

Abstract
Many replication events are involved in the influenza A virus life cycle, and they are accomplished by different virus proteins with specific functions. However, because the size of the influenza virus genome is limited, the virus uses different mechanisms to express multiple viral proteins from a single gene segment. The M2 and NS2 proteins are produced by splicing, and several novel influenza A virus proteins, such as PB1-F2, PB1-N40, and PA-X, have recently been identified. Here, we identified novel PA-related proteins in influenza A virus-infected cells. These newly identified proteins are translated from the 11th and 13th in-frame AUG codons in the PA mRNA and are, therefore, N-terminally truncated forms of PA, which we named PA-N155 and PA-N182, respectively. The 11th and 13th AUG codons are highly conserved among influenza A viruses, and the PA-N155 and PA-N182 proteins were detected in cells infected with various influenza A viruses isolated from different host species, suggesting the expression of these N-truncated PAs is universal in nature among influenza A viruses. These N-truncated PAs did not show polymerase activity when expressed together with PB1 and PB2; however, mutant viruses lacking the N-truncated PAs replicated more slowly in cell culture and had lower pathogenicity in mice than did wild-type virus. These results suggest that these novel PA-related proteins likely possess important functions in the replication cycle of influenza A virus.
INTRODUCTION
Influenza A virus is a pathogen of a wide range of avian and mammalian species, including humans. It is an enveloped negative-strand RNA virus whose genome comprises eight viral RNA (vRNA) segments. Initially, each of the eight vRNA segments was shown to encode a single virus polypeptide, PB2, PB1, PA, HA, NP, NA, M1, or NS1 (1, 2). Later, transcripts of the M and NS genes were found to be spliced to produce M2 and NS2 as splicing variants (3, 4). Thus, until recently, the influenza viral genome was thought to encode 10 viral proteins in total (5).
However, since the beginning of the 21st century, several novel influenza A virus proteins have been identified. In 2001, a viral protein, PB1-F2, was discovered as a second polypeptide made from the PB1 mRNA (6). PB1-F2 is translated from the fourth AUG codon in an alternative reading frame of PB1, and its expression is thought to occur by leaky ribosomal scanning of the three upstream AUG codons. PB1-F2 modulates the host response to influenza A virus infection and is a virulence factor of influenza A virus (6–10). N-terminally truncated forms of PB1-F2 are also synthesized from AUG codons further downstream (i.e., the seventh, eighth, and ninth AUGs) of the PB1-F2 start codon (6, 7). PB1-N40 was then identified as a third major polypeptide synthesized from the PB1 mRNA (11). PB1-N40, an N-terminal 39-amino-acid-truncated form of PB1, is translated from the fifth AUG codon that is in-frame with the PB1 start codon, most likely as a result of leaky ribosomal scanning. Although virus that lacks PB1-N40 shows slower replication kinetics, the functions of the protein remain unclear. More recently, the novel influenza A virus protein PA-X was discovered. PA-X is a ribosomal frame-shifting product that comprises the N-terminal domain of PA (191 amino acids) with a short C-terminal domain (61 amino acids) encoded by an alternative reading frame of PA (12). PA-X modulates the host response and viral virulence. Further as-yet-unidentified viral proteins important for efficient virus replication may be found.
Early reports by Akkina et al. (13, 14) suggested that influenza A viruses express proteins that are reactive with an antibody to PA but are smaller than authentic PA; the nature of these proteins has remained unclear. Therefore, here, we focused on the PA gene and examined whether unidentified PA-related proteins are translated from the PA mRNA. The PA protein, which is a subunit of the RNA polymerase complex, is the main product of the PA gene and possesses endonuclease, cap binding, and promoter binding activities (15–18). We found small proteins produced from the PA segment and identified the translation initiation codons of these proteins on the PA mRNA by use of mutational analysis. We also evaluated the importance of these novel PA-related proteins for the replication of influenza A virus.
MATERIALS AND METHODS
Cells and viruses.
Human embryonic kidney 293T cells, 293 cells, and African green monkey kidney Vero E6 cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, penicillin-streptomycin, and amphotericin B. Madin-Darby canine kidney (MDCK) cells were maintained in Eagle's minimal essential medium containing 5% newborn calf serum and antibiotics. All cells were maintained at 37°C under 5% CO2. Influenza A/WSN/33 (H1N1; referred to as WSN), A/California/04/2009 (H1N1; Cal04), A/Yokohama/2017/2003 (H3N2; Yokohama), and A/swine/Ontario/41848/97 (H3N2; Sw/Ontario) were propagated in MDCK cells. A/duck/Mongolia/301/2001 (H3N2; Dk/Mongolia) was propagated in 10-day-old embryonated chicken eggs.
Antibodies.
Mouse anti-A/Puerto Rico/8/34 (PR8)-PA monoclonal antibodies 9/1, 37/6, 39/2, 65/4, 185/1, and 58/1 and mouse anti-PR8-PB1 monoclonal antibody 45/10 were available in our laboratory (19). Anti-PA 58/1 recognizes the N-terminal region of the PA protein (amino acid positions 101 to 107), whereas the other five anti-PA antibodies recognize other regions of PA (19). Rabbit monospecific antiserum to the WSN-PA protein (20) was also used.
