues. This Hε-templated DNA synthesis reaction with Mn2+, unlike that with Mg2+, is partially sensitive to PFA after primer transfer to DR1, suggesting a difference in conformation between HP-Mg2+ and HP-Mn2+. The “A” in parenthesis denotes the fact that the initiating nt could be a dA (when dG is absent) in the Mn2+ reaction, due to misincorporation or skipping at the 1st template position of Hε in vitro. Dashed lines denote PFA resistance of the indicated reactions, and the solid lines indicate PFA sensitivity. The different shapes and shadings of HP denote the different conformations adopted by the protein during the various DNA synthesis reactions. See the text for details.
Limited exonuclease I digestion verified the de novo synthesis of DNA homopolymers by HP in vitro.
The results presented so far strongly indicated that the protein-primed DNA synthesis by HP in vitro in the absence of Hε represented a novel terminal transferase activity. However, there remained an alternative explanation that HP, as purified, was already attached covalently to an array of heterogeneous DNA strands (homo- and/or hetero-oligomers and -polymers) as a result of protein-primed DNA synthesis in vivo and that this heterogeneous array of HP-linked DNA species was extended in vitro in a limited fashion. Although this possibility was made unlikely by our recent finding that in vivo (in cell) protein-primed DNA synthesis is strictly dependent on Hε coexpression in cells (27), we decided to use a more direct assay to verify that de novo DNA synthesis was occurring in vitro, rather than limited extension of DNA already synthesized in vivo.
To formally exclude this alternative possibility, we treated the Tdp2-released DNA oligomers from the TTP priming reaction with Exo I in a limited digestion reaction. Exo I removes nucleotides one at a time from the 3′ end of single-stranded DNA (ssDNA). If only the 3′ end of a preexisting (unlabeled) DNA oligomer was labeled with [α-32P]TTP in the in vitro reaction, then the limited digestion would lead to a uniform decrease in the intensities of all DNA species, independent of their lengths, and there would be a buildup only of labeled TMP. On the other hand, Exo I digestion of a uniformly labeled DNA substrate, as would be produced by de novo DNA synthesis in vitro, would generate a very different digestion pattern, as the longer DNA molecules would be converted to shorter but still labeled fragments, causing a buildup of smaller DNA species at the expense of longer ones. As a control for the former DNA substrate, a 10-bp DNA ladder was labeled at the 3′ end with a single [α-32P]dAMP by Taq DNA polymerase and heat denatured to produce ssDNA substrates for Exo I. Limited Exo I digestion of this ladder produced a uniform decrease in the labeled signals of all DNA fragments and a buildup of only [α-32P]dAMP molecules, as anticipated (Fig. 5, lanes 2 and 3). In contrast, Exo I treatment of the second control, a population of uniformly labeled DNA molecules that were produced in a DP Mn2+ priming reaction mixture containing all four labeled dNTPs (42, 44), produced the expected buildup of smaller DNA products concomitant with a decrease of the longer DNA species (Fig. 5, lanes 4 and 5). The expected buildup of the smaller DNA species and the single dNMPs following Exo I treatment was less than anticipated from the loss of the longer DNA species, possibly due to further degradation of the Exo I-released dNMPs and thus the apparent net loss of the radiolabel on urea-PAGE. This was also supported by the less-than-expected buildup of dAMP from the DNA ladder. However, the Exo I digestion pattern could clearly differentiate the uniformly labeled DP-linked DNA species from the end-labeled DNA markers. When Tdp2-released HP priming products were similarly treated with limited Exo I digestion, there was also a buildup of smaller DNA species at the expense of the longer ones, just like the uniformly labeled DP control products and distinct from the end-labeled DNA markers (Fig. 5, lanes 6 and 7), thus signifying that the DNA species released by Tdp2 in the HP priming reaction were indeed de novo-synthesized and uniformly labeled TMP homo-oligomers and -polymers.
Fig 5.
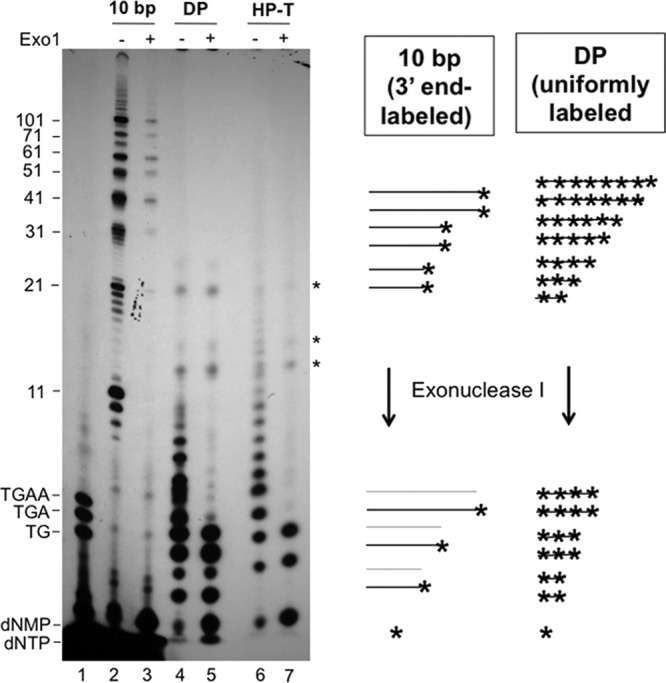
Analysis of protein-primed DNA synthesis products by limited Exo I digestion to differentiate in vitro end labeling of DNA synthesized in vivo versus de novo in vitro initiation and extension (i.e., uniform labeling). The 10-bp marker (Invitrogen) was 3′-end labeled with [α-32P]dATP by Taq polymerase (see Materials and Methods) in a terminal transferase reaction, thus creating 32P-labeled 11-nt, 21-nt, etc., products (lanes 2 and 3). DP was primed in TMnNK buffer in the presence of all four [α-32P]dNTPs, creating uniformly labeled DNA products that were then released by Tdp2 (lanes 4 and 5). HP was primed with [α-32P]TTP, and the HP DNA synthesis products were released by Tdp2 (lanes 6 and 7). All DNA species were then mock treated (−) (even-numbered lanes) or treated with Exo I (+) (odd-numbered lanes, except for lane 1) before resolution by urea-PAGE. The 5′-end-labeled DNA oligonucleotides (dTG, dTGA, and dTGAA) were loaded in lane 1. The positions of the DNA markers are indicated, as are the positions of the dNMPs and dNTPs. *, unknown species resistant to Exo I, apparently bound to protein A/G beads (in both the HP and control DP reactions). These could represent labeled RNAs or peptide fragments. The diagram to the right depicts the expected results from the Exo I digestion of the control, 3′-end-labeled 10-bp DNA markers and the uniformly labeled DP priming products. The asterisks denote the radiolabels at the 3′ ends of or uniformly distributed throughout the DNA molecules. The gray lines denote the DNA molecules that were no longer radiolabeled after removal of the 3′-end nucleotide by Exo I.
