

Abstract
Venezuelan equine encephalitis virus (VEEV) is a reemerging virus that causes a severe and often fatal disease in equids and humans. In spite of a continuous public health threat, to date, no vaccines or antiviral drugs have been developed for human use. Experimental vaccines demonstrate either poor efficiency or severe adverse effects. In this study, we developed a new strategy of alphavirus modification aimed at making these viruses capable of replication and efficient induction of the immune response without causing a progressive infection, which might lead to disease development. To achieve this, we developed a pseudoinfectious virus (PIV) version of VEEV. VEE PIV mimics natural viral infection in that it efficiently replicates its genome, expresses all of the viral structural proteins, and releases viral particles at levels similar to those found in wild-type VEEV-infected cells. However, the mutations introduced into the capsid protein make this protein almost incapable of packaging the PIV genome, and most of the released virions lack genetic material and do not produce a spreading infection. Thus, VEE PIV mimics viral infection in terms of antigen production but is safer due to its inability to incorporate the viral genome into released virions. These genome-free virions are referred to as virus-like particles (VLPs). Importantly, the capsid-specific mutations introduced make the PIV a very strong inducer of the innate immune response and add self-adjuvant characteristics to the designed virus. This unique strategy of virus modification can be applied for vaccine development against other alphaviruses.
INTRODUCTION
The Alphavirus genus of the Togaviridae family contains a wide variety of viruses that circulate on all continents and are transmitted primarily by mosquito vectors between vertebrate hosts (1). This group of positive-strand RNA viruses contains a number of important human and animal pathogens (2). In vertebrates, alphaviruses cause acute infections characterized by high-titer viremia and development of diseases with various severities (3). The New World alphaviruses, such as Venezuelan (VEEV), eastern (EEEV), and western (WEEV) equine encephalitis viruses, represent the most significant public health threat. They are present in the South, Central, and North Americas and cause periodic, extensive equine epizootics and epidemics of encephalitis with frequent lethal outcomes and neurological sequelae (4). VEEV is an infectious agent of particular importance, because it can replicate to very high titers in many commonly used cell lines, is stable in lyophilized form, and is very infectious in aerosol form. These characteristics make VEEV “user friendly,” and it was previously investigated for use as a biological warfare agent.
To date, no effective antivirals have been developed against VEEV, EEEV, or WEEV infections. The currently available live experimental vaccine, the VEEV TC-83 strain, was developed more than 4 decades ago by serial passaging of the virulent, subtype IAB Trinidad donkey (TrD) VEEV strain in guinea pig heart cell cultures (5). Currently, TC-83 is still the only VEEV strain available for vaccination of laboratory workers and military personnel. More than 8,000 humans have been vaccinated with this live vaccine during the past 4 decades (5–7), and nearly 40% of vaccinees developed a disease with some symptoms similar to those normally observed during natural VEEV infection (5). Residual virulence of the TC-83 strain has also been detected in mice. This virus is uniformly lethal for the C3H/HeN mouse strain after intracerebral inoculation and produces a clinical illness lasting for at least 14 days in BALB/c mice (8). The attenuated phenotype of TC-83 relies on two point mutations. The first is located in the 5′-untranslated region (5′UTR) of the genome, and the second is found in the glycoprotein E2 gene (9). Therefore, a high probability of reversion to a more virulent phenotype during replication in vivo exists. Moreover, the ability of VEEV TC-83 to persist in mosquito vectors increases the possibility of reversion and further virus evolution to a more pathogenic phenotype (10). A formalin-inactivated VEEV TC-83 vaccine, C-84 (7), while safer, requires numerous boosters to induce and maintain a detectable level of neutralizing antibodies. Recently, a new promising vaccine candidate was described. The VEEV TrD strain was attenuated by introducing lethal mutations into the E3/E2 furin-specific cleavage site of an infectious cDNA clone, followed by selection of a pseudorevertant containing a second-site suppressor mutation in the E1 glycoprotein (11). The latter virus is attenuated, but residual neurovirulence can be detected in 6-day-old mice (12). The design of chimeric alphaviruses which encode the replicative machinery of Sindbis virus (SINV), a poorly replicating virus in humans, as well as the structural proteins of VEEV, represents another interesting approach (2). However, the possibility of their replication in immunocompromised individuals cannot be ruled out. Thus, both current live attenuated and inactivated VEEV vaccines and vaccine candidates demonstrate significant drawbacks which make their application for human vaccination questionable.
