

Abstract
Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin1) protein is known as a regulator which recognizes phosphorylated Ser/Thr-Pro motifs and increases the rate of cis and trans amide isomer interconversion, thereby altering the conformation of its substrates. We found that Pin1 knockdown using short hairpin RNA (shRNA) technology resulted in strong suppression of productive Epstein-Barr virus (EBV) DNA replication. We further identified the EBV DNA polymerase catalytic subunit, BALF5, as a Pin1 substrate in glutathione S-transferase (GST) pulldown and immunoprecipitation assays. Lambda protein phosphatase treatment abolished the binding of BALF5 to Pin1, and mutation analysis of BALF5 revealed that replacement of the Thr178 residue by Ala (BALF5 T178A) disrupted the interaction with Pin1. To further test the effects of Pin1 in the context of virus infection, we constructed a BALF5-deficient recombinant virus. Exogenous supply of wild-type BALF5 in HEK293 cells with knockout recombinant EBV allowed efficient synthesis of viral genome DNA, but BALF5 T178A could not provide support as efficiently as wild-type BALF5. In conclusion, we found that EBV DNA polymerase BALF5 subunit interacts with Pin1 through BALF5 Thr178 in a phosphorylation-dependent manner. Pin1 might modulate EBV DNA polymerase conformation for efficient, productive viral DNA replication.
INTRODUCTION
The Epstein-Barr virus (EBV) is a human gammaherpesvirus that mainly infects and establishes latent infection in B lymphocytes, but it also can infect other types of cells, such as NK, T, and epithelial cells.
EBV has both a latent state and a lytic replicative cycle in the nuclei of EBV-infected cells (1). During the latent phase of the EBV life cycle, the EBV genome is maintained as a circular plasmid molecule, which is amplified once in S phase by cellular DNA replication machinery. However, a small percentage of infected cells switch from the latent stage into the lytic cycle, which is triggered by the expression of an immediate-early protein, BZLF1, to produce progeny viruses. This type of activation contributes to the development and maintenance of human cancers (2, 3), suggesting that the EBV switching mechanism is also a key determinant of EBV pathogenesis. After induction of productive viral replication, the EBV genome is amplified 100- to 1,000-fold by viral replication machinery composed of BALF5 DNA polymerase (Pol), BMRF1 polymerase processivity factor, BALF2 single-stranded DNA-binding protein, and BBLF4-BSLF1-BBLF2/BBLF3 (BBLF2/3) helicase-primase complex via a rolling-circle mechanism in discrete sites in nuclei, called replication compartments (4, 5). BALF5 possesses intrinsic DNA polymerase and 3′-to-5′ exonuclease activities (6) and forms a complex with the BMRF1 polymerase accessory protein to exhibit high polymerase processivity (7). The DNA polymerase and exonuclease domains are highly conserved among a variety of DNA polymerases (8, 9). Unlike the case of the eukaryotic chromosomal replication apparatus, the EBV DNA Pol holoenzyme is used in the synthesis of both leading and lagging strands at the replication fork (6).
The peptidyl-prolyl bond has a low rate of spontaneous cis-trans isomerization. This is frequently a limiting step for protein folding and usually requires an isomerase to catalyze the process. Phosphorylation on a serine or threonine residue preceding proline (pSer/Thr-Pro) is a key regulatory mechanism, and the conformation of certain phosphorylated Ser/Thr-Pro bonds is regulated specifically by the prolyl isomerase Pin1 (10). The WW domain of Pin1 binds only to specific pSer/Thr-Pro motifs, which are isomerized by the peptidyl-prolyl isomerase (PPIase) domain to induce conformational changes in proteins (11). In this way Pin1 regulates various protein functions, including protein stability, catalytic activity, phosphorylation status, protein-protein interactions, and/or subcellular localization (11–14). Pin1 works in concert with protein kinases that phosphorylate Ser/Thr-Pro motifs, and protein phosphatases, in turn, can also be regulators of the process (15). Pin1 has a pivotal role in a variety of biological processes such as cell cycle control (16), and its deregulation contributes to various pathological conditions, most notably cancer (11, 12, 17, 18). Pin1 is overexpressed in various human cancers, contributing to centrosome amplification, chromosome instability, and tumor development in vitro and in vivo and correlating with poor clinical outcomes (10, 19–22). In contrast, inhibition of Pin1 suppresses tumorigenesis in vitro (23) and prevents cancer development induced by overexpression of oncogenes such as Neu or Ras (24) or by knockout of tumor suppressors such as p53 (25) in mice.
Thus, Pin1 has key roles in control of cellular functions. However, its significance for EBV replication has yet to be clarified in detail. In this study, we show that Pin1 interacts with EBV DNA polymerase BALF5 and modulates productive viral DNA replication. Because there is a very limited number of anti-EBV drugs developed or being developed to date, including acyclic nucleoside analogs, such as acyclovir, and kinase inhibitors, such as maribavir (26, 27), a search for an effective molecular target has been needed. Pin1 may be a potential target for development of novel antiviral drugs.
MATERIALS AND METHODS
Cell culture and reagents.
HEK293T and HEK293 EBV-bacterial artificial chromosome (BAC) cells were maintained in Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% fetal bovine serum. B95-8 cells and an EBV-negative cell clone derived from Akata cells [Akata(−)] were cultured in RPMI medium supplemented with 10% fetal bovine serum. To induce lytic EBV replication, tetradecanoyl phorbol acetate (TPA), A23187, and sodium butyrate were added to the culture medium at final concentrations of 20 ng/ml, 1 μM, and 5 mM, respectively.
Antibodies.