Requirement of RNA for Mn2+-dependent HP priming in vitro.
The above results all indicated that the Mn2+-dependent protein-primed DNA synthesis in vitro reflected a novel, template-independent transferase activity of HP. To further test the requirement for RNA, not just the viral Hε RNA, for this apparent transferase activity, we treated the purified HP with RNase before conducting the priming reaction in vitro. Surprisingly, RNase pretreatment drastically reduced the priming activity whether Hε was coexpressed or not (Fig. 6A, lanes 2 and 4), indicating a requirement for some RNA (unrelated to the authentic viral Hε RNA template and possibly of host origin) for efficient transferase activity. As discussed later, RNA most likely facilitated the Mn2+-dependent HP transferase activity by serving as a scaffold to maintain the HP polymerase activity or as an allosteric activator of HP rather than as a true template.
Fig 6.
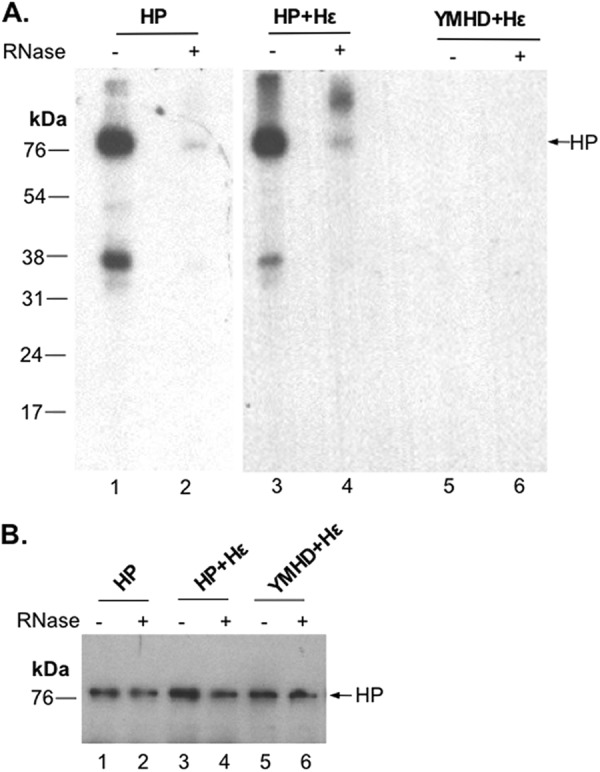
RNA requirement for in vitro HP priming. HP or the YMHD mutant with Hε coexpression (lanes 3 to 6) or HP alone (lanes 1 and 2) was mock treated (odd-numbered lanes) or treated with RNase A (even-numbered lanes). Subsequently, all samples were assayed for priming activity in TMnNK with [α-32P]TTP. Labeled HP was detected by autoradiography after SDS-PAGE (A), and HP protein levels were measured by Western blotting using an anti-FLAG antibody (B).
HP transferase activity in vitro was maintained at near-physiological Mn2+ concentrations.
As a first step to test if the HP protein-primed transferase activity detected in vitro would be relevant in vivo, we decided to titrate the Mn2+ concentrations used in the in vitro priming reaction mixture down to near-physiological levels (0.7 μM free Mn2+ and 34.4 μM total Mn2+ in hepatocytes) (51, 52). HP priming was reduced in a dose-responsive manner as the Mn2+ concentration was decreased from 1 mM to 1 μM but was still clearly detectable (by SDS-PAGE analysis) with 10 μM Mn2+ (Fig. 7A, lane 7). The urea-PAGE analysis of DNAs released by Tdp2 was less sensitive in detecting the in vitro-primed products but was able to detect a DNA ladder attached to HP with the same pattern, albeit at a reduced level, at 100 μM Mn2+ as that at 1 mM Mn2+ (Fig. 7B, lane 5). Because hepatocytes also contain approximately 0.5 mM Mg2+ (53–55), we decided to test if the presence of 0.5 mM Mg2+ would affect the in vitro Hε-independent priming activity of HP in the presence of Mn2+. It was clear that Hε-independent protein priming was still detectable at a Mn2+ concentration of 10 μM in the presence of 0.5 mM Mg2+ (Fig. 7C, lane 1), whereas Mg2+ alone did not support priming without Hε (Fig. 7C, lane 3) (27).
