

Abstract
Herpes simplex virus 1 (HSV-1) is a neurotropic virus that travels long distances through cells using the microtubule network. Its 125-nm-diameter capsid is a large cargo which efficiently recruits molecular motors for movement. Upon entry, capsids reach the centrosome by minus-end-directed transport. From there, they are believed to reach the nucleus by plus-end-directed transport. Plus-end-directed transport is also important during egress, when capsids leave the nucleus to reach the site of envelopment in the cytoplasm. Although capsid interactions with dynein and kinesins have been described in vitro, the actual composition of the cellular machinery recruited by herpesviruses for capsid transport in infected cells remains unknown. Here, we identify the spectraplakin protein, dystonin/BPAG1, an important cytoskeleton cross-linker involved in microtubule-based transport, as a binding partner of the HSV-1 protein pUL37, which has been implicated in capsid transport. Viral replication is delayed in dystonin-depleted cells, and, using video microscopy of living infected cells, we show that dystonin depletion strongly inhibits capsid movement in the cytoplasm during egress. This study provides new insights into the cellular requirements for HSV-1 capsid transport and identifies dystonin as a nonmotor protein part of the transport machinery.
INTRODUCTION
Microtubules (MTs) are long, polarized structures made of heterodimers of alpha and beta tubulin. They are nucleated from microtubule organizing centers (MTOCs), the nature of which may differ according to cell type (reviewed in reference 1). The most common MTOC is the centrosome, from where nascent MTs grow by polymerization of tubulin at their plus ends. The minus ends are thought to be anchored and stabilized at the centrosome by specific proteins (2). In general, MTs radiate from the centrosome to the cell periphery.
Microtubule motors such as cytoplasmic dynein or kinesins are involved in trafficking and transport of various cargoes, such as protein complexes, vesicles, or even organelles along microtubules. It has become progressively apparent that motor-cargo interactions are often indirect in nature, with adaptor proteins or lipids linking the molecular motor to its cargo (reviewed in reference 3).
Herpes simplex virus 1 (HSV-1) is a neurotropic human virus, well known for its ability to traffic efficiently within cells, especially sensory neurons (4, 5), using the MT network (6, 7). It is a large, double-stranded DNA virus. The genome is packaged in an icosahedral capsid which is surrounded by tegument, a proteinaceous layer that is characteristic of herpesviruses, and an outer envelope. On entry, capsids migrate from the plasma membrane to the nuclear membrane (retrograde transport), where they release the viral genome into the nucleus. Newly formed capsids exiting the nucleus are assumed to travel from the nucleus to sites of secondary envelopment through plus-end-directed transport (anterograde transport). In neurons, this potentially implies traveling long distances between the cell body and the axon terminus during both entry and egress. Therefore, efficient recruitment of the cellular trafficking machinery is required. HSV-1 tegumented capsids were shown to bind to the molecular motors dynein and kinesin-1 and -2 in vitro (8), and the dynein complex was shown to be necessary for capsid transport in infected cells (9). Kinesin-1 is also present on enveloped capsids undergoing anterograde transport in neuronal processes (10). However, although numerous interactions have been described between HSV-1 proteins and molecular motors, as yet, none have been demonstrated as functionally relevant in infected cells, and the actual composition of the cellular transport machinery recruited by herpesvirus capsids remains unknown (5, 11). To date, the best documented viral candidates for a role in capsid transport are the tegument proteins pUL36 and pUL37 (12). Unlike the majority of tegument proteins, these proteins, which interact with each other, have been reported to remain attached, at least in part, to capsids in transit to the nucleus (4, 13). The same is true for the related pseudorabies herpesvirus (PrV) (14, 15). In addition, it was demonstrated that in their absence, intracellular transport of PrV capsids is either severely impaired (pUL37) or totally absent (pUL36) (16, 17).
To unravel cellular factors involved in herpes capsid trafficking, we used pUL37 as bait in a yeast two-hybrid screen and identified the protein dystonin (DST; or BPAG1) as a binding partner. Dystonin is a giant protein which belongs to the conserved spectraplakin superfamily of proteins and, as such, contains numerous spectrin repeats and a plakin domain (reviewed in references 18, 19, and 20). Additionally, it may have an actin-binding domain (AB) and an MT-binding domain (MTBD) (Fig. 1A), depending on the isoform. Four major isoforms have been identified to date, with different cell specificities. Dystonin e (2,611 residues; sizes relate to the murine form of dystonin) is found in epithelial cells, whereas dystonin a (5,379 residues) is predominantly neuronal and dystonin b (7393 residues) is mostly muscular (21). Isoform n refers to the originally described neuronal dystonin (BPAG1n) (22), but it is still unclear whether this isoform is actually produced (21). Determining the molecular mechanism of action of dystonin has proved to be challenging, mostly because of its large size and the variety of isoforms. It has been shown to be necessary for stabilizing MTs in neurones (23), and one isoform was reported to be essential for retrograde transport in neuronal cells through its interaction with the p150glued subunit of dynactin, a cofactor of the dynein motor (24). Recently, it was also shown to function during anterograde transport of secretory vesicles (25).
Fig 1.

pUL37 interacts with the plakin domain of dystonin. (A) A yeast two-hybrid (Y2H) screen was set up using the LexA-pUL37 HSV-1 construct as bait and a cDNA library isolated from differentiated PC12 cells (rat neuroblastoma) as prey. pUL37 is shown on top, with the domain interacting with dystonin in light gray (residues 578 to 899, see panel B). A simplified domain map of the neuronal isoform of murine dystonin (isoform a) is depicted below pUL37. Note that dystonin is not drawn to scale compared to pUL37 and that although the plakin domain is common to isoforms a, b, and e of dystonin, only isoform a is shown here. CH, calponin homology domains; EF, EF hands; GAS2, GAS2 domain; AB, actin-binding domain; MTBD, MT-binding domain. Based on reference 20. The domain of dystonin interacting with pUL37 (526 to 851) is shown. (B) Different truncations of pUL37 (black lines, left) were fused to the LexA DNA-binding domain and tested for Y2H interaction with pGAD-dystonin, which contains the plakin domain of dystonin obtained from the initial Y2H screen, fused to the GAL4 activation domain (AD). pGAD alone was used as a negative control. The interaction was evaluated by quantification of β-galactosidase activity in liquid yeast cultures by an optical density at 420 nm (OD420) (right). Bars represent standard deviations of the mean. (C, D) Coimmunoprecipitation of HA-pUL37 and myc-dystonin. Vero cells were cotransfected with plasmids coding for HA-pUL37 or HA-pUL32 and the 526-to-851 region of rat dystonin (myc-dystonin) and were lysed 16 h later. Following immunoprecipitation with anti-myc A14 (C) or anti-HA Y11 (D) antibodies, cell extracts (inputs) and immune complexes (IP) were separated by SDS-PAGE and analyzed by Western blotting using anti-HA F7 to reveal the presence of HA-pUL37 and HA-pUL32 and using anti-myc 9E10 to reveal the presence of myc-dystonin. The coimmunoprecipitations between pUL37 and dystonin were carried out in duplicate for each set of conditions.