Rabbit anti-BZLF1, -BMRF1, -BALF2, and -BALF5 antibodies were as reported previously (28). An anti-EBV EA-D-p52/50 (BMRF1 gene product) protein-specific mouse monoclonal antibody, clone R3, was purchased from Chemicon Inc. Anti-Pin1 (H-123 and G-8) and anti-α/β-tubulin (2148) antibodies were purchased from Santa Cruz Biotechnology, Inc., and Cell Signaling, respectively. Horseradish peroxidase (HRP)-linked goat antibodies to rabbit IgG were from Amersham Biosciences.
shRNA and siRNA.
Knockdown of Pin1 with short hairpin (shRNA) was carried out as described previously (29). As a control, we targeted the luciferase gene (designated shluc). Oligonucleotide sequences for the Pin1 shRNA (shPin1) were 5′-GATCCGCCGAGTGTACTACTTCAATTCAAGAGATTGAAGTAGTACACTCGGCTTTTTTAT-3′ (shPin1 for) and 5′-CGATAAAAAAGCCGAGTGTACTACTTCAATCTCTTGAATTGAAGTAGTACACTCGGCG-3′ (shPin1 rev), and the sequence for shluc was as noted previously (29). Duplexes of 21-nucleotide small interfering RNAs (siRNAs) were synthesized and annealed (Gene Design, Inc.). The sense and antisense sequences of the duplex were 5′-GCCAUUUGAAGACGCCUCGdTdT-3′ and 5′-CGAGGCGUCUUCAAAUGGCdTdT-3′ for Pin1 and 5′-GCAGAGCUGGUUUAGUGAAdTdT-3′ and 5′-UUCACUAAACCAGCUCUGCdTdT-3′ for the control siRNA.
Measurement of the viral genome by qRT-PCR.
Cells were harvested at the time indicated in the figure legends and lysed with 200 μl of PCR lysis buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 0.001% Triton X-100, and 0.001% SDS). After treatment with 25 μg of proteinase K at 50°C for 2 h, samples were boiled at 95°C for 10 min. Quantitative real-time PCR (qRT-PCR) was performed in 10 μl of solution containing 1 μM each forward and reverse primer, 5 μl of FastStart Universal Probe Master (Rox) (containing 6-carboxy-X-rhodamine [ROX] dye; Roche Applied Science), 0.5 μl of eukaryotic 18S rRNA endogenous control (Applied Biosystems), and 1 μl of prepared sample DNA in PCR lysis buffer. The intensity of ROX dye was used to compensate for volume fluctuations among the tubes. PCR included 2 min at 50°C and 10 min at 95°C and then 40 cycles at 95°C for 15 s, followed by 1 min at 60°C. Immediately after the PCR, we carried out dissociation curve analysis and confirmed the specificity of each PCR product. A standard curve was constructed using serial dilutions of DNA and was used to quantify the amount of DNA. Primers and a probe for detection of the viral genome were designed using Primer Express (Applied Biosystems) within the BALF2-coding region. The sequences were as follows: 5′-GCCCGTCCGGTTGTCA-3′ (forward primer), 5′-AATATCTGGTTGTTGCCGTTGA-3′ (reverse primer), and 5′-FAM-CTGCCAGTGACCATCAACAAGTACACGG-TAMRA-3′ (probe; where FAM is 6-carboxyfluorescein and TAMRA is tetramethyl rhodamine).
GST pulldown assays.
For bacterial expression of glutathione S-transferase (GST)-tagged Pin1 (wild type [WT] or the W34A mutant), Escherichia coli strain DH5α was transformed with the pGEX expression vector for each protein (10). Expression of GST fusion proteins was induced by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG; 0.5 mM), followed by incubation at 25°C for 4 h. GST pulldown assays were conducted as described previously (10). In brief, B95-8 or HEK293 cell proteins were lysed in GST lysis buffer at 4°C. After sonication and centrifugation (at 20,000 × g for 10 min at 4°C), the supernatant was preincubated with glutathione-Sepharose beads (GE Healthcare) for 30 min at 4°C. Afterwards, the supernatant was mixed with 50 μg of GST fusion protein and glutathione beads for 1 h at 4°C with rotation. The beads were then washed with GST lysis buffer five times and subjected to immunoblotting.
Transfection and IP.
Cells were transfected with appropriate plasmids using Lipofectamine 2000 reagent (Invitrogen) or by electroporation using a Microporator (Disital Bio). The total amounts of plasmid DNAs were standardized by addition of an empty vector. For immunoprecipitation (IP), cells were solubilized in 200 μl of 0.5% Nonidet P-40 buffer (10 mM Tris-HCl [pH 7.5], 100 mM NaCl, 0.5% Nonidet P-40, and protease and phosphatase inhibitor mixture). Cell extracts were then diluted with 800 μl of lysis buffer and precleared with protein G-Sepharose (GE Healthcare). Supernatants were then mixed with protein G-Sepharose and antibody and then incubated at 4°C for 4 h with rotation. Immunocomplexes were washed five times with the same buffer. Samples were subjected to SDS-PAGE, followed by immunoblotting with the antibodies indicated in the figures and figure legends. We used TrueBlot anti-rabbit IgG HRP-conjugated antibodies (eBioscience) as the secondary antibody to eliminate the immunoglobulin heavy chain/light chain-specific band.
BALF5 expression plasmid and mutagenesis.
The expression vector for BALF5 was made by inserting the BALF5 open reading frame into the EcoRI/HindIII site of pcDNA3.1 (Invitrogen). Mutant vectors were generated by a PCR-based method using the following primers: TTCCATGTCTACGACATACTC (BALF5Δ1 For), ACACTTGGGAATGAGACGC (BALF5Δ1 Rev), AAGGTCACGCGCCGTTCCATT (BALF5Δ2 For), GAGGACTGCAAACTCCACGTC (BALF5Δ2 Rev), AGAAGAGCACAGGCTAGCC (BALF5Δ3 For), TTGTAGAATCCGGACAGGGG (BALF5Δ3 Rev), CTTCGAGTCATCTACGGGGAC (BALF5Δ4 For), GATGGAGAGGCAGGGAAAG (BALF5Δ4 Rev), GCCCCCTGCCGGGTCTCGG (BALF5-T178A For), and CCTGCGGTCGAAGGTGCTGG (BALF5-T178A Rev). The DNA sequence of each vector was confirmed by DNA sequencing using the following primers: GCATCGTCATCAAGCTACTG (BALF5-1), AGCTCGAGTACGACTGTGAG (BALF5-2), CACATCTACAGCATCAACCC (BALF5-3), GATCCGCGTGTTCTCCTGC (BALF5-4), CCTTCTTGGCTAGTCTGTTG (BALF5-5), and TCCTGCCTGATGCTGATTAC (BALF5-6).