The use of subviral particles (SVPs) or virus-like particles (VLPs) represents a new and promising direction for alphavirus vaccine design. These particles are assembled in the absence of the viral genomic RNA in cells expressing all of the viral structural proteins, produced from DNA-based vectors. Structural proteins of chikungunya virus (CHIKV) form VLPs with the same architecture as that of wild-type (wt) virions, but lacking the viral genome, and thus do not require chemical inactivation before use for vaccination (13). However, production of such VLPs appears to be costly.
In our recent studies, we endeavored to develop a new type of vaccine candidate against flavivirus infections. These vaccine candidates, termed pseudoinfectious viruses (PIVs), combined the safety of inactivated vaccines and the high immunogenicity of live attenuated viruses. Replication of PIV genomes encoding either defective capsid protein or prM proteins led to an efficient release of noninfectious SVPs which were unable to induce a spreading infection (14–17). Viral RNA replication levels and expression of flavivirus nonstructural and structural proteins during intracellular replication of PIV genomes closely mimicked those observed during a natural infection. PIV replication in vivo induced activation of both innate and adaptive immune responses (14, 17). Thus, application of the accumulated knowledge regarding the molecular mechanisms of flavivirus replication and particle formation resulted in development of a new type of vaccine candidate.
Based on our success with flavivirus PIV development, we applied a similar strategy for the design of pseudoinfectious alphaviruses. These pseudoinfectious viruses were also expected to be capable of mimicking natural infection in terms of synthesis, compartmentalization, and presentation of viral antigens. However, they were required to induce only a single round of infection and produce immunogenic VLPs containing packaged PIV genomes at a very low concentration, which was insufficient for developing spreading infection. In this study, we developed VEE PIVs which demonstrated very efficient intracellular viral genome replication and expression of viral proteins. VEE PIVs' glycoproteins were expressed at the plasma membrane and were efficiently released together with the assembled nucleocapsid in VLP form. Replication of VEE PIVs induced a high level of type I interferon (IFN) release and, unlike wt VEEV, did not interfere with induction of interferon-stimulated genes (ISGs). To deliver VEE PIV genomes into cells, we designed in vitro systems for genome packaging into infectious virions and also demonstrated that delivery of VEE PIV genomes as plasmid-encoded cDNA induces their efficient replication and VLP release.
MATERIALS AND METHODS
Cell cultures.
BHK-21 cells were kindly provided by Paul Olivo (Washington University, St. Louis, MO). NIH 3T3 cells were obtained from the American Type Culture Collection (Manassas, VA). These cell lines were maintained at 37°C in alpha minimum essential medium (αMEM) supplemented with 10% fetal bovine serum (FBS) and vitamins.
Plasmid constructs.
Standard recombinant DNA techniques were applied for the construction of plasmids. The modified capsid gene was assembled using oligonucleotides designed to change 26 basic amino acids (aa) at the N terminus to glycines, alanines, serines, and asparagines. A PCR fragment containing the modified capsid gene was cloned into pVEEV/GFP (18) to replace the wt capsid sequence. The resulting plasmid was named pVEEV/CRK−/GFP. To generate pVEEV/CRK2−/GFP and pVEEV/CRK4−/GFP, an additional 2 and 4 mutations, respectively, were introduced into the CP111/126 peptide coding sequence of pVEEV/CRK−/GFP by PCR-based mutagenesis. The pH/C plasmid, encoding a helper RNA in which the capsid gene was placed under the control of a subgenomic promoter, is described elsewhere (19). The pH/2A-C1 plasmid contained a helper construct genome encoding a noncytopathic capsid (18) and lacked the subgenomic promoter (see Fig. 8A). In this plasmid, a 228-nucleotide (nt) sequence from the 5′ terminus of the VEEV TC-83 genome was fused with the foot-and-mouth disease virus (FMDV) 2A protease-encoding sequence, followed by VEEV capsid and green fluorescent protein (GFP) genes and the VEEV 3′UTR. To increase capsid protein translation, all of the AUG codons in the H/2A-C1 cassette were placed in frame with the capsid gene by deleting nt 129 and 181 from the sequence encoding the amino terminus of VEEV nsP1. pH1234/2A-C1-Cherry encoded a self-replicating helper RNA which we termed a passaging helper. This helper contained all of the nonstructural VEEV genes, and the entire H/2A-C1 sequence was placed under the control of the subgenomic promoter (see Fig. 8A). The fluorescent protein Cherry coding sequence was cloned in frame with the capsid gene to visualize helper replication. Plasmids for the DNA-based delivery of PIV genomes were designed by cloning of the VEEV/CRK4−/GFP and VEEV/CRK4− genomic cDNA sequences under the control of the cytomegalovirus (CMV) promoter into high-copy-number plasmids. To optimize synthesis of the correct RNA transcripts, the hepatitis delta virus ribozyme sequence was cloned immediately downstream of the VEEV poly(A) sequence. The pVEEV/C1/GFP, pVEErep/Cwt/GFP, and chimeric Csin-veev-encoding plasmids are described elsewhere (18, 20, 21). Plasmids with viral genomes lacking either the E1 or E2 glycoprotein gene (see Fig. 7 for details) were designed using standard PCR techniques. In all of the plasmids, cDNAs of VEE PIV, VEEV replicon, and helper genomes (except those containing cDNAs of viral genomes under the control of the CMV promoter) were placed under the control of the SP6 RNA polymerase promoter. The sequences of the recombinant genomes and details of cloning procedures can be provided upon request.
