

Abstract
The tropism of herpes simplex virus (HSV-1) for human sensory neurons infected in vivo was examined using dorsal root ganglion (DRG) xenografts maintained in mice with severe combined immunodeficiency (SCID). In contrast to the HSV-1 lytic infectious cycle in vitro, replication of the HSV-1 F strain was restricted in human DRG neurons despite the absence of adaptive immune responses in SCID mice, allowing the establishment of neuronal latency. At 12 days after DRG inoculation, 26.2% of human neurons expressed HSV-1 protein and 13.1% expressed latency-associated transcripts (LAT). Some infected neurons showed cytopathic changes, but HSV-1, unlike varicella-zoster virus (VZV), only rarely infected satellite cells and did not induce fusion of neuronal and satellite cell plasma membranes. Cell-free enveloped HSV-1 virions were observed, indicating productive infection. A recombinant HSV-1-expressing luciferase exhibited less virulence than HSV-1 F in the SCID mouse host, enabling analysis of infection in human DRG xenografts for a 61-day interval. At 12 days after inoculation, 4.2% of neurons expressed HSV-1 proteins; frequencies increased to 32.1% at 33 days but declined to 20.8% by 61 days. Frequencies of LAT-positive neurons were 1.2% at 12 days and increased to 40.2% at 33 days. LAT expression remained at 37% at 61 days, in contrast to the decline in neurons expressing viral proteins. These observations show that the progression of HSV-1 infection is highly restricted in human DRG, and HSV-1 genome silencing occurs in human neurons infected in vivo as a consequence of virus-host cell interactions and does not require adaptive immune control.
INTRODUCTION
The simplex viruses, herpes simplex virus types 1 and 2 (HSV-1 and -2), are ubiquitous human viral pathogens that may cause mild to severe mucocutaneous infections (1). In rare instances, these viruses cause meningoencephalitis. The prevalence of HSV-1 infection among adults ranges from 50 to 90%. Primary HSV infection is acquired by inoculation of mucosal epithelium and abraded skin. Local replication facilitates retrograde axonal transport of HSV virions within innervating cutaneous sensory nerve fibers to the corresponding neuronal cell bodies in the peripheral dorsal root ganglia (DRG) or trigeminal ganglia, where latency is established. Although most primary infections are asymptomatic, HSV-1 and HSV-2 have a dynamic relationship with the human host, and transmission to other susceptible individuals is ensured by frequent episodes of reactivation; these episodes are also usually asymptomatic but release infectious virus into oral and genital tract secretions. Notably, the many hundreds of HSV reactivations from latency that occur in sensory ganglia over the lifetime of the infected individual seldom result in ganglionitis, peripheral neuropathy, or spread to the central nervous system (2–4).
Lytic HSV-1 infection, which produces infectious progeny virions, involves transcription of more than 80 viral genes in a highly ordered process governed by sequential derepression of viral transcription factors encoded by immediate-early genes (5). Persistent HSV-1 infection of neurons is characterized by presence of the HSV-1 genome in a silenced, unproductive state from which it may periodically initiate lytic infection (reviewed in reference 1). Stable, nonpolyadenylated RNAs, known as latency-associated transcripts (LAT), that result from differential splicing of the LAT intron accumulate in the nuclei of neurons that harbor nonreplicating HSV-1 or HSV-2. How LAT contributes to latency is not well understood. LAT-encoded proteins have not been reliably detected in latently infected neurons (6). Functions attributed to LAT include inhibitory effects on the critical immediate-early gene product ICP0 (7), promoting heterochromatin silencing of HSV-1 genes (8), antiapoptotic functions (9), and effects on the establishment of HSV-1 latency in mice (10) and reactivation in rabbits, as shown using LAT-null mutants (11, 12).
While humans are the only natural host, both HSV-1 and HSV-2 infect across species barriers. Rodent, rabbit, and nonhuman neuronal culture systems have been used extensively to investigate HSV-1 neuropathogenesis (10, 13–18), but many questions regarding tropism and virulence in human neural tissues remain unanswered. Our objective was to examine HSV-1 infection of human sensory ganglia in vivo using a model that we developed to investigate the neuropathogenesis of varicella-zoster virus (VZV), a highly human-restricted alphaherpesvirus (19–21), that has the potential to add substantially to observations from nonhuman studies. This model involves experimental infection of human DRG xenografts maintained in mice with severe combined immunodeficiency (SCID). The model has allowed us to elucidate key aspects of VZV replication in human sensory ganglia, such as the capacity of the virus to induce fusion of neuronal and satellite cell membranes in neuron-satellite cell complexes and to persist in a nonreplicating state in neurons without adaptive immune regulation, which is lacking in SCID mice (19, 22).
Our aims in this study were to define the course of events that follows HSV-1 inoculation of DRG xenografts, identify ganglionic cell types permissive for HSV-1 replication, and determine whether HSV-1 achieves latency, based on LAT expression. We observed that HSV-1 is restricted in human DRG xenografts despite the absence of adaptive immune responses in SCID mice. Infection was lytic in some neurons, but LAT expression also occurred in neurons shortly after infection and the frequency of LAT-positive neurons relative to that of HSV-1 protein-positive neurons increased over time. Although both HSV-1 and VZV establish latency within sensory neurons following primary infection, the neuropathogenesis of HSV-1 differed significantly from that of VZV when human neurons were infected within their natural host tissue microenvironment in vivo. HSV-1 infection of satellite cells was rarely detected, and neuron-satellite cell fusion was not observed in infected DRG.
MATERIALS AND METHODS
Construction and infection of human DRG xenografts in SCID mice.
DRG xenotransplantation into SCID mice was done as previously described (19). Briefly, a single DRG (1 to 2.5 mm3) was inserted under the left renal capsule of a sedated male C.B.-17 scid/scid mouse (Taconic Farms, Germantown, NY). After engraftment (>8 weeks), DRG were surgically exposed and inoculated with HSV-1 by a single direct injection of 10 μl containing 10,000 PFU. At the designated time point, DRG xenografts were recovered immediately from euthanized mice and fixed with 4% paraformaldehyde. Human fetal tissue was provided by Advanced Bioscience Resources (Alameda, CA) in compliance with state and federal regulations. Animal procedures complied with the Animal Welfare Act and were approved by the Stanford University Administrative Panel on Laboratory Animal Care.
Construction and characterization of an HSV-1 F strain expressing luciferase (R8411 mutant).
The HSV-1 F strain expressing luciferase (R8411) under the control of the ICP27 promoter was a gift from Bernard Roizman (University of Chicago) and was constructed using an HSV-1 bacterial artificial chromosome (BAC) system described in Horsburgh et al. (23). Briefly, the gene for firefly luciferase was amplified by PCR, and the product was ligated after SpeI digestion into pRB5260, between the ICP27 promoter and UL21/UL22 poly(A) signal. The ICP27-luciferase-poly(A) fragment was inserted between UL3 and UL4 to generate plasmid pRB8538. A HindII fragment containing the insert was cloned into the gene replacement vector pKO5 to generate pKO8538. pKO8538 was transformed into HSV BAC (tk-) to select for recombinant BAC8411. Recombinant BAC8411 DNA was cotransfected with pRB4867 into Vero cells (24). R8411 was selected on 143tk- cells in medium containing hypoxanthine, aminopterin, and thymidine.
