

Abstract
Immediate-early 3 (IE3) gene products are required to activate early (E)-stage gene expression of murine cytomegaloviruses (MCMV). The first early gene activated by IE3 is the M112-113 gene (also called E1), although a complete understanding of the activation mechanism is still lacking. In this paper, we identify a 10-bp cis-regulating motif upstream of the M112-113 TATA box as important for IE3 activation of M112-113 expression. Results from DNA affinity assays and chromatin immunoprecipitation assays show that the association of IE3 with the M112-113 gene promoter was eliminated by deletion of the 10-bp DNA sequence, now named IE3AM (for IE3 activating motif). In addition, IE3 interacts with TATA box binding protein (TBP), a core protein of TFIID (transcription initiation) complexes. Finally, we created an IE3AM-deleted MCMV (MCMVdIE3AM) using a bacterial artificial chromosome system. The mutant virus can still replicate in NIH 3T3 cells but at a significantly lower level. The defectiveness of the MCMVdIE3AM infection can be rescued in an M112-113-complemented cell line. Our results suggest that the interactions of IE3 with IE3AM and with TBP stabilize the TFIID complex at the M112-113 promoter such that M112-113 gene expression can be activated and/or enhanced.
INTRODUCTION
Human cytomegalovirus (HCMV) is an opportunistic agent causing serious diseases in immunocompromised individuals and in newborns (1). CMV gene expression occurs immediately after CMV genomic DNA enters the nucleus and relies on cellular gene expression and splicing machineries (1). Murine CMV (MCMV) is often used as a model system to study infection by HCMV and other CMVs due to their significant similarities in terms of their biological, genetic, and pathogenic properties (2). Accumulated studies have demonstrated that the immediate-early 3 (IE3) protein of the major immediate-early (MIE) gene is essential for MCMV replication (3–5). We recently demonstrated that IE3 has a strong effect on early gene regulation and is critical to the formation of viral prereplication compartments (pre-RC) via interactions with viral and cellular proteins (6, 7). However, the detailed mechanisms used by MCMV IE3 to regulate viral early gene expression and to counter cellular defenses still remain enigmatic. Identifying the trans- and cis-regulating elements that interact with IE3, ascertaining how IE3 regulates early gene expression, and understanding the biological importance of the IE3 targeting various elements in viral replication will shed light on early gene regulation of CMV infection and aid in developing antiviral strategies against viral replication.
The first gene expressed at the early stage (i.e., after the immediate-early stage) might be the one encoded by M112-113 (E1, or early protein 1) (8). M112-113 proteins are detectable at almost the same time as IE3, but M112-113 gene expression is upregulated strongly by IE3 (6). Compared to HCMV genes, M112-113 is the equivalent of HCMV UL112-113, and MCMV IE3 is the counterpart to HCMV IE2 (9). The M112-113 gene consists of three exons, and alternative splicing creates at least 4 proteins (33, 36, 38, and 87 kDa) (8, 10). UL112-113 is 1 of the 11 core proteins that are essential for HCMV DNA replication (11, 12), and it interacts with the lytic origin of replication (13) and forms prereplication domains by recruiting a diversity of viral and cellular proteins that are important for DNA replication (14, 15).
The M112-113 gene has drawn a lot of attention recently because of its important biological functions. First, the M112-113 gene is silent during latency and might be the first activated gene when switching to lytic infection (2). Therefore, M112-113 might be critical for progression of viral reactivation to viral particle formation. Second, the M112-113 promoter was specifically activated in neuronal cells (not in nonneuronal cells) in a transgenic mouse strain, which implied that M112-113 is important for CMV persistent infection in brain (16). Third, UL112-113 (the HCMV homolog) was found to be able to reactivate Kaposi's sarcoma-associated herpesvirus from latency (17), which suggested a clinical importance of CMV in mixed herpesviral infection. In addition, it was recently found that M112-113 was important for MCMV to productively infect human cells (18), which also raises a clinical concern with regard to the possibility of MCMV infection in human populations. Furthermore, M112-113 has evolved functions to inhibit IE3's repressive effects on the major immediate-early promoter (MIEP) so that MCMV can more efficiently express its genes and replicate (7). In a transfection system using M112-113-expressing plasmid where the M112-113 gene is under the control of its own promoter, we found that M112-113 proteins can be expressed at a low level without IE3, but IE3 enhanced M112-113 gene expression to a much higher level (7).
So far we know little about M112-113 gene regulation, except the observation that IE3 can strongly enhance M112-113 gene production (3, 5, 7). Earlier studies of HCMV UL112-113 gene regulation by IE2 (an equivalent of MCMV IE3) have indicated that a minimal DNA sequence containing an ATF/CREB binding motif is important for IE2 to activate UL112-113 promoter activity (19). Further, it was confirmed that IE2 mediated the transactivation of the UL112-113 promoter via interactions with CREB and CREB binding protein (CBP) (20, 21). It was reported that there exist three binding sites for IE2 upstream of the UL112-113 promoter, but those binding sites were found not to be critical for IE2 to activate UL112-113 gene activation (20, 22). More interestingly, Rodems and coworkers discovered that cis elements containing the ATF/CREB binding motif and IE86 binding motif differentially activate the UL112-113 promoter at early and late times in HCMV infection (21). HCMV and MCMV share many biological characteristics, and M112-113 and UL112-113 have similar gene structures (23), but M112-113 lacks the ATF/CREB binding motif. Therefore, the model of IE2 activation of UL112-113 in HMCV might not simply be applied to the mechanism of IE3 activation of M112-113, and it is significant to explore the mechanism(s) IE3 uses to activate the M112-113 gene. Since it is evident that IE3 activates M112-113 gene expression in the system of cotransfection or infection, we are curious to know whether the activation of M112-113 production by IE3 occurs at the transcriptional or translational level. If it happens at the transcriptional level, we wish to discover the specific DNA in the M112-113 promoter region that interacts with IE3. In the present study, we identified a 10-bp DNA sequence that precedes the TATA box of the M112-113 gene, associates with IE3, and is not only essential for IE3 to activate M112-113 gene expression but also is important for MCMV replication.
MATERIALS AND METHODS
Tissue culture.
NIH 3T3 cells (ATCC) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 1% penicillin-streptomycin. An NIH 3T3-based cell line (NIH 3T3e1) that constitutively expresses M112-113 that was not upregulated by transiently expressed IE3 was kindly provided by J. Kerry (7, 10).
Antibodies.