Protein analyses.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting were performed according to standard procedures. Plasmid-transfected cells were lysed in SDS-PAGE sample buffer and separated on a 7.5% PAGE Tris-glycine gel (Cosmobio, Tokyo, Japan). The blots were probed with a mixture of five mouse anti-PA antibodies (9/1, 37/6, 39/2, 65/4, and 185/1) and a Vectastain ABC kit (Vector Laboratories, CA) and visualized by using immunostaining with HRP-1000 (Konica Minolta, Tokyo, Japan).
Radiolabeling and immunoprecipitation.
MDCK cells were infected with each influenza virus at a multiplicity of infection (MOI) of 1 to 10. At 3 h postinfection (p.i.), the cells were radiolabeled for 3 h in [35S]methionine/cysteine-containing medium. The labeled cells were lysed in lysis buffer (10 mM Tris-HCl [pH 7.5], 100 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate, 2 mM EDTA, and Complete Mini protease inhibitor mixture [Roche, Basel, Switzerland]). After clarification by centrifugation to remove cell debris, the supernatants were treated with 1% SDS and incubated at 37°C for 40 min to dissociate the three polymerase proteins from each other. After the SDS treatment, the cell lysates were diluted in lysis buffer that did not contain SDS (SDS−) to bring the SDS concentration to 0.1%; they were incubated with a mixture of five mouse anti-PA monoclonal antibodies or with rabbit anti-PA antiserum for 14 h at 4°C. Protein G magnet beads (NEB, MA) were then added to each sample, and the samples were incubated for 3 h at 4°C. The antigen-antibody-protein G-magnet complexes were washed four times with SDS− lysis buffer, lysed in SDS sample buffer, and then analyzed by means of SDS-PAGE, followed by visualization by autoradiography.
Virulence in mice.
To determine the 50% mouse lethal dose (MLD50) (i.e., the dose required to kill 50% of the infected mice) of N-truncated PA-deficient viruses, groups (n = 3 per group) of 6-week-old female BALB/c mice (Japan SLC, Shizuoka, Japan) were anesthetized with isoflurane and infected intranasally with 50 μl of serial 10-fold dilutions of viruses. The mice were monitored daily for clinical signs of infection for 14 days p.i. MLD50 values were calculated by using the method of Reed and Muench (21).
To compare the replication of N-truncated PA-deficient viruses in the lungs of mice, groups (n = 10 per group) of mice were intranasally infected with 102 PFU of each virus and euthanized on days 3 and 6 p.i. Lung tissues were collected to titrate virus infectivity with MDCK cells.
Mini-genome assays.
To assess the contributions of the viral PA mutants to polymerase activity, a luciferase activity-based mini-genome assay was performed as described previously (22) by using the following plasmids: expression plasmids for WSN-PB2, -PB1, and -NP (50 ng each); pPolI-NP(0)luc2(0) (2.5 ng), which expresses the firefly luciferase gene between the noncoding regions of the WSN-NP gene; and pGL4.74[hRluc/TK] (5 ng; Promega, WI). The luciferase activity in the plasmid-transfected 293 cells was quantified by using the Dual-Luciferase Reporter Assay System (Promega, WI) at 24 h or 40 h posttransfection (p.t.).
Immunofluorescence and confocal microscopy.
293 cells grown on glass-bottom dishes were transfected with expression plasmids for PB1 and PA or mutant PA. The cells were fixed at 24 h p.t. with 4% paraformaldehyde for 1 h, permeabilized with phosphate-buffered saline (PBS) containing 0.1% Triton X-100, and incubated with anti-PB1 and anti-PA antibodies for 1 h at room temperature. After being washed with PBS, they were incubated with Alexa Fluor 594 goat anti-mouse immunoglobulin G (IgG) (Invitrogen/Molecular Probes, OR) and Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen/Molecular Probes, OR) or Alexa Fluor 488 goat anti-mouse IgG (Invitrogen/Molecular Probes, OR). Nuclei were stained with Hoechst 33342 (Invitrogen, CA). Microscopic observation was performed with an LSM510 microscope (Carl Zeiss, Oberkochen, Germany). The z-stack image was reconstructed with five images through 3.54-μm z thickness by using the Zeiss LSM Image Browser.
RESULTS
Identification of novel viral proteins translated from PA mRNA.
To seek as-yet-unidentified viral proteins expressed from the PA gene of influenza A virus, we transfected pPolI-PA, the plasmid for the expression of PA vRNA, from WSN (H1N1) into 293T cells, along with pCAGGS-WSN-PB2, -PB1, -PA, and -NP, to provide polymerases and the NP protein. Forty-eight hours later, the cell lysates were analyzed by Western blotting using a mixture of five anti-PA monoclonal antibodies. Two bands, migrating to approximately 62 and 60 kDa, that were reactive with the anti-PA antibodies were detected, in addition to a strong signal for the full-length PA (83 kDa) (Fig. 1A, third lane from the left).
Fig 1.

Expression of N-truncated PAs in plasmid-transfected cells. (A) 293T cells were transfected with a plasmid for the expression of PA vRNA or PA mutant vRNA (pPolI-PA or PA mutants) in combination with expression plasmids encoding WSN-PB1, -PB2, -NP (1 μg each), and -PA (0.2 μg); an expression plasmid, pCAGGS-PA; or N-truncated PAs (1 μg). Forty-eight hours later, total cell lysates were analyzed by Western blotting for the detection of the full-length PA and the N-truncated PAs by using a mixture of anti-PA monoclonal antibodies. (B) Schematic representation of N-truncated forms of PA. N-truncated PAs are translated from the indicated AUG codons at amino acid positions 155 and 182.