Fig 8.
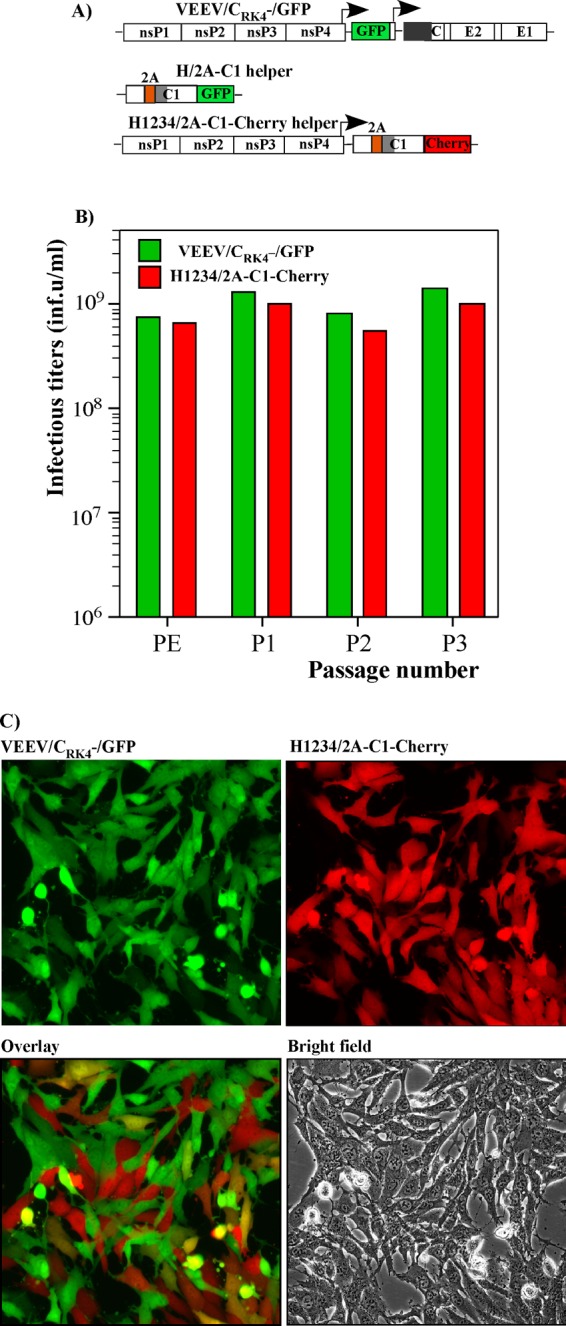
VEE PIV can be passaged as a two-component-genome virus. (A) Schematic representations of VEE PIV and helper genomes. (B) In vitro-synthesized VEEV/CRK4−/GFP and helper H1234/2A-C1-Cherry RNAs were electroporated into BHK-21 cells, and titers of the resultant particles, containing the indicated genomes (PE), were determined as described in Materials and Methods. Passaging was performed by infecting cells at an MOI of 2 infectious units/cell with virus harvested after electroporation or harvested at the previous passage. Media were harvested at 48 h postinfection. Titers of the particles containing PIV or helper genomes were determined as described in Materials and Methods. (C) Representative figures showing PIV and helper genome replication, as indicated by GFP and Cherry marker protein expression, 24 h after infection at an MOI of 2 infectious units/cell.
Fig 7.