The antibodies used for Western blotting (WB) are listed and their dilutions are shown below. Monoclonal antibody against tubulin (T-9026) was purchased from Sigma-Aldrich (1:1,000 for WB; St. Louis, MO); monoclonal anti-enhanced green fluorescent protein (EGFP; sc-9966) was purchased from Santa Cruz Biotechnology Inc. (1:1,000 for WB to probe EGFP; Santa Cruz, CA); monoclonal antibodies against MCMV IE1 and M112-113 (1:200 for WB) proteins were provided by Stipan Jonjic (Croatia) (7, 24); monoclonal antibodies against M44 and M25 (1:250 for WB) were generous gifts from J. Shanley (University of Connecticut) (25, 26). Rabbit anti-TATA binding protein (TBP) (1:100 for WB; P100971) and rabbit anti-TBP-associated protein 15 (TAF15) (1:100 for WB; ARP30111) were purchased from Aviva System Biology Corporation (San Diego, CA).
Plasmids and molecular cloning.
Plasmid pBB5.5, containing the whole M112-113 gene and promoter, was generously provided by M. Messerle (5, 7, 8); pET (expressing GFP) and pgfie3 (expressing GFP-tagged IE3) were reported previously (6). A diagram of part of the M112-113 gene and the constructed M112-113-expressing vectors are shown in Fig. 1A. pBB5.5 consists of a 2.76-kb DNA sequence before the first amino acid code (ATG) and 2.8 kb of the M112-113 coding sequence (including exons and introns). Using this plasmid as the template, we performed PCR and generated a series of DNA fragments containing the M112-113 gene with different sizes of upstream DNA sequence (Fig. 1A). Primers were designed to include XbaI and PstI restriction sites at the ends (Table 1). The PCR-amplified DNA fragments were cloned into XbaI and PstI sites of pUC18. To further map the minimal size of DNA motif that is required for IE3 to activate M112-113 gene expression, we designed primers (Table 2) and performed PCR using pTATA1 as the template (Fig. 1A) and generated three mutations, each with a different DNA sequence before the TATA box: pdl7A, pdl7B, and pdl10C (Fig. 2A).
Fig 1.

Mapping out the minimal size in M112-113 gene promoter that is essential for IE3 to activate M112-113 gene expression. (A) Diagram of the deletional mutation of the M112-113 gene; pBB5.5 contains the whole gene of M112-113, including promoter and open reading frame (ORF). Numbers on the left side of each construct stand for the length upstream from the initiation site of the M112-113 gene in the plasmid named on the left side. ORF112-113 stands for the open reading frame of M112-113. (B and C) Western blots to examine M112-113 gene production. NIH 3T3 cells were harvested 24 h after cotransfection of the plasmids indicated on the top of each picture. IE3 (expressed from pgfpie3) and GFP (expressed from pET) were detected by anti-GFP antibody, while M112-113 was examined by anti-M112-113 antibody. Tubulin was used as a sample loading control. (D) Real-time RT-PCR to examine M112-113 gene expression. Total RNAs were isolated from NIH 3T3 cells after 24 h of cotransfection of M112-113-expressing plasmids (as indicated) with pgfpie3 or pET. The CT values first were normalized to that of beta-actin, and then comparisons were made between pgfpie3 and pET groups to calculate the ΔΔCT. Relative quantity was shown as R = 2−ΔΔCT.
Table 1.
Primers used for PCR to generate different sizes of upstream DNA of the M112-113 gene
| Primera | Sequence |
|---|---|
| pBB4.5 | TATCTAGAGAGAATAGAGCGAAAATGATTTACC |
| pBB3.8 | TATCTAGAAGTGATCTCTCGCTTCGACCGAC |
| pBB3.0 | TGTCTAGAGCTCGACCGCGCGTCCGGAGCGCC |
| pTATA1 | GGCGTGTCTAGAGCGGGTAGCGAGAAGAACG |
| pTATA1/2 | GGCGTGTCTAGACCGCAGGTTATAACAACGTCAT |
| pTATA2 | GGCGTGTCTAGAATTAAGAATGGGCGTGATG |
| pTATA2/3 | GGCGTGTCTAGAGGCGTGATGCAGACTT |
| pTATA3 | GGCGTGTCTAGACAGACTTTATAAATCGCAAG |
| pTATA4 | GGCGTGTCTAGATGCTGATAGTTCCTGCGTCG |
| pTATA5 | GGCGTGTCTAGACCGGTGAAAAGACCTTCGTTCG |
| Reverse | TGGGGGCTGCAGGCTCAGAAGAAGGAATAC |
The forward primers were named according to the plasmid names that are shown in Fig. 1A, and they have XbaI sites (italics) at their ends. The reverse primer used for constructing the M112-113-expressing plasmids is the same for each pair and has a PstI site (italics) at its end.
Table 2.
Primers used for PCR to generate minimal mutations in the upstream DNA of the M112-113 gene TATA boxa
| Primer | Sequence |
|---|---|
| pdl10bp | GGCGTGTCTAGAGCGGGTAGCGAGAAGAACGCCGGAGACCGCAGGTTATAACAACGTCATGCATAAGGCGTGATGCAGACTTTATA |
| pdl7A | GGCGTGTCTAGAGCGGGTAGCGAGAAGAACGCCGGAGACCGCAGGTTATAACAACGTCATGCATAAATTAAGAATGGGCAGACTTTATAAATCGC |
| pdl7B | GGCGTGTCTAGAGCGGGTAGCGAGAAGAACGCCGGAGACCGCAGGTTATAACAACGTCATGCATAAATTAAGAATGGGCGTGATTTATAAATCGCAAGCCGG |
| Reverseb | TGGGGGCTGCAGGCTCAGAAGAAGGAATAC |
PCR was used to make a more detailed mutation in front of the TATA box, using pTATA1 as the template.
The reverse primer is the same for all three mutations.
Fig 2.

Identification of a DNA motif that is required for IE3 to activate M112-113 gene expression. (A) Partial DNA sequence before the TATA box (boxed) in the M112-113-expressing vectors. Black lines represent the DNA sequences deleted. (B) Western blot assay to examine the activation of the M112-113 gene by IE3. The experimental methods are the same as those for Fig. 1B and C. (C) Real-time RT-PCR to examine the activation of the M112-113 gene by IE3. The experimental methods are the same as those for Fig. 1D.
RNA isolation, treatment with DNase I (RNase free), and real-time reverse transcription-PCR (RT-PCR).
Total RNA was isolated using TRI reagent (Ambion, Inc., Austin, TX) and treated with DNase I (RNase-free; 18047-019; Invitrogen) according to the manufacturer's instructions. DNase I was inactivated by extraction of the RNA sample with phenol and chloroform. PCR was performed using primers to amplify the M112-113 gene.