We speculated that translation initiations at downstream AUG codons are involved in the expression of the additional smaller PA-related proteins, as is the case with PB1-F2 and PB1-N40 from the PB1 gene (6, 11). Based on their molecular weights and the presence of Kozak initiation consensus sequences, we hypothesized that the 11th AUG codon at amino acid position 155 and the 13th AUG codon at amino acid position 182 on the PA gene were used as the initiation codons for these additional PA gene products.
First, we mutated these two AUG codons to CUA, which encodes leucine. The smaller PA-related proteins expressed from the PA mutant plasmids were analyzed by Western blotting using a mixture of anti-PA monoclonal antibodies (Fig. 1A). When the CUA mutation was introduced on the 11th AUG codon (PA-M155L), a full-length PA and a smaller protein were detected, but the upper, smaller PA-related protein was undetectable. For the PA-M182L construct, which contains the AUG-to-CUA mutation on the 13th codon, the lower, smaller PA-related protein was undetectable. For the PA-DM construct, which possesses the AUG-to-CUA mutation at both the 11th and 13th codons, neither smaller PA-related protein was detected. To further confirm that the 11th and 13th AUG codons are the initiation codons for these smaller PA-related proteins, we made a plasmid encoding a PA-N155 protein, which is an N-terminal 154-amino-acid-truncated form of PA, and a plasmid encoding a PA-N182 protein, which is an N-terminal 181-amino-acid-truncated form of PA (pCAGGS-PA-N155 and pCAGGS-PA-N182) (Fig. 1B), and analyzed them by Western blotting. These N-truncated PAs comigrated with the smaller proteins expressed from the PA gene. In addition, transfection of an expression plasmid encoding PA (pCAGGS-PA) also produced these smaller PA-related proteins, although the band for PA-N182 is weak due to the low expression level. These results indicate that, in addition to authentic PA protein, N-terminally truncated forms of PA gene products, PA-N155 and PA-N182, are produced from the 11th and 13th AUG codons, respectively, in cells transfected with plasmids carrying PA genes (Fig. 1B).
Detection of N-truncated PAs in influenza A virus-infected cells.
Next, we investigated whether PA-N155 and PA-N182 are detectable in virus-infected cells. The WSN PA mutant viruses, M155L, M182L, and DM, were generated by use of reverse genetics. These viruses possess replacements of methionine (AUG) with leucine (CUA) at amino acid position 155 (M155L), 182 (M182L), or both (DM). MDCK cells were infected with wild-type WSN or with each of the PA mutant viruses and then radiolabeled with [35S]methionine/cysteine. The cell lysates were treated with 1% SDS to disrupt noncovalent interactions among the polymerase subunits and then immunoprecipitated with a mixture of five anti-PA monoclonal antibodies, followed by SDS-PAGE and autoradiography. Two smaller PA-related proteins, in addition to the full-length PA, were detected in WSN-infected cells (Fig. 2A). One smaller protein was not detected in each of the M155L- and M182L-infected cells. The DM-infected cells expressed neither of these two proteins. In addition to the two smaller proteins and the wild type, a fourth band was detected in WSN- and PA mutant-infected cells (indicated by the asterisk in Fig. 2A). Although the nature of this protein remains unknown, the signal intensity of this fourth band was weaker in a non-SDS-treated sample (right lane), indicating that the fourth protein may be a cleavage product of PA. These results indicate that both N-truncated PAs, PA-N155 and PA-N182, are synthesized in WSN virus-infected cells.
Fig 2.

Detection of N-truncated PAs in virus-infected cells. MDCK cells were infected with WSN and PA mutant viruses (A) or several different influenza A virus strains (B) and at 3 h p.i. radiolabeled for 3 h in [35S]methionine/cysteine-containing medium. After cell lysis, the supernatants were immunoprecipitated (IP) with a mixture of mouse anti-PA monoclonal antibodies (MAbs) or rabbit anti-PA antiserum and analyzed by SDS-PAGE, followed by visualization by autoradiography. The asterisk indicates a fourth band whose nature remains unknown.
To evaluate the importance of PA-N155 and PA-N182 in influenza viruses in general, we analyzed the conservation of the 11th and 13th AUG codons at amino acid positions 155 and 182, respectively, among influenza viruses by using publically accessible PA genes in GenBank. While these AUG codons were not conserved among influenza B and C viruses (17, 23), the AUG codon at position 155 was highly conserved among influenza A viruses, being absent in only 10 strains of the 11,023 PA sequences of influenza A virus examined (Table 1). The AUG codon at position 182 was also well conserved, being absent in 79 strains of the 11,023 PA sequences examined (Table 2). The viruses possessing non-AUG codons at position 182 were mainly avian and swine H9N2 viruses (Table 2), indicating that these viruses could spread at least among these hosts and that the AUG at position 182 may be dispensable for the viruses. In contrast, viruses possessing non-AUG codons at position 155 were isolated only sporadically, suggesting that the viruses could not spread efficiently. In addition, we found no strain that lacked both AUG codons at positions 155 and 182 in the PA gene. These results suggest that these AUG codons, especially that at position 155, might be important in the evolution of influenza A virus.
Table 1.