Field-inversion gel electrophoresis (FIGE) was performed on restriction endonuclease-digested nucleocapsid DNA, prepared from HSV-1 F strain- and R8411-infected Vero cell monolayers, using the method described by Szpara et al. (25). One microgram of nucleocapsid DNA was digested for 2 h with ClaI, HindIII, and NsiI and run overnight using a FIGE mapper (Bio-Rad; program 2.). For immunoblot analysis, Vero infected cell monolayers were lysed in radioimmunoprecipitation (RIPA) buffer and run on a 7.5% gel. Blotting was performed using rabbit anti-HSV-1 thymidine kinase (Santa Cruz Biotechnology), rabbit anti-VP16 (a gift from Nancy Sawtell, University of Cincinnati), mouse anti-HSV-1 ICP5 (Santa Cruz Biotechnology), and mouse anti-tubulin (Sigma), using methods previously described (21). Growth curves were performed on human embryonic lung fibroblasts (HELF) grown to confluence on 6-well plates. Wells were infected using a multiplicity of infection (MOI) of 1 or 7. After 1 h adsorption, the inoculum was removed and medium containing 1.25% carboxymethylcellulose was overlaid. At various time points after infection (hours 1, 5, 9, 14, and 20), cells and media were harvested from separate wells and subjected to three freeze-thaw cycles, and then viral titers were determined by a plaque assay on Vero cells. For plaque assays, samples were added to Vero cell wells in triplicate, allowed to grow for 3 days, and stained using an immunohistochemical method with alkaline phosphatase detection using Fast Red substrate.
Preparation of HSV-1 inocula.
HSV-1 inocula were prepared from infected Vero cells by high-speed centrifugation of the supernatant and monolayer (17,000 × g for 10 min at 4°C), followed by pulse sonication (Sonic Dismembrator 550; Fisher Scientific) for 1 min (5 s on, 5 s rest) and low-speed centrifugation (800 × g for 5 min); a plaque assay was used to determine the titer, and aliquots were stored at −80°C. Each aliquot used to infect DRG had the titer redetermined at the time of inoculation.
Drug intervention.
Valacyclovir was administered by oral gavage (100 μl, 50 mg/kg of body weight). The oral suspension of valacyclovir was prepared by grinding Valtrex caplets (GlaxoSmithKline, Research Triangle Park, NC) using a pestle and mortar and thorough mixing with Ora Plus suspending vehicle (Paddock Laboratories, Inc., Minneapolis, MN). Mice were gavaged upon fully awakening from surgery following DRG infection (4 h later) and then once daily.
Live animal imaging.
Mice were given luciferin (3 mg D-luciferin; Xenogen) 5 min prior to imaging under isoflurane anesthesia using an IVIS Spectra imaging system (Stanford Small Animal Imaging Facility). Bioluminescence values were measured from the region of interest (ROI) over the site of the xenograft and over the surrounding area and calculated as photon flux (photons/s/cm2) using Living Image Software (version 3.2; Xenogen, Alameda, CA). Values were corrected for background by subtracting an average background value from the measurement of signal intensity in the ROI. Areas of peak pixel intensity were determined using Living Image Software; pixels were included in the ROI if the pixel intensity was greater than the threshold percentage (50%). Values were normalized for binning, exposure time, and area (cm2) using Living Image Software and analyzed using GraphPad Prism (version 5.0).
Viral and cellular protein expression.
DRG xenografts were fixed in 4% paraformaldehyde and paraffin embedded into tissue blocks. Paraffin-embedded samples were serially sectioned, generating approximately 150 5-μm sections/DRG. Approximately every tenth slide was stained with hematoxylin and eosin (H&E) to assess morphology and identify regions containing islets of neuron-satellite cell complexes. Enzyme immunohistochemistry and confocal immunofluorescence microscopy were performed as previously described using a standard avidin-biotin-peroxidase method with 3,3′-diaminobenzidine (DAB) substrate (Vector Laboratories, Burlingame, CA) or fluorescent secondary antibodies used at dilutions recommended by the manufacturer (Invitrogen, Carlsbad, CA). For each staining condition, a slide without any primary antibody applied was stained and examined in parallel. An uninfected negative-control slide and a known positive slide were included in each staining run. A hematoxylin blue nuclear counterstain was applied after DAB chromogen development (Vector Laboratories, Burlingame, CA). Alexa 594 goat anti-mouse and anti-rabbit and Alexa 488 goat anti-rabbit and anti-mouse secondary antibodies (Invitrogen, Carlsbad, CA) were used for confocal immunofluorescence microscopy along with a Hoechst nuclear stain (Sigma-Aldrich, St. Louis, MO). Slides were examined using an AxioPlan 2 LSM 510 confocal microscope (Zeiss, New York, NY). In some cases, antigen retrieval using the citrate buffer/pressure cooker method was performed on paraffin-embedded sections prior to staining. Staining experiments to assess satellite cell tropism and fusion were performed on multiple tissue sections (6 to 8 sections) for each DRG, representing 700 to 900 neurons examined.
HSV-1-infected neurons were detected with a pan-HSV rabbit polyclonal antibody (AXL237; Accurate Chemical & Scientific Corp., Westbury, NY), a mouse monoclonal antibody to ICP4 (Abcam, Cambridge, MA), and a rabbit anti-VP16 antibody. Monoclonal antibody or rabbit polyclonal antibody to neural cell adhesion molecule (NCAM) and a pan-neurofilament marker were obtained from EMD Millipore (Billerica, MA). The rabbit antibody to synaptophysin was obtained from Invitrogen, and the rabbit antibody to herpesvirus entry mediator (HVEM) (anti-TNFRS14) was obtained from Abcam.
Detection of LAT by RNA in situ hybridization.
Paraffin sections were dewaxed and rehydrated through graded alcohols and then water for RNA in situ hybridization. Slides were permeabilized with proteinase K and acetylated prior to hybridization. Prehybridization, hybridization, and probe preparation were as described by Margolis et al. using labeled riboprobes for the HSV-1 stable LAT intron prepared from the plasmid pATD-19 (26). The digoxigenin-labeled probe was detected after incubation with anti-DIG-alkaline phosphatase (Roche) using naphthol/Fast Red (Sigma-Aldrich), which has fluorescent properties when viewed using a HeNe 543 spectrometer or nitroblue tetrazolium and 5-bromo-4-chloro-3-indolylphosphate (NBT/BCIP; Roche). A red nuclear counterstain was used following NBT/BCIP development (Vector); a blue nuclear stain (Hoechst) was used for immunofluorescence.
Quantitative analysis of HSV-1 protein and LAT expression.
DRG xenografts were collected for examination at various time points days after infection. Tissues from each time point were embedded into one to two paraffin blocks (∼3 DRG per block), and the entire block was serially sectioned to generate approximately 150 slides containing two sections, each with a 5-μm thickness, per slide. Every tenth slide was stained with H&E to identify regions of interest for each individual DRG that contained at least 20 neurons/section. Viral protein expression was assessed by immunohistochemistry using a rabbit polyclonal antibody that recognizes multiple HSV-1-infected cell proteins; LAT transcripts were detected by RNA in situ hybridization. Six to eight slides were tested for the presence of HSV-1 protein or LAT RNA, with every tenth slide from each block selected for staining. The adjacent section was stained in parallel without the primary antibody, and this slide was used to determine the total number of neurons per section. Sections from uninfected DRG were included in each run as a negative control. DRG contain clusters of neurons within axon bundles, and so the number of neurons per section was variable (range, 0 to 430 neurons per tissue section). However, since each DRG section contained ∼120 neuronal cell bodies on average and multiple sections were tested, approximately 700 to 900 neurons were examined for each DRG xenograft. All DRG sections were examined under code. Statistical analyses were performed using Prism 5 for Mac, version 5.0d.