Using live-cell imaging and RNA silencing, we investigated the relevance of the pUL37-dystonin interaction to intracytoplasmic transport of HSV-1 capsids.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney (Vero), 293T, baby hamster kidney (BHK), and human fetal foreskin fibroblast (HFFF2) cells were grown at 37°C in Dulbecco's modified Eagle medium (DMEM; PAA Laboratories) supplemented with 8% fetal calf serum (FCS). For live-cell microscopy studies, cells were grown on 35-mm MaTek glass-bottomed petri dishes.
Wild-type (WT) HSV-1 (strain 17+) and vSR27-VP26GFP (where GFP is green fluorescent protein) were propagated on BHK cells infected at 0.01 PFU per cell, and virions were concentrated from supernatant medium by centrifugation at 15,000 × g for 2 h.
The UL37-null mutant of HSV-1 (FRΔUL37-VP26GFP) was grown on the complementing cell line 80C02 (26, 27). Cells were infected at 0.01 PFU/cell, and 3 days later virions were concentrated from supernatant medium by centrifugation at 15,000 × g for 2 h.
vSR27-VP26GFP was obtained by cotransfecting BHK cells with the SR27 BACmid, containing the full-length genome of HSV-1 17+ (provided by C. Cunningham) and with the plasmid pK26GFP (provided by P. Desai), which encodes a VP26-GFP fusion protein (28). Progeny virus was serially diluted, and a GFP-positive plaque was isolated and grown to high titer.
The vUL35RFP1D1 virus has a wild-type background (17syn+), except that it encodes a VP26 capsid protein fused at its N terminus to the monomeric red fluorescent protein (mRFP) (26).
vUL37GFP-VP26RFP was obtained by coinfecting BHK cells with vUL35RFP1D1 and with vUL37GFP, a virus encoding a fusion of GFP to the C terminus of pUL37 (10). Progeny virus was collected and serially diluted on fresh cells. Plaques exhibiting both GFP and RFP fluorescence were selected and purified through five rounds of plaque purification.
Plasmids.
pCDNA3.1-myc and pCDNA3.1-hemagluttinin (HA) vectors were described in reference 29. pHA-UL32 was provided by E. Palmer. UL37 was amplified by PCR from HSV-1 BAC SR27 and cloned into the EcoRI-XbaI sites of pCDNA3.1-HA (pHA-UL37) or the EcoRI-BamHI sites of the pLexA vector (pLexA-UL37), in frame with the respective tag. The pUL37-interacting fragment of dystonin was amplified by PCR from the clone isolated in the yeast two-hybrid screen and cloned into the EcoRI-XbaI sites of pcDNA3.1-myc, yielding pMyc-dystonin. Plasmid pGFP-hEB3 was a kind gift of John Victor Small (Institute of Molecular Biotechnology, Vienna, Austria).
Antibodies.
The following antibodies were used. Rabbit antibodies Y11 and monoclonal antibody (MAb) F7 against a defined epitope (HA) from the influenza hemagglutinin and rabbit antibody A14 against c-myc were obtained from Santa Cruz Biotechnology. MAb 9E10 against c-myc, mouse anti-alpha-tubulin clone DM1A, and rabbit polyclonal anti-DST antibody against human were obtained from Sigma. Capsids were visualized using the rabbit PTNC antibody (29). Alexa Fluor 488- or 633-conjugated goat anti-mouse (GAM488 or GAM633) antibodies and Alexa Fluor 568-conjugated goat anti-rabbit antibody (GAR568) were obtained from Molecular Probes, and tetramethyl rhodamine isocyanate (TRITC)-conjugated phalloïdin and horseradish peroxidase-conjugated goat anti-mouse antibody were from Sigma.
Fluorescence microscopy.
All immunofluorescence with tubulin staining was done as follows. Cells were fixed in a mix of 3.7% formaldehyde and 0.1% Triton X-100 in PEM buffer (100 mM PIPES, 5 mM EGTA, 2 mM MgCl2, pH 6.8) for 5 min at room temperature as described in reference 30. Microtubules were stained using MAb DM1A against alpha-tubulin (Sigma) and GAM488 or GAM633 antibody (90 min each incubation). Actin fibers were stained using TRITC-phalloidin.
Samples were mounted in ImmuMount (Thermo) containing 1 μg/ml 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma) for DNA staining. All samples were visualized using a Zeiss Axio Observer Z1 microscope using a 63× Plan apochromat oil-immersion objective (numerical aperture [NA] of 1.4; Zeiss).
Live-cell microscopy.
HFFF2 cells were infected with 1 PFU/cell of virus for 1 h at room temperature and incubated for 24 h at 37°C before live-cell imaging.
At 37°C, the distance traveled by capsids moving at ∼2 μm/s on average is ∼1.3 μm in the minimum acquisition window of 600 ms allowed by our equipment. These conditions would allow tracking of only small numbers of capsid moving in a large space. Under normal infection conditions, numerous moving capsids are present in a small space, making it very difficult to identify and track efficiently capsids which have traveled ∼1 μm between each frame. Therefore, all live-cell microscopy was recorded at room temperature (∼25°C). Under these conditions, capsid motion is slower, allowing more efficient tracking of a greater number of capsids per movie. Movies were recorded at a rate of 1 frame/s for 55 to 60 s, with an average exposure time of 200 ms.
Capsid tracking.
Movies were converted to an .avi file format using the AxioVision software (release 4.8.1; Zeiss) and imported into ImageJ (version 1.43u). Capsids were tracked using the Particles Detector&Tracker plug-in (version 1.5) (31). Depending on the quality of each individual movie, detection parameters were set as follows: radius = 3 or 5, cutoff = 3.0 or 0.0, percentile = 0.6 to 1.5%; linking parameters were: link range = 2 and displacement = 10 to 20. Capsid motion was analyzed using the coordinates provided by the software to calculate the distance to origin. Capsid tracking starts as the capsid enters the field of view and stops as it leaves it or when it moves out of focus. The directionality of each trajectory was estimated according to the respective positions of the nucleus and of the plasma membrane (see Fig. 6).
Fig 6.

Impact of dystonin reduction on capsid transport during egress. HFFF2 cells transduced with shControl (A and B and E to H) or shDyst1 (C and D) were infected with one PFU/cell of vSR27-VP26GFP (A to F) or vFRΔ37-VP26GFP (G and H). Cytoplasmic capsid movements were monitored by live-cell imaging at 24 h postinfection, at a rate of one frame per second. Results are plotted as the distance to origin (in μm) for every individual capsid for each frame taken (A, C, E, and G). The premature truncation of some lines is due to the capsids moving out of the field of view. The slope of each line indicates capsid speed. A representative cell with all capsid trajectories from one movie per condition is shown (B, D, F, and H). Dashed boxes show the area displayed in the corresponding movie (see Movies S1, S2, S3, and S4, respectively). N, nucleus. Note the absence (B, F) or presence (arrowheads in D, H) of capsid aggregates. (I) Summary of the maximum distances to origin from the data shown in panels A to H as percentages of cells in categories of distance to origin. (J) Every moving capsid was tracked individually, and its directionality was estimated according to positions of the nucleus and the plasma membrane. “Both or none” indicates either a capsid having opposite directionalities within the same run or the absence of clear directionality. Scale bars, 10 μm.
shRNA.