Fig 7.

Protein-primed DNA synthesis activity of HP at low Mn2+ concentrations. Purified HP was washed twice before priming in TNK buffer with individual protease inhibitors and 10 mM β-ME to remove any EDTA present in the FLAG lysis buffer and then primed in the presence of [α-32P]TTP and TMnNK buffer with the indicated concentrations of Mn2+. (A) Primed HP was visualized by SDS-PAGE and subsequent autoradiography. Lanes 5 to 8 represent a longer exposure of lanes 1 to 4. (B) HP priming products were mock treated (even-numbered lanes) or treated with Tdp2 (odd-numbered lanes, except for lane 1). The Tdp2-released nucleotides/DNA were resolved by urea-PAGE and visualized by autoradiography. The 5′-labeled 10-nt marker (Invitrogen) and DNA oligonucleotides (dTG, dTGA, and dTGAA) were loaded in lane 1, and their migration positions are indicated, as are the positions of TMP and TTP. (C) HP was incubated in a priming reaction mixture with the indicated concentrations of Mn2+ and/or Mg2+. Primed HP was visualized by SDS-PAGE and subsequent autoradiography.
HP displayed a conformational transition during the Mn2+-dependent, Hε-independent, and protein-primed transferase reaction in vitro.
It has long been proposed that during protein-primed viral DNA synthesis by the hepadnavirus polymerase, a conformational change occurs in the polymerase following the synthesis of a short DNA oligonucleotide that is templated by the viral ε RNA, which dissociates the polymerase from the ε RNA and facilitates the transfer of the nascent (−)-DNA–polymerase complex to the 3′ end of pgRNA to continue (−)-DNA synthesis (31, 40). Interestingly, this putative conformational change of the polymerase is thought to be manifested as a difference in its sensitivity to inhibition by the pyrophosphate analog PFA, which inhibits DP- and HP-catalyzed DNA strand elongation subsequent to protein priming (as in the so-called endogenous polymerase reaction) but does not inhibit DP or HP priming (12, 31). However, the exact transition point from PFA resistance to sensitivity during DP or HP DNA synthesis has never been defined. Our ability now to directly detect the very short DNA species attached to HP following Tdp2 cleavage afforded the opportunity to address this issue.
During Hε- and Mg2+-dependent authentic protein priming (27), PFA indeed had no effect on either priming initiation (i.e., the covalent attachment of dGMP to HP) (Fig. 8B, lane 2) or polymerization (the subsequent addition of the 2nd and 3rd nt) (Fig. 8B, lanes 3 and 5, and D, lanes 2 and 4), as anticipated. Furthermore, PFA also did not seem to have any inhibitory effect on the addition of the 4th, 5th, or 6th nt (Fig. 8D, lanes 2 and 4) in these reactions, suggesting that the gain of PFA sensitivity by HP was not acquired until sometime after minus-strand primer transfer (see Discussion). Interestingly, PFA showed a dramatic inhibitory effect on the Hε-independent synthesis of DNA strands longer than 4 nt by HP in the presence of Mn2+ (Fig. 8A, lane 3, and C, lane 4). In sharp contrast, the addition of the first 3 nt to HP was not inhibited by PFA, and the addition of the 4th nt was only partially inhibited (Fig. 8C, lane 4), indicating a rather sharp transition from PFA resistance to sensitivity during the addition of the 4th and 5th nt in the Mn2+-dependent and Hε-independent DNA synthesis.
We then tested the effect of PFA on the incorporation of dATP by HP purified with Hε coexpression in the presence of Mn2+ (Fig. 8E and F). Because we were anticipating the production in these dATP incorporation reactions of hetero-oligomers containing the initiating dG plus a stretch of consecutive dA residues due to the initiation of Hε-templated protein priming in vivo (27) (see Discussion), as well as the dA homopolymers produced in vitro, we included both dGAn and dAn oligonucleotides as markers so that we could differentiate the two different types of DNA products expected from these reactions. Indeed, the strong DNA signals (ca. 7-mer to 10-mer) could be resolved into closely spaced doublets, with the lower species comigrating with the dAn homo-oligomers and the upper species comigrating with the dGAn oligonucleotide marker. Since no dGTP, i.e., the initiating substrate in the Hε-templated DNA synthesis, was included in these reaction mixtures in vitro, we attributed the production of the dA homo-oligomers to misincorporation in vitro (dA instead of dG) as the 1st nt attached to HP or skipping of the 1st nt altogether. This was plausible because Mn2+ is well known to decrease the fidelity of the hepadnavirus polymerase (42). It was apparent that the transition from PFA resistance to sensitivity in dAMP incorporation was not as abrupt as in the case of TMP incorporation described above (Fig. 8C, lane 4). The synthesis of the 3-mer (dAAA or dGA) appeared to be insensitive to PFA, that of the 4-mer to 8-mer partially sensitive, and that of still longer species almost completely inhibited by PFA (Fig. 8F, lane 6).
To compare the HP transferase activity to that of a standard terminal transferase, we treated a commercial calf thymus TdT with PFA and found that PFA only marginally inhibited this transferase and that this weak inhibition was not selective in terms of short versus longer DNA products (Fig. 8G, lane 4), in contrast to its effect on HP.