To quantitatively examine the mRNA level of M112-113 in M112-113-transfected cells, real-time RT-PCR was undertaken using the QuantiTect SYBR green RT-PCR kit (Qiagen, Valencia, CA). A total of 1 μg of total RNA and 0.2 μM sense and antisense primers were used in a final 25-μl volume of master mix. A reverse transcription step of 20 min at 50°C was included prior to PCR. PCRs consisted of 50 cycles with optimal conditions of 94°C for 20 s, 50°C for 1 min, 72°C for 30 s, and an optimized collection data step at 80°C for 5 s. Fluorescence captured at 80°C was determined to be lacking signal generated by primer dimers. All samples were run in triplicate; data were collected and recorded by the iCycler iQ software (Bio-Rad) and expressed as a function of the cycle threshold (CT), which represents the number of cycles at which the fluorescent intensity of the SYBR green dye is significantly greater than the background fluorescence. The CT is directly correlated to the log10 copy number of the RNA standards. RNA copies were extrapolated from standard curves (CT versus log10 copy number) representing at least seven serial dilutions of standard RNA (101 to 107 copies/μl). RNA standards were used as calibrators to the relative quantification of product generated in the exponential phase of the amplification curve for real-time RT-PCR. The results were accepted for standard curves with correlation coefficients greater than 0.95. A melting temperature curve analysis was obtained by measuring the fluorescence during a period of warming from 60 to 95°C after the amplification cycles.
ChIP assay and real-time PCR.
NIH 3T3 cells were cotransfected with pBB5.5 and pgfpie3 for 24 h and were fixed with 1% formaldehyde. A chromatin immunoprecipitation (ChIP) assay was performed using the EZChIP kit (Upstate-Millipore, Billerica, MA) according to the manufacturer's protocol. The amount of DNA subjected to ChIP by antibodies (normal IgG and anti-GFP) was examined by real-time PCR per 25-μl reaction mix using the primers shown in Table 4, which were designed according to reference 27. All samples were analyzed in triplicate using SYBR green 2× master mix (Applied Biosystems, Foster City, CA) on an Applied Biosystems 7900HT fast real-time PCR system. After an initial denaturation incubation of 5 min at 95°C, 45 cycles of 3-step cycling were performed with an annealing temperature of 60°C. Melting curve analysis was then performed to verify product specificity. The relative ratios of CT of DNA subjected to ChIP with each antibody to DNA subjected to ChIP with normal IgG were determined by examining the change in threshold cycle number (ΔCT), with standard deviations calculated to account for variance in signals of DNA subjected to ChIP with both IgG and antibody. The ChIP DNA signals were compared to those for input DNA and are reported as a percentage.
Table 4.
Primers used for PCR to amplify the DNA in ChIP assay
| Primer | Sequence |
|
|---|---|---|
| Forward | Reverse | |
| Promoter A | CTC ACC TGA GCC TGC T | CCG TCC GTC CAT GCG CCC AT |
| Promoter B | GGG GTT CCT TCG GCC GA | AAA TAA CAC GGA CGA GCG |
| Promoter C | AGT GAT CTC TCG CTT CGA | CCG CCT CGT TAC GCA CGT |
| Promoter D | GCT CGA CAC GAG CTA CCG T | CGA GGC CTT CGC CGA A |
| Promoter E | TAC GGT GCG ACG GAC G | GTC GAA TGG CGC TCC GGA |
| Promoter F | ACA TTA CTG GCC TCG GCG | CGA ACG AAG GTC TTT TCA |
| Promoter G | TGC TGA TAG TTC CTG CGT | AGA CTG CAT GAT GGT CG |
| Promoter H | ACT GCA CGA ACG GCG AGG | CAG GAT GAC TTG CAG CAA |
| Promoter I | GCC GGG GTC CGA CGG CA | TTC GAA TCT CTT ACG TC |
DNA affinity chromatography.
DNA affinity purification for detecting DNA binding protein with Western blotting was described by Atanasiu et al. (28) with minimal modification. DNA fragments with biotin label were made from pTATA1 or pdl10C by PCR using the primers 5′ GCG GGT AGC GAG AAG AA 3′ (forward) and Biotin-5′CGA ACG AAG GTC TTT TCA (reverse). The biotin-labeled DNA fragments were then purified using a Qiaquick PCR purification kit (Qiagen) and checked for purity and quantity on a 1.2% agarose gel. The biotin-labeled DNA fragments were then incubated with Dynabeads M-280 streptavidin beads (Invitrogen). After washing off the free DNA, the DNA-bead mixtures were incubated with nuclear extract (NE) prepared from MCMVE5gfp-infected NIH 3T3 cells (6) by the Dignam method (29), with purified GFPIE3 from MCMVE5gfp-infected NIH 3T3 cells, or with TATA binding protein (TBP). After washing the protein-DNA-bead mixtures, we performed Western blotting to detect the protein bound to the DNA-bead mixtures using anti-GFP, anti-TBP, anti-IE1, and anti-TAF15 antibodies.
Purification of TBP and GFPIE3.
MCMVE5gfp-infected NIH 3T3 cells were collected at 24 h postinfection (hpi) and washed with phosphate-buffered saline (PBS) one time. The cells were lysed with 0.025 M Tris, 0.15 M NaCl, 0.001 M EDTA, 1% NP-40, 5% glycerol, pH 7.4 (lysis buffer). The whole-cell lysate was applied to react with antibody against TBP or GFPIE3. The protein-antibody immune complex was captured with protein G-agarose beads. After washing the beads 3 times with the lysis buffer, the beads were eluted with low-pH elution buffer (0.1 M glycine-HCl at pH 2.5 to 3.0). The eluate was neutralized soon after the elution with 10% (vol/vol) 1 M Tris (pH 9.5), and at this point it contained the purified TBP or GFPIE3.
Generation of MCMVs deleted of IE3AM.
MCMV bacmid DNA (Sm3frE5gfp) was made from the bacmid Sm3fr (30) by tagging GFP to the C terminus of IE3 (end of exon 5) (6). Using Sm3frE5gfp as the template, we further mutated DNA sequence in the region upstream of the M112-113 gene. The minimal DNA motif identified as important for IE3 to activate and interact with E1 promoter DNA is named IE3AM (for IE3 activating motif). The seamless bacterial artificial chromosome (BAC) system using galK as the selection marker (6, 31) was utilized to construct the mutant and the revertant, and the primers for the mutagenesis are summarized in Table 3. First, the DNA sequence, including the M112-113 promoter, was replaced with the galK gene by PCR using primers TATA1galK_Fw and TATA1_galK_Rv (Table 3). The galK gene then was replaced by TATA1 (DNA from pTATA1) and dl10C (DNA from pTATAdl10C) by PCR using primers TATA1_Fw and TATA1_Rv (Table 3). The resultant BAC DNAs were named Sm3frE5gfrpdlIE3AM and Sm3frE5gfpRevIE3AM, respectively. To prepare the viruses from the BAC DNAs, the BAC DNAs were transfected into NIH 3T3 cells. Positive viral plaques were picked and propagated in NIH 3T3 cells to make larger volumes of viral stock.