The use of lentiviruses expressing short hairpin RNA (shRNA) has been described previously (29, 32). Silencing of dystonin was done by using two different 19-nt-long sequences, GTGTTGAAAGCCATTTAGA (shDyst1) or GTTGCACAGATTAGAGAGT (shDyst2). These sequences (positions 1897 and 2027, respectively, on the murine open reading frame) correspond to the plakin domain of human and murine dystonin and are specific for all known isoforms of dystonin. They are not conserved within sequences of other known plakin proteins. shControl corresponds to a sequence specific to the luciferase gene (GTGCGTTGCTAGTACCAAC) for all experiments. Silencing efficiency was routinely assessed by real-time reverse transcription (RT)-PCR from total RNA before every experiment. Only cells with a silencing rate of 70% or more were used.
Reverse transcription and real-time PCR analysis.
Total RNA was isolated using TRI reagent (Molecular Research Center) according to the manufacturer's protocol. Reverse transcription experiments were performed with 1 μg of total RNA in a total volume of 20 μl using Transcriptor reverse transcriptase (Roche). PCRs were performed in triplicate using a LightCycler capillar machine (Roche). For each reaction, a 1:400 final dilution of the reverse transcription product was used with a 0.4 μM final concentration of each primer in SYBR green I master mixture (Roche). PCR conditions were one step of denaturation (8 min at 95°C) followed by 45 cycles (each cycle consisted of 10 s at 95°C, 10 s at 60°C, and 10 s at 72°C). Gene expression was normalized to expression of GAPDH. The following primers were used for real-time PCR analysis (forward primer listed first and reverse primer second): human dystonin (5′-CCAGCCCGGTTAACTATTGA-3′/5′-TGGCAGAGCTGTAAGATCCA-3′) and human GAPDH (5′-GAGTCCACTGGCGTCTTCAC-3′/5′-TTCACACCCATGACGAACAT-3′).
A PCR control was also performed from the same cDNA samples using the following primers: 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ (GAPDH, forward and reverse, respectively, human and murine), 5′-AGCAAAGGACGCATACTGAC-3′ (dystonin, forward, human only), 5′-AGCAAAGGACGCATGCTGAC-3′ (dystonin, forward, murine only), and 5′-CTTGGCAAATCTGAGCCCCA-3′ (dystonin, reverse, human and murine). The PCR products were run on a 1% agarose gel.
Single-step virus growth.
Replicate 35-mm dishes of HFFF2 cells, expressing shControl, shDyst1, or no shRNA, were infected with 5 PFU/cell of HSV-1 strain 17+ (WT). After 1 h at 37°C, the cells were washed at low pH to remove residual input infectivity and overlaid with 1 ml of DMEM, and incubation was continued at 37°C. At 6, 12, 24, and 48 h after infection, the cells were harvested by scraping into the medium and pelleted, and the progeny virus was titrated from the supernatant on Vero cells. For growth in the presence of nocodazole, the drug (10 μM) was added onto the cells 1 h before infection and kept all the time of infection (see “Nocodazole” in Fig. 4B) or not at all and was replaced by dimethyl sulfoxide (DMSO). Viral titers were determined as described above.
Fig 4.
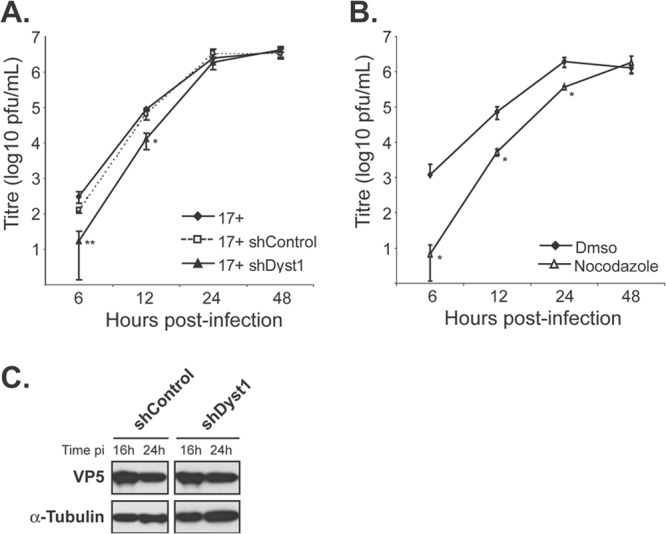
Growth kinetics of HSV-1 in the absence of dystonin or of MTs. Single-step growth curves of HSV-1 WT virus (17+) in control cells or cells silenced for dystonin (A) or in the presence or absence of nocodazole (B). (A) HFFF2 cells were transduced with shControl or shDyst1 shRNAs or untreated before being infected with 5 PFU/cell of HSV-1 17+. One hour after infection, the viral suspension was removed and cells were acid washed. At 6, 12, 24, or 48 h after infection, cells were scraped into the medium and pelleted. The supernatant was harvested and the titer of progeny virus was determined on Vero cells. (B) HFFF2 cells were incubated with 10 μM nocodazole 1 h before infection and all the time of infection (nocodazole) or not at all (DMSO). Infection and titration were done as described for panel A. Experiments were done in triplicate. Error bars indicate standard deviations of the mean (SDM). Asterisks indicate statistical differences with control conditions (17+ in untreated cells in panel A and DMSO condition in panel B) as determined by a paired Student t test (*, P < 0.05; **, P < 0.01). (C) Western blot analysis of lysates obtained from shControl or shDyst1 cells infected with 5 PFU/cell of 17+ for the indicated times (16 h or 24 h pi). The progress of infection was visualized using the DM165 antibody directed against the major capsid protein VP5, and alpha-tubulin levels were monitored as loading controls using the DM1A antibody.
Yeast two-hybrid assay.
The yeast two-hybrid system used in this study has been described previously (33). L40 yeasts were transformed with pLexA-UL37 HSV1 using lithium acetate, and transformants were selected by their ability to grow on tryptophane-depleted agar plates. These were then transformed with a cDNA library from differentiated PC12 cells cloned in pGAD-GH (kindly provided by H. Raux, VMS, CNRS, Gif-sur-Yvette) and screened as described in reference 33. The control for specificity was the lack of interaction between the putative pUL37-binding proteins and control proteins (lamin and LexA domain alone). A total of 127,000 transformants were obtained, and 27 were positive for specific interaction with pUL37 after screening and control. Liquid β-galactosidase assays were conducted as described in reference 33.
Immunoprecipitation.
Coimmunoprecipitations were done as described previously (29), except that only the cytoplasmic fraction was kept for analysis.
Secondary structure prediction.