DISCUSSION
We have discovered an Hε-independent, protein-primed DNA synthesis activity by the HBV polymerase purified from human cells (Fig. 9). This activity was dependent on Mn2+ as the metal ion cofactor. HP used each of the four dNTPs for DNA synthesis in this reaction but clearly favored TTP as a substrate. DNA synthesis under these conditions was highly distributive, leading to the production of DNA homo-oligomers and -polymers (when only a single dNTP was present) and hetero-oligomers and -polymers (when more than one dNTP was present) ranging from a single nt to over 100 nt long, in single-nt increments. As with Hε-dependent protein priming, DNA synthesis in the Hε-independent reaction was carried out by the HP DNA polymerase active site, and the products were covalently attached to HP, predominantly at the authentic priming site, Y63, in the TP domain of HP.
We have presented several lines of evidence to support the notion that the Hε-independent and Mn2+-dependent DNA synthesis uncovered here was likely due to a novel, protein-primed and template-independent terminal transferase activity of HP. First, protein-primed DNA synthesis in the presence of Mn2+ was clearly independent of the viral RNA template Hε. Second, each of the four deoxynucleotides could be used individually to synthesize a homo-oligomeric and -polymeric stretch of DNA. Third, when more than one dNTP substrate was provided, HP was able to use a variety of nucleotides to synthesize DNA hetero-oligomers and -polymers, including the ability to extend preexisting homo-oligomers with another, different nucleotide substrate (Fig. 4). The preference for TTP as a substrate might have been considered evidence for some poly(A)-containing mRNA(s) acting as a template for the DNA synthesis observed, except that when TTP and the other three dNTPs were all present, HP synthesized hetero-oligomers and -polymers instead of T homo-oligomers and -polymers (Fig. 2D). Although we cannot completely exclude the possibility that the purified HP was associated with a heterogeneous populations of RNA molecules that could all be utilized to template the synthesis of the various DNA products observed, this is considered very unlikely. The HP-associated RNAs would need to contain consecutive stretches of each of the four different ribonucleotides to account for the production of all four different DNA homopolymers, as well as heteromeric template sequences to account for the synthesis of the heteropolymeric DNAs (Fig. 2 and 4). Fourth, Hε-independent DNA synthesis was highly distributive, a known characteristic of bona fide terminal transferases (56, 57). It is important to note that the HP transferase activity detected here was distinct from the low-fidelity DNA synthesis by a truncated DP (designated MiniRT2) with Mn2+ in that the HP transferase activity was completely independent of the viral Hε template and could produce DNA strands of >100 nt long, while DP (MiniRT2) DNA synthesis, even with Mn2+, was still largely templated by its cognate viral RNA, Dε, and was limited to short DNA strands of 10 nt at most (Fig. 5) (42). It remains to be determined if these discrepancies between HP (full length) and DP (truncated MiniRT2) are due to the truncation of DP or reflect an intrinsic difference between these two related enzymes.
Transferase activity is defined as the template-independent addition of nucleotides to a primer, in the form of nucleic acid molecules such as ssDNA (57). Mn2+-stimulated transferase activity of template-dependent polymerases is not without precedence. For example, telomerase, another specialized RT that shares a number of interesting properties with hepadnavirus polymerases, including the presence of a specific RNA (the telomerase RNA [TR]) as an integral component of the enzyme complex and as the specific template, was recently shown to display a transferase-like activity that is independent of TR and is evident only in the presence of Mn2+ (58). There are several novel aspects of our findings that differentiate our findings from previous reports of transferase activities. First, HP was able to synthesize homopolymeric and heteropolymeric DNAs of more than 100 nt. Although it has previously been shown that many viral and cellular template-dependent polymerases (including RTs) have terminal transferase activity, they can carry out only very limited DNA synthesis (addition of 1 to 5 nt to a primer in a template-independent fashion) (59–62). The HP transferase activity uncovered here is thus much more robust and is similar to that of a bona fide terminal transferase such as TdT. Second, HP is different from TdT in a number of interesting aspects. HP and TdT differ in their nucleotide preferences (TTP for HP and dGTP, dCTP, and TTP over dATP for TdT) (56, 57, 63). Also, in contrast to TdT, HP cannot utilize exogenous ssDNA or double-stranded DNA (dsDNA) as a primer and depends strictly on HP itself as a protein primer (our unpublished data). This apparent “primer commitment” is analogous to the well-known phenomenon of template commitment exhibited by hepadnavirus polymerases (7). Moreover, whereas TdT can extend only from primers of at least 3 nt long (57), HP can initiate DNA synthesis de novo by using its own Y63 residue as a primer. Little sequence homology is evident between HP and TdT. Structurally, how HP accommodates ssDNA products during the template-independent transferase reaction, as opposed to the double-stranded template-primer complex during template-directed DNA synthesis, remains to be understood but presumably involves different HP conformations induced by Mn2+ versus Mg2+. We note that a protein-primed transferase activity has been identified, tentatively, for one other polymerase, an RT-like protein carried by some bacteria (64). Interestingly, this bacterial protein, like HP, is able to synthesize DNAs of up to several hundred nt long. In contrast to HP, however, the bacterial protein is also able to utilize exogenous primers in addition to using a protein (possibly itself) as a putative primer.