Genetic manipulation of EBV-BAC DNA.
EBV-BAC DNA was provided by W. Hammerschmidt (30). Homologous recombination was undertaken in E. coli as described previously (28, 29, 31) with the following oligonucleotide primers: 5′-TGTGTGAACGTGTTTGGGCAGCAGGCCTACTTCTACGCCAGCGCGCCTCAGGGTCTGGACGGCCTGGTGATGATGGCGGGATC-3′ (Neo/stFor), 5′-CGCGTGGCATCCACGTTGGCCTCAAAGATCCGACACCCGTGCTTGTCTTGCCACGTGTCAGAAGAACTCGTCAAGAAGG-3′ (Neo/stRev), 5′-AAGCCCTCTGGACTTCCATG-3′ (Transfer vector For), and 5′-CATTGTCCAGGACAAAGCGG-3′ (Transfer vector Rev). Electroporation was performed using a Gene Pulser III (Bio-Rad), and purification of EBV-BAC DNA was achieved with NucleoBond Bac100 (Macherey-Nagel, Germany).
RESULTS
Productive EBV DNA replication is strongly suppressed by knockdown of Pin1.
To determine whether Pin1 might influence productive EBV replication, we first carried out knockdown experiments. In HEK293 EBV-BAC cells featuring EBV latent infection, Pin1 expression was suppressed by shRNA (shPin1) transduction (Fig. 1A). An shRNA against the luciferase gene served as the control. Cells were transfected with empty vector pcDNA3 or pBZLF1, an expression vector for BZLF1, the molecular switch from latent to lytic infection of EBV. After 24 h, levels of viral and cellular proteins were examined by immunoblotting (Fig. 1A). In the control HEK293 EBV-BAC shluc cells, BZLF1 transfection induced expression of viral genes such as BALF5, BALF2, and BMRF1, as expected, and knockdown of Pin1 (shPin1) had little effect on the expression levels.
Fig 1.
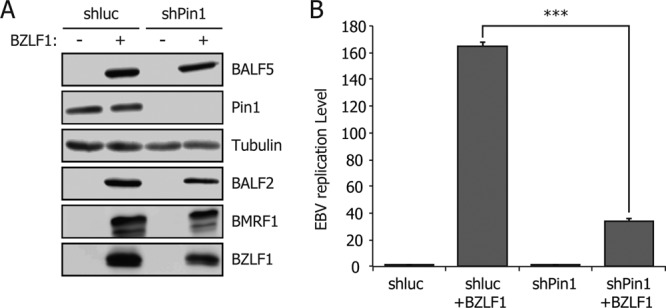
Knockdown of Pin1 decreases the level of EBV viral replication. (A) HEK293 EBV-BAC cells, transduced with control shRNA (shluc) or shRNA for Pin1 (shPin1), were transfected with 50 ng of BZLF1 expression vector or empty vector (pcDNA3). After 24 h, aliquots of cells were harvested and subjected to immunoblotting with the indicated antibodies. (B) Remaining cells transfected in panel A were subjected to qRT-PCR assays 60 h after transfection. The amount of EBV viral DNA was quantified and standardized with an 18S ribosome probe. Each bar represents the mean and standard deviation of three independent transfections and quantifications. ***, P < 0.002.
We next checked the levels of viral DNA by qRT-PCR (Fig. 1B). The amount of synthesized EBV DNA was drastically and significantly reduced in Pin1-depleted cells upon induction compared to that in control cells, while levels of intrinsic, latent EBV genome copy numbers were comparable (Fig. 1B). Similar results were also obtained in lymphocytes (data not shown).
Pin1 interacts with EBV DNA polymerase BALF5.
Since Pin1 was shown to contribute to EBV lytic replication (Fig. 1B), we next examined if certain EBV proteins could interact with Pin1 by GST pulldown assay. Whole-cell lysate from B95-8 cells treated with TPA-A23187-butyrate was incubated with purified GST-Pin1, GST-Pin1 W34A or GST alone expressed in bacteria. The W34A mutant of Pin1 served as a negative control because it cannot bind to the Ser/Thr-Pro motif of the substrates due to the mutation in its WW domain. We found that the viral DNA polymerase catalytic subunit, BALF5, was specifically and repeatedly coprecipitated with GST-Pin1 but not with GST alone or GST-Pin1 W34A (Fig. 2A). Other than BALF5, viral factors, such as BZLF1, BMRF1, BBLF4, BBLF2/3, and BSLF1, were not copurified with Pin1 in our GST pulldown assays (data not shown).
Fig 2.

Pin1 interacts with EBV DNA polymerase BALF5. (A) GST-Pin1 binds to BALF5. Proteins from B95-8 cells, induced with TPA, A23187, and sodium butyrate for 24 h, were harvested and lysed in GST lysis buffer. GST pulldown assays were carried out using GST, GST-Pin1, or GST-Pin1 W34A. Pin1 W34A cannot bind with target proteins because of the mutation in its WW domain. (B) Phosphorylation-dependent association of Pin1. A GST pulldown assay was carried out as described for panel A except that the B95-8 cell lysate was incubated with lambda protein phosphatase (PP) (New England BioLabs) for 30 min at 30°C as indicated on the figure. (C) Immunoprecipitation assays confirmed the interaction. Cell proteins from lytic B95-8 lysate were subjected to immunoprecipitation using anti-BALF5 antibodies or normal IgG. The precipitates were then immunoblotted using anti-BALF5 or -Pin1 antibodies. α, anti. Arrowhead indicates size of Pin1. (D) Exogenously overexpressed BALF5 can bind to Pin1. Cell proteins from HEK293T cells transfected with BALF5 expression vector were subjected to GST pulldown assay. IB, immunoblotting; CBB, Coomassie brilliant blue. k, kilodaltons.