Table 3.
Primers used for PCR to generate DNA fragments to produce MCMV mutationsa
| Primer | Sequence |
|---|---|
| TATA1_galK_Fw | GCGGGTAGCGAGAAGAACGCCGGAGACCGCAGGTTATAACAACGTCATGCATCCTGTTGACAATTAATCATCGGCA |
| TATA1_galK_Rv | CGAACGAAGGTCTTTTCACCGGTCGCGACGAGAAAAGTGGGCACGTAATCTACCGTCAGCACTGTCCTGCTCCTT |
| TATA1_Fw | GCGGGTAGCGAGAAGAACG |
| TATA1_Rv | CGAACGAAGGTCTTTTCA |
The PCR was used to make MCMV BAC with the original DNA and IE3AM-deleted DNA. The original DNA was subjected to PCR using pTATA1 as the template; the dIE3AM BAC was made from pdl10C.
Immunoblot analysis.
Proteins were separated by SDS-PAGE in 7.5% polyacrylamide gels (10 to 20 μg loaded in each lane), transferred to nitrocellulose membranes (Amersham Inc., Piscataway, NJ), and blocked with 5% nonfat milk for 60 min at room temperature. Membranes were incubated overnight at 4°C with primary antibody, followed by an incubation with horseradish peroxidase-coupled secondary antibody. For regular WB, we used secondary antibody from Amersham; for the detection of protein in the immunoprecipitation, we used secondary antibodies from TrueBlot ULTRA (18-8817 for mouse or 18-8816 for rabbit; eBioscience). Detection was accomplished with enhanced chemiluminescence (Pierce, Rockford, IL) according to standard methods. Membranes were stripped with stripping buffer (100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.8), washed with PBS–0.1% Tween 20, and reprobed with new primary antibodies to detect additional proteins.
Coimmunoprecipitation.
Antibodies were coupled to protein G-Sepharose beads (Amersham Pharmacia Biotech AB, Sweden) according to the manufacturer's instructions. After a wash with PBS–0.1% bovine serum albumin, beads were incubated overnight at 4°C with clarified extracts, washed again in PBS–0.1% bovine serum albumin, and resuspended in a mixture of PBS and 2× Laemmli buffer (20 μl of each). After heating at 95°C for 5 min, beads were removed by centrifugation and supernatants were analyzed by SDS-PAGE and immunoblotting.
Plaque formation unit assay.
Viral titers were determined by plaque assay, largely as described previously (6) but with a slight modification. Supernatants containing serially diluted virus particles were added to confluent NIH 3T3 cell monolayers in 6-well plates. After adsorption for 2 h, medium was removed and cells were washed twice with serum-free DMEM and overlaid with phenol-free DMEM containing 5% FCS, 0.5% low-melting-point agarose (GIBCO), and 1% penicillin-streptomycin. The number of plaques that were stained red was counted and calculated as that in 1 ml, which is PFU per ml. Mean PFU numbers were determined after averaging results from different dilutions.
RESULTS
A short DNA sequence mapped out in the upstream of the M112-113 promoter region was found to be essential and sufficient for IE3 activation of M112-113 gene expression.
Although it has been reported that IE3 activates M112-113 gene expression (4–7, 10), it still remains unclear how M112-113 gene expression is regulated by IE3. In the present study, we hypothesize that IE3 interacts with a cis-regulating element to activate M112-113 gene expression. To examine this hypothesis, we made a series of deletion mutations based on the M112-113M112-113-expressing plasmid, pBB5.5, that contains the full-length M112-113 gene, including the promoter and open reading frame (ORF). The diagrams of M112-113 gene structure and deletion mutations are shown in Fig. 1A. First, we cotransfected M112-113-expressing plasmid with pgfpie3 (GFP-tagged IE3) or with pET (expressing GFP; used to control the input DNA in cotransfection) into NIH 3T3 cells, then M112-113 expression levels were examined at the protein level by Western blotting (Fig. 1B and C) and at the mRNA level by real-time RT-PCR (Fig. 1D). Whether or not the M112-113 gene was activated by IE3 was determined by the comparison of M112-113 levels in the cotransfection of pgfpie3 with M112-113-expressing plasmid to that in the cotransfection of pET with M112-113-expressing plasmid. As can be seen in Fig. 1C, M112-113 expression from pTATA2/3, pTATA3, and pTATA4 was no longer activated by IE3, which was consistent with the results of real-time RT-PCR shown in Fig. 1D. Therefore, the minimal DNA sequence of the M112-113 gene that is required for IE3 activation is mapped to only 25 bp before the TATA box.
A 10-bp DNA motif is essential for IE3's activity on M112-113 gene promoter.
To further narrow down a smaller cis-acting element that is essential for IE3 to activate M112-113 gene expression, we made more deletion mutations based on pTATA1. First, we deleted 10 bp of DNA at the beginning of pTATA2, resulting in pdl10C; we then deleted a 7-bp DNA fragment after the 10-bp DNA, making pdl7B; finally, we deleted the 7 bp right before the TATA box to construct pdl7A. Figure 2A shows the M112-113 promoter DNA sequences that are absent from pdl10C, pdl7B, and pdl7A. After cotransfection of M112-113 expressing wild-type and deleted plasmids with pET or with pgfpie3 in NIH 3T3 cells, the M112-113 gene expression levels were examined by Western blotting (Fig. 2B) and real-time RT-PCR (Fig. 2C). We found that M112-113 expression was not activated by IE3 in pdl10C in which the 10-bp DNA sequence was deleted from pTATA1.
The short DNA motif (10 bp) is associated with IE3.
The effects of HCMV IE2 on UL112-113 gene expression depend on the association of IE2 with a consensus ATF/CREB sequence in the UL112-113 gene (21). However, no consensus ATF/CREB binding sequence in M112-113 of the NIH 3T3 cells was found by gene DNA alignment analysis. Therefore, it might be that the 10-bp motif interacts with IE3. Therefore, we performed a DNA ChIP assay to determine whether IE3 interacts with the M112-113 DNA. NIH 3T3 cells were cotransfected with pBB5.5 and pgfpie3 for 24 h, fixed with 1% formaldehyde, and sonicated to shear the DNA to ∼200 to 300 bp. The DNA-GFPIE3 complexes were pulled down with anti-GFP antibody (normal IgG was used as a control). After degrading proteins with protease K, the purified DNA samples (input, IgG control, and anti-GFP pulldown; Fig. 3A) were used as the template for regular PCR (Fig. 3A) and real-time PCR (Fig. 3B). Nine pairs of continuous primers covering the whole M112-113 promoter region (promoters A to I; Table 4) were used to amplify the DNA that had been subjected to ChIP. The PCR products were all strongly visualized by ethidium bromide in an agarose gel for input samples (Fig. 3A). Real-time PCR was performed, and the PCR products were normalized to IgG samples and calculated as percent input. As shown in Fig. 3B, the PCR using primer F generated the most DNA product, containing the 10-bp DNA sequence. As a control, we also cotransfected pBB5.5 with pET (GFP-expressing plasmid) into NIH 3T3 cells and followed the same procedures using anti-GFP antibody. The ChIP DNA samples were amplified by PCR and yielded virtually no product (Fig. 3A, IgG). Therefore, we show that IE3 can interact with the M112-113 gene promoter DNA that contains the 10-bp motif.