The sequence of pUL37 from HSV-1 strain 17+ (34) was analyzed by two secondary structure prediction algorithms, APSSP2 (http://imtech.res.in/raghava/apssp/) and SOPMA (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl? page=npsa_sopma.html) to determine regions of the protein which were predicted to be unfolded. These were used to select suitable positions at which to truncate the protein.
RESULTS
pUL37 interacts with the plakin domain of dystonin.
To identify cellular proteins interacting with pUL37, a cDNA library prepared from differentiated PC12 cells, a rat neuronal cell line, was screened by yeast two-hybrid (Y2H) assay using pLexA-UL37 as bait. Out of 127,000 transformants, 27 were positive for interaction as assessed by β-galactosidase staining and growth on selective medium. Among these 27 clones representing 17 different genes, we identified a sequence corresponding to dystonin/BPAG1. This protein was of particular interest for several reasons: (i) its ability to bind MTs (23, 35), (ii) its role in retrograde and anterograde transport (24, 25), and (iii) it is expressed in tissues relevant to infection by HSV-1, namely, epithelial and neuronal cells (21). In particular, dystonin-null mice develop severe sensory nerve degeneration (36), highlighting its role in the natural target cell type of HSV-1, the sensory neuron.
The fragment of dystonin that we isolated from the Y2H screen encompasses residues 526 to 851 on isoform a and represents part of the plakin domain of the protein which is common to all dystonin isoforms (Fig. 1A).
To map the domain of pUL37 that interacts with dystonin, we cloned different truncations of pUL37 as LexA fusions and tested them against the 526-to-851 fragment of dystonin isolated from the Y2H screen (Fig. 1B). The boundaries of each truncation were chosen to be in regions of the protein expected to be unfolded, as determined by secondary structure prediction algorithms. Fragments 517 to 1123, 1 to 899, and 578 to 899 were shown to interact with dystonin in Y2H as assessed by liquid β-galactosidase assay (Fig. 1B). Surprisingly, we were unable to detect any interaction with fragment 517 to 899, while fragment 1 to 899 gave a stronger β-galactosidase signal (around 3-fold higher) than wild-type UL37 or any of the other positive fragments. Presumably, amino acids 517 to 578 fold in such a way as to mask the interaction site or disrupt its folding when present in the 517-to-899 fragment, but this effect is prevented in longer proteins or when these sequences are deleted. No interaction could be observed with fragments shorter than 578 to 899. These results map the dystonin interaction domain between residues 578 and 899 of pUL37.
We attempted to confirm the interaction in mammalian cells by coimmunoprecipitation. Vero cells were cotransfected with pHA-UL37, encoding full-length pUL37 fused to an N-terminal HA tag, and with pMyc-dystonin, encoding the 526-to-851 fragment of dystonin fused to an N-terminal myc tag. As a control, pHA-UL32, encoding a full-length, unrelated herpesviral protein, pUL32, fused to an N-terminal HA tag, was used in place of pHA-UL37. Twenty-four hours after transfection, cells were lysed and pUL37 or pUL32 were immunoprecipitated with an HA-specific antibody. The immunoprecipitates were analyzed by Western blotting using a myc-specific antibody to reveal the presence of myc-dystonin. In pHA-UL37 immunoprecipitates, myc-dystonin could be readily detected, but it was absent from pHA-UL32 immunoprecipitates (Fig. 1C). Similar results were obtained when the converse experiment was carried out. Thus, HA-pUL37 could be immunoprecipitated with myc-dystonin, whereas HA-pUL32 could not (Fig. 1D). These results validate the interaction between pUL37 and dystonin in mammalian cells.
Based on these results, we conclude that pUL37 interacts with the plakin domain of dystonin and that residues 578 to 899 of pUL37 are sufficient for this interaction.
Reduction of dystonin levels in HFFF2 cells and cellular localization of dystonin.
In order to identify a possible role for dystonin in the HSV-1 replication cycle, silencing experiments were set up using small-hairpin RNA (shRNA) technology. HFFF2 cells were transduced with lentivirus vectors expressing two different shRNAs against sequences that are common to all known isoforms of human dystonin or with a control lentivirus (henceforward referred to as shDyst1, shDyst2, and shControl cells, respectively). As it proved difficult to detect endogenous dystonin in cells by Western blotting, due to its large size and relatively low abundance, the efficiency of RNA silencing was assessed at the transcript level and by immunofluorescence. RT-PCR assays using a set of universal primers for the known isoforms of dystonin confirmed that dystonin mRNA could be detected in our cell line (Fig. 2A). Upon RNA silencing in HFFF2 cells, the amount of dystonin transcripts decreased by ∼80% and ∼60% in shDyst1 and shDyst2 cells, respectively, compared to the control, as assessed by quantitative RT-PCR (Fig. 2B).
Fig 2.
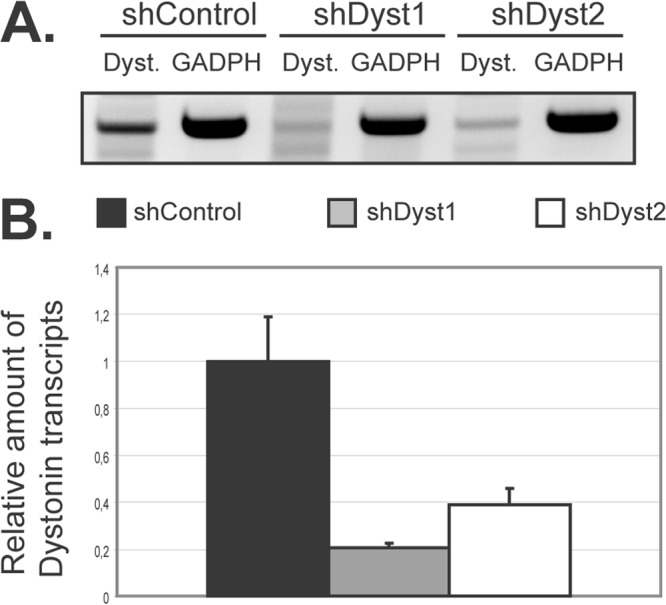
Silencing of dystonin in HFFF2 cells as assessed by quantitative reverse transcription-PCR (qRT-PCR). (A) Dystonin mRNA levels in silenced cells. HFFF2 cells transduced by two different shRNAs targeting dystonin (shDyst1 and shDyst2) or by an irrelevant shRNA (shControl) were lysed 3 days postransduction. RT-PCR was performed on the extracted total mRNA, and the resulting cDNA was used as a template for a PCR using primers specific for the plakin domain of dystonin (Dyst.) or for GAPDH as a loading control. The PCR product was visualized on an agarose gel (A) or quantified by real-time qPCR where shControl transcript levels were defined as 1 (B). GAPDH transcripts were used as a standard for quantification.