To test if the interaction was phosphorylation dependent, an aliquot of the lysate was treated with lambda phosphatase while the rest was left untreated (Fig. 2B). Dephosphorylation by the phosphatase diminished the interaction (Fig. 2B), suggesting that the two proteins associate in a phosphorylation-dependent manner (Fig. 2B).
Immunoprecipitation assays were next conducted in order to confirm the association between endogenous Pin1 and BALF5 in B95-8 cells. Pin1 was coimmunoprecipitated with BALF5, as expected (Fig. 2C).
For the experiments shown in Fig. 2A to C, we collected viral proteins from EBV-positive B95-8 cells after induction of lytic replication and demonstrated that Pin1 interacts with BALF5 in a phosphorylation-dependent manner. Since EBV encodes a protein kinase, BGLF4, we then examined whether phosphorylation of BALF5 by the viral kinase BGLF4 was necessary for the Pin1-BALF5 association. To this end, EBV-negative HEK293T cells were transfected with a expression vector plasmid for BALF5, and at 24 h posttransfection, GST pulldown assays were carried out using the lysate (Fig. 2D). Because the interaction between BALF5 and Pin1 was reproduced in an overexpression system, the viral kinase is apparently not a prerequisite for the interaction, and cellular kinases, such as mitogen-activated protein kinases (MAPKs) or cyclin-dependent kinases (CDKs), may be sufficient to mediate the association (17).
Identification of the Ser/Thr-Pro motif in BALF5 required for association with Pin1.
Since phospho-Ser/Thr-Pro is the binding motif for Pin1, we searched for its presence in BALF5 protein. As shown in Fig. 3A (top panel), BALF5 has seven Ser/Thr-Pro motifs. In order to map the domain in BALF5 important for the interaction, we generated a series of BALF5 truncation mutants (Fig. 3A). There are four Ser-Pro (Ser90, Ser578, Ser726, and Ser736) and three Thr-Pro (Thr35, Thr178, and Thr605) motifs in the EBV BALF5 protein. We prepared mutants designated BALF5Δ1, BALF5Δ2, BALF5Δ3, and BALF5Δ4, featuring deletion of amino acids 34 to 93, 167 to 185, 578 to 607, and 704 to 748, respectively. HEK293T cells were transfected with BALF5 wild type (WT) or its mutants, and at 24 h posttransfection whole-cell extracts were subjected to GST pulldown assays. Among the truncation mutants, BALF5Δ2 exhibited attenuated association with GST-Pin1, whereas other mutants showed comparable binding ability with the wild type (Fig. 3B). As the truncated BALF5Δ2 mutant lacks BALF5 Thr178-Pro, the motif is suggested to be a potential candidate Pin1 binding site. Pulldown of BALF5Δ1 with GST-Pin1 appeared strong, but we believe the result was just an artifact simply because the input level of the protein was also high.
Fig 3.

Pin1 interacts with BALF5 Thr178. (A) Scheme of BALF5 truncated mutants, featuring deletion of amino acids 34 to 93 (BALF5Δ1), 167 to 185 (BALF5Δ2), 578 to 607 (BALF5Δ3), and 704 to 748 (BALF5Δ4). Primers used for constructing these mutants are listed in Materials and Methods. (B) Cell proteins lysed from HEK293T cells, transfected with the WT BALF5 expression vector or its derivatives shown in panel A, were subjected to GST pulldown assay.
Considering BALF5 T178 as the Pin1 binding motif, we next constructed an alanine substitution mutant, designated BALF5 T178A. BALF5 WT or BALF5 T178A was expressed in HEK293T cells, and then cell lysates were subjected to GST pulldown assays (Fig. 4A). Levels of copurified BALF5 T178A with GST-Pin1 were markedly lower than with the wild type, as expected (Fig. 4A). Furthermore, immunoprecipitation assays also confirmed that the T178A mutation diminished the BALF5 association with Pin1 (Fig. 4B). Therefore, we conclude that BALF5 Thr178-Pro is the Pin1 binding target.
Fig 4.
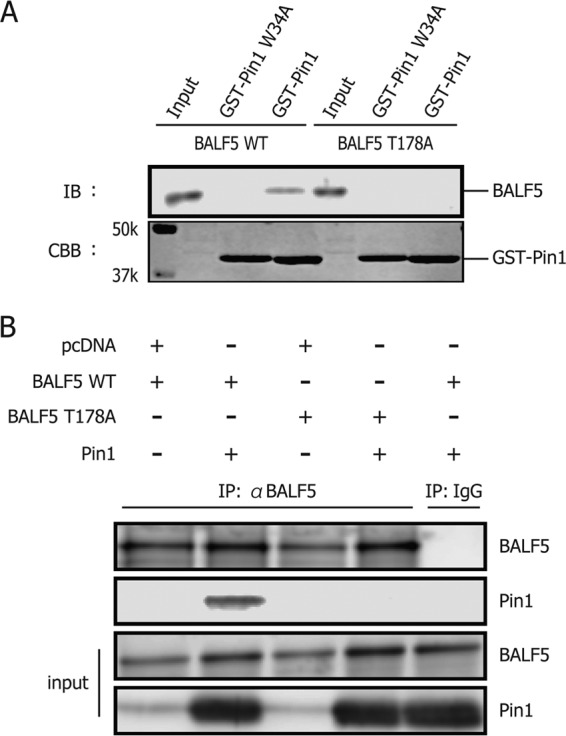
BALF5 T178A diminishes binding ability to Pin1. (A) Cell lysates from HEK293T cells, transfected with BALF5 wild type or T178A, were subjected to GST pulldown assays. (B) HEK293T cells were transfected with expression vectors for BALF5 wild type or T178A, with or without the expression vector for Pin1, as indicated. Cell proteins were lysed and subjected to immunoprecipitation using anti-BALF5 antibodies or normal IgG. The precipitates were then immunoblotted using anti-BALF5 or -Pin1 antibodies.