Transmission electron microscopy.
Transmission electron microscopy (TEM) of DRG xenografts was done as previously described (21, 22). Briefly, DRG xenografts were inoculated with 10,000 PFU/implant HSV-1 F strain. At 18 days after infection, mice were euthanized and DRG were removed and fixed in 2% paraformaldehyde and 2% glutaraldehyde in phosphate buffer (0.1 M; pH 7.2). The fixed tissue was then postfixed with 1% osmium tetroxide (2 h) and then incubated in 1% aqueous uranyl acetate overnight, followed by dehydration in a series of increasing ethanol concentrations with a final propylene oxide step. After embedding and polymerization in epoxy resin Embed812 (Electron Microscopy Sciences, Hatfield, PA), semithin (300-nm) sections were stained with toluidine blue to screen for areas within the DRG xenograft that exhibited cytopathic changes suggestive of HSV infection. Of these areas, ultrathin (60-nm) sections were prepared using a diamond knife (Diatome, Hatfield, PA) and an ultramicrotome (Ultracut; Leica). Ultrathin sections were stained with 3.5% aqueous uranyl acetate for 10 min and 0.2% lead citrate for 3 min and analyzed using a Jeol 1230 transmission electron microscope at 80 kV; digital photographs were taken with a Gatan Multiscan 701 digital camera.
RESULTS
Human neurons in DRG xenografts express HSV-1 entry receptor and neuron-specific proteins.
Human DRG xenografts were confirmed to retain a typical ganglionic architecture, with islets of neuronal cell bodies and their surrounding satellite cells that comprise the neuron-satellite cell complex, contained within a nerve-collagen fiber matrix (Fig. 1A), as previously reported (19). In addition, most neurons in DRG xenografts were found to have herpesvirus entry mediator A (HVEM) on cell membranes (Fig. 1B). As shown in Fig. 1C, DRG neurons expressed human neural cell adhesion molecule (NCAM), which demarcates the boundary between the neuronal cytoplasm and encapsulating satellite glial cells (SGCs), and synaptophysin, a synaptic vesicle protein. Neuronal axons identified by a pan-neurofilament marker were found in the xenografts and contained synaptophysin within axonal synaptic vesicles, indicating that the motor structures involved in the directional movement of cargo within individual nerve fibers used by HSV-1 for retrograde transport to the neuronal cell nucleus were present (Fig. 1D). Myelinating Schwann cells wrapping DRG axons were also observed (Fig. 1D; S, Schwann cell nuclei).
Fig 1.

Histological appearance and expression of neural proteins in human dorsal root ganglion (DRG) xenografts maintained in SCID mice. (A) Tissue section from a human DRG xenograft at 20 weeks after implantation, stained with hematoxylin and eosin. (B) Tissue section from a DRG xenograft stained with antibody to herpes viral entry mediator (HVEM)/Alexa Fluor 488 (green) and with Hoechst nuclear stain (blue); inset panel is a control section stained with secondary antibody only. (C) DRG neuron-satellite cell complexes in a DRG xenograft section stained with anti-neural cell adhesion molecule (NCAM)/Alexa Fluor 594 (red) to define neuronal cell membranes and anti-synaptophysin/Alexa Fluor 488 (green) to detect this axonal synaptic vesicle protein in cytoplasmic vesicles; nuclei are stained with Hoechst (blue). Panels on the right are single-channel images. (D) DRG axons stained with antibody to neurofilament-M protein/Alexa Fluor 594 (red) and anti-synaptophysin/Alexa Fluor 488 (green); nuclei are stained with Hoechst (blue). S, Schwann cell nucleus. Panels on the right are single-channel images.
Infection of human DRG xenografts with the HSV-1 F strain.
DRG xenografts in 12 mice were inoculated with 10,000 PFU/implant HSV-1 F strain. DRG were recovered from mice at 12 and 18 days after infection, at which point mice were moribund, warranting euthanasia. When histopathologic changes were examined by H&E staining of tissue sections, the DRG xenografts retained an organotypic architecture, with the exception of debris fields in discrete areas containing infiltrating mouse host cells, presumably macrophages in these SCID mice (Fig. 2A, thick arrow). However, axons proximal to the debris (Fig. 2A, thin arrow) and densely packed neuronal cell bodies within nearby areas (Fig. 2B) had normal morphology. A few neurons exhibited a cytopathic effect (Fig. 2B, arrow).
Fig 2.

Effects of HSV-1 infection on cellular morphology and tissue architecture of human DRG xenografts. (A and B) Tissue sections from HSV-1 F strain-infected DRG stained with hematoxylin and eosin (H&E) were examined for pathological changes at 12 days after infection. Thick arrows in panel A indicate a cellular infiltrate, and the thin arrow denotes axons. The arrow in panel B indicates necrotic-appearing neurons. (C to J) Tissue sections from HSV-1 R8411-infected DRG at 12 days (C and D), 16 days (E and F), 33 days (G and H), and 61 days (I and J) after infection. Arrows in panel F indicate neurons with evidence of chromatolysis, and those in panel H indicate necrotic-appearing neurons. The arrow in panel J indicates Nageotte nodules characteristic of areas of neuronal cell loss. Panels on the left are shown at a magnification of ×50; panels on the right are at a magnification of ×200.
HSV-1 F strain exhibits tropism for neurons but not satellite cells in DRG xenografts and does not induce fusion of neurons and satellite cell plasma membranes.
High-resolution confocal microscopy was used to assess HSV-1 infection, tropism for satellite cells, and whether HSV-1 induced fusion of plasma membranes separating the neuron and satellite cells within neuron-satellite cell complexes. VP16 and ICP4 were prominently expressed and localized to neuronal cell nuclei and cytoplasms (Fig. 3A). Fewer than 2% of HSV-1-positive neurons were encapsulated by one or more satellite cells expressing HSV-1 protein (Fig. 3A, arrow). HSV-1 protein expression in satellite cells was always accompanied by positive neuronal staining. Continuity of the neuronal membrane was assessed by staining with antibody to NCAM, which is a reliable marker for the fusion of neuronal and satellite cell membranes induced by VZV, as validated by TEM (22). NCAM staining was evenly distributed along the inner margin of the neuronal cell cytoplasm and between neurons and encapsulating satellite cells (Fig. 3B). These observations and the absence of HSV-1 protein expression in satellite cells, with very rare exceptions, indicate that HSV-1 infection is highly restricted to DRG neurons.
Fig 3.

Localization of HSV-1 proteins and LAT in human DRG neurons. Tissue sections from HSV-1 F strain-infected DRG 12 days after infection (A to C) or R8411-infected DRG (D to F) at 21, 61, and 33 days after infection, respectively. (A) Stained with rabbit anti-VP16/Alexa Fluor 488 (green) and mouse anti-ICP4 (red) monoclonal antibody. (B, D, and E) Stained with rabbit anti-HSV/Alexa Fluor 488 (green) and mouse anti-NCAM/Alexa Fluor 594 (red). NCAM (red staining in panels B, D, and E) is expressed on neuronal cell membranes and demarcates boundaries between the neuronal cell cytoplasm and encapsulating satellite cells; nuclei (blue) are stained with Hoechst. (C and F) Dual LAT RNA in situ hybridization using Fast Red substrate, detected with a HeNe 543 spectrometer (red), and immunostaining using anti-HSV-1/Alexa Fluor 488 (green). Single LAT-positive neurons (solid thin arrow), single HSV antigen-positive neurons (solid thick arrow), and dual LAT-/antigen-positive neurons (dashed arrow) were observed. Images from individual 488 and 594 channels are shown for each panel.