Influenza A virus strains lacking the AUG codon at amino acid position 155 of PA in GenBank
| Host | Virus strain | Subtype | Position 155 |
Accession no. | |
|---|---|---|---|---|---|
| Codon | Amino acid | ||||
| Human | A/Waikato/102/2003 | H3N2 | AUA | I | CY012093 |
| A/New York/392/2004 | H3N2 | AUA | I | CY002069 | |
| A/Azerbaijan/011–162/2006 | H5N1 | CUG | L | EU146863 | |
| Avian | A/unknown/New York/Sg-00359/2001 | H7N2 | CUG | L | CY036320 |
| A/chicken/Yokohama/aq134/2002 | H9N2 | CUG | L | AB256713 | |
| A/goose/Yunnan/3798/2006 | H5N1 | CUA | L | CY030904 | |
| A/chicken/Iran/SR110/2007a | H9N2 | AAG | K | EU157931 | |
| A/duck/Korea/112–25/2008 | H6N1 | GUG | V | GQ414839 | |
| Swine | A/Swine/Minnesota/55551/00 | H1N2 | AUA | I | AF455718 |
| A/swine/Cotes d'Armor/016007/2005 | H1N1 | CUG | L | AM921757 | |
A note in the database indicates that the sequence data for the region around amino acid position 155 might not be correct.
Table 2.
Influenza A virus strains lacking the AUG codon at amino acid position 182 of PA in GenBank
| Host | Subtype | No. of strains | Position 182 |
|
|---|---|---|---|---|
| Codon(s) (no. of strains) | Amino acid (no. of strains) | |||
| Human | H1N1 | 2 | UUG (2) | L (2) |
| H3N2 | 2 | UUG (1), CUG (1) | L (2) | |
| Avian | H5N1 | 1 | CUG (1) | L (1) |
| H5N2 | 1 | GUG (1) | V (1) | |
| H9N2 | 63 | CUG (57), CUA (4), UUA (2), GUG (1) | L (63) | |
| Swine | H1N1 | 2 | UUG (2) | L (2) |
| H9N2 | 7 | CUG (6), UUG (1) | L (7) | |
| Canine | H3N2 | 1 | CUG (1) | L (1) |
Accordingly, we investigated whether the expression of PA-N155 and PA-N182 is common among influenza A viruses. MDCK cells were infected with a human-pandemic H1N1 2009 virus, a seasonal H3N2 virus, a swine virus, and a duck virus, in addition to the WSN strain. The radiolabeled cell lysates were immunoprecipitated with anti-PA monoclonal antibodies or anti-PA antiserum as described above. The PA and N-truncated PAs expressed in WSN-infected cells were immunoprecipitated by anti-PA antiserum, as well as by anti-PA monoclonal antibodies (Fig. 2B). In all viruses tested, we detected smaller PA-related proteins whose mobilities were slightly different among viruses but similar to those of PA-N155 and PA-N182 expressed in WSN-infected cells (Fig. 2B); similarly, the mobilities of the full-length PA proteins differed among the viral strains. An unidentified fourth short band was also detected in all samples (indicated by the asterisk), which was likely identical to the fourth band detected in Fig. 2A (asterisk). In addition to these proteins, several other bands were detected, although the nature of these bands is unknown. These results suggest that the expression of N-truncated PAs is not limited to the WSN strain but is a universal feature of influenza A viruses.
Growth properties of N-truncated PA-deficient viruses.
To assess the importance of these N-truncated forms of PA in virus replication, we compared the growth properties of mutant viruses that were unable to express the N-truncated PAs with those of wild-type WSN in MDCK cells (Fig. 3). The M182L mutant replicated as well as wild-type WSN. In contrast, the titers of the M155L mutant, which lacks PA-N155, and the DM mutant, which lacks both PA-N155 and PA-N182, were significantly lower than that of wild-type WSN at every time point (P < 0.05). The differences between these mutants and the wild type were particularly remarkable at the early time points (24, 28, and 32 h p.i.); for example, the titers of the M155L and DM mutants were about 1.1 and 1.5 log10 units lower than that of the wild type at 24 h p.i., respectively, indicating that the M155L and DM mutant viruses, which lack the expression of PA-N155, showed slower replication kinetics in cells. These results suggest that even though the N-truncated PAs are not essential for replication, PA-N155 possesses some functions that are important for efficient virus replication.
Fig 3.

Growth kinetics of PA mutant viruses lacking N-truncated PAs in cell culture. MDCK cells were infected with wild-type WSN or mutant viruses lacking N-truncated PAs at a multiplicity of infection of 0.0005. At different time points postinfection, virus titers in the supernatants of the infected cells were determined by means of plaque assays in MDCK cells. The mean titers from triplicate independent cultures ± standard deviations (SD) are shown.
Pathogenicity of N-truncated PA-deficient viruses in mice.
To assess whether the N-truncated forms of PA are important for viral pathogenicity in vivo, we compared the MLD50s of the PA mutant viruses with that of wild-type WSN. Although the pathogenicity of the M182L mutant was similar to that of the wild-type virus, the M155L and DM mutants were attenuated compared with the wild-type virus (the MLD50 value was approximately 1.5 log10 units higher than that of the wild-type virus) (Table 3). In addition, the virus titers in the lungs of mice infected with the M155L and DM mutants were more than 1.0 log10 unit lower than that of wild-type WSN on day 3, although the differences in virus titers between the PA mutants and WSN on day 6 were smaller than on day 3 (Fig. 4). These results suggest that PA-N155 likely plays important roles in virus pathogenicity in animals.