The protein-primed transferase activity of HP appeared to be stimulated strongly by a bound RNA(s) (Fig. 9). As indicated above, we believe that the bound RNA does not serve as a template for DNA synthesis by HP. Rather, we suggest that the bound RNA may serve to induce and maintain an active HP conformation. Indeed, the authentic viral RNA template for protein-primed viral DNA synthesis, Hε, is also thought to be an integral component of the enzymatically active HP-Hε complex and to induce structural changes in the polymerase that are necessary to activate HP's enzymatic function (15, 23–25, 65, 66). Similar to this structural or allosteric role of Hε, the HP-bound cellular or viral (non-Hε) RNA might have stabilized the HP in an active configuration that allowed for the protein-primed transferase activity. Interestingly, it has been proposed that the Hε-independent protein priming activity detected with HP purified from insect cells, in the presence of Mg2+ in this case, may also be dependent on an unknown cellular RNA (13).
While Hε was clearly not required for the Mn2+-stimulated HP transferase activity, its presence did modify HP behavior in this reaction, and it was most likely used as a template for DNA synthesis in vitro in the presence of Mn2+ as well as Mg2+ (Fig. 9). The Hε RNA template we expressed in the cells also contained additional 5′ sequences, including a copy of DR1 (27), to which the nascent HP-dGAA complex can be transferred (27, 31, 67). The RNA sequence around DR1 (3′-UUUuuc-5′; the residues denoted in lowercase anneal with the dGAA residues attached to HP) (Fig. 9) could allow the synthesis of DNA oligomers with up to five dA residues following a template switch from Hε to DR1. Our previous results suggest that in the absence of the viral capsids, limited primer translocation followed by DNA synthesis from DR1 could occur in the cell before HP purification (27). These DNA oligomers produced in vivo were apparently extended by HP in vitro in the presence of dATP alone to produce the dGAn oligomers detected, using the bound Hε (including DR1) RNA as the template. The addition of more than five dA residues (up to ca. 10) could have resulted from limited HP slippage and reannealing facilitated by the presence of the five consecutive rU residues around DR1 (19).
The resistance to PFA inhibition during the synthesis of the nascent 3- to 4-nt DNA oligomer in the Hε-independent HP transferase reaction is reminiscent of the HP behavior during Hε-dependent authentic protein priming, which produces the Hε-templated dGAA oligomer in a PFA-insensitive manner (Fig. 9). Our results here further demonstrated a sharp transition of HP from PFA resistance to sensitivity during the addition of the 4th and 5th nt to the nascent DNA strand, and this HP conformational transition was independent of Hε (or any template). These results suggest that this conformational change early during DNA synthesis is an intrinsic property of HP. Interestingly, DNA synthesis in the presence of Hε and Mg2+ remained resistant to PFA inhibition for at least a short time after primer translocation to DR1, suggesting that Hε could delay the acquirement of PFA sensitivity to a later stage in viral DNA synthesis (after the addition of the 6th nt). Thus, the putative HP conformational change that dissociates it from Hε is apparently not (at least not immediately) manifested as a change in PFA sensitivity as has been assumed. When Mg2+ was replaced by Mn2+, the Hε- and DR1-templated priming reaction showed partial sensitivity to PFA between the addition of the 4th and 8th nt, and the addition of the 9th nt and beyond became almost completely sensitive to PFA. We suggest that the presence of the nascent DNA oligomer (as short as 3 to 4 nt with the nontemplated reaction but somewhat longer for Hε-templated DNA synthesis) in the polymerase active site per se induces this conformational change, independent of any template, that leads to a gain of PFA sensitivity (likely corresponding to a “generic” elongation mode of DNA synthesis). In fact, distinct phases of initiation (defined by the synthesis of short, ca. 10-nt DNA or RNA strands) and subsequent elongation seem to be characteristic of DNA or RNA synthesis for all polynucleotide polymerases (68–72). The influence of Hε and metal ions on the timing of PFA sensitivity is also consistent with the known effects of the viral RNA and metal ions on HP structure and function (24, 25, 42). The Hε-dependent and -independent protein priming assays we have now developed should facilitate the dissection of this important early conformational change that HP undergoes during protein-primed DNA synthesis and help efforts to target this for developing novel HP inhibitors for antiviral therapy. Importantly, the utility of PFA as a sensitive detector of polymerase conformations was verified by recent structural studies using a chimeric DNA polymerase derived from a cytomegalovirus (73).
The biological significance of the protein-primed transferase activity of HP uncovered here for either the virus or host remains to be determined. Intriguingly, the putative protein-primed transferase activity of the aforementioned bacterial RT-like protein may be required for defense against bacteriophage infection (64), suggesting that the HP protein-primed transferase activity may also have an as yet uncovered or uncharacterized function(s) in vivo. The free Mn2+ concentration in rat hepatocytes was reported to be 0.7 μM, and the total Mn2+ concentration is as high as 34.4 μM (51, 52). Given that we were unable to detect the HP transferase activity in vitro at 1 μM Mn2+, it is unlikely that this activity is present at any significant levels in vivo. However, this by no means excludes the possibility that the transferase activity may manifest itself sometimes during the decades of chronic HBV infections in some of the hundreds of millions infected individuals. Furthermore, as the cellular concentration of Mn2+ might be different depending on the cell type and subcellular compartment and the free and bound Mn2+ pools may exchange, it is possible that local concentrations of Mn2+ in certain subcellular compartments in some cells might be substantially higher so as to support significant HP transferase activity. Indeed, at least one DNA polymerase has been shown to prefer Mn2+ over Mg2+ as a cofactor in vivo (74). In addition, the Hε-independent, protein-primed DNA synthesis detected in vitro using HP expressed in insect cells (13) and in Xenopus oocytes (75) may also represent, at least in part, a protein-primed transferase activity similar to that described here. Indeed, the insect cell-expressed HP, like the HP purified from human cells here, displays a similar preference for TTP for in vitro DNA synthesis (12, 13). In this case, this activity can be detected in the presence of Mg2+, raising the possibility that the HP transferase activity may not be dependent completely on Mn2+ and can in certain cellular environments manifest itself also in the presence of Mg2+ as a result of altered HP folding.