Localization of HSV-1 protein and latency-associated transcripts in HSV-1 F strain-infected human DRG neurons.
Dual confocal immunofluorescence for HSV-1 protein using the rabbit anti-HSV-1 polyclonal antibody and LAT RNA in situ hybridization was done to determine whether human DRG neurons in xenografts contain LAT (Fig. 3C). Single HSV-1 protein- and LAT-positive neurons were readily observed (Fig. 3C, thick arrow and thin arrow, respectively). Neurons that contained both HSV-1 protein and LAT were observed as well (Fig. 3C, dashed arrow). LAT was not detected in satellite cells or in any other nonneuronal supportive cells in DRG xenografts.
Quantitation of the frequency of neurons expressing HSV-1 protein and LAT in HSV-1 F-infected DRG.
Quantitative immunostaining was performed to determine the proportions of DRG neurons that expressed HSV-1 protein and LAT. Viral protein expression was assessed by immunohistochemistry using a rabbit polyclonal antibody that recognizes multiple HSV-1-infected cell proteins; LAT transcripts were detected by RNA in situ hybridization. The numbers of HSV-1 protein- and LAT-positive cells were determined by examining approximately 700 to 900 neuronal cell bodies for each DRG xenograft (6 to 8 sections/DRG). Representative sections are shown after staining for HSV-1 protein and for LAT (Fig. 4A and B, left panels) with their respective negative controls (Fig. 4A and B, right panels). HSV-1 protein was expressed in 26.2% ± 1.9% of neurons; 13.1% ± 2.4% of neurons contained LAT (Fig. 4C).
Fig 4.

Quantitation of HSV-1 proteins and LAT in human DRG neurons. (A to C) HSV-1 F strain-infected DRG xenografts at 12 days after infection. (A) Representative detection of HSV-1 LAT by RNA in situ hybridization using a digoxigenin (DIG)-labeled riboprobe and anti-DIG antibody detection with NBT/BCIP (black), counterstained with nuclear red (left panel) and a control in which the riboprobe was omitted (right panel). (B) Representative detection of HSV-1 proteins using anti-HSV antibody with DAB (brown) detection, counterstained with hematoxylin (left) and the control with the primary antibody omitted (right). The arrow indicates a positive neuron. (C) Quantitation of the number of neurons expressing HSV-1 proteins or LAT RNA; HSV-1 protein was detected in 26.2% ± 1.9% of neurons, and LAT was detected in 13.1% ± 2.4% of neurons. (D to F) HSV-1 R8411-infected DRG xenografts. (D) (left) Representative detection of HSV-1 LAT by RNA in situ hybridization at 33 days after infection; (right) no-probe negative control. (E) (left) Representative detection of HSV-1 proteins using anti-HSV antibody at 16 days after infection; (right) negative control. (F) Quantitation of the number of neurons expressing HSV-1 proteins or LAT RNA at 12, 16, 33, and 61 days after infection; each value represents analysis of ∼700 to 900 neurons per DRG.
Analysis of HSV-1 F strain-infected DRG by transmission electron microscopy.
DRG xenografts infected with HSV-1 F strain and recovered at 18 days after infection were examined by transmission electron microscopy (TEM). As observed in tissue sections from 12 days after infection that were stained with H&E, neurons contained within the DRG xenograft in areas away from the debris fields had normal tissue architecture when thick sections were examined by toluidine blue staining. Only a few neurons exhibited cytopathic changes in discrete localized regions when examined at low magnification using light microscopy. Analysis of ultrathin sections by TEM revealed marginated hyperdense chromatin and HSV-1 particles within cell nuclei in these regions (Fig. 5A). Most virions were sequestered near electron-dense regions within the nucleus (Fig. 5B, dashed arrow). Enveloped HSV-1 virions were present in cytoplasmic vesicles (Fig. 5B, solid arrow), and extracellular virions were observed within interstitial spaces, identified by the presence of collagen fibers (Fig. 5C, arrow).
Fig 5.
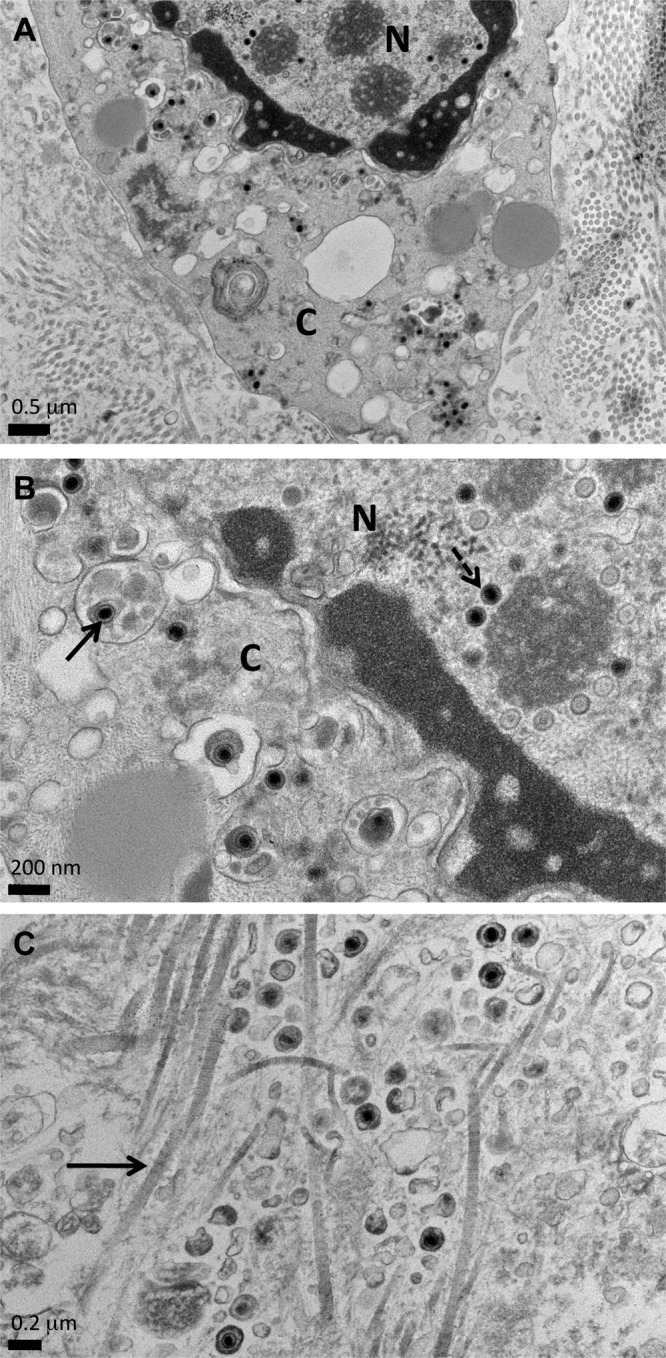
Analysis of HSV-1-infected DRG by transmission electron microscopy. Ultrathin tissue sections prepared from DRG xenografts recovered at 18 days after inoculation with HSV-1 F strain were examined by TEM. (A) HSV-1 virions were observed within cell nuclei and the cytoplasm in small, localized regions. N, nucleus; C, cytoplasm. (B) Higher-magnification image showing HSV-1 particles in the nucleus (dashed arrow) and enveloped particles within cytoplasmic vesicles (solid arrow). N, nucleus; C, cytoplasm. (C) Higher-magnification image showing enveloped HSV-1 particles in interstitial spaces, identified by collagen fibers (solid arrow), indicating productive infection. Scale bars are shown at the lower left of each image.