Fig 3.

Ten-bp DNA motif mediates the interaction of IE3 with TBP and stabilizes TFIID complexes. (A and B) DNA ChIP assay to examine IE3-DNA interaction. (A) M112-113 gene structure is shown at the top; input DNA was amplified to show the total DNA in the samples, and IgG-incubated samples were used as a negative control. In the bottom panel, ChIP DNA using anti-GFP shows that IE3-bound DNA is the strongest in the region (F) where the 10-bp motif resides. (B) Real-time PCR was performed to show the percentage anti-GFP ChIP DNA compared to the amount of the input. Results are consistent with those of regular PCR shown in A. (C) DNA affinity assay to ascertain direct interaction of IE3 with the M112-113 promoter with or without the 10-bp DNA. (Left side) DNA affinity assay with the whole nuclear extract (NE). Mock indicates the NE prepared from uninfected NIH 3T3 cells. Input was NE made from MCMV-infected (MCMV Inf.) NIH 3T3 cells. (Right side) DNA affinity assay with purified proteins (Puri. Pr.) of TBP (upper) and GFPIE3 (lower). (D) Coimmunoprecipitation assay to determine the interaction of TBP with IE3. Nuclear extracts were prepared from NIH 3T3 cells infected with MCMVE5gfp at a multiplicity of infection of 1 and incubated with antibodies as indicated at the top of the panel. The pulled-down proteins were detected by Western blotting using antibodies shown on the right side. For the detection of protein in the immunoprecipitation, we used secondary antibodies from TrueBlot ULTRA (18-8817 for mouse or 18-8816 for rabbit; eBioscience) to avoid the heavy chain.
To further confirm that IE3 can interact with the M112-113 promoter and to know whether the 10-bp motif is required for IE3 to interact with the M112-113 promoter, we performed DNA affinity assays. First, we used primers (5′ GCG GGT AGC GAG AAG AA 3′ [forward] and Biotin-5′CGA ACG AAG GTC TTT TCA [reverse]) to perform PCR with pTATA1 as the template to generate a probe that contains the 10-bp motif or with pdl10C as the template to produce a probe that does not contain the 10-bp motif. The probes were then conjugated to Dynabeads M-280 streptavidin beads (Invitrogen), and the DNA-bead mixtures were reacted with nuclear extracts made from MCMVE5gfp-infected NIH 3T3 cells, with purified TBP, or with purified GFPIE3. After several washing steps, the DNA-protein-avidin bead mixtures were eluted with Western blot loading buffer. The proteins were detected by Western blot assay as shown in Fig. 3C. Panels on the left side of Fig. 3C show the DNA affinity assay using nuclear extract, and the right side shows the DNA affinity assay using purified TBP (upper) or GFPIE3 (lower). The results demonstrate that (i) the 10-bp motif was required for IE3 to interact with the M112-113 gene promoter, because the purified IE3 can only be pulled down by the probe containing the 10-bp sequence (Fig. 3C, right, upper); (ii) in the assay using purified TBP, TBP binding to the promoter was not affected by the 10-bp motif (Fig. 3C, right, lower); (iii) in the assay using NE, TBP and TBP-associated protein 15 (TAF15) bound to the promoter and the protein-DNA interactions were reduced by deletion of the 10-bp motif (Fig. 3C, left); and (iv) the observation that IE3 can still be pulled down by the DNA probe without the 10-bp sequence suggests that IE3 interacts with TBP (Fig. 3C, left). In summary, the 10-bp DNA sequence is demonstrated not only to be essential for IE3 to activate M112-113 gene expression but also to be required for IE3 to interact with the M112-113 promoter region. Therefore, the 10-bp DNA motif was designated IE3 activating motif (IE3AM).
Although IE3 interaction with the M112-113 promoter depends on the 10-bp motif, IE3 can still be weakly pulled down by the DNA without the 10-bp motif, perhaps through interaction with TBP. If this is the case, then it should be possible to coimmunoprecipitate IE3 together with TBP. To test our speculation, we performed a coimmunoprecipitation assay using anti-TBP, anti-GFP (for GFPIE3), and anti-IE1 antibodies. As can be seen in Fig. 3D, IE3 was found to interact with TBP, but neither interacted with IE1. Therefore, IE3 interacts not only with the M112-113 DNA but also with core proteins of TFIID complexes, TBP. Since the 10-bp motif is very close to the TATA box, the simultaneous interaction of IE3 with the IE3AM DNA and TBP might contribute to stabilization of TBP-cored TFIID complexes, which is critical for the activity of the promoter.
The IE3AM in the M112-113 gene is important for MCMV replication.
′To test the importance of the IE3AM for MCMV replication, we removed the IE3AM using the bacmid Sm3frE5gfp (6), resulting in bacmid SM3frE5gfp_dIE3AM, and also created a rescued bacmid, SM3frE5gfp_dIE3AMRQ. After transfecting the bacmids into NIH 3T3e1 cells that stably express M112-113 proteins, we made viruses MCMVE5gfpdIE3AM and MCMVE5gfpdIE3AMRQ. The viruses were confirmed by sequencing the whole M112-113 gene region (Fig. 4A).
Fig 4.