BALF5 T178 is important for viral DNA polymerase activity.
Our experiments indicated only one Pin1 binding site in the BALF5 amino acid sequence. To further extend and verify the finding, recombinant EBV with BALF5 deletion was prepared. As shown in Fig. 5A, part of the BALF5 sequence encompassing the Pin1 binding site (Thr178) was replaced with a marker cassette (for neomycin resistance and streptomycin sensitivity [Neo/st]). Integrity of the BAC DNA was checked by BamHI digestion, followed by electrophoresis to confirm that the recombinant viruses did not carry obvious deletions or insertions. The BamHI-digested A fragment of EBV-BAC BALF5Δ (Fig. 5B, filled arrowhead) migrated more slowly than that of the wild type (open arrow), as expected, since the Neo/st marker cassette was inserted into the fragment (Fig. 5B).
Fig 5.
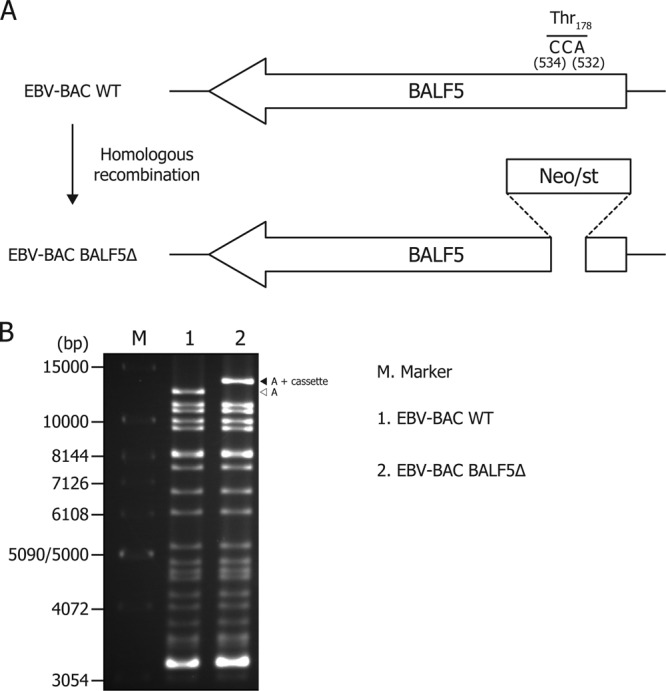
Scheme of EBV-BAC BALF5Δ construction. (A) Schematic arrangement of the recombination of the EBV genome using the neomycin-resistance and streptomycin-sensitivity genes (Neo/st) arranged in tandem. The sequence around the Pin1 binding site of BALF5 (Thr178) was replaced with the Neo/st cassette. (B) Electrophoresis of the recombinant viruses. The recombinant EBV genomes were digested with BamHI and separated in an agarose gel. A, the A fragment of BamHI-digested EBV-BAC.
Recombinant EBV-BAC DNA was introduced into a virus-producing cell line, HEK293, followed by hygromycin selection, to establish cell lines in which the EBV-BAC genome was maintained as an episome. More than 10 cell colonies from each recombinant virus were obtained, and viral protein expression levels in the presence and absence of BZLF1 induction were examined (data not shown). As the result, we obtained BALF5 knockout EBV-BAC cells (EBV-BAC BALF5Δ) which exhibited a typical nature, i.e., viral lytic protein expression was restricted without BZLF1 and efficiently induced by BZLF1.
In the BALF5 knockout cell line, exogenous expression of BZLF1 led to induction of early genes, such as BALF2 and BMRF1, but failed to produce BALF5, in line with expectations (Fig. 6A, BZLF1). Because of the lack of BALF5, the DNA Pol catalytic subunit, the virus could not amplify viral DNA even after induction with BZLF1 (Fig. 6C, BZLF1). Next, in order to compare the efficiencies of complementation, HEK293 EBV-BAC BALF5Δ cells were transfected with either BALF5 WT or BALF5 T178A expression vectors in addition to BZLF1. Exogenous supply of BALF5 WT restored the replication and increased viral DNA levels by 14.6-fold (Fig. 6C, BZLF1+BALF5 WT). On the other hand, the BALF5 T178A mutant increased the viral DNA only 4.54-fold (Fig. 6C, BZLF1+BALF5 T178A), when the mutant BALF5 protein expression level was equivalent to that of the wild type (Fig. 6A and B). In addition, we measured viral particles produced from the cells (Fig. 6D). Culture supernatant from the cells were collected and cocultured with naive Akata(−) cells. Since the recombinant EBVs used here encode green fluorescent protein (GFP), Akata(−) cells infected with EBV become GFP positive. When wild-type BALF5 was transfected with BZLF1, 759 infectious particles were obtained per ml of supernatant on average. Although the DNA replication levels (Fig. 6C) and viral yield (Fig. 6D) were slightly weak, we assume that this experimental condition is still physiologically relevant. The T178A mutation of BALF5 caused a slight decrease in the viral yield (Fig. 6D). These results indicate that the T178 residue of BALF5 is needed for efficient lytic replication of the EBV genome and suggest that optimal activity of the DNA polymerase is mediated through the interaction with Pin1.
Fig 6.