Using an antibody directed against human dystonin (anti-DST antibody), the protein could be detected in uninfected HFFF2 shControl cells, where it was localized throughout the cytoplasm in spots or short lines, mostly alongside and at the end of MTs (Fig. 3A). In shDyst1 cells, however, no specific fluorescence could be detected, confirming that the silencing was efficient at the protein level (Fig. 3A). As reported previously, the distribution of MTs in shControl cells that had been infected for 16 h was less well organized (37). However, dystonin localization appeared unaffected by infection (Fig. 3B). The localization pattern of dystonin is typical of a protein linked to the plus end of MTs. To check whether this was the case, HFFF2 cells were transfected with a plasmid expressing human end-binding protein 3 (EB3), a plus-end-binding protein (38), fused to GFP. As shown in Fig. 3C, dystonin short lines colocalized with EB3 “comets” at the plus ends of MTs, thereby confirming that dystonin can localize to MT plus ends.
Fig 3.

Dystonin localization in HFFF2 cells. (A) shControl or shDyst1 cells were fixed, and the distributions of dystonin (red) and alpha-tubulin (green) were determined using antibodies DST and GAR568 and DM1A and GAM488, respectively. Identical exposure times were used to collect images of shControl and shDyst1 cells. (B) shControl cells were infected with vUL35RFP1D1, a virus that encodes a VP26 capsid protein fused to the mRFP. Sixteen hours postinfection, cells were fixed and stained as described above, except that secondary antibodies were GAR488 and GAM647. Infected cells (i) were distinguished from noninfected cells (ni) through mRFP fluorescence (not shown). (C) HFFF2 cells were transfected with pGFP-hEB3, which encodes GFP fused to the plus-end-binding protein EB3 or with pEGFP-C1 as a control. Twenty-four hours after transfection, cells were fixed and immunostained for dystonin as described for panel A (red). GFP-EB3 and GFP are visualized through autofluorescence (green). An area from each cell image (dashed box) is enlarged. Scale bars, 20 μm.
Effect of dystonin reduction on growth of HSV-1.
To establish whether reducing the level of dystonin transcripts affected HSV-1 replication, single-step growth analysis of WT HSV-1 was carried out in shDyst1, shControl, or untreated HFFF2 cells. At 6 h, 12 h, 24 h, and 48 h postinfection, cells were scraped into the medium and pelleted. The supernatant was harvested, and the titer of progeny virus was determined on Vero cells (Fig. 4A). This showed that silencing of dystonin led to a significant delay in the production of infectious virus, compared to both the untreated and shControl cells, although by 24 h after infection, the amount of virus produced under all 3 conditions was similar. We compared these growth kinetics with those obtained under conditions where the MT network was disrupted by nocodazole (Fig. 4B). The time course of virus production when the MT network was disrupted by nocodazole was very similar to that seen following dystonin depletion. Thus, virus production was delayed but eventually attained equivalent levels to that seen in control cells. Similar behavior has been reported previously for cells treated with nocodazole (37). To show that the delay in infection following dystonin depletion was not a result of nonspecific inhibition of viral gene expression, levels of late protein synthesis were determined by examining the major capsid protein VP5. Western blotting confirmed that VP5 expression at 16 h and 24 h postinfection was unaffected by treatment of cells with shDyst1 (Fig. 4C). These results showed that the effects of reducing the amount of dystonin and disrupting the MT network were similar and suggested a possible involvement of dystonin in virus transport.
Association of capsids with endogenous dystonin in infected cells.
The possibility that dystonin is involved in transport during virus egress was examined by looking for association of capsids with endogenous dystonin. HFFF2 cells were infected with 1 PFU/cell of vSR27-VP26GFP or vFRΔ37-VP26GFP for 16 h, and capsid localization relative to dystonin was monitored. As shown in Fig. 5, capsids could be observed colocalizing with dystonin spots in vVP26GFP-infected cells, albeit at low frequency. In contrast, we were unable to see any colocalization between individual capsids and dystonin in cells infected with the UL37-null mutant vFRΔ37-VP26GFP. These data indicate that HSV-1 capsids can recruit dystonin during infection and that this requires pUL37. However, the low frequency of colocalization between dystonin and capsids may indicate that this interaction is transient or intermittent.
Fig 5.
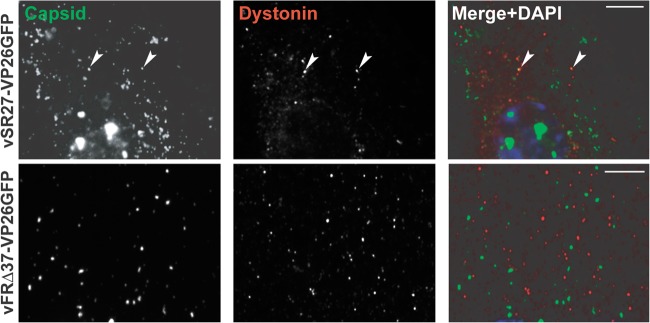
Capsid association with dystonin. HFFF2 cells were infected with 1 PFU/cell of vSR27-VP26GFP or vFRΔ37-VP26GFP. Sixteen hours later, cells were fixed and endogenous dystonin was visualized using specific antibodies DST and GAR568 (red). Capsids were visualized through autofluorescence (green). Arrowheads indicate capsids colocalizing with dystonin spots. Scale bars, 5 μm.
Role of dystonin in capsid movement during egress.
Given the demonstrated role of dystonin in anterograde transport of vesicles (25), its preferred localization at plus ends of MTs (Fig. 3), and its association with capsids, we postulated that dystonin might be involved in capsid transport during egress. To verify this, shControl and shDyst1 HFFF2 cells were infected at 37°C with 1 PFU/cell of vSR27-VP26GFP (a virus expressing a GFP-tagged capsid protein), and 24 h postinfection, capsid movements in the cytoplasm were monitored by live-cell microscopy. Live-cell imaging was carried out at room temperature (∼25°C), because at 37°C the large number of capsids moved too fast to be tracked efficiently, thus limiting both the reliability of the tracking and the number of capsids that could be analyzed (see Materials and Methods). However, the large effect of dystonin depletion on capsid transport as described in Fig. 6 was also observed at 37°C (data not shown). shControl cells contained numerous capsids moving throughout the cytoplasm (Fig. 6A and B; see also Movie S1 in the supplemental material). Movement was both anterograde (away from the nucleus; 50% of capsids) and retrograde (35% of capsids) (Fig. 6J). The average maximum distance to origin (defined as the capsid position at the start of recording) was 5.6 μm, with a peak at more than 19 μm for the duration of the acquisition (around 1 min, Fig. 6A and I). The average maximum speed observed was 0.95 μm/s. This was in sharp contrast with what was seen in shDyst1 cells, where capsids showed no or limited motion (Fig. 6C and D; see also Movie S2 in the supplemental material). The average maximum distance to origin was 0.54 μm (Fig. 6C and I), and the average maximum speed was 0.19 μm/s. The few moving capsids had essentially retrograde movement (Fig. 6J). This behavior was very similar to that observed in cells where the MT network was disrupted by treatment with 10 μM nocodazole (Fig. 6E, F, and I; see also Movie S3 in the supplemental material).