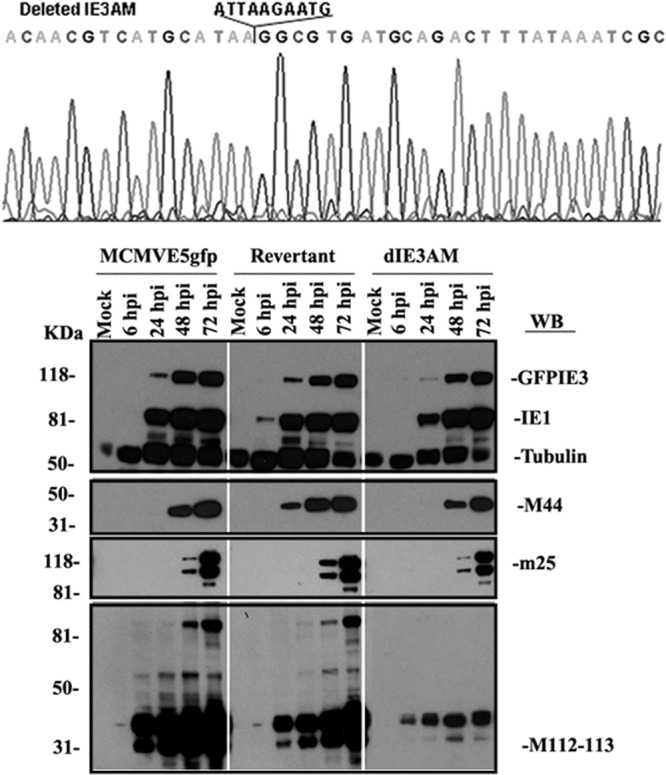
Comparative protein production of MCMVE5gfp, MCMVE5gfpdIE3AM, and MCMVE5gfpdIE3AMRQ. (Upper) The viral DNAs of MCMVE5gfpdIE3AM and MCMVE5gfpdIE3AMRQ were isolated and sequenced for the whole M112-113 region, confirming that IE3AM is removed from MCMVE5gfpdIE3AM and restored in MCMVE5gfpdIE3AMRQ, and that the DNA sequences of the whole region are correct. (Lower) Three viruses were used to infect NIH 3T3 cells, and the cells were harvested at the indicated time points. Viral proteins were detected by Western blotting using antibodies against the viral proteins shown on the right side (IE1, GFPIE3, m25, M44, and M112-113). Tubulin was used for controlling sample loading.
We wanted to determine any changes in viral protein production when IE3AM is deleted. As shown in Fig. 4B, we collected samples at four time points after infection and performed Western blotting. We examined the production of several viral proteins, including IE proteins (IE1 and GFP-tagged IE3), early proteins (M112-113 and M44), and late protein (m25). Although IE1 and IE3 can be produced from MCMVE5gfpdIE3AM at a level similar to that from MCMVE5gfp or MCMVE5gfpdIE3AMRQ, the levels of early and late protein production were significantly decreased. Consistent with the results from the cotransfection system shown in Fig. 2B, deletion of IE3AM resulted in the inability of IE3 to activate M112-113 production, although it still has a basal expression. Therefore, this experiment showed that the IE3AM is important for viral early and late protein production.
We next examined whether the IE3BAM is important for viral replication using a PFU assay. The viruses (MCMVE5gfp, MCMVE5gfpdIE3AM, and MCMVE5gfpdIE3AMRQ) were used to infect NIH 3T3 cells at an MOI of 0.5. As shown in Fig. 5A, deletion of IE3AM resulted in slower and decreased viral growth. However, complementation of M112-113 using NIH 3T3e1 cells that stably express M112-113 can completely restore the defectiveness caused by deletion of the IE3AM (Fig. 5B). Our results suggest that M112-113 gene products are important but not required for MCMV replication, and that activation of M112-113 by IE3 enhances not only some early and late viral protein production but also the efficiency of MCMV infection.
Fig 5.
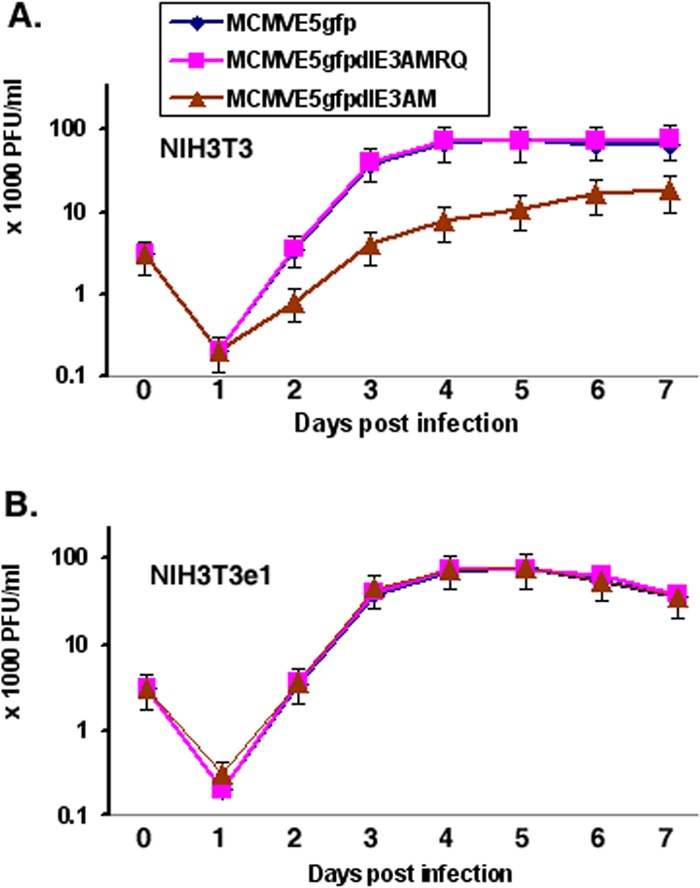
Viral PFU assay. (A) Viral replication ability in NIH 3T3 cells. MCMVE5gfp, MCMVE5gfpdIE3AM, or MCMVE5gfpdIE3AMRQ was used to infect NIH 3T3 cells, and the cells were collected on the days postinfection indicated. After three cycles of freezing and thawing, the cell-medium mixture was centrifuged and the supernatant was used for PFU assays. Each experiment was performed independently three times, and the mean number of PFU was obtained by averaging the numbers from different dilutions. (B) The same as A, but infection was carried out in the NIH 3T3e1 cell line.
DISCUSSION
CMVs are large, double-stranded DNA viruses whose genomes are ∼230 kb in size, and they are highly adapted to their respective hosts. CMV replication in host cells is a well-defined, sequential process: entry into cells, immediate-early (IE) gene expression, early gene expression, DNA replication, late gene expression, and viral production (1). Many viral gene products function through interaction with cellular proteins or other viral proteins at different stages of viral infection. Interactions start once the viruses contact cells and involve many processes, including cell signal transduction, cell cycle control, and cellular defensive responses; in addition, CMVs usurp cellular functional machineries, thus leading to replicative success (32). It is believed that the molecular interactions between CMV and host cells at different stages determine the fates of infected viruses: productive replication, latency, or being cleared out of cells. Understanding the complicated interplay between viruses and host cells is critical not only for elucidating the pathogenesis of viruses but also for developing strategies against viral infections. For example, chemical arrays have been used to screen for small molecules to interfere with viral replication by targeting particular molecular interactions between viruses and cells (33). Studies of the early essential gene products of CMV may lead to novel therapeutic strategies against CMV-associated diseases that might avoid the toxicities of DNA replication inhibitors.