Characterization of an HSV-1 F strain expressing luciferase (R8411).
To examine HSV-1 replication in human DRG over an extended interval, we utilized the HSV-1 recombinant virus R8411, an F strain BAC-derived mutant in which the immediate-early ICP27 gene promoter drives firefly luciferase expression (Fig. 6A). The final construct was examined by Southern blotting in the lab of origin to verify that no adventitious mutations were introduced. R8411 exhibits reduced replication in cultured human fibroblasts (Fig. 6B) compared to that of the HSV-1 F strain, consistent with a previous report (27). Although the reason for the attenuated phenotype of R8411 is unknown, insertion of the ICP27-luciferase reporter gene cassette between genes UL3 and UL4 was verified by sequencing, which confirmed that no adventitious mutations were introduced into this site during mutagenesis (data not shown). No differences in accumulation of HSV-1 proteins of different kinetic classes were observed by immunoblotting (Fig. 6C), and expression of the viral thymidine kinase, which is absent in HSV BAC, was restored in recombinant R8411. Global analysis of restriction endonuclease digestion patterns of purified nucleocapsid DNA revealed some differences between R8411 and HSV-1 F strain, as expected for lab-passaged virus (Fig. 6D).
Fig 6.
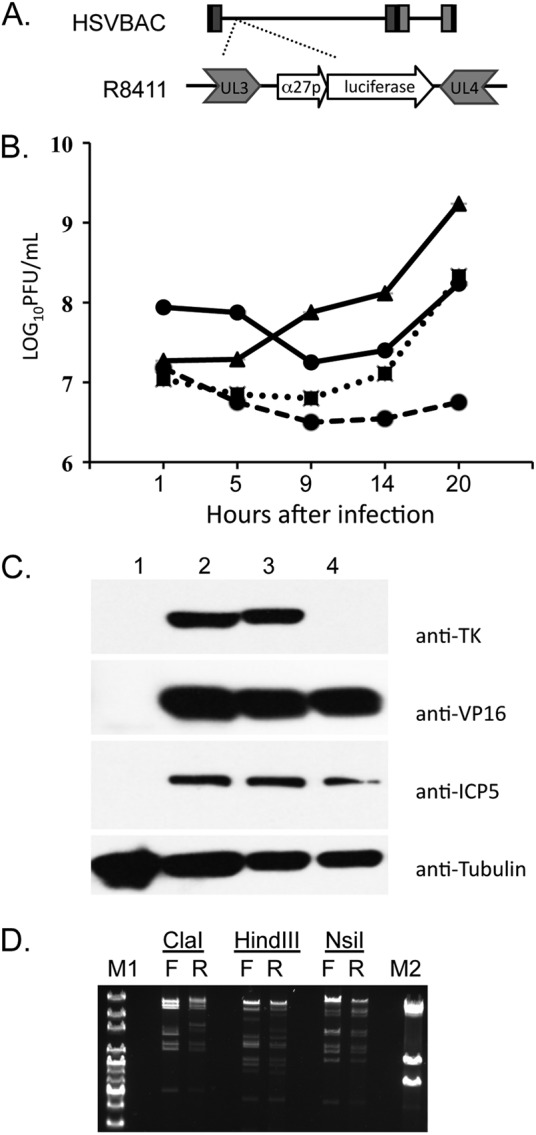
Characterization of R8411. (A) Diagram of the ICP27 promoter-luciferase cassette inserted between UL3 and UL4 of HSV F strain BAC to generate R8411 virus. (B) Growth curve comparing the HSV F strain with an MOI of 1 (triangle, solid line), HSV BAC with an MOI of 1 (square, dotted line), R8411 with an MOI of 1 (circle, dashed line), and R8411 with an MOI of 7 (circle, solid line) in human fibroblasts. (C) Immunoblot analysis of uninfected cells (lane 1), R8411-infected cells (lane 2), HSV-1 F strain-infected cells (lane 3), and HSV BAC-infected cells (lane 4). Blots were probed using antibodies to HSV-1 thymidine kinase (TK), VP16, ICP5, and cellular tubulin. (D) Restriction endonuclease-digested nucleocapsid DNA from HSV F strain (F)- and R8411 (R)-infected cells was analyzed by field-inversion gel electrophoresis (FIGE). One microgram of nucleocapsid DNA was digested for 2 h with ClaI, HindIII, and NsiI and run overnight using a FIGE mapper. M1, 1-kb plus extended ladder; M2, λ phage ladder.
Replication of HSV-1 F strain expressing luciferase (R8411) in DRG xenografts.
Replication of R8411 in DRG xenografts of four mice (10,000 PFU/implant) was monitored using bioluminescence as a correlate by imaging each mouse 24 h after inoculation to measure photon flux and then daily. Uninfected xenografted mice (n = 4) were injected with luciferin and imaged in parallel to establish background bioluminescence (Fig. 7A and 8A). The median photon flux peaked during the first 3 days after DRG inoculation, and the signal was detected over the abdomen as well as in the region of the DRG xenograft during this initial phase (Fig. 7A and 8B). Photon flux values then declined, and the area of detection became smaller; by day 9, the signal was localized to the region over the DRG xenograft (Fig. 7A). Bioluminescence remained restricted to the DRG xenograft area over a 33-day period and ranged from 106 to 107 photon flux units (Fig. 7A), values which were significantly higher than values in uninfected mice (Fig. 7A; dashed line). The median bioluminescence values did not change over time but fluctuated somewhat within individual DRG (Fig. 8B). Unlike the morbidity observed in mice with DRG xenografts infected with the HSV-1 F strain, only one of four mice infected with R8411 demonstrated signs of HSV-1 infection beyond the human tissue xenograft, developing ataxia at day 24. The DRG xenograft and a large segment of the kidney proximal to the point of xenograft attachment were recovered from this animal and imaged ex vivo. Bioluminescence was detected within the DRG xenograft (Fig. 8C, tissue on left) but not in the adjacent murine kidney (Fig. 8C). The other three animals showed no evidence of HSV-1 spread from the human DRG xenograft (Fig. 8B).
Fig 7.

Kinetics of infection of DRG xenografts inoculated with HSV-1 F strain expressing luciferase (R8411). (A) Photon flux (photons/s/cm2) values within the region of interest are shown for individual mice (black circles) that were imaged daily for 33 days; the vertical lines indicate means and standard errors of the means. (B) Photon flux values within the region of interest are shown for individual HSV-1-infected mice treated with valacyclovir during the 33-day imaging interval; the vertical lines indicate means and standard errors of the mean. (C) Photon flux values within the region of interest are shown for individual mice (black circles) imaged every 3 to 6 days for 61 days; the vertical lines indicate means and standard errors of the means. The number of mice imaged decreases over the study period because DRG xenografts were recovered from 3 to 5 mice for histological evaluation at 12, 16, 21, and 33 days. The dashed lines in panels A to C show the average background photon flux values in mock-infected control mice injected with luciferin.