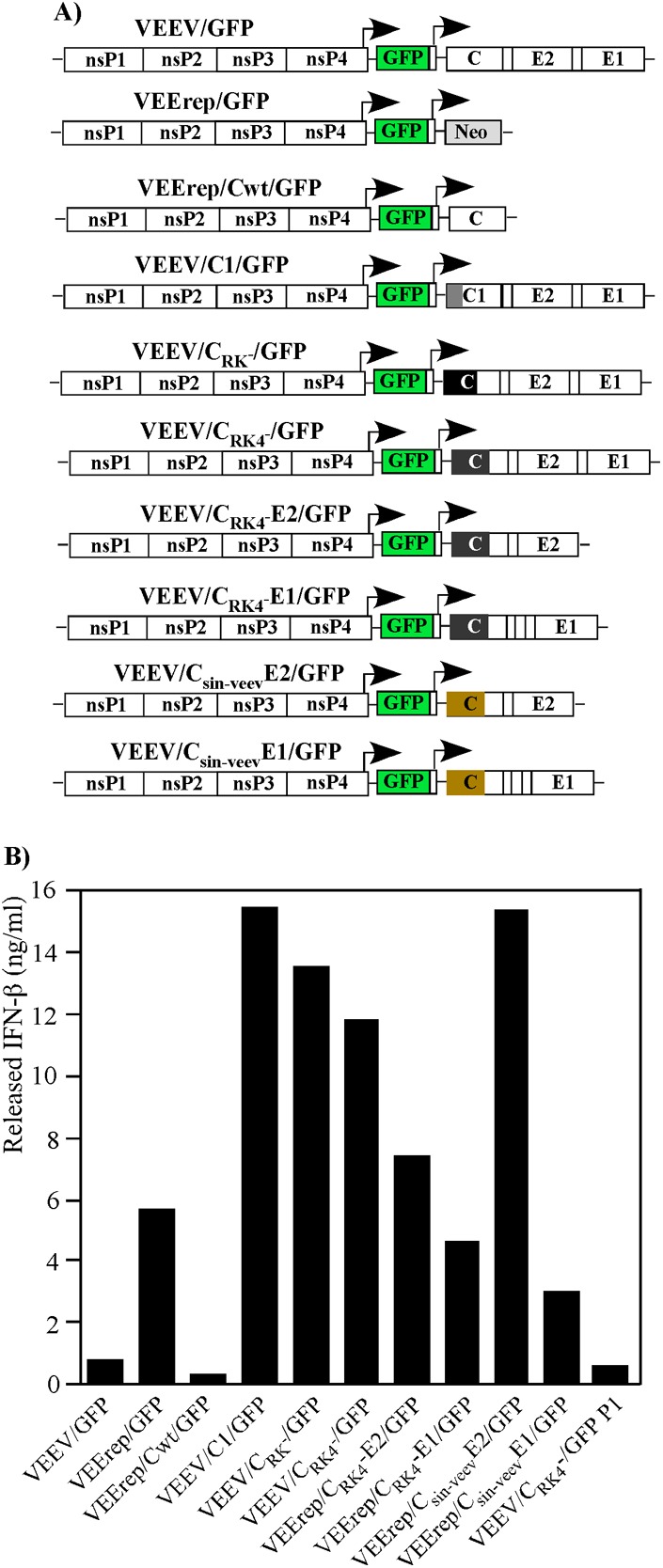
VEE PIVs are potent inducers of IFN-β. (A) Schematic representations of viral, replicon, and PIV genomes used in the experiment. (B) NIH 3T3 cells (5 × 105) in 6-well Costar plates were infected with the indicated viruses or packaged replicon and PIV genomes at an MOI of 20 infectious units/cell. Media were harvested at 20 h postinfection, and concentrations of released IFN-β were determined as described in Materials and Methods. “VEEV/CRK4−/GFP P1” indicates infection of cells with genome-free particles released from VEEV/CRK4−/GFP-infected cells (see the text for details).
In vitro RNA transcription and electroporation.
Plasmids were purified by centrifugation in CsCl gradients. Before the transcription reaction, the viral and replicon genome-carrying plasmids were linearized using a unique restriction site located downstream of the poly(A) sequence. RNAs were synthesized by SP6 RNA polymerase in the presence of a cap analog according to the manufacturer's recommendations (Invitrogen). The yield and integrity of the transcripts were analyzed by gel electrophoresis under nondenaturing conditions. Aliquots of transcription reaction mixtures were used for electroporation without additional purification. Electroporation of BHK-21 cells was performed under previously described conditions (22). Packaging of the defective viral genomes was performed by coelectroporating their in vitro-synthesized RNA and helper RNA genomes. Viral stocks were harvested within 24 h postelectroporation.