One possible consequence of the HP transferase activity for the virus might be the expansion of its genetic diversity. Genetic variation of HBV is limited due to the compact nature of the genome and the lack of noncoding regions, although the host response and antiviral treatment can select for mutant viruses (76). Insertions of 3 or 4 consecutive dG into the viral envelope gene region have been reported (77). Furthermore, the HBV genome can vary from 3,182 to 3,248 bp in length, and genotype G strains harbor a unique 36-nt insertion in the core gene region (76). The mechanisms for generating these insertion events are currently unknown. It is conceivable that the HP transferase activity, among other possibilities, could contribute to these insertion events. Obviously, the HP transferase activity must be kept at very low levels to maintain viral genome integrity, consistent with the rather low activity observed at low (near-physiological) Mn2+ concentrations. For the host cell, the Hε-independent HP transferase activity could contribute to cellular alterations. It has been reported that unpackaged HP or DP can accumulate in cells replicating HBV or DHBV (78–80) and could have DNA synthesis activity in the absence of its cognate ε RNA (42; this report). Also, DP can initiate DNA synthesis not only in cis (i.e., using itself as the primer) but also in trans, using multiple fragments derived from DP (34, 44), and both DP (43, 44) and HP (this report) can initiate DNA synthesis from sites other than the authentic Y residue in their respective TP domains (cryptic site priming). It is thus tempting to speculate that HP could modify host protein function, albeit at a low frequency, by covalent nucleotide or DNA attachment, potentially contributing to viral pathogenesis, especially during the long course of chronic HBV infections. Indeed, protein nucleotidylation is gaining more attention as one of the important posttranslational regulatory mechanisms of cellular functions (81).
ACKNOWLEDGMENTS
We thank Christina Adams for excellent technical assistance and Rajeev Boregowda for supplying the purified DHBV MiniRT2 protein.
This work was supported by a Public Health Service grant (R01 AI074982 to J.H.) from the National Institutes of Health. S.A.J. was supported by a Viruses and Cancer training grant (2 T32 CA60395) from the National Cancer Institute.
Footnotes
Published ahead of print 19 December 2012
REFERENCES
- 1. Lee W. 1997. Hepatitis B virus infection. N. Engl. J. Med. 337:1733–1745 [DOI] [PubMed] [Google Scholar]
- 2. Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Seeger C, Mason WS, Zoulim F. 2007. Hepadnaviruses, p 2977–3029 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4. Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415 [DOI] [PubMed] [Google Scholar]
- 5. Chang LJ, Hirsch RC, Ganem D, Varmus HE. 1990. Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase. J. Virol. 64:5553–5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hu J, Seeger C. 1996. Expression and characterization of hepadnavirus reverse transcriptases. Methods Enzymol. 275:195–208 [DOI] [PubMed] [Google Scholar]
- 7. Radziwill G, Tucker W, Schaller H. 1990. Mutational analysis of the hepatitis B virus P gene product: domain structure and RNase H activity. J. Virol. 64:613–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bartenschlager R, Schaller H. 1988. The amino-terminal domain of the hepadnaviral P-gene encodes the terminal protein (genome-linked protein) believed to prime reverse transcription. EMBO J. 7:4185–4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Y, Robinson WS, Marion PL. 1994. Selected mutations of the duck hepatitis B virus P gene RNase H domain affect both RNA packaging and priming of minus-strand DNA synthesis. J. Virol. 68:5232–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Toh H, Hayashida H, Miyata T. 1983. Sequence homology between retroviral reverse transcriptase and putative polymerases of hepatitis B virus and cauliflower mosaic virus. Nature 305:827–829 [DOI] [PubMed] [Google Scholar]
- 11. Xiong Y, Eickbush TH. 1990. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 9:3353–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lanford RE, Notvall L, Beames B. 1995. Nucleotide priming and reverse transcriptase activity of hepatitis B virus polymerase expressed in insect cells. J. Virol. 69:4431–4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lanford RE, Notvall L, Lee H, Beames B. 1997. Transcomplementation of nucleotide priming and reverse transcription between independently expressed TP and RT domains of the hepatitis B virus reverse transcriptase. J. Virol. 71:2996–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bartenschlager R, Junker-Niepmann M, Schaller H. 1990. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J. Virol. 64:5324–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bartenschlager R, Schaller H. 1992. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 11:3413–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fallows DA, Goff SP. 1995. Mutations in the epsilon sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J. Virol. 69:3067–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu J, Boyer M. 2006. Hepatitis B virus reverse transcriptase and epsilon RNA sequences required for specific interaction in vitro. J. Virol. 80:2141–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu J, Lin L. 2009. RNA-protein interactions in hepadnavirus reverse transcription. Front. Biosci. 14:1606–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nassal M, Rieger A. 1996. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J. Virol. 70:2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tavis JE, Perri S, Ganem D. 1994. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J. Virol. 68:3536–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang GH, Seeger C. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 67:6507–6512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beck J, Nassal M. 1998. Formation of a functional hepatitis B virus replication initiation complex involves a major structural alteration in the RNA template. Mol. Cell. Biol. 18:6265–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beck J, Nassal M. 1997. Sequence- and structure-specific determinants in the interaction between the RNA encapsidation signal and reverse transcriptase of avian hepatitis B viruses. J. Virol. 71:4971–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tavis JE, Ganem D. 1996. Evidence for activation of the hepatitis B virus polymerase by binding of its RNA template. J. Virol. 70:5741–5750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tavis JE, Massey B, Gong Y. 1998. The duck hepatitis B virus polymerase is activated by its RNA packaging signal, epsilon. J. Virol. 72:5789–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu J, Seeger C. 1997. RNA signals that control DNA replication in hepadnaviruses. Semin. Virol. 8:205–211 [Google Scholar]
- 27. Jones SA, Boregowda R, Spratt TE, Hu J. 2012. In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J. Virol. 86:5134–5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rieger A, Nassal M. 1996. Specific hepatitis B virus minus-strand DNA synthesis requires only the 5′ encapsidation signal and the 3′-proximal direct repeat DR1. J. Virol. 70:585–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Junker-Niepmann M, Bartenschlager R, Schaller H. 1990. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 9:3389–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J. Virol. 78:13122–13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang GH, Seeger C. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663–670 [DOI] [PubMed] [Google Scholar]
- 32. Weber M, Bronsema V, Bartos H, Bosserhoff A, Bartenschlager R, Schaller H. 1994. Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J. Virol. 68:2994–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zoulim F, Seeger C. 1994. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J. Virol. 68:6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boregowda R, Adams C, Hu J. 2012. TP-RT domain interactions of duck hepatitis B virus reverse transcriptase in cis and in trans during protein-primed initiation of DNA synthesis in vitro. J. Virol. 86:6522–6536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu J, Anselmo D. 2000. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J. Virol. 74:11447–11455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu J, Seeger C. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 93:1060–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu J, Toft D, Anselmo D, Wang X. 2002. In vitro reconstitution of functional hepadnavirus reverse transcriptase with cellular chaperone proteins. J. Virol. 76:269–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu J, Toft DO, Seeger C. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 16:59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stahl M, Beck J, Nassal M. 2007. Chaperones activate hepadnavirus reverse transcriptase by transiently exposing a C-proximal region in the terminal protein domain that contributes to epsilon RNA binding. J. Virol. 81:13354–13364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang X, Hu J. 2002. Distinct requirement for two stages of protein-primed initiation of reverse transcription in hepadnaviruses. J. Virol. 76:5857–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang X, Qian X, Guo HC, Hu J. 2003. Heat shock protein 90-independent activation of truncated hepadnavirus reverse transcriptase. J. Virol. 77:4471–4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin L, Wan F, Hu J. 2008. Functional and structural dynamics of hepadnavirus reverse transcriptase during protein-primed initiation of reverse transcription: effects of metal ions. J. Virol. 82:5703–5714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beck J, Nassal M. 2011. A Tyr residue in the reverse transcriptase domain can mimic the protein-priming Tyr residue in the terminal protein domain of a hepadnavirus P protein. J. Virol. 85:7742–7753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Boregowda RK, Lin L, Zhu Q, Tian F, Hu J. 2011. Cryptic protein priming sites in two different domains of duck hepatitis B virus reverse transcriptase for initiating DNA synthesis in vitro. J. Virol. 85:7754–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Urban M, McMillan DJ, Canning G, Newell A, Brown E, Mills JS, Jupp R. 1998. In vitro activity of hepatitis B virus polymerase: requirement for distinct metal ions and the viral epsilon stem-loop. J. Gen. Virol. 79:1121–1131 [DOI] [PubMed] [Google Scholar]
- 46. Kim HY, Park GS, Kim EG, Kang SH, Shin HJ, Park S, Kim KH. 2004. Oligomer synthesis by priming deficient polymerase in hepatitis B virus core particle. Virology 322:22–30 [DOI] [PubMed] [Google Scholar]
- 47. Nguyen DH, Gummuluru S, Hu J. 2007. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 81:4465–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin L, Hu J. 2008. Inhibition of hepadnavirus reverse transcriptase-epsilon RNA interaction by porphyrin compounds. J. Virol. 82:2305–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW. 2009. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 461:674–678 [DOI] [PubMed] [Google Scholar]
- 50. Zeng Z, Cortes-Ledesma F, El Khamisy SF, Caldecott KW. 2011. TDP2/TTRAP is the major 5′-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J. Biol. Chem. 286:403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ash DE, Schramm VL. 1982. Determination of free and bound manganese(II) in hepatocytes from fed and fasted rats. J. Biol. Chem. 257:9261–9264 [PubMed] [Google Scholar]
- 52. Bolton EC, Mildvan AS, Boeke JD. 2002. Inhibition of reverse transcription in vivo by elevated manganese ion concentration. Mol. Cell 9:879–889 [DOI] [PubMed] [Google Scholar]