Fig 8.

Imaging panels from SCID mice with human DRG xenografts infected with the HSV-1 F strain expressing luciferase (R8411). Daily photon flux values (color bar on the left side of panel B) in 4 uninfected mice (A; 1 day after infection) and 4 mice infected with HSV expressing luciferase (B; daily images of mice infected with R8411 during the 33-day imaging experiment). (C) DRG xenograft (left side) and kidney (right side) from mouse number 1 imaged ex vivo after euthanasia at 24 days.
A second cohort of five HSV-infected mice was administered valacyclovir by oral gavage upon recovery after DRG inoculation and daily for the 33-day imaging experiment. Bioluminescence values were significantly reduced compared with those of untreated HSV-1-infected mice at day 10 and subsequently over the 33-day period (Fig. 7B). One valacyclovir-treated mouse died on day 18.
To extend our observations using R8411, DRG xenografts in a large cohort of mice (n = 22) were inoculated with 10,000 PFU/xenograft and imaging was done every 3 to 6 days for 61 days (Fig. 7C). Four uninfected animals were imaged in parallel to establish average background photon flux values (Fig. 7C; dashed line). Again, bioluminescence was restricted to the region of the DRG xenograft within 8 days. On days 12, 16, 21, and 33 after infection, 3 to 5 mice from the R8411-infected cohort were euthanized to recover DRG xenografts for tissue analyses, reducing the number of animals imaged at the later time points. However, photon flux remained within a narrow range of 106 to 107 photon flux units across the 61-day period and was significantly higher than values in uninfected mice. Only one of 22 animals developed neurological symptoms (ataxia at day 33).
Effects of HSV-1 R8411 infection on DRG cellular morphology and tissue architecture.
When histopathologic changes were examined in tissue sections of R8411-infected DRG xenografts recovered 12 days after inoculation, the DRG xenografts were similar in appearance to uninfected DRG. The debris field and cellular infiltrate observed after infection with HSV-1 F strain were less prominent. DRG neurons rarely exhibited a cytopathic effect (Fig. 2D). When examined at 21 days after infection, most neurons appeared intact (Fig. 2E), although a few had evidence of neuronal chromatolysis, which is a reversible cell body reaction to axonal damage characterized by a swollen cell body, nuclear condensation, and granular Nissl substance (Fig. 2F, arrows). Neuronal damage became prominent by day 33 after infection (Fig. 2G), with some areas showing many large, necrotic-appearing neurons when examined at high magnification (Fig. 2H, arrows). At day 61, many normal neurons remained in the DRG but were located within regions containing Nageotte nodules, which are compact areas of satellite cell proliferation that indicate neuronal cell loss (Fig. 2I to J, arrow).
Effects of HSV-1 R8411 infection on satellite cell tropism, fusion, and LAT expression.
Tropism for satellite cells, fusion, and LAT expression in DRG xenografts inoculated with R8411 were examined by confocal immunofluorescent microscopy. When examined at 21 and 33 days after infection, R8411 infection did not alter the restriction from satellite cells or induce fusion of plasma membranes separating the neuron and encapsulating satellite cells within neuron-satellite cell complexes (Fig. 3D). Similar to observations using F strain, HSV-1 proteins were rarely observed in satellite cells and only in satellite cells encapsulating a neuron coexpressing HSV-1 protein. Intact NCAM staining was apparent along the inner margin of the neuronal cell cytoplasm. Singular HSV-1 protein- and LAT-positive neurons were readily observed (Fig. 3F, solid arrow). Neurons that contained both HSV-1 protein and LAT were observed as well (Fig. 3F, dashed arrow). At 61 days after infection, HSV-1 protein was readily detected within intact, normal-appearing neurons contained within HSV-1-negative satellite cells, but staining was also observed within small cell clusters, which resemble Nageotte nodules (Fig. 3E, solid arrow), and in axons (Fig. 3E, dashed arrow).
Quantitation of the frequency of neurons expressing HSV-1 protein and LAT in R8411-infected DRG.
The proportions of DRG neurons expressing HSV-1 protein or LAT were determined by quantitative immunohistochemistry using a rabbit anti-HSV antibody and LAT RNA in situ hybridization. Representative sections are shown after staining for LAT (day 33) and HSV-1 protein (day 16) (Fig. 4D and E, left panels) with their respective negative controls (Fig. 4D and E, right panels). At 12 days, 4.3% ± 0.9% of neurons expressed HSV-1 protein and 1.2% ± 0.3% of neurons contained LAT (Fig. 4F). By day 16, 13.4% ± 1.8% of neurons expressed HSV-1 protein and 20.3% ± 3.5% of neurons contained LAT; by day 33, 32.1% ± 2.6% were HSV-1 protein positive and 40.1% ± 5.8% were LAT positive. Between days 33 and 61, the proportion of neurons that expressed HSV-1 protein declined to 20.8% ± 1.4% whereas LAT was detected in 37% ± 3.1% of neurons. Overall, neurons expressing HSV-1 protein were less frequent than those containing detectable LAT at all time points except day 12, the earliest time point examined, and decreased significantly compared to the frequency of LAT-positive cells over the 61-day interval (P < 0.01; day 61).
DISCUSSION
The capacities for neuroinvasion, the establishment of latency in sensory ganglia, and the potential for reactivation are the primary mechanisms through which human alphaherpesviruses persist in the human population and may cause serious disease in the human host. However, current knowledge about virus-host cell interactions within intact human neural tissues infected in vivo is limited to examination of autopsy specimens. This study of HSV-1 infection of human DRG xenografts in SCID mice was designed to address basic questions about events in HSV-1 neuropathogenesis, taking advantage of the model that we established to investigate VZV neurotropism (19).
These experiments demonstrated that human neurons in DRG xenografts were permissive for HSV-1 entry, which resulted in either production of HSV-1 proteins associated with lytic infection or HSV-1 genome silencing and expression of latency-associated transcripts. HSV-1 genome silencing occurred independently of adaptive immune control (28) and did not require replication at a peripheral mucosal or cutaneous site or entry into distal axons (29). These observations support a paradigm in which HSV-1 gene silencing occurs in human neurons infected in vivo as a consequence of intrinsic virus-host cell interactions and local production of antiviral mediators, such as interferons (IFN). Work by others suggests a role for alpha interferon (IFN-α) in suppression of HSV-1 lytic replication (30–33).
The observation that only 26.2% of neurons in DRG xenografts infected with HSV-1 F strain expressed viral proteins at 12 days after infection differs markedly from the rapid spread of VZV during the acute stage of DRG xenograft infection (19). The limited ganglionic spread of HSV-1 compared with VZV may result from the inability of HSV to productively infect satellite cells or to induce fusion of plasma membranes within the neuron-satellite cell complex, both of which occur in VZV-infected DRG. VZV-induced membrane fusion provides a mechanism for viral spread to satellite cells surrounding an infected neuron and secondarily to satellite cells that are part of an adjacent neuron-satellite cell complex, thereby enhancing opportunities for access to neuronal axons that transport VZV to skin during reactivation. The failure of HSV-1 to productively infect satellite cells suggests an intrinsic barrier to satellite cell entry, replication in satellite cells, or both which is independent of adaptive immune control. This evidence of disparate mechanisms for HSV and VZV neuropathogenesis is consistent with their clinical differences. Since satellite cells are essential for neuronal homeostasis, destruction of satellite cells within neuron-satellite cell complexes by VZV helps to explain the severity of neuropathic pain and the risk of prolonged postherpetic neuralgia associated with herpes zoster, which is rarely observed with HSV-1 reactivation.