An interesting feature of the shDyst1 cells was that many of the capsids formed small aggregates (arrowheads in Fig. 6D), although this was not the reason for the lack of movement, as tracking was carried out on individual capsids. Capsid aggregation is typically observed in cells infected with a virus lacking the UL37 gene (26, 27, 39, 40). Furthermore, capsid motion was shown to be very limited in cells infected with a UL37-null mutant of PrV (17). Indeed, HFFF2 cells infected with a UL37-null virus (vFRΔ37-VP26GFP) showed very similar behavior to shDyst1 cells infected with the parental virus. Both contained capsid aggregates, while individual capsids displayed essentially no motion (Fig. 6G to J; see also Movie S4 in the supplemental material). This similarity in behavior suggests that the effect of reducing dystonin levels is mimicking the absence of pUL37. This phenotype is different from what was observed in nocodazole-treated HFFF2 cells infected with vSR27-VP26GFP, where capsid motion was also absent, but capsids did not aggregate (Fig. 6E and F).
To determine whether the capsid aggregation and lack of mobility in shDyst1 cells could be caused by lack of pUL37, we checked whether pUL37 was present on capsids in dystonin-depleted cells. shControl or shDyst1 cells were infected for 30 h with vUL37GFP-VP26RFP, a virus with GFP fused to the C terminus of pUL37 and mRFP fused to the N terminus of the capsid protein VP26. As shown in Fig. 7, pUL37 was associated with capsids in both control and dystonin-depleted cells, confirming that the lack of capsid movement in the latter was not due to a lack of pUL37 on these capsids.
Fig 7.
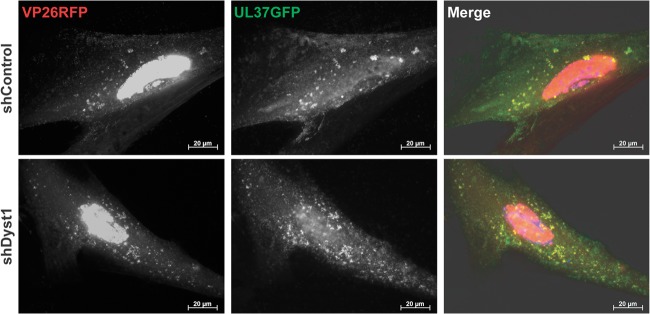
Association of pUL37 to capsids in shControl and shDyst1 cells. shControl or shDyst1 HFFF2 cells were infected with 5 PFU/cell of vUL37GFP-VP26RFP for 30 h before fixation. pUL37 was visualized through direct GFP fluorescence (green) and capsids through direct RFP fluorescence (red). Scale bars, 20 μm.
These results demonstrate that dystonin has a major role in capsid trafficking during egress.
Effect of dystonin silencing and of HSV-1 infection on microtubule and actin networks.
HSV-1 has been reported to cause reorganization of the MT network at late stages of infection (37). In addition, dystonin can stabilize MTs by cross-linking them to the actin cytoskeleton (23). Therefore, a possible explanation for the effects we observed is that the combination of dystonin silencing and infection by HSV-1 destabilized the cytoskeleton, thereby preventing efficient capsid transport during egress.
To determine whether this was the case, shControl or shDyst1 HFFF2 cells were mock infected or infected with vSR27-VP26GFP for 24 h and then stained to reveal MTs and F-actin. As shown in Fig. 8A, treatment with 10 μM nocodazole for 1 h, which prevents capsid transport (Fig. 6E and I), caused most of the MTs to depolymerize, leaving only a small number of twisted filaments. In contrast, dystonin silencing did not disrupt the MT network in mock-infected cells, but it appeared to result in a denser pattern of MTs at the vicinity of the nucleus than that in shControl cells (Fig. 8B and C). Infection had a noticeable effect on the MT network in both shDyst1 and shControl cells, resulting in a more disorganized, less nuclear-centric arrangement, although the higher density of MTs observed in mock-infected shDyst1 cells was still apparent in infected shDyst1 cells (Fig. 8C).
Fig 8.

Effect of nocodazole, infection, and dystonin silencing on MT and actin networks. (A) HFFF2 cells were incubated with 10 μM nocodazole for 1 h before being fixed and stained with MAb DM1A against alpha-tubulin and GAM488 (pseudocolored in red) and with TRITC-conjugated phalloidin to label actin (blue). Nuclei were visualized with DAPI (white). shControl (B) or shDyst1 HFFF2 (C) cells were either mock infected or infected with 5 PFU/cell of vSR27-VP26GFP. Twenty-four hours later, cells were fixed and stained with MAb DM1A against alpha-tubulin and GAM633 (red) and with TRITC-conjugated phalloidin to label actin (blue). Capsids were visible through direct GFP fluorescence (green). N, nucleus. Scale bars, 20 μm.
Therefore, although the combination of dystonin silencing and infection may lead to a local reorganization of the MTs close to the nucleus, it does not cause the disruption of the MT network that is observed in cells treated with nocodazole.
Since dystonin localizes at plus-end tips of MTs (Fig. 3C), we looked at possible effects of dystonin depletion on MT plus-end dynamics. shControl or shDyst1 HFFF2 cells were transfected with a plasmid encoding EB3-GFP, and MT plus ends were tracked by live-cell microscopy (see Movie S5 in the supplemental material). This showed that MT plus-end dynamics were unaffected by dystonin depletion.
In summary, the striking reduction in capsid movement during egress observed in shDyst1 cells (Fig. 6) does not appear to result from disruption of the MT network or from an effect on MT plus-end dynamics.
DISCUSSION
In this paper, our aim was to identify cellular factors that could account for the efficient capsid trafficking seen in HSV-1-infected cells. Although it is clear that microtubules and their associated molecular motors are involved (6, 7, 9), the intermediates between the viral capsid and these motors, if any, were unknown. The inner tegument proteins pUL36 and pUL37 are strongly suspected as having important roles in capsid transport. For example, although pUL37 is dispensable for release of viral DNA into the nucleus of HSV-1-infected cells (27), PrV capsid translocation to the nucleus (16) and capsid trafficking during egress (17) are both impaired in its absence. Furthermore, in vitro studies have shown that HSV-1 capsids which are associated with inner tegument proteins bind to dynein, dynactin, and kinesin-1, whereas untegumented capsids do not (8). However, whether capsid transport requires direct interactions between motor proteins and particle components is unknown, and the role such interactions play in transport within infected cells remains to be determined.
The cellular protein dystonin is an important element in cytoskeleton organization in skin, muscle, and neuron cells. It belongs to the plakin family (18) and, as such, contains a plakin domain, which is common to all known isoforms of dystonin and which, as we show here, is targeted by pUL37. It is possible that pUL37 may bind to other members of the plakin family, such as the closely related protein MACF/ACF7 (41, 42), although we have not examined this. One isoform of dystonin (BPAG1n4) was shown to have a role in retrograde transport in neurons through interactions with the p150glued subunit of dynactin (a cofactor of the dynein motor [24]) and retrolinkin, a component of vesicle membranes (43). More recently, dystonin has also been shown to be involved in anterograde transport of vesicles (25). The identification of dystonin as a binding partner for pUL37 in our Y2H assay therefore suggested a plausible role for this interaction in trafficking of HSV-1 capsids. Efficient pUL37-dependent transport of capsids is necessary during egress for acquisition of the outer tegument and subsequent envelopment (17, 26, 39, 40), and capsid trafficking during egress was severely inhibited by dystonin shRNA treatment. The consequence for capsid transport of reducing the level of dystonin was equivalent to that seen in the absence of pUL37 or when the MT network was disrupted by the addition of nocodazole (Fig. 6).