Table 3.
Comparison of pathogenicities of PA mutant viruses lacking N-truncated PAs in mice
| Virus | MLD50 (log10 PFU)a |
|---|---|
| WSN | 3.8 |
| M155L | 5.3 |
| M182L | 4.0 |
| DM | 5.5 |
Groups (n = 3 per group) of 6-week-old female BALB/c mice were anesthetized with isoflurane and infected intranasally with 50 μl of serial 10-fold dilutions of viruses, thereby creating doses ranging from 102 to 106 PFU. The mice were monitored daily for clinical signs of infection for 14 days p.i. MLD50 values were calculated by using the method of Reed and Muench (21).
Fig 4.
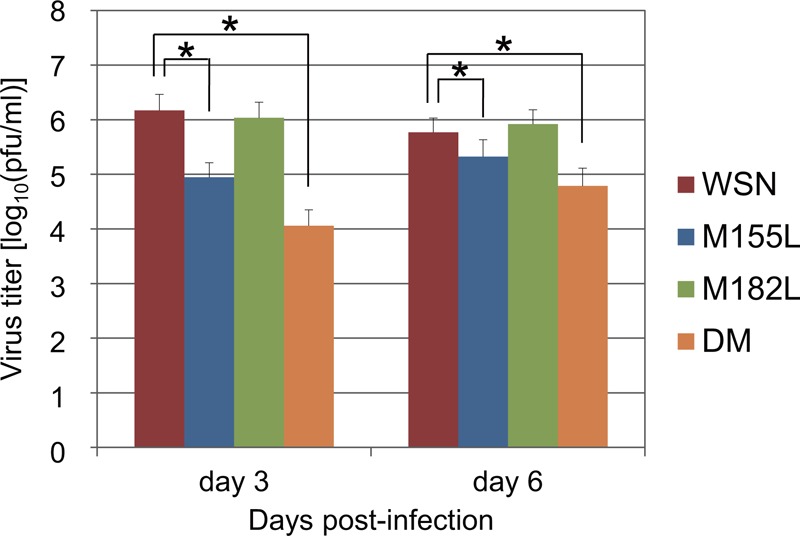
Replication of PA mutant viruses lacking N-truncated PAs in the lungs of mice. Eight groups (n = 10 per group) of 6-week-old female BALB/c mice were infected intranasally with 102 PFU of virus, and 3 and 6 days later, lungs were collected for virus titration. Each bar represents the mean titer for each virus. The data are the mean titer and the SD for each virus. An asterisk indicates that the mean titer of the PA mutant virus was significantly lower than that of the wild-type WSN (P < 0.05). Detection limit, 2 log10 PFU/g tissue.
Polymerase functions of mutant PA proteins.
To understand the mechanisms behind the reduced growth of the mutant viruses M155L and DM, we tested the effects of the PA mutations on polymerase activity. 293 cells were cotransfected with a plasmid encoding wild-type PA or each mutant PA protein, along with plasmids for the expression of PB2, PB1, NP, and a vRNA replicon possessing the luciferase gene. At 24 and 40 h p.t., luciferase activity was measured (Fig. 5). All PA mutants, PA-M155L, PA-M182L, and PA-DM, supported viral RNA replication and transcription at levels similar to that of wild-type PA at both 24 and 40 h p.t. These results indicate that the expression levels of the N-truncated PAs have little effect on viral polymerase activity. They also suggest that the reduced replication and lower pathogenicity of the PA mutant viruses, M155L and DM, were not the result of reduced polymerase activity of the mutant PAs, at least as detected by the above-described mini-genome assay.
Fig 5.

Polymerase activities of mutant PAs lacking the expression of N-truncated PAs. 293 cells were transfected with expression plasmids encoding PB2, PB1, NP, and a vRNA replicon possessing a firefly luciferase gene between the noncoding regions of WSN-NP vRNA, along with the indicated PA- or PA mutant-expressing plasmid. Twenty-four or 40 h later, the luciferase activity was quantified. The data are the means ± SD of eight independent experiments plotted as the percent activity with wild-type PA for each experiment.
Functions of N-truncated PA proteins.
Since N-truncated PAs lack the regions required for endonuclease activity (24, 25), they should not be functionally equivalent to full-length PA. We therefore tested whether N-truncated PAs support viral polymerase activity in the mini-genome assay, as described above. Only background levels of luciferase activity were detected in the absence of the PA protein (empty vector), and both PA-N155 and PA-N182 also produced only background levels of luciferase (Fig. 6), indicating that the N-truncated PAs lack polymerase activity. These results also suggest that PA-N155, which was shown to be important for viral replication in vitro and in vivo, is likely involved in viral replication steps other than the transcription and replication of the viral genome.
Fig 6.

Functions of N-truncated PAs. (A) Polymerase activities of N-truncated PAs. 293 cells were transfected with expression plasmids encoding PB2, PB1, and NP and a vRNA replicon possessing a firefly luciferase gene, along with the indicated PA- or N-truncated PA-expressing plasmid. Twenty-four hours later, the luciferase activity was quantified. The data shown here are representative of three independent experiments. An asterisk indicates that the activity of the N-truncated PA was significantly lower than that of wild-type PA (P < 0.05). (B) Viral growth kinetics of PA mutant viruses that lack N-truncated PAs in Vero E6 cells. Vero E6 cells were infected with wild-type WSN or mutant viruses lacking N-truncated PAs at a multiplicity of infection of 0.005. At different time points postinfection, virus titers in the supernatants of the infected cells were determined by means of plaque assays. The mean titers from triplicate independent cultures ± standard deviations are shown.