Titers of VEEV/GFP were determined by a standard plaque assay on BHK-21 cells (23). The infectious titers of noncytopathic viruses and replicons were determined by infecting BHK-21 cells (5 × 105 cells/well) in 6-well Costar plates with 10-fold dilutions of the samples and counting the number of GFP- or Cherry-positive cells.
Virus concentration.
BHK-21 cells were infected at a multiplicity of infection (MOI) of 20 infectious units/cell for 1 h, washed with phosphate-buffered saline (PBS), and overlaid with a complete medium. At 6 h postinfection, cells were washed three times with PBS to remove the FBS and then overlaid with serum-free medium (VP-SFM; Gibco). Media were harvested at 20 h postinfection, the pH in the media was stabilized by adding HEPES buffer (pH 7.5) to 0.005 M, and possible cell debris was pelleted by centrifugation at 4,500 × g for 20 min. Particles were concentrated either by ultracentrifugation or using centrifugal Ultracel-100K filters (Millipore). In many experiments, particles were pelleted at 50,000 rpm for 1 h at 4°C in a TLA-55 rotor in a TL-100 tabletop ultracentrifuge (Beckman). Concentrated or pelleted samples were analyzed by SDS-PAGE followed by either Coomassie staining or Western blotting. Western blots were performed using anti-VEEV TC-83 antibodies (a generous gift from R. Tesh [University of Texas Medical Branch at Galveston]) followed by treatment of the membranes with infrared dye-labeled secondary antibodies. For quantitative analysis, the membranes were scanned on a Li-Cor imager.
Viral particle densities were compared by ultracentrifugation of the concentrated samples of VEEV/GFP and VLPs released from VEE PIV-infected cells in linear 10 to 45% sucrose gradients prepared with PBS. Centrifugation was performed in an SW-40 rotor at 34,000 rpm for 4 h at 4°C. Gradients were fractionated, collected fractions were diluted with PBS and ultracentrifuged at 50,000 rpm for 1 h at 4°C in a TLA-55 rotor in a TL-100 tabletop ultracentrifuge (Beckman), and the presence of the particles in the fraction was analyzed by SDS-PAGE followed by Western blotting.
EM.
A 5-μl droplet of virus suspension was placed onto a freshly glow-discharged, 200-mesh copper electron microscope grid covered with a thin carbon film. After ∼30 s, the grid was blotted dry, stained for ∼60 s with 1% uranyl acetate, blotted again, and allowed to dry. Images were acquired using a Jeol JEM 3200FS electron microscope in the cryo-electron microscopy (cryo-EM) facility of Indiana University at Bloomington.
RT-qPCR.
For protein and RNA analyses, viral particles were pelleted from 1 ml of harvested medium. Samples for RNA analysis also contained 0.2 ml of a SINV stock of known concentration. It was added prior to centrifugation and used for normalization of the data and to control the quality of RNA isolation. RNAs from pelleted samples were isolated using an RNeasy Mini kit (Qiagen). cDNAs were synthesized using equal aliquots of isolated RNAs and a QuantyTect reverse transcription (RT) kit (Qiagen), using random and virus-specific reverse primers. VEEV nsP2- and E1-specific primers were used to quantify genomic and total viral RNAs. SINV nsP2-specific primers were used to quantify SINV genomic RNA in the samples. Quantitative PCR (qPCR) was performed using SsoFast EvaGreen supermix (Bio-Rad) in a CFX96 real-time PCR detection system (Bio-Rad) for 40 cycles, with 2 steps per cycle. Each step lasted 5 s: a denaturing step at 98°C and an annealing/extension step at 60°C. The specificity of qPCR was confirmed by analysis of the melting temperature of the amplified products. Results of quantification were normalized to the amount of SINV RNA in the same sample. The fold difference in RNA concentration was calculated using the ΔΔCT method. Each qPCR was performed in triplicate, and the mean and standard deviation (SD) were calculated.
Particle stability analysis.