It is significant that despite the almost total block on capsid movement during virus egress resulting either from depletion of dystonin or from MT disruption, production of infectious virus eventually reached normal levels (Fig. 4). However, in both cases, virus production was delayed. This is consistent with previous studies which have shown that MT transport is important for increasing the efficiency of infection (37) but that alternative transport mechanisms for capsids exist if the MT network is disabled, for example, the actin cytoskeleton (17). It is also possible that some MTs are stabilized by infection and therefore more resistant to nocodazole treatment, as was shown previously (44).
During egress, it is likely that the greater part of capsid movement from the nucleus to the site of secondary envelopment occurs through plus-end-directed transport. Therefore, the inhibition of capsid transport during egress following reduction of dystonin levels (Fig. 6) is probably indicative of a role for the dystonin-pUL37 interaction in plus-end-directed transport. It is notable, therefore, that our analysis of dystonin distribution within cells showed that it preferentially colocalized with the MT plus-end-binding protein, EB3 (Fig. 3). This is consistent with the presence of an SxIP EB1-binding motif (45) in the microtubule-binding domain of dystonin (MTBD in Fig. 1). Interestingly, a recent study demonstrated that Shot (the Drosophila orthologue of spectraplakin proteins) also associates with the plus end of MTs through an interaction with the plus-end-binding protein EB1 (46). We are currently investigating whether plus-end-directed transport of capsids is also affected during virus entry.
In the course of this study, the interaction domain of pUL37 with dystonin was mapped to residues 578 to 899 (Fig. 1). We were not able to investigate the importance of the interaction in virus infection by deleting this region, as it is known to be important for the essential interaction of pUL37 with pUL36 (47, 48). Therefore, we attempted to define the interaction domain more accurately by random mutagenesis. However, although we were able to identify residues which abolished the interaction of full-length pUL37 with dystonin (but not with pUL36) in Y2H, these were in regions of the protein important for other interactions, indicating that this portion of pUL37 contains multiple binding sites (data not shown).
A well-described phenotype of UL37-null mutants of HSV-1 and PrV is the aggregation of capsids in the cytoplasm of infected cells (26, 27, 39, 40). This phenotype is very similar to what we observed in dystonin-depleted cells, where aggregation occurred even though pUL37 was present on the capsids (Fig. 7). Therefore, preventing the interaction of pUL37 and dystonin appears to mimic the effect of removing pUL37 altogether, indicating the importance of this interaction for pUL37 function.
The mechanism of action of the pUL37-dystonin interaction remains to be elucidated. Dystonin appears to have several functions in the cell. It has been shown to interact with the MT-associated protein 1 (MAP1) to support MT stability through tubulin acetylation and is also important for maintaining TGN integrity (25). However, we did not observe any obvious defect in MT organization or stability in our shDyst cells (Fig. 8), and although TGN fragmentation occurred in shDyst cells (data not shown), it is also a common consequence of HSV-1 infection (37). Therefore, these effects appear unlikely to be the cause of the defect in capsid transport observed in dystonin-depleted cells. A more intriguing possibility is that dystonin acts as a linker between HSV-1 capsids and molecular motors, as was suggested to be the case with neuronal vesicles (24, 43). By nature, this interaction would be expected to be transient and could explain the low frequency of colocalization observed between capsids and endogenous dystonin in infected cells (Fig. 5). Such a linker role would account for both the effects of dystonin depletion described in this paper and for the properties of UL37-null mutants. Further investigation into the mechanism of action of the pUL37-dystonin interaction would shed light not only on herpesvirus capsid transport but also on the role of dystonin in cellular mechanisms of transport.
Supplementary Material
ACKNOWLEDGMENTS
We thank Y. Gaudin for critical reading of the manuscript. We are grateful to H. Raux for advice with the yeast two-hybrid screen and the gift of the PC12 cDNA library. We are indebted to P. Desai (John Hopkins University) for the plasmid pK26GFP, to John Victor Small (Institute of Molecular Biotechnology, Vienna, Austria) for pGFP-hEB3, and to the following from the CVR, Glasgow: R. Everett for silencing plasmids, E. Palmer for the plasmid pCMV10-HA-UL32, and V. Preston and C. Cunnigham for BACmid SR27.
Footnotes
Published ahead of print 26 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02676-12.
REFERENCES
- 1. Bartolini F, Gundersen GG. 2006. Generation of noncentrosomal microtubule arrays. J. Cell Sci. 119:4155–4163 [DOI] [PubMed] [Google Scholar]
- 2. Bornens M. 2002. Centrosome composition and microtubule anchoring mechanisms. Curr. Opin. Cell Biol. 14:25–34 [DOI] [PubMed] [Google Scholar]
- 3. Caviston JP, Holzbaur EL. 2006. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 16:530–537 [DOI] [PubMed] [Google Scholar]
- 4. Antinone SE, Smith GA. 2010. Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J. Virol. 84:1504–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lyman MG, Enquist LW. 2009. Herpesvirus interactions with the host cytoskeleton. J. Virol. 83:2058–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mabit H, Nakano MY, Prank U, Saam B, Dohner K, Sodeik B, Greber UF. 2002. Intact microtubules support adenovirus and herpes simplex virus infections. J. Virol. 76:9962–9971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sodeik B, Ebersold MW, Helenius A. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, Karger A, Sodeik B. 2010. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 6:e1000991 doi:10.1371/journal.ppat.1000991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dohner K, Wolfstein A, Prank U, Echeverri C, Dujardin D, Vallee R, Sodeik B. 2002. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 13:2795–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miranda-Saksena M, Boadle RA, Aggarwal A, Tijono B, Rixon FJ, Diefenbach RJ, Cunningham AL. 2009. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J. Virol. 83:3187–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dohner K, Nagel CH, Sodeik B. 2005. Viral stop-and-go along microtubules: taking a ride with dynein and kinesins. Trends Microbiol. 13:320–327 [DOI] [PubMed] [Google Scholar]
- 12. Wolfstein A, Nagel CH, Radtke K, Dohner K, Allan VJ, Sodeik B. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227–237 [DOI] [PubMed] [Google Scholar]