We then investigated whether PA-N155 was an antagonist of the antiviral response induced by type I interferon by comparing the growth properties of mutant viruses that lack the N-truncated PAs with those of wild-type WSN in Vero E6 cells, which do not produce type I interferon. As is true for virus growth in MDCK cells, the M182L mutant replicated as well as wild-type WSN (Fig. 6B). The titers of the M155L and DM mutants were significantly lower than that of wild-type WSN at 12, 24, and 72 h p.i. in M155L and at 12, 24, and 48 h p.i. in DM (P < 0.05). These results indicate that mutant viruses that lack PA-N155 exhibit slower replication kinetics in Vero E6 cells, suggesting that PA-N155 is not an antagonist of the antiviral response induced by type I interferon.
We then examined the intracellular distribution of the N-truncated PAs. The PA protein has two nuclear localization signals (NLSs), which are located at amino acid positions 124 to 139 and 186 to 247 (26). Since both of the N-truncated PAs lack the former NLS, we hypothesized that the N-truncated PAs would show a localization pattern different from that of full-length PA. By using an immunofluorescence assay, we examined the intracellular localization of PA-N155 and PA-N182 and compared it with that of full-length PA. To distinguish full-length PA from the N-truncated PAs expressed from the plasmid encoding PA (pCAGGS-PA) (Fig. 1A), we used both the anti-PA monoclonal antibody 58/1, which recognizes the N-terminal region of the PA protein (amino acid positions 101 to 107), and anti-PA antiserum, which recognizes both full-length PA and N-truncated PA (Fig. 2B).
When cells transfected with pCAGGS-PA were treated with the anti-PA monoclonal antibody 58/1, signals were detected in both the cytoplasm and the nucleus (data not shown). Cells transfected with pCAGGS-PA-N155 or pCAGGS-PA-N182 did not react with the antibody. These results indicate that full-length PA localized to both the cytoplasm and the nucleus. When we used the anti-PA antiserum, positive signals were detected in both the cytoplasm and the nuclei of cells transfected with pCAGGS-PA, and there was no apparent difference in PA detection between the monoclonal antibody and the antiserum (data not shown). The N-truncated PAs were also located in both the cytoplasm and the nuclei of the plasmid-transfected cells, and there was no significant difference in localization between full-length PA and N-truncated PAs (data not shown).
We then investigated the intracellular localization of the N-truncated PAs in the presence of PB1, since PA and PB1 interact with each other in the cytoplasm and are transported into the nucleus as a heterodimer (27–29) and the N-truncated PAs possess the regions responsible for binding to PB1 (located at positions 601 to 692 of PA) (30, 31). In the absence of PA, PB1 was detected predominantly in the cytoplasm, whereas when PA was coexpressed, PB1 was largely found in the nucleus (data not shown). PA was predominantly detected in the nucleus in the presence of PB1, although it localized to both the cytoplasm and the nucleus in the absence of PB1 (data not shown), confirming that PA interacts with PB1 and together they are transported into the nucleus. The N-truncated PAs showed localization patterns similar to that of PA; the N-truncated PAs were predominantly found in the nucleus, as was PB1 in cells expressing both proteins. These results indicate that PA-N155 and PA-N182 interact with PB1 and are transported into the nucleus.
DISCUSSION
Here, we identified two novel influenza virus proteins, PA-N155 and PA-N182, which are N-truncated forms of PA. Given that the translation initiation codons for these proteins are highly conserved among influenza A viruses, it is possible that expression of these N-truncated PAs is a universal feature of influenza A viruses. This concept is supported by early reports by Akkina et al. (13, 14) that various influenza A virus strains (i.e., all 18 strains belonging to H1 to H13) express shorter PA-related polypeptides of 62 and 60 kDa, although the nature of these polypeptides remained unclear in those reports. The molecular weights of the polypeptides described by Akkina et al. are similar to those of the N-truncated PAs identified in this study, supporting the idea that these N-truncated PAs are commonly expressed in virus-infected cells. The universal expression of these N-truncated PAs among influenza A viruses strongly suggests their importance in the life cycle of influenza A viruses.
The mutant viruses M155L and DM, which lack expression of PA-N155, showed slower replication kinetics in cells and were less pathogenic in mice than wild-type virus (Fig. 3 and 4). These mutant viruses possess a point mutation of methionine to leucine at position 155 in PA. The reason why we replaced methionine with leucine at this position is that 5 out of 10 influenza A virus strains lacking methionine in this position possess leucine (Table 1), indicating that the leucine mutation occurs naturally. It should be noted that mutations near position 155 affect some of the functions of PA. The E154G mutation, which arose spontaneously during the cDNA cloning of the PA gene, completely abolished polymerase activity and proteolysis activity (32, 33). A mutation at amino acid position 157, which is a potential phosphorylation site, also diminished polymerase activity and proteolysis activity (34–36). Therefore, we were concerned that the point mutation at position 155 would disrupt some PA functions and in so doing impair virus replication and pathogenicity. However, both PA-M155L and PA-DM supported viral polymerase activity (Fig. 5), suggesting that the methionine at position 155 is not essential for this function. Therefore, the reduction in virus replication and pathogenicity of the M155L and DM mutants is unlikely to involve polymerase functions, but rather, may be due to the lack of expression of PA-N155.