For temperature stability analysis, 0.2 ml of viral particle or VLP sample was incubated at 56°C for 1 h. The same volume of virus was treated with 1% Triton X-100 for 15 min on ice or, for combinational treatment, incubated at 56°C for 1 h and then treated with 1% Triton X-100. Immediately after treatment, 0.3 ml of ice-cold VP-SFM was added to each tube. The material remaining after treatment was pelleted by ultracentrifugation through 0.6 ml of 25% sucrose in PBS in a TLA-55 rotor (50,000 rpm, 4°C, 1 h) and analyzed by SDS-PAGE followed by Western blotting, using the VEEV-specific antibodies described above.
IFN-β measurement.
NIH 3T3 cells were infected with different viruses or packaged replicons at an MOI of 20 infectious units/cell. Media were harvested at 20 h postinfection, and the pH in the media was stabilized by adding HEPES buffer, pH 7.5, to 0.01 M. Concentrations of IFN-β in the samples were measured with a VeriKine mouse interferon beta enzyme-linked immunosorbent assay (ELISA) kit (PBL Interferon Source) according to the manufacturer's recommendations.
DNA transfections.
Equal amounts of plasmid DNA (5 μg) were transfected into subconfluent BHK-21 cells in 35-mm dishes by use of Lipofectamine 2000 reagent according to the manufacturer's instructions (Invitrogen). At 4 h posttransfection, cells were washed with PBS and overlaid with 2 ml of VP-SFM (Gibco). At 24 h posttransfection, the supernatants were harvested, and VLPs were pelleted in a TLA-55 rotor (50,000 rpm, 4°C, 1 h). They were analyzed as described above and in the corresponding figure legends.
RESULTS
Mutations in RNA-binding domain of VEEV capsid protein have deleterious effects on release of infectious virus.
The VEEV capsid protein contains a number of amino acid sequences whose functions appear to be similar to those previously defined for capsid proteins of other alphaviruses. The carboxy-terminal domain (aa 127 to 275) of the VEEV capsid protein functions as a protease and plays a critical role in assembly of the icosahedral nucleocapsid (Fig. 1) (3). The amino-terminal domain (aa 1 to 126) contains an aa sequence characterized by a high concentration of positively charged amino acids, which are likely to be required for viral RNA packaging into nucleocapsids (Fig. 1) (3). This domain does not have a well-defined secondary structure, but it contains a peptide which is predicted to form a short alpha helix (Helix I). This helix, as previously demonstrated, is essential for dimerization of the SINV capsid protein during nucleocapsid assembly (24, 25). In the case of VEEV, Helix I also serves as a supraphysiological nuclear export signal (supraNES) (26). The supraNES and the downstream nuclear localization signal (NLS) determine the VEEV capsid's function in regulation of nucleocytoplasmic trafficking and, ultimately, in inhibition of cellular transcription (20, 26). Another important sequence is the conserved peptide CP111/126, which is located between the positively charged aa sequence and the protease domain. In the case of SINV, this sequence was shown to determine the specificity of viral genomic RNA packaging (27).
Fig 1.
Schematic representations of viral genomes encoding wt and mutated capsid proteins. The position and sequence of the VEEV Helix I/supraNES are indicated by the red box and red letters. The position and sequence of the VEEV NLS are indicated by the blue box and blue letters. The position and sequence of the conserved peptide CP111/126 are indicated by black boxes. The sequences of the amino termini of the wt and mutated capsid proteins are aligned, and the introduced mutations are indicated. The arrows above the genome diagrams indicate the positions of the subgenomic promoters.
Using this knowledge about the alphavirus capsid protein structure, we developed a cDNA clone of the VEEV genome encoding 26 mutations in the amino-terminal fragment of the capsid protein. These mutations changed most of the positively charged amino acids to alanines, serines, asparagines, and glycines (Fig. 1). The frequencies of the mutated codons were designed to be similar to their frequencies in the wt capsid coding sequence. Some of the introduced mutations destroyed the capsid-specific NLS and the supraNES-NLS connecting peptide (Fig. 1). Thus, these mutations were expected to affect the capsid's ability to inhibit cellular transcription. Due to the importance of the previously described Helix I/supraNES function in nucleocapsid assembly (25), this peptide was left unmodified. The high level of CP111/126 conservation among the distantly related alphaviruses and its specific location in the VEEV nucleocapsid (28) suggest the possibility that this peptide plays a role not only in capsid-specific interaction with RNA packaging signals but also in other processes of particle assembly. Therefore, in the initially designed construct, no mutations were made in CP111/126.