Murine cytomegalovirus (MCMV) infection in mouse is the most used and, so far, the best small-animal model for studies of human CMV (HCMV), because the viruses have similar genomic structures, infectious cycles, and pathogeneses (2, 34, 35). MCMV and HCMV present a similar biological interaction with host cells at the early stage of the infection. In this stage, MIE (major immediate-early) gene products, such as IE3 of MCMV or the HCMV homolog IE2, are needed to shut off host gene expression and DNA replication to cause cell cycle arrest (36–38). Both HCMV and MCMV MIE genes are expressed independently of other instances of de novo viral gene expression (4). The IE gene transcription unit is activated by tegument proteins, possibly through sequestration of Daxx and histone deacetylases (HDACs) (39, 40), and the transcript is differentially spliced. Translation of the major spliced MIE transcripts produces two most abundant proteins: IE1 and IE2 in HCMV or IE1 and IE3 in MCMV (3–5, 41). For both CMVs, IE1 is formed by splicing exons 1 to 4, and MCMV IE3 (or HCMV IE2) is formed by splicing exons 1 to 3 plus exon 5 (4). These spliced products appear to be necessary for the activation of early promoters in the tightly regulated transcription cascade (3, 5, 42, 43). IE3 is an essential gene for viral replication and serves as an activator of early and late MCMV genes. One of the genes targeted by IE3 is M112-113.
Due to similarities in gene structure of M112-113 of MCMV to that of UL112-113 of HCMV and of the function of IE3 of MCMV to that of IE2 of HCMV, one can speculate that the model of how IE2 activates UL112-113 in HCMV can be applied to the mechanism that IE3 uses to activate the M112-113 gene in MCMV. However, analysis of the DNA sequence of M112-113 reveals no significant homology between M112-113 and UL112-113. More surprisingly, no CREB/ATF binding site that exists in UL112-113 was found in the M112-113 gene. Therefore, we wanted to know the MCMV mechanism by which IE3 activates M112-113. As shown in Fig. 1 and 2, we identified a 10-bp DNA motif that is essential for IE3 to activate M112-113 gene expression. We then found that the short motif is important for IE3 to interact with the M112-113 promoter region by DNA affinity chromatography and DNA ChIP assay. Therefore, we designated the motif IE3AM (for IE3 activating motif).
Initiation of transcription of a gene from a core promoter region by polymerase II requires the assembly of several initiation factors to form preinitiation complex (44). Assembly of the initiation complexes has been thought to be nucleated exclusively by the sequence-specific binding of the TFIID transcription factor complex, which is composed of the TATA binding protein (TBP) and TBP-associated factors (TAFs) associated with the different promoters (45). The preinitiation complexes might vary within different promoters, but the core components are TBP, TAFs, and RNA polymerase II (46). For the M112-113 promoter, the TATA box ensures a basal expression of the M112-113 gene, but the IE3AM is needed for IE3 to enhance expression. The IE3AM is so close to the TATA box that the DNA-bound IE3 must have an effect on the TFIID complex. IE3 can also interact with TBP directly, as shown by coimmunoprecipitation assay. We hypothesize that IE3 can interact with IE3AM and TBP simultaneously to clamp the TFIID complex onto the M112-113 gene promoter, which activates M112-113 gene expression. If the IE3AM is eliminated, the IE3-TFIID complexes will not be stabilized and the M112-113 promoter will be less activated. However, free TBP can still bind to the M112-113 TATA box to form TFIID complex; therefore, M112-113 still has basal expression even when the IE3AM was deleted, as shown in Fig. 4.
In summary, our data imply that the mechanism MCMV IE3 uses to activate the M112-113 gene differs from that of HCMV IE2. A 10-bp motif named the IE3AM is identified to be essential for IE3 to interact with M112-113 promoter and enhance M112-113 gene expression. IE3 interacts with TBP, so it is likely that IE3 functions as a mediator of TFIID complex assembly or as a factor to hold the TFIID complex on the M112-113 promoter.
ACKNOWLEDGMENTS
We thank J. Kerry, S. Jonjic, M. Messerle, and J. Shanley for donating reagents.
This study was supported by a pilot grant from the National Center for Research Resources (G12 RR003050) and the National Institute on Minority Health and Health Disparities (8G12MD007579-27) from the National Institutes of Health (Q.T.), an American Cancer Society grant (117448-RSG-09-289-01-MPC) (Q.T.), and NIH/NCRR U54RR022762 (Q.T.).
We acknowledge the PSM Molecular Biology Core Laboratory for instrument support. We acknowledge Andrew Boileau (Saba University School of Medicine) for critical reading of the manuscript.
Footnotes
Published ahead of print 19 December 2012
REFERENCES
- 1. Mocarski ES, Jr, Shenk T, Pass RF. 2006. Cytomegaloviruses, 5th ed Lippincott Williams &Wilkins, Philadelphia, PA [Google Scholar]
- 2. Reddehase MJ, Simon CO, Seckert CK, Lemmermann N, Grzimek NK. 2008. Murine model of cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 325:315–331 [DOI] [PubMed] [Google Scholar]
- 3. Angulo A, Ghazal P, Messerle M. 2000. The major immediate-early gene ie3 of mouse cytomegalovirus is essential for viral growth. J. Virol. 74:11129–11136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Keil GM, Ebeling-Keil A, Koszinowski UH. 1987. Immediate-early genes of murine cytomegalovirus: location, transcripts, and translation products. J. Virol. 61:526–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Messerle M, Buhler B, Keil GM, Koszinowski UH. 1992. Structural organization, expression, and functional characterization of the murine cytomegalovirus immediate-early gene 3. J. Virol. 66:27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martinez FP, Cosme RS, Tang Q. 2010. Murine cytomegalovirus major immediate-early protein 3 interacts with cellular and viral proteins in viral DNA replication compartments and is important for early gene activation. J. Gen. Virol. 91:2664–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang Q, Li L, Maul GG. 2005. Mouse cytomegalovirus early M112/113 proteins control the repressive effect of IE3 on the major immediate-early promoter. J. Virol. 79:257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buhler B, Keil GM, Weiland F, Koszinowski UH. 1990. Characterization of the murine cytomegalovirus early transcription unit e1 that is induced by immediate-early proteins. J. Virol. 64:1907–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White EA, Spector DH. 2007. Early viral gene expression and function. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed). Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 10. Ciocco-Schmitt GM, Karabekian Z, Godfrey EW, Stenberg RM, Campbell AE, Kerry JA. 2002. Identification and characterization of novel murine cytomegalovirus M112-113 (e1) gene products. Virology 294:199–208 [DOI] [PubMed] [Google Scholar]