On the other hand, the mutant virus that lacks PA-N182 replicated as efficiently as wild-type virus. What, then, are the differences between PA-N155 and PA-N182? Between amino acid positions 155 and 182 of PA, there are potential sites for phosphorylation by casein kinase II at positions 157 and 162. A point mutation at position 157 (T157A) or 162 (T162A) of PA, which eliminates the phosphorylation site, reduces both the proteolysis-inducing activity and the viral genome replication activity of PA (36). However, expression of the N-truncated PAs had little effect on viral polymerase activity (Fig. 5), and PA-N155 possesses no proteolysis activity (32, 33), suggesting that phosphorylation at positions 157 and 162 in PA-N155 is not involved in viral transcription and replication or the proteolysis-inducing activity of PA.
The cRNA promoter binding site is also located between amino acid positions 155 and 182 on PA (at position 163 to 178) (18), implying that PA-N155 could contribute to vRNA replication. However, the expression of PA-N155 had little effect on polymerase activity (Fig. 5), and therefore, it is unlikely that PA-N155 contributes to viral genome replication via the cRNA promoter binding site. Taken together, our results support a role for PA-N155 in virus replication, although the exact role remains unclear. Further work is necessary to understand the contributions of PA-N155 to the virus replication cycle.
The first AUG codon in the PA mRNA possesses a Kozak consensus sequence that is important for efficient initiation of translation (37, 38). However, translation is also initiated downstream at the 11th and 13th AUG codons in the PA mRNA. The mechanistic basis for the initiation of translation at these downstream AUG codons remains unclear. In addition, the natures of the several PA-related proteins that were detected in influenza A virus-infected cells also remain unknown (Fig. 2B). These results suggest that translation is initiated at AUG codons downstream of those used for known influenza A virus proteins in all viral gene segments, leading to the production of N-terminally truncated viral proteins. Further work on the identification of novel influenza A virus proteins and elucidation of their functions should contribute to a better understanding of the influenza virus replication cycle.
ACKNOWLEDGMENTS
We thank Susan Watson for editing the manuscript.
This work was supported by a Grant-in-Aid for Specially Promoted Research; by the Japan Initiative for Global Research Network on Infectious Diseases from the Ministry of Education, Culture, Sports, Science, and Technology; by grants-in-aid from the Ministry of Health, Labor, and Welfare of Japan; by ERATO (Japan Science and Technology Agency); and by Public Health Service research grants from the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print 12 December 2012
REFERENCES
- 1. Sugiura A. 1975. Introduction-historical review, p 171–213 In Kilbourne ED. (ed), The influenza viruses and influenza. Academic Press, New York, NY [Google Scholar]
- 2. Palese P. 1977. The genes of influenza virus. Cell 10:1–10 [DOI] [PubMed] [Google Scholar]
- 3. Lamb RA, Lai CJ. 1980. Sequence of interrupted and uninterrupted mRNAs and cloned DNA coding for the two overlapping nonstructural proteins of influenza virus. Cell 21:475–485 [DOI] [PubMed] [Google Scholar]
- 4. Lamb RA, Lai CJ, Choppin PW. 1981. Sequences of mRNAs derived from genome RNA segment 7 of influenza virus: colinear and interrupted mRNAs code for overlapping proteins. Proc. Natl. Acad. Sci. U. S. A. 78:4170–4174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lamb RA. 1983. The influenza virus RNA segments and their encoded proteins, p 26–69 In Palese P, Kingsbury DW. (ed), Genetics of influenza viruses. Springer, New York, NY [Google Scholar]
- 6. Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O'Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW. 2001. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 7:1306–1312 [DOI] [PubMed] [Google Scholar]
- 7. Zamarin D, Ortigoza MB, Palese P. 2006. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J. Virol. 80:7976–7983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McAuley JL, Hornung F, Boyd KL, Smith AM, McKeon R, Bennink J, Yewdell JW, McCullers JA. 2007. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2:240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. 2007. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 3:1414–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Conenello GM, Tisoncik JR, Rosenzweig E, Varga ZT, Palese P, Katze MG. 2011. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J. Virol. 85:652–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wise HM, Foeglein A, Sun J, Dalton RM, Patel S, Howard W, Anderson EC, Barclay WS, Digard P. 2009. A complicated message: identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J. Virol. 83:8021–8031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, Dalton RM, Lo A, Efstathiou S, Atkins JF, Firth AE, Taubenberger JK, Digard P. 2012. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akkina RK. 1990. Antigenic reactivity and electrophoretic migrational heterogeneity of the three polymerase proteins of type A human and animal influenza viruses. Arch. Virol. 111:187–197 [DOI] [PubMed] [Google Scholar]