The presence of LAT-positive neurons (13.1%) within 12 days of DRG inoculation suggests that some neurons may be inherently more likely to maintain HSV-1 gene repression than others. A preponderance of observations support the hypothesis that some neuronal subtypes are differentially permissive for HSV-1 lytic replication (34–37). Human DRG neurons are heterogeneous; subtype specification may be based on morphological, electrophysiological, or neurochemical criteria (38). In a murine neuronal culture system, TrkA-immunoreactive neurons that also express a carbohydrate antigen recognized by the rodent-specific A5 monoclonal antibody (Developmental Studies Hybridoma Bank, Iowa City, IA) have been identified as reservoirs of HSV-1 latency (14). Further implicating this neuronal subtype in HSV-1 latency is the observation that nerve growth factor (NGF) deprivation of latently infected murine neuronal cultures is a potent inducer of reactivation (18). Whether NGF-sensitive TrkA-immunoreactive neurons are the reservoir for human HSV-1 latency, as has been demonstrated in mice, will be addressed in future investigations using this DRG xenograft model.
The presence of dual HSV-1 protein-/LAT-positive neurons suggests that (i) neurons that were initially nonpermissive for productive infection can undergo lytic gene expression in the presence of accumulated LAT, (ii) in some neurons, LAT and lytic genes are concomitantly expressed, or (iii) derepression of HSV-1 genomes is not invariably lytic. In murine models, lytic genes may be expressed in the absence of infectious viral progeny, a situation that has been described as spontaneous molecular reactivation (39); however, others have suggested that limited immediate-early viral gene expression may be a component of latency (10). Given the cytopathic changes observed in dual HSV-1 protein-/LAT-positive neurons, a situation in which neurons that were initially nonpermissive succumb to lytic replication, and so, in the classic sense, are “reactivating,” seems most likely. Thus, HSV-1 infection of human DRG xenografts recapitulates the three possible outcomes of neuronal infection, (i) lytic replication, (ii) latency, and (iii) reactivation.
Although restricted for spread in human neurons in DRG xenografts, the significant morbidity in HSV-1 F strain-infected mice was a barrier to time course experiments. However, we found that using the R8411 luciferase virus permitted a prolonged assessment of viral infection of human sensory ganglia. The changing pattern of HSV-1 protein and LAT expression over time in DRG neurons infected with R8411 raises some intriguing questions. Although infection was controlled in DRG xenografts, the frequency of HSV-1 infection of human neurons, judged by HSV-1 protein and LAT expression, was significantly higher at 8 weeks after DRG inoculation than has been observed in murine models of HSV-1 ganglion infection after footpad or ocular inoculation, even when using the attenuated R8411 strain. In the ocular model, mice that survive have no apparent replicating virus in DRG or trigeminal ganglia four to six weeks after infection (10, 36, 40, 41). However, the frequency of infected ganglion cells in murine models is much lower than the estimate of 2 to 10.5% from human autopsy studies (42). The different outcomes of infection of human and mouse ganglia in vivo are likely to reflect adaptations of HSV-1 to its natural host. In murine models, functional adaptive immune cells are important for control and clearance of HSV-1 replication in the central nervous system (CNS) (43–45), but LAT-positive HSV antigen-negative neurons are detected 2 days after corneal scarification in the mouse trigeminal ganglion, before the arrival of HSV-specific T cells (40). Whether HSV-1-specific adaptive immunity functions to further restrict viral spread in human sensory ganglia is not known, although T cells that recognize viral peptides are detected in latently infected ganglia obtained at autopsy (46).
This study showed that the SCID mouse model of neuropathogenesis reveals unique characteristics of HSV-1 interactions with human neurons in vivo. Experiments using recombinant virus demonstrate its potential relevance for testing live attenuated HSV-1 vaccine candidates. Experiments using valacyclovir, a widely used antiviral drug treatment for HSV-1 infection, demonstrate its potential value for testing antiviral drugs. Understanding HSV-1 infection of human neurons in peripheral nerve ganglia is important to further our understanding of HSV-1 neuroinvasion and neurovirulence in the human host.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institute of Allergy and Infectious Diseases (AI081994 and AI20459).
We thank Phillip Sung, Jaya Rajamani, Preeti Sikka, and Michelle Lai (Stanford University) and Shaniya Khan (University of Iowa) for technical assistance and Tim Doyle (Small Animal Imaging Facility, Stanford University). We also thank Todd Margolis, Andrea Bertke, and Aye-Aye Ma (University of California, San Francisco, CA) for valuable technical assistance and discussions.
Footnotes
Published ahead of print 26 December 2012
REFERENCES
- 1. Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2502–2601 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Wolters Kluwer, Lippincott, Williams, and Wilkins, Philadelphia, PA [Google Scholar]
- 2. Sawtell NM. 1998. The probability of in vivo reactivation of herpes simplex virus type 1 increases with the number of latently infected neurons in the ganglia. J. Virol. 72:6888–6892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spruance SL, Overall JC, Jr, Kern ER, Krueger GG, Pliam V, Miller W. 1977. The natural history of recurrent herpes simplex labialis: implications for antiviral therapy. N. Engl. J. Med. 297:69–75 [DOI] [PubMed] [Google Scholar]
- 4. Whitley R. (ed). 2001. Herpes simplex viruses, 4th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 5. Roizman B, Zhou G, Du T. 2011. Checkpoints in productive and latent infections with herpes simplex virus 1: conceptualization of the issues. J. Neurovirol. 17:512–517 [DOI] [PubMed] [Google Scholar]
- 6. Henderson G, Jaber T, Carpenter D, Wechsler SL, Jones C. 2009. Identification of herpes simplex virus type 1 proteins encoded within the first 1.5 kb of the latency-associated transcript. J. Neurovirol. 15:439–448 [DOI] [PubMed] [Google Scholar]
- 7. Maillet S, Naas T, Crepin S, Roque-Afonso AM, Lafay F, Efstathiou S, Labetoulle M. 2006. Herpes simplex virus type 1 latently infected neurons differentially express latency-associated and ICP0 transcripts. J. Virol. 80:9310–9321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J. Virol. 63:2893–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 287:1500–1503 [DOI] [PubMed] [Google Scholar]
- 10. Sawtell NM. 2005. Detection and quantification of the rare latently infected cell undergoing herpes simplex virus transcriptional activation in the nervous system in vivo. Methods Mol. Biol. 292:57–72 [DOI] [PubMed] [Google Scholar]