The synthesized fragment, containing replacements of the majority of the Lys and Arg residues in the amino-terminal region of the capsid protein, was cloned into the VEEV genome to replace the wt counterpart (Fig. 1). Replication of the new VEEV variant, VEEV/CRK−/GFP, was anticipated to be incapable of inducing a spreading infection (i.e., a pseudoinfectious virus [PIV]) due to numerous mutations in the RNA-binding domain. Moreover, the designed virus was expected to be noncytopathic. Therefore, we cloned the GFP gene under the control of one of the subgenomic promoters, and in the following experiments, GFP expression from both VEEV/CRK−/GFP and the control, parental VEEV/GFP encoding the wt capsid, was used to assess the number of infected cells, determine virus titers, and monitor the spread of infection.
The in vitro-synthesized RNAs of VEEV/CRK−/GFP and control VEEV/GFP (Fig. 2A) were electroporated into BHK-21 cells, and aliquots of the medium were harvested at the times indicated in Fig. 2B. Titers of VEEV/GFP were determined in a standard plaque assay. VEEV/CRK−/GFP, in contrast, was unable to develop plaques. Its titers were determined by infecting naïve BHK-21 cells with different dilutions of harvested samples and evaluating the number of GFP-positive cells at 6 h postinfection. In all of the numerous experiments, infectious titers of VEEV/CRK−/GFP after electroporation were 5 to 6 orders of magnitude lower than those of VEEV/GFP. The presence of infectious virus at such low concentrations and the inability to develop spreading infection indicated that either VEEV/CRK−/GFP was unable to form virions or the released particles contained almost no viral genomes.
Fig 2.
Replicating VEE PIV genomes produce infectious virus very inefficiently, and wt capsid protein can trans-complement the defect in genome packaging. (A) Schematic representations of the viral, PIV, and helper genomes. (B) Release of infectious virus particles from cells electroporated with in vitro-synthesized viral and PIV genomic RNAs (∼4 μg) or coelectroporated with PIV and helper RNAs (∼4 μg of each). Media were harvested at the indicated times, and infectious titers were determined as described in Materials and Methods.
VEEV capsid protein supplied in trans can complement the defect in RNA packaging.
To test whether the capsid supplied in trans could overcome the VEEV/CRK−/GFP defect in RNA packaging, we coelectroporated cells with VEEV/CRK−/GFP and the previously designed H/C helper RNA (Fig. 2A) (19). This helper RNA contained only the 5′- and 3′-terminal sequences required for RNA replication and carried a full-length VEEV capsid gene under the control of the subgenomic promoter. All of the viral nonstructural genes and most of the structural protein genes were deleted. As we previously described, due to the lack of a packaging signal, the H/C helper RNA was packaged very inefficiently into virions (19). Coelectroporation of the VEEV/CRK−/GFP genome with H/C helper RNA resulted in a dramatic increase of packaged PIV genome titers, making them comparable to those of VEEV/GFP (Fig. 2B). Therefore, in the next set of experiments, we applied capsid-encoding helpers to prepare high-titer samples of virions containing this and other defective VEEV genomes and used them for delivery of PIV genomes into cells. The initial stocks were prepared using the H/C helper, but it was later replaced with more advanced versions (see the following sections for details).
A VEEV encoding a capsid protein with a mutated RNA-binding domain forms VLPs.
Next, we analyzed whether VEEV/CRK−/GFP was able to produce VLPs during intracellular replication. BHK-21 cells were infected with VEEV/CRK−/GFP packaged into infectious virions by use of capsid-encoding helper RNA. The medium samples were harvested at different times postinfection, and infectious titers were determined as described above, by infecting naïve cells with different sample dilutions. The titers of VEEV/CRK−/GFP remained a few orders of magnitude below those of the control, VEEV/GFP (Fig. 3A). However, when the samples harvested at 24 h postinfection were analyzed for the presence of viral particles, the results were dramatically different. After concentrating the samples (see Materials and Methods for details), we recovered almost as much VEEV capsid-, E2-, and E1-containing material from the VEEV/CRK−/GFP samples as from those harvested from VEEV/GFP-infected cells (Fig. 3B). The detected structural proteins were found to be components of the released assembled virions and not the result of cell destruction, as cells infected with the packaged VEEV/CRK−/GFP genome did not develop cytopathic effect (CPE). In this and other numerous experiments, VEEV/GFP usually produced only ∼2-fold more virions than VEEV/CRK−/GFP, indicating that despite extensive mutations, the defective capsid could still efficiently assemble into a nucleocapsid, and ultimately into virions.
Fig 3.