Significance of Pin1 binding to BALF5 Thr178 for viral replication. (A) HEK293 EBV-BAC BALF5Δ cells were transfected with 50 ng of BZLF1 and 10 ng of either BALF5 WT or BALF5 T178A expression vector using a Microporator (Digital Bio). Aliquots of cells were harvested at 24 h after transfection and subjected to immunoblotting with indicated antibodies. (B) The samples from the third and fourth lanes of the experiment shown in panel A were diluted and loaded as indicated, followed by immunoblotting with anti-BALF5 and -tubulin antibodies. The remaining cells were harvested at 36 h after transfection and subjected to qRT-PCR (C). Each bar represents the mean and standard deviation for the viral DNA level after normalization, calculated from three independent samples. **, P < 0.005. (D) Culture supernatants from HEK293 EBV-BAC BALF5Δ cells, transfected in the same fashion as described for panel C and followed by 3 days of incubation, were collected and used to infect naive Akata(−) cells. Viral load in the medium was determined by fluorescence-activated cell sorting analysis and is shown as the number of GFP-positive units per milliliter. (E and F) HEK293 EBV-BAC BALF5Δ cells were transfected with siRNA against Pin1 (siPin1) or a control siRNA (siControl). After 2 days, the cells were then transfected with expression vectors for BZLF1, BALF5 WT, BALF5 T178A, and/or the empty vector pcDNA, as indicated. Samples were harvested at 24 h after transfection of plasmids and subjected to immunoblotting with the indicated antibodies (E). The remaining cells were harvested at 36 h after transfection of plasmids for qRT-PCR (F).
To further verify the conclusion, we lastly tested if knockdown of Pin1 could influence EBV replication under this condition, too. Transfection of siRNA to HEK293 EBV-BAC BALF5Δ cells caused a considerable decrease in Pin1 levels when levels of other markers like BALF5, BZLF1, and tubulin remained unchanged (Fig. 6E). Viral replication levels this time reached 17.8-fold with BZLF1 plus BALF5 (wild type) and control siRNA, but Pin1 knockdown resulted in only a 12.2-fold increase (Fig. 6F). Although the difference in the results shown in Fig. 6F was less remarkable than the difference shown in Fig. 1B, we speculate that this reduction level by siPin1 is convincing enough because knockdown of Pin1 here (Fig. 6E) was not as complete as that in the experiment shown in Fig. 1A.
DISCUSSION
In this study, we obtained evidence for the first time of an interaction between the EBV lytic protein BALF5 and the cellular regulator Pin1. The results documented show clear involvement of the Pin1 protein in efficient EBV lytic replication. Initially, Pin1 was identified as a key regulator from knockdown experiments since silencing of Pin1 resulted in significant suppression of the viral replication level (Fig. 1).
We first speculated that knockdown of Pin1 might directly influence viral lytic gene transcription because Pin1 reportedly regulates RNA polymerase II activity (32–34). Pin1 also impacts cellular signaling through factors such as Akt (35), c-Jun (36, 37), and p65/NF-κB (38). In fact, shPin1 might have slightly decreased EBV early gene expression (Fig. 1A) although we assume the levels are comparable.
Then, we found that the EBV DNA polymerase BALF5 interacted directly with Pin1, as demonstrated by GST pulldown (Fig. 2A), and that the interaction is dependent on phosphorylation of BALF5 Thr178 (Fig. 2B and 4). Results of immunoprecipitation assays also supported this conclusion (Fig. 2C and 4B). Although the association of Pin1 with BALF5 (Fig. 2 to 4) and its influence on viral DNA replication (Fig. 6) are clear, we cannot preclude the possibility that there may be other Pin1 substrates besides BALF5 that affect EBV genome amplification as Pin1 has a large number of substrates (12). Thus, the search for other Pin1 targets is still under way. In addition, it must also be noted that because there are a number of cellular target proteins that are up-/downregulated by Pin1, we cannot ignore the possibility that some of these cellular target proteins may cause even adverse effects on EBV replication. But as a whole, Pin1 clearly upregulates EBV lytic replication (Fig. 1), at least partly through the action of BALF5 (Fig. 6).
In this paper, we identified Pin1 interaction with the EBV DNA polymerase BALF5 enzyme at Thr178. The polymerase contains conserved domains, including polymerase catalytic and exonuclease domains at the C terminus, but the sequence around Thr178 is not conserved. We have no concrete idea of how Pin1 modulates BALF5 function, but it is possible that the N-terminal domain of BALF5, including Thr178, may somehow regulate its C-terminal functional domains by altering the structure of the protein in a subtle way. Further studies are required to gain an understanding of the molecular mechanism of how Pin1 regulates BALF5 enzymatic activity.
To our knowledge, EBV BALF5 is the only Pin1 target so far identified, not just in EBV but among all herpesvirus genes. Milbradt and others reported data suggesting that Pin1 may be involved in reorganization of nuclear lamin after phosphorylation by the human cytomegalovirus (HCMV)-encoded protein kinase UL97 (39). Elsewhere, Peloponese and others showed that Pin1 binds to the Tax protein of human T cell leukemia virus 1 and regulates Tax-induced NF-κB activation (40). Jeong and others demonstrated that Pin1 prolongs the Tax protein half-life by suppressing ubiquitination and proteasome-dependent degradation (41). Furthermore, Pin1 increased stability of the hepatitis B virus oncoprotein X and enhanced transactivation and cell proliferation (42). Hepatitis C virus replication is regulated by Pin1, probably through binding to NS5A/NS5B (43). Human immunodeficiency virus type 1 genome integration (44) and capsid uncoating (45) are also regulated by Pin1. Although exact roles of Pin1 in controlling herpesvirus replication remain elusive, it has already been proposed as an important modulator of viral proteins and a unique target for antiviral therapy (46).
A number of studies suggest that Pin1 has a role in tumorigenesis as it is overexpressed in a number of human cancers (47, 48). Since we determined that Pin1 is a positive regulator of EBV lytic replication, EBV-positive cancer tissue may be an efficient site for producing novel virus particles. While further studies are required to clarify the underlying mechanisms, Pin1 clearly warrants attention as a novel target for potential antiviral/cancer drug development.