- 11. Pari GS, Anders DG. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 67:6979–6988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sarisky RT, Hayward GS. 1996. Evidence that the UL84 gene product of human cytomegalovirus is essential for promoting oriLyt-dependent DNA replication and formation of replication compartments in cotransfection assays. J. Virol. 70:7398–7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kagele D, Rossetto CC, Tarrant MT, Pari GS. 2012. Analysis of the interactions of viral and cellular factors with human cytomegalovirus lytic origin of replication, oriLyt. Virology 424:106–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ahn JH, Jang WJ, Hayward GS. 1999. The human cytomegalovirus IE2 and UL112-113 proteins accumulate in viral DNA replication compartments that initiate from the periphery of promyelocytic leukemia protein-associated nuclear bodies (PODs or ND10). J. Virol. 73:10458–10471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park MY, Kim YE, Seo MR, Lee JR, Lee CH, Ahn JH. 2006. Interactions among four proteins encoded by the human cytomegalovirus UL112-113 region regulate their intranuclear targeting and the recruitment of UL44 to prereplication foci. J. Virol. 80:2718–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arai Y, Ishiwata M, Baba S, Kawasaki H, Kosugi I, Li RY, Tsuchida T, Miura K, Tsutsui Y. 2003. Neuron-specific activation of murine cytomegalovirus early gene e1 promoter in transgenic mice. Am. J. Pathol. 163:643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wells R, Stensland L, Vieira J. 2009. The human cytomegalovirus UL112-113 locus can activate the full Kaposi's sarcoma-associated herpesvirus lytic replication cycle. J. Virol. 83:4695–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schumacher U, Handke W, Jurak I, Brune W. Mutations in the M112/M113 coding region facilitate murine cytomegalovirus replication in human cells. J. Virol. 84:7994–8006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schwartz R, Helmich B, Spector DH. 1996. CREB and CREB-binding proteins play an important role in the IE2 86-kilodalton protein-mediated transactivation of the human cytomegalovirus 2.2-kilobase RNA promoter. J. Virol. 70:6955–6966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lang D, Gebert S, Arlt H, Stamminger T. 1995. Functional interaction between the human cytomegalovirus 86-kilodalton IE2 protein and the cellular transcription factor CREB. J. Virol. 69:6030–6037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodems SM, Clark CL, Spector DH. 1998. Separate DNA elements containing ATF/CREB and IE86 binding sites differentially regulate the human cytomegalovirus UL112-113 promoter at early and late times in the infection. J. Virol. 72:2697–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arlt H, Lang D, Gebert S, Stamminger T. 1994. Identification of binding sites for the 86-kilodalton IE2 protein of human cytomegalovirus within an IE2-responsive viral early promoter. J. Virol. 68:4117–4125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spector DH. 1996. Activation and regulation of human cytomegalovirus early genes. Intervirology 39:361–377 [DOI] [PubMed] [Google Scholar]
- 24. Hengel H, Lucin P, Jonjic S, Ruppert T, Koszinowski UH. 1994. Restoration of cytomegalovirus antigen presentation by gamma interferon combats viral escape. J. Virol. 68:289–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loh LC, Keeler VD, Shanley JD. 1999. Sequence requirements for the nuclear localization of the murine cytomegalovirus M44 gene product pp50. Virology 259:43–59 [DOI] [PubMed] [Google Scholar]
- 26. Wu CA, Carlson ME, Henry SC, Shanley JD. 1999. The murine cytomegalovirus M25 open reading frame encodes a component of the tegument. Virology 262:265–276 [DOI] [PubMed] [Google Scholar]
- 27. Ellison TJ, Izumiya Y, Izumiya C, Luciw PA, Kung HJ. 2009. A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi's sarcoma-associated herpesvirus. Virology 387:76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Atanasiu C, Lezina L, Lieberman PM. 2005. DNA affinity purification of Epstein-Barr virus OriP-binding proteins. Methods Mol. Biol. 292:267–276 [DOI] [PubMed] [Google Scholar]
- 29. Dignam JD, Lebovitz RM, Roeder RG. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wagner M, Jonjic S, Koszinowski UH, Messerle M. 1999. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 73:7056–7060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Warden C, Tang Q, Zhu H. 2011. Herpesvirus BACs: past, present, and future. J. Biomed. Biotechnol. 2011:124595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fortunato EA, McElroy AK, Sanchez I, Spector DH. 2000. Exploitation of cellular signaling and regulatory pathways by human cytomegalovirus. Trends Microbiol. 8:111–119 [DOI] [PubMed] [Google Scholar]
- 33. Loregian A, Coen DM. 2006. Selective anti-cytomegalovirus compounds discovered by screening for inhibitors of subunit interactions of the viral polymerase. Chem. Biol. 13:191–200 [DOI] [PubMed] [Google Scholar]
- 34. Stinski MF, Isomura H. 2008. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med. Microbiol. Immunol. 197:223–231 [DOI] [PubMed] [Google Scholar]
- 35. Stinski MF, Petrik DT. 2008. Functional roles of the human cytomegalovirus essential IE86 protein. Curr. Top. Microbiol. Immunol. 325:133–152 [DOI] [PubMed] [Google Scholar]
- 36. Salvant BS, Fortunato EA, Spector DH. 1998. Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J. Virol. 72:3729–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wiebusch L, Hagemeier C. 1999. Human cytomegalovirus 86-kilodalton IE2 protein blocks cell cycle progression in G(1). J. Virol. 73:9274–9283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wiebusch L, Neuwirth A, Grabenhenrich L, Voigt S, Hagemeier C. 2008. Cell cycle-independent expression of immediate-early gene 3 results in G1 and G2 arrest in murine cytomegalovirus-infected cells. J. Virol. 82:10188–10198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tang Q, Maul GG. 2003. Mouse cytomegalovirus immediate-early protein 1 binds with host cell repressors to relieve suppressive effects on viral transcription and replication during lytic infection. J. Virol. 77:1357–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Taylor RT, Bresnahan WA. 2005. Human cytomegalovirus immediate-early 2 gene expression blocks virus-induced beta interferon production. J. Virol. 79:3873–3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stenberg RM. 1996. The human cytomegalovirus major immediate-early gene. Intervirology 39:343–349 [DOI] [PubMed] [Google Scholar]
- 42. Ghazal P, Visser AE, Gustems M, Garcia R, Borst EM, Sullivan K, Messerle M, Angulo A. 2005. Elimination of ie1 significantly attenuates murine cytomegalovirus virulence but does not alter replicative capacity in cell culture. J. Virol. 79:7182–7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mendelson M, Monard S, Sissons P, Sinclair J. 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 77(Pt 12):3099–3102 [DOI] [PubMed] [Google Scholar]
- 44. Vannini A, Cramer P. 2012. Conservation between the RNA polymerase I, II, and III transcription initiation machineries. Mol. Cell 45:439–446 [DOI] [PubMed] [Google Scholar]
- 45. Papai G, Weil PA, Schultz P. 2011. New insights into the function of transcription factor TFIID from recent structural studies. Curr. Opin. Genet. Dev. 21:219–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee TI, Young RA. 2000. Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet. 34:77–137 [DOI] [PubMed] [Google Scholar]