- 13. Aggarwal A, Miranda-Saksena M, Boadle RA, Kelly BJ, Diefenbach RJ, Alam W, Cunningham AL. 2012. Ultrastructural visualization of individual tegument protein dissociation during entry of herpes simplex virus 1 into human and rat dorsal root ganglion neurons. J. Virol. 86:6123–6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Granzow H, Klupp BG, Mettenleiter TC. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 79:3200–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luxton GW, Haverlock S, Coller KE, Antinone SE, Pincetic A, Smith GA. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. U. S. A. 102:5832–5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krautwald M, Fuchs W, Klupp BG, Mettenleiter TC. 2009. Translocation of incoming pseudorabies virus capsids to the cell nucleus is delayed in the absence of tegument protein pUL37. J. Virol. 83:3389–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80:201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leung CL, Green KJ, Liem RK. 2002. Plakins: a family of versatile cytolinker proteins. Trends Cell Biol. 12:37–45 [DOI] [PubMed] [Google Scholar]
- 19. Roper K, Gregory SL, Brown NH. 2002. The ‘spectraplakins’: cytoskeletal giants with characteristics of both spectrin and plakin families. J. Cell Sci. 115:4215–4225 [DOI] [PubMed] [Google Scholar]
- 20. Young KG, Kothary R. 2007. Dystonin/Bpag1—a link to what? Cell Motil. Cytoskeleton 64:897–905 [DOI] [PubMed] [Google Scholar]
- 21. Leung CL, Zheng M, Prater SM, Liem RK. 2001. The BPAG1 locus: alternative splicing produces multiple isoforms with distinct cytoskeletal linker domains, including predominant isoforms in neurons and muscles. J. Cell Biol. 154:691–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brown A, Bernier G, Mathieu M, Rossant J, Kothary R. 1995. The mouse dystonia musculorum gene is a neural isoform of bullous pemphigoid antigen 1. Nat. Genet. 10:301–306 [DOI] [PubMed] [Google Scholar]
- 23. Yang Y, Bauer C, Strasser G, Wollman R, Julien JP, Fuchs E. 1999. Integrators of the cytoskeleton that stabilize microtubules. Cell 98:229–238 [DOI] [PubMed] [Google Scholar]
- 24. Liu JJ, Ding J, Kowal AS, Nardine T, Allen E, Delcroix JD, Wu C, Mobley W, Fuchs E, Yang Y. 2003. BPAG1n4 is essential for retrograde axonal transport in sensory neurons. J. Cell Biol. 163:223–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ryan SD, Bhanot K, Ferrier A, De Repentigny Y, Chu A, Blais A, Kothary R. 2012. Microtubule stability, Golgi organization, and transport flux require dystonin-a2-MAP1B interaction. J. Cell Biol. 196:727–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pasdeloup D, Beilstein F, Roberts AP, McElwee M, McNab D, Rixon FJ. 2010. Inner tegument protein pUL37 of herpes simplex virus type 1 is involved in directing capsids to the trans-Golgi network for envelopment. J. Gen. Virol. 91:2145–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roberts AP, Abaitua F, O'Hare P, McNab D, Rixon FJ, Pasdeloup D. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J. Virol. 83:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Desai P, Person S. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563–7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J. Virol. 83:6610–6623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vielkind U, Swierenga SH. 1989. A simple fixation procedure for immunofluorescent detection of different cytoskeletal components within the same cell. Histochemistry 91:81–88 [DOI] [PubMed] [Google Scholar]
- 31. Sbalzarini IF, Koumoutsakos P. 2005. Feature point tracking and trajectory analysis for video imaging in cell biology. J. Struct. Biol. 151:182–195 [DOI] [PubMed] [Google Scholar]
- 32. Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80:7995–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Raux H, Flamand A, Blondel D. 2000. Interaction of the rabies virus P protein with the LC8 dynein light chain. J. Virol. 74:10212–10216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D, Perry LJ, Scott JE, Taylor P. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69(Part 7):1531–1574 [DOI] [PubMed] [Google Scholar]
- 35. Kapur M, Wang W, Maloney MT, Millan I, Lundin VF, Tran TA, Yang Y. 2012. Calcium tips the balance: a microtubule plus end to lattice binding switch operates in the carboxyl terminus of BPAG1n4. EMBO Rep. 13:1021–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo L, Degenstein L, Dowling J, Yu QC, Wollmann R, Perman B, Fuchs E. 1995. Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell 81:233–243 [DOI] [PubMed] [Google Scholar]
- 37. Avitabile E, Di Gaeta S, Torrisi MR, Ward PL, Roizman B, Campadelli-Fiume G. 1995. Redistribution of microtubules and Golgi apparatus in herpes simplex virus-infected cells and their role in viral exocytosis. J. Virol. 69:7472–7482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nakagawa H, Koyama K, Murata Y, Morito M, Akiyama T, Nakamura Y. 2000. EB3, a novel member of the EB1 family preferentially expressed in the central nervous system, binds to a CNS-specific APC homologue. Oncogene 19:210–216 [DOI] [PubMed] [Google Scholar]
- 39. Desai P, Sexton GL, McCaffery JM, Person S. 2001. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J. Virol. 75:10259–10271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klupp BG, Granzow H, Mundt E, Mettenleiter TC. 2001. Pseudorabies virus UL37 gene product is involved in secondary envelopment. J. Virol. 75:8927–8936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kodama A, Karakesisoglou I, Wong E, Vaezi A, Fuchs E. 2003. ACF7: an essential integrator of microtubule dynamics. Cell 115:343–354 [DOI] [PubMed] [Google Scholar]
- 42. Leung CL, Sun D, Zheng M, Knowles DR, Liem RK. 1999. Microtubule actin cross-linking factor (MACF): a hybrid of dystonin and dystrophin that can interact with the actin and microtubule cytoskeletons. J. Cell Biol. 147:1275–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu JJ, Ding J, Wu C, Bhagavatula P, Cui B, Chu S, Mobley WC, Yang Y. 2007. Retrolinkin, a membrane protein, plays an important role in retrograde axonal transport. Proc. Natl. Acad. Sci. U. S. A. 104:2223–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Elliott G, O'Hare P. 1998. Herpes simplex virus type 1 tegument protein VP22 induces the stabilization and hyperacetylation of microtubules. J. Virol. 72:6448–6455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Honnappa S, Gouveia SM, Weisbrich A, Damberger FF, Bhavesh NS, Jawhari H, Grigoriev I, van Rijssel FJ, Buey RM, Lawera A, Jelesarov I, Winkler FK, Wuthrich K, Akhmanova A, Steinmetz MO. 2009. An EB1-binding motif acts as a microtubule tip localization signal. Cell 138:366–376 [DOI] [PubMed] [Google Scholar]
- 46. Alves-Silva J, Sanchez-Soriano N, Beaven R, Klein M, Parkin J, Millard TH, Bellen HJ, Venken KJ, Ballestrem C, Kammerer RA, Prokop A. 2012. Spectraplakins promote microtubule-mediated axonal growth by functioning as structural microtubule-associated proteins and EB1-dependent +TIPs (tip interacting proteins). J. Neurosci. 32:9143–9158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bucks MA, Murphy MA, O'Regan KJ, Courtney RJ. 2011. Identification of interaction domains within the UL37 tegument protein of herpes simplex virus type 1. Virology 416:42–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kelly BJ, Mijatov B, Fraefel C, Cunningham AL, Diefenbach RJ. 2012. Identification of a single amino acid residue which is critical for the interaction between HSV-1 inner tegument proteins pUL36 and pUL37. Virology 422:308–316 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.