- 11. Devi-Rao GB, Bloom DC, Stevens JG, Wagner EK. 1994. Herpes simplex virus type 1 DNA replication and gene expression during explant-induced reactivation of latently infected murine sensory ganglia. J. Virol. 68:1271–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wagner EK, Bloom DC. 1997. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev. 10:419–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arthur JL, Scarpini CG, Connor V, Lachmann RH, Tolkovsky AM, Efstathiou S. 2001. Herpes simplex virus type 1 promoter activity during latency establishment, maintenance, and reactivation in primary dorsal root neurons in vitro. J. Virol. 75:3885–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, Margolis TP. 2011. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J. Virol. 85:6669–6677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. 2010. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8:320–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hill JM, Garza HH, Jr, Helmy MF, Cook SD, Osborne PA, Johnson EM, Jr, Thompson HW, Green LC, O'Callaghan RJ, Gebhardt BM. 1997. Nerve growth factor antibody stimulates reactivation of ocular herpes simplex virus type 1 in latently infected rabbits. J. Neurovirol. 3:206–211 [DOI] [PubMed] [Google Scholar]
- 17. Webre JM, Hill JM, Nolan NM, Clement C, McFerrin HE, Bhattacharjee PS, Hsia V, Neumann DM, Foster, Lukiw WJ, Thompson HW. 2012. Rabbit and mouse models of HSV-1 latency, reactivation, and recurrent eye diseases. J. Biomed. Biotechnol. 2012:612316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilcox CL, Johnson EM., Jr 1987. Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J. Virol. 61:2311–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zerboni L, Ku CC, Jones CD, Zehnder JL, Arvin AM. 2005. Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc. Natl. Acad. Sci. U. S. A. 102:6490–6495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zerboni L, Reichelt M, Arvin A. 2010. Varicella-zoster virus neurotropism in SCID mouse-human dorsal root ganglia xenografts. Curr. Top. Microbiol. Immunol. 342:255–276 [DOI] [PubMed] [Google Scholar]
- 21. Zerboni L, Reichelt M, Jones CD, Zehnder JL, Ito H, Arvin AM. 2007. Aberrant infection and persistence of varicella-zoster virus in human dorsal root ganglia in vivo in the absence of glycoprotein I. Proc. Natl. Acad. Sci. U. S. A. 104:14086–14091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reichelt M, Zerboni L, Arvin AM. 2008. Mechanisms of varicella-zoster virus neuropathogenesis in human dorsal root ganglia. J. Virol. 82:3971–3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horsburgh BC, Hubinette MM, Tufaro F. 1999. Genetic manipulation of herpes simplex virus using bacterial artificial chromosomes. Methods Enzymol. 306:337–352 [DOI] [PubMed] [Google Scholar]
- 24. He B, Chou J, Liebermann DA, Hoffman B, Roizman B. 1996. The carboxyl terminus of the murine MyD116 gene substitutes for the corresponding domain of the gamma(1)34.5 gene of herpes simplex virus to preclude the premature shutoff of total protein synthesis in infected human cells. J. Virol. 70:84–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szpara ML, Tafuri YR, Enquist LW. 2011. Preparation of viral DNA from nucleocapsids. J. Vis. Exp. 2011:e3151 doi:10.3791/3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Margolis TP, Imai Y, Yang L, Vallas V, Krause PR. 2007. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J. Virol. 81:1872–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Markovitz NS, Roizman B. 2000. Replication-competent herpes simplex viral vectors for cancer therapy. Adv. Virus Res. 55:409–424 [DOI] [PubMed] [Google Scholar]
- 28. Gesser RM, Valyi-Nagy T, Fraser NW. 1994. Restricted herpes simplex virus type 1 gene expression within sensory neurons in the absence of functional B and T lymphocytes. Virology 200:791–795 [DOI] [PubMed] [Google Scholar]
- 29. Hafezi W, Lorentzen EU, Eing BR, Muller M, King NJ, Klupp B, Mettenleiter TC, Kuhn JE. 2012. Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog. 8:e1002679 doi:10.1371/journal.ppat.1002679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davido DJ, Leib DA. 1996. Role of cis-acting sequences of the ICPO promoter of herpes simplex virus type 1 in viral pathogenesis, latency and reactivation. J. Gen. Virol. 77(Part 8):1853–1863 [DOI] [PubMed] [Google Scholar]
- 31. De Regge N, Van Opdenbosch N, Nauwynck HJ, Efstathiou S, Favoreel HW. 2010. Interferon alpha induces establishment of alphaherpesvirus latency in sensory neurons in vitro. PLoS One 5:e13076 doi:10.1371/journal.pone.0013076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Halford WP, Weisend C, Grace J, Soboleski M, Carr DJ, Balliet JW, Imai Y, Margolis TP, Gebhardt BM. 2006. ICP0 antagonizes Stat 1-dependent repression of herpes simplex virus: implications for the regulation of viral latency. Virol. J. 3:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sainz B, Jr, Halford WP. 2002. Alpha/beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1. J. Virol. 76:11541–11550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bertke AS, Patel A, Krause PR. 2007. Herpes simplex virus latency-associated transcript sequence downstream of the promoter influences type-specific reactivation and viral neurotropism. J. Virol. 81:6605–6613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. LaVail JH, Johnson WE, Spencer LC. 1993. Immunohistochemical identification of trigeminal ganglion neurons that innervate the mouse cornea: relevance to intercellular spread of herpes simplex virus. J. Comp. Neurol. 327:133–140 [DOI] [PubMed] [Google Scholar]
- 36. Sawtell NM, Thompson RL. 1992. Herpes simplex virus type 1 latency-associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J. Virol. 66:2157–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang L, Voytek CC, Margolis TP. 2000. Immunohistochemical analysis of primary sensory neurons latently infected with herpes simplex virus type 1. J. Virol. 74:209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mu X, Silos-Santiago I, Carroll SL, Snider WD. 1993. Neurotrophin receptor genes are expressed in distinct patterns in developing dorsal root ganglia. J. Neurosci. 13:4029–4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. 2002. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc. Natl. Acad. Sci. U. S. A. 99:978–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Margolis TP, Dawson CR, LaVail JH. 1992. Herpes simplex viral infection of the mouse trigeminal ganglion. Immunohistochemical analysis of cell populations. Invest. Ophthalmol. Vis. Sci. 33:259–267 [PubMed] [Google Scholar]
- 41. Margolis TP, Elfman FL, Leib D, Pakpour N, Apakupakul K, Imai Y, Voytek C. 2007. Spontaneous reactivation of herpes simplex virus type 1 in latently infected murine sensory ganglia. J. Virol. 81:11069–11074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang K, Lau TY, Morales M, Mont EK, Straus SE. 2005. Laser-capture microdissection: refining estimates of the quantity and distribution of latent herpes simplex virus 1 and varicella-zoster virus DNA in human trigeminal Ganglia at the single-cell level. J. Virol. 79:14079–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Minagawa H, Sakuma S, Mohri S, Mori R, Watanabe T. 1988. Herpes simplex virus type 1 infection in mice with severe combined immunodeficiency (SCID). Arch. Virol. 103:73–82 [DOI] [PubMed] [Google Scholar]
- 44. Minagawa H, Yanagi Y. 2000. Latent herpes simplex virus-1 infection in SCID mice transferred with immune CD4+T cells: a new model for latency. Arch. Virol. 145:2259–2272 [DOI] [PubMed] [Google Scholar]
- 45. Valyi-Nagy T, Deshmane SL, Raengsakulrach B, Nicosia M, Gesser RM, Wysocka M, Dillner A, Fraser NW. 1992. Herpes simplex virus type 1 mutant strain in1814 establishes a unique, slowly progressing infection in SCID mice. J. Virol. 66:7336–7345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verjans GM, Hintzen RQ, van Dun JM, Poot A, Milikan JC, Laman JD, Langerak AW, Kinchington PR, Osterhaus AD. 2007. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc. Natl. Acad. Sci. U. S. A. 104:3496–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]