- 14. Akkina RK, Richardson JC, Aguilera MC, Yang CM. 1991. Heterogeneous forms of polymerase proteins exist in influenza A virus-infected cells. Virus Res. 19:17–30 [DOI] [PubMed] [Google Scholar]
- 15. Lee MT, Bishop K, Medcalf L, Elton D, Digard P, Tiley L. 2002. Definition of the minimal viral components required for the initiation of unprimed RNA synthesis by influenza virus RNA polymerase. Nucleic Acids Res. 30:429–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fodor E, Crow M, Mingay LJ, Deng T, Sharps J, Fechter P, Brownlee GG. 2002. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol. 76:8989–9001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hara K, Schmidt FI, Crow M, Brownlee GG. 2006. Amino acid residues in the N-terminal region of the PA subunit of influenza A virus RNA polymerase play a critical role in protein stability, endonuclease activity, cap binding, and virion RNA promoter binding. J. Virol. 80:7789–7798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maier HJ, Kashiwagi T, Hara K, Brownlee GG. 2008. Differential role of the influenza A virus polymerase PA subunit for vRNA and cRNA promoter binding. Virology 370:194–204 [DOI] [PubMed] [Google Scholar]
- 19. Hatta M, Asano Y, Masunaga K, Ito T, Okazaki K, Toyoda T, Kawaoka Y, Ishihama A, Kida H. 2000. Epitope mapping of the influenza A virus RNA polymerase PA using monoclonal antibodies. Arch. Virol. 145:895–903 [DOI] [PubMed] [Google Scholar]
- 20. Akkina RK, Chambers TM, Londo DR, Nayak DP. 1987. Intracellular localization of the viral polymerase proteins in cells infected with influenza virus and cells expressing PB1 protein from cloned cDNA. J. Virol. 61:2217–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Hyg. (London) 27:493–497 [Google Scholar]
- 22. Ozawa M, Fujii K, Muramoto Y, Yamada S, Yamayoshi S, Takada A, Goto H, Horimoto T, Kawaoka Y. 2007. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J. Virol. 81:30–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Akoto-Amanfu E, Sivasubramanian N, Nayak DP. 1987. Primary structure of the polymerase acidic (PA) gene of an influenza B virus (B/Sing/222/79). Virology 159:147–153 [DOI] [PubMed] [Google Scholar]
- 24. Dias A, Bouvier D, Crépin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW. 2009. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458:914–918 [DOI] [PubMed] [Google Scholar]
- 25. Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, Fodor E, Rao Z, Liu Y. 2009. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 458:909–913 [DOI] [PubMed] [Google Scholar]
- 26. Nieto A, de la Luna S, Bárcena J, Portela A, Ortín J. 1994. Complex structure of the nuclear translocation signal of influenza virus polymerase PA subunit. J. Gen. Virol. 75:29–36 [DOI] [PubMed] [Google Scholar]
- 27. Nieto A, de la Luna S, Bárcena J, Portela A, Valcárcel J, Melero JA, Ortín J. 1992. Nuclear transport of influenza virus polymerase PA protein. Virus Res. 24:65–75 [DOI] [PubMed] [Google Scholar]
- 28. Fodor E, Smith M. 2004. The PA subunit is required for efficient nuclear accumulation of the PB1 subunit of the influenza A virus RNA polymerase complex. J. Virol. 78:9144–9153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deng T, Sharps J, Fodor E, Brownlee GG. 2005. In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J. Virol. 79:8669–8674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zürcher T, de la Luna S, Sanz-Ezquerro JJ, Nieto A, Ortín J. 1996. Mutational analysis of the influenza virus A/Victoria/3/75 PA protein: studies of interaction with PB1 protein and identification of a dominant negative mutant. J. Gen. Virol. 77:1745–1749 [DOI] [PubMed] [Google Scholar]
- 31. Toyoda T, Adyshev DM, Kobayashi M, Iwata A, Ishihama A. 1996. Molecular assembly of the influenza virus RNA polymerase: determination of the subunit-subunit contact sites. J. Gen. Virol. 77:2149–2157 [DOI] [PubMed] [Google Scholar]
- 32. Sanz-Ezquerro JJ, de la Luna S, Ortín J, Nieto A. 1995. Individual expression of influenza virus PA protein induces degradation of coexpressed proteins. J. Virol. 69:2420–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sanz-Ezquerro JJ, Zürcher T, de la Luna S, Ortín J, Nieto A. 1996. The amino-terminal one-third of the influenza virus PA protein is responsible for the induction of proteolysis. J. Virol. 70:1905–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sanz-Ezquerro JJ, Fernández Santarén J, Sierra T, Aragón T, Ortega J, Ortín J, Smith GL, Nieto A. 1998. The PA influenza virus polymerase subunit is a phosphorylated protein. J. Gen. Virol. 79:471–478 [DOI] [PubMed] [Google Scholar]
- 35. Perales B, Sanz-Ezquerro JJ, Gastaminza P, Ortega J, Santarén JF, Ortín J, Nieto A. 2000. The replication activity of influenza virus polymerase is linked to the capacity of the PA subunit to induce proteolysis. J. Virol. 74:1307–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huarte M, Falcón A, Nakaya Y, Ortín J, GarcíA-Sastre A, Nieto A. 2003. Threonine 157 of influenza virus PA polymerase subunit modulates RNA replication in infectious viruses. J. Virol. 77:6007–6013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kozak M. 1986. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 44:283–292 [DOI] [PubMed] [Google Scholar]
- 38. Maeda Y, Goto H, Horimoto T, Takada A, Kawaoka Y. 2004. Biological significance of the U residue at the −3 position of the mRNA sequences of influenza A viral segments PB1 and NA. Virus Res. 100:153–157 [DOI] [PubMed] [Google Scholar]