ACKNOWLEDGMENTS
We thank W. Hammerschmidt, H. J. Delecluse, and S. Tsuzuki for providing the EBV-BAC system, HEK293 cells, and shRNA technology, respectively. We also express our appreciation to C. Noda and T. Gamano for preliminary experiments and technical assistance.
This work was supported by grants-in-aid for Scientific Research from the Ministry of Education, Science, Sports, Culture and Technology (numbers 23390118 and 23114512 to T.T. and numbers 22790448 and 24590566 to T.M.), the Ministry of Health, Labor and Welfare (to T.T. and H.K.), and partly by the Takeda Science Foundation (to T.T. and T.M.).
Footnotes
Published ahead of print 5 December 2012
REFERENCES
- 1. Tsurumi T, Fujita M, Kudoh A. 2005. Latent and lytic Epstein-Barr virus replication strategies. Rev. Med. Virol. 15:3–15 [DOI] [PubMed] [Google Scholar]
- 2. Feng WH, Cohen JI, Fischer S, Li L, Sneller M, Goldbach-Mansky R, Raab-Traub N, Delecluse HJ, Kenney SC. 2004. Reactivation of latent Epstein-Barr virus by methotrexate: a potential contributor to methotrexate-associated lymphomas. J. Natl. Cancer Inst. 96:1691–1702 [DOI] [PubMed] [Google Scholar]
- 3. Joab I, Nicolas JC, Schwaab G, de-The G, Clausse B, Perricaudet M, Zeng Y. 1991. Detection of anti-Epstein-Barr-virus transactivator (ZEBRA) antibodies in sera from patients with nasopharyngeal carcinoma. Int. J. Cancer 48:647–649 [DOI] [PubMed] [Google Scholar]
- 4. Daikoku T, Kudoh A, Fujita M, Sugaya Y, Isomura H, Shirata N, Tsurumi T. 2005. Architecture of replication compartments formed during Epstein-Barr virus lytic replication. J. Virol. 79:3409–3418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fixman ED, Hayward GS, Hayward SD. 1995. Replication of Epstein-Barr virus oriLyt: lack of a dedicated virally encoded origin-binding protein and dependence on Zta in cotransfection assays. J. Virol. 69:2998–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsurumi T, Daikoku T, Nishiyama Y. 1994. Further characterization of the interaction between the Epstein-Barr virus DNA polymerase catalytic subunit and its accessory subunit with regard to the 3′-to-5′ exonucleolytic activity and stability of initiation complex at primer terminus. J. Virol. 68:3354–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsurumi T. 1993. Purification and characterization of the DNA-binding activity of the Epstein-Barr virus DNA polymerase accessory protein BMRF1 gene products, as expressed in insect cells by using the baculovirus system. J. Virol. 67:1681–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bernad A, Blanco L, Lazaro JM, Martin G, Salas M. 1989. A conserved 3′–5′ exonuclease active site in prokaryotic and eukaryotic DNA polymerases. Cell 59:219–228 [DOI] [PubMed] [Google Scholar]
- 9. Bernad A, Zaballos A, Salas M, Blanco L. 1987. Structural and functional relationships between prokaryotic and eukaryotic DNA polymerases. EMBO J. 6:4219–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP. 2001. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat. Cell Biol. 3:793–801 [DOI] [PubMed] [Google Scholar]
- 11. Lu KP, Zhou XZ. 2007. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8:904–916 [DOI] [PubMed] [Google Scholar]
- 12. Liou YC, Zhou XZ, Lu KP. 2011. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 36:501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, Zhou XZ, Lu KP. 2012. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell 149:232–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wulf G, Finn G, Suizu F, Lu KP. 2005. Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nat. Cell Biol. 7:435–441 [DOI] [PubMed] [Google Scholar]
- 15. Yeh ES, Means AR. 2007. PIN1, the cell cycle and cancer. Nat. Rev. Cancer 7:381–388 [DOI] [PubMed] [Google Scholar]
- 16. Winkler KE, Swenson KI, Kornbluth S, Means AR. 2000. Requirement of the prolyl isomerase Pin1 for the replication checkpoint. Science 287:1644–1647 [DOI] [PubMed] [Google Scholar]
- 17. Lu KP. 2004. Pinning down cell signaling, cancer and Alzheimer's disease. Trends Biochem. Sci. 29:200–209 [DOI] [PubMed] [Google Scholar]
- 18. Tun-Kyi A, Finn G, Greenwood A, Nowak M, Lee TH, Asara JM, Tsokos GC, Fitzgerald K, Israel E, Li X, Exley M, Nicholson LK, Lu KP. 2011. Essential role for the prolyl isomerase Pin1 in Toll-like receptor signaling and type I interferon-mediated immunity. Nat. Immunol. 12:733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ayala G, Wang D, Wulf G, Frolov A, Li R, Sowadski J, Wheeler TM, Lu KP, Bao L. 2003. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 63:6244–6251 [PubMed] [Google Scholar]
- 20. Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang DG. 2004. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am. J. Pathol. 164:1727–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee KY, Lee JW, Nam HJ, Shim JH, Song Y, Kang KW. 2011. PI3-kinase/p38 kinase-dependent E2F1 activation is critical for Pin1 induction in tamoxifen-resistant breast cancer cells. Mol. Cells 32:107–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suizu F, Ryo A, Wulf G, Lim J, Lu KP. 2006. Pin1 regulates centrosome duplication, and its overexpression induces centrosome amplification, chromosome instability, and oncogenesis. Mol. Cell. Biol. 26:1463–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ryo A, Liou YC, Wulf G, Nakamura M, Lee SW, Lu KP. 2002. PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol. Cell. Biol. 22:5281–5295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wulf G, Garg P, Liou YC, Iglehart D, Lu KP. 2004. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 23:3397–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takahashi K, Akiyama H, Shimazaki K, Uchida C, Akiyama-Okunuki H, Tomita M, Fukumoto M, Uchida T. 2007. Ablation of a peptidyl prolyl isomerase Pin1 from p53-null mice accelerated thymic hyperplasia by increasing the level of the intracellular form of Notch1. Oncogene 26:3835–3845 [DOI] [PubMed] [Google Scholar]