- 53. Corkey BE, Duszynski J, Rich TL, Matschinsky B, Williamson JR. 1986. Regulation of free and bound magnesium in rat hepatocytes and isolated mitochondria. J. Biol. Chem. 261:2567–2574 [PubMed] [Google Scholar]
- 54. Raju B, Murphy E, Levy LA, Hall RD, London RE. 1989. A fluorescent indicator for measuring cytosolic free magnesium. Am. J. Physiol. 256:C540–C548 [DOI] [PubMed] [Google Scholar]
- 55. Romani A, Scarpa A. 1992. Regulation of cell magnesium. Arch. Biochem. Biophys. 298:1–12 [DOI] [PubMed] [Google Scholar]
- 56. Berdis AJ, McCutcheon D. 2007. The use of non-natural nucleotides to probe template-independent DNA synthesis. Chembiochem 8:1399–1408 [DOI] [PubMed] [Google Scholar]
- 57. Motea EA, Berdis AJ. 2010. Terminal deoxynucleotidyl transferase: the story of a misguided DNA polymerase. Biochim. Biophys. Acta 1804:1151–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lue NF, Bosoy D, Moriarty TJ, Autexier C, Altman B, Leng S. 2005. Telomerase can act as a template- and RNA-independent terminal transferase. Proc. Natl. Acad. Sci. U. S. A. 102:9778–9783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Clark JM. 1988. Novel non-templated nucleotide addition reactions catalyzed by procaryotic and eucaryotic DNA polymerases. Nucleic Acids Res. 16:9677–9686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Patel PH, Preston BD. 1994. Marked infidelity of human immunodeficiency virus type 1 reverse transcriptase at RNA and DNA template ends. Proc. Natl. Acad. Sci. U. S. A. 91:549–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Poranen MM, Koivunen MR, Bamford DH. 2008. Nontemplated terminal nucleotidyltransferase activity of double-stranded RNA bacteriophage phi6 RNA-dependent RNA polymerase. J. Virol. 82:9254–9264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ranjith-Kumar CT, Gajewski J, Gutshall L, Maley D, Sarisky RT, Kao CC. 2001. Terminal nucleotidyl transferase activity of recombinant Flaviviridae RNA-dependent RNA polymerases: implication for viral RNA synthesis. J. Virol. 75:8615–8623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Modak MJ. 1978. Biochemistry of terminal deoxynucleotidyltransferase: mechanism of inhibition by adenosine 5′-triphosphate. Biochemistry 17:3116–3120 [DOI] [PubMed] [Google Scholar]
- 64. Wang C, Villion M, Semper C, Coros C, Moineau S, Zimmerly S. 2011. A reverse transcriptase-related protein mediates phage resistance and polymerizes untemplated DNA in vitro. Nucleic Acids Res. 39:7620–7629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pollack JR, Ganem D. 1993. An RNA stem-loop structure directs hepatitis B virus genomic RNA encapsidation. J. Virol. 67:3254–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pollack JR, Ganem D. 1994. Site-specific RNA binding by a hepatitis B virus reverse transcriptase initiates two distinct reactions: RNA packaging and DNA synthesis. J. Virol. 68:5579–5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Abraham TM, Loeb DD. 2006. Base pairing between the 5′ half of epsilon and a cis-acting sequence, phi, makes a contribution to the synthesis of minus-strand DNA for human hepatitis B virus. J. Virol. 80:4380–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Goldman SR, Ebright RH, Nickels BE. 2009. Direct detection of abortive RNA transcripts in vivo. Science 324:927–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hieb AR, Baran S, Goodrich JA, Kugel JF. 2006. An 8 nt RNA triggers a rate-limiting shift of RNA polymerase II complexes into elongation. EMBO J. 25:3100–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kamtekar S, Berman AJ, Wang J, Lazaro JM, de Vega M, Blanco L, Salas M, Steitz TA. 2006. The phi29 DNA polymerase:protein-primer structure suggests a model for the initiation to elongation transition. EMBO J. 25:1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nechaev S, Adelman K. 2011. Pol II waiting in the starting gates: regulating the transition from transcription initiation into productive elongation. Biochim. Biophys. Acta 1809:34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Steitz TA. 2006. Visualizing polynucleotide polymerase machines at work. EMBO J. 25:3458–3468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zahn KE, Tchesnokov EP, Gotte M, Doublie S. 2011. Phosphonoformic acid inhibits viral replication by trapping the closed form of the DNA polymerase. J. Biol. Chem. 286:25246–25255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Blanca G, Shevelev I, Ramadan K, Villani G, Spadari S, Hubscher U, Maga G. 2003. Human DNA polymerase lambda diverged in evolution from DNA polymerase beta toward specific Mn(++) dependence: a kinetic and thermodynamic study. Biochemistry 42:7467–7476 [DOI] [PubMed] [Google Scholar]
- 75. Seifer M, Standring DN. 1993. Recombinant human hepatitis B virus reverse transcriptase is active in the absence of the nucleocapsid or the viral replication origin, DR1. J. Virol. 67:4513–4520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kay A, Zoulim F. 2007. Hepatitis B virus genetic variability and evolution. Virus Res. 127:164–176 [DOI] [PubMed] [Google Scholar]
- 77. Hou J, Karayiannis P, Waters J, Luo K, Liang C, Thomas HC. 1995. A unique insertion in the S gene of surface antigen-negative hepatitis B virus Chinese carriers. Hepatology 21:273–278 [DOI] [PubMed] [Google Scholar]
- 78. Cao F, Tavis JE. 2004. Detection and characterization of cytoplasmic hepatitis B virus reverse transcriptase. J. Gen. Virol. 85:3353–3360 [DOI] [PubMed] [Google Scholar]
- 79. Yao E, Gong Y, Chen N, Tavis JE. 2000. The majority of duck hepatitis B virus reverse transcriptase in cells is nonencapsidated and is bound to a cytoplasmic structure. J. Virol. 74:8648–8657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yao E, Tavis JE. 2004. Localization of duck hepatitis B virus polymerase within cells. Methods Mol. Med. 95:281–293 [DOI] [PubMed] [Google Scholar]
- 81. Itzen A, Blankenfeldt W, Goody RS. 2011. Adenylylation: renaissance of a forgotten post-translational modification. Trends Biochem. Sci. 36:221–228 [DOI] [PubMed] [Google Scholar]