- 26. Gershburg E, Hong K, Pagano JS. 2004. Effects of maribavir and selected indolocarbazoles on Epstein-Barr virus protein kinase BGLF4 and on viral lytic replication. Antimicrob. Agents Chemother. 48:1900–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang FZ, Roy D, Gershburg E, Whitehurst CB, Dittmer DP, Pagano JS. 2009. Maribavir inhibits Epstein-Barr virus transcription in addition to viral DNA replication. J. Virol. 83:12108–12117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murata T, Isomura H, Yamashita Y, Toyama S, Sato Y, Nakayama S, Kudoh A, Iwahori S, Kanda T, Tsurumi T. 2009. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology 389:75–81 [DOI] [PubMed] [Google Scholar]
- 29. Noda C, Murata T, Kanda T, Yoshiyama H, Sugimoto A, Kawashima D, Saito S, Isomura H, Tsurumi T. 2011. Identification and characterization of CCAAT enhancer-binding protein (C/EBP) as a transcriptional activator for Epstein-Barr virus oncogene latent membrane protein 1. J. Biol. Chem. 286:42524–42533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. U. S. A. 95:8245–8250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Isomura H, Stinski MF, Kudoh A, Murata T, Nakayama S, Sato Y, Iwahori S, Tsurumi T. 2008. Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J. Virol. 82:1638–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Palancade B, Marshall NF, Tremeau-Bravard A, Bensaude O, Dahmus ME, Dubois MF. 2004. Dephosphorylation of RNA polymerase II by CTD-phosphatase FCP1 is inhibited by phospho-CTD associating proteins. J. Mol. Biol. 335:415–424 [DOI] [PubMed] [Google Scholar]
- 33. Xu YX, Hirose Y, Zhou XZ, Lu KP, Manley JL. 2003. Pin1 modulates the structure and function of human RNA polymerase II. Genes Dev. 17:2765–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu YX, Manley JL. 2007. Pin1 modulates RNA polymerase II activity during the transcription cycle. Genes Dev. 21:2950–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liao Y, Wei Y, Zhou X, Yang JY, Dai C, Chen YJ, Agarwal NK, Sarbassov D, Shi D, Yu D, Hung MC. 2009. Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene 28:2436–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pulikkan JA, Dengler V, Peer Zada AA, Kawasaki A, Geletu M, Pasalic Z, Bohlander SK, Ryo A, Tenen DG, Behre G. 2010. Elevated PIN1 expression by C/EBPα-p30 blocks C/EBPα-induced granulocytic differentiation through c-Jun in AML. Leukemia 24:914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, Lu KP. 2001. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 20:3459–3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP. 2003. Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 12:1413–1426 [DOI] [PubMed] [Google Scholar]
- 39. Milbradt J, Webel R, Auerochs S, Sticht H, Marschall M. 2010. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 285:13979–13989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peloponese JM, Jr, Yasunaga J, Kinjo T, Watashi K, Jeang KT. 2009. Peptidylproline cis-trans-isomerase Pin1 interacts with human T-cell leukemia virus type 1 Tax and modulates its activation of NF-κB. J. Virol. 83:3238–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jeong SJ, Ryo A, Yamamoto N. 2009. The prolyl isomerase Pin1 stabilizes the human T-cell leukemia virus type 1 (HTLV-1) Tax oncoprotein and promotes malignant transformation. Biochem. Biophys. Res. Commun. 381:294–299 [DOI] [PubMed] [Google Scholar]
- 42. Pang R, Lee TK, Poon RT, Fan ST, Wong KB, Kwong YL, Tse E. 2007. Pin1 interacts with a specific serine-proline motif of hepatitis B virus X-protein to enhance hepatocarcinogenesis. Gastroenterology 132:1088–1103 [DOI] [PubMed] [Google Scholar]
- 43. Lim YS, Tran HT, Park SJ, Yim SA, Hwang SB. 2011. Peptidyl-prolyl isomerase Pin1 is a cellular factor required for hepatitis C virus propagation. J. Virol. 85:8777–8788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Manganaro L, Lusic M, Gutierrez MI, Cereseto A, Del Sal G, Giacca M. 2010. Concerted action of cellular JNK and Pin1 restricts HIV-1 genome integration to activated CD4+ T lymphocytes. Nat. Med. 16:329–333 [DOI] [PubMed] [Google Scholar]
- 45. Misumi S, Inoue M, Dochi T, Kishimoto N, Hasegawa N, Takamune N, Shoji S. 2010. Uncoating of human immunodeficiency virus type 1 requires prolyl isomerase Pin1. J. Biol. Chem. 285:25185–25195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kojima Y, Ryo A. 2010. Pinning down viral proteins: a new prototype for virus-host cell interaction. Front. Microbiol. 1:107 doi:10.3389/fmicb.2010.00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ryo A, Liou YC, Lu KP, Wulf G. 2003. Prolyl isomerase Pin1: a catalyst for oncogenesis and a potential therapeutic target in cancer. J. Cell Sci. 116:773–783 [DOI] [PubMed] [Google Scholar]
- 48. Ryo A, Uemura H, Ishiguro H, Saitoh T, Yamaguchi A, Perrem K, Kubota Y, Lu KP, Aoki I. 2005. Stable suppression of tumorigenicity by Pin1-targeted RNA interference in prostate cancer. Clin. Cancer Res. 11:7523–7531 [DOI] [PubMed] [Google Scholar]