Cells containing replicating VEE PIV genomes produce noninfectious VLPs. (A) Subconfluent BHK-21 cells in 6-well Costar plates were infected with viral or PIV genome-containing viral particles, generated by coelectroporation of VEEV/CRK−/GFP and H/C helper genomes into the cells, at an MOI of 20 infectious units/cell. Media were replaced at the indicated times postinfection, and infectious titers were determined as described in Materials and Methods. (B) Analysis of viral particles released from cells coelectroporated with in vitro-synthesized VEEV/CRK−/GFP PIV and H/C helper RNAs or infected at an MOI of 20 infectious units/cell with particles containing the packaged PIV genome or with VEEV/GFP. Media were harvested at 24 h postelectroporation or -infection, and particles were pelleted by ultracentrifugation and analyzed by SDS-10% PAGE followed by Coomassie blue staining. Each line on the gel contains an aliquot corresponding to 3 ml of harvested medium. (C) Analysis of the density of particles released by cells either infected by virus or infected by PIV genome-containing particles. Analysis was performed by ultracentrifugation of sucrose density gradients as described in Materials and Methods. (D) The genome-containing wt viral particles and VLPs formed by the replicating VEEV/CRK−/GFP PIV genome were concentrated from serum-free medium by use of centrifugal Ultracel-100K filters (see Materials and Methods for details). These samples were used directly for negative staining and EM analysis (see Materials and Methods for details).
In additional experiments, we analyzed pelleted material by centrifugation of sucrose density gradients. Opalescent bands were readily detectable in gradients with the samples harvested from either VEEV/GFP-infected cells or those infected with packaged VEEV/CRK−/GFP genomes (Fig. 3C). However, the particles released from VEEV/CRK−/GFP-infected cells demonstrated a detectably lower density (Fig. 3C), which was most likely the result of a lack of packaged RNA or its presence at a very low concentration. The presence of viral particles in the sucrose gradient-purified samples was additionally confirmed by EM (Fig. 3D). The particles released from the cells infected with packaged VEEV/CRK−/GFP contained VLPs of a size and architecture similar to those of VEEV/GFP genome-containing virus. Thus, the profound difference between infectious titers and particle release strongly indicates that the expression of a capsid lacking positively charged amino acids leads to very inefficient packaging of viral genomes into released VLPs.
Additional mutations in CP111/126 do not affect VLP assembly but abrogate PIV evolution.
The data described above demonstrated that mutations in the RNA-binding domain of the VEEV capsid had a deleterious effect on its ability to form infectious virions. This defect was most likely due to the inability of the defective capsid protein to package viral genomes. However, despite the introduction of 26 Arg- and Lys-specific mutations, we detected virus evolution that led to noticeable virus spread in some of the samples of the serially passaged VEEV/CRK−/GFP variant. The titers of the evolved variants remained 3 orders of magnitude lower than those of VEEV/GFP but could approach 107 infectious units/ml (Fig. 4B). Sequencing of the evolved virus genomes revealed the presence of adaptive mutations in the capsid and ns polyprotein coding sequences. None of the identified mutations led to the appearance of positively charged amino acids. This discovery led to the opening of another line of investigation into the RNA packaging mechanism and virus evolution (data not shown). However, these results are not within the scope of this study.
Fig 4.
Additional mutations in CP111/126 prevent further PIV evolution. (A) Schematic representations of the PIV genome, capsid domain structure, and additional mutations introduced into CP111/126. The introduced mutations are indicated by green letters. (B) Starting stocks of PIV genome-containing viral particles were prepared by electroporation of BHK-21 cells with in vitro-synthesized PIV and helper RNAs. Titers were measured at 24 h postelectroporation (PE samples). Blind passaging of VLPs was performed by infecting 2 × 106 naïve BHK-21 cells in 100-mm dishes with 100 μl of samples harvested after electroporation or 10 μl of medium harvested from the previous passage. Titers were determined at 48 h postinfection. The dashed line indicates the limit of detection.
To exclude the possibility of VEE PIV evolution, additional modifications were introduced into the VEEV/CRK−/GFP-specific capsid protein. These modifications were made in the conserved peptide CP111/126, which is predicted to have an alpha-helical folding (28). We were interested in preserving its potential role in nucleocapsid assembly and thus mutated only the positively charged amino acids in CP111/126 to alanines (Fig. 4A). The newly designed VEEV mutants VEEV/CRK2−/GFP and VEEV/CRK4−/GFP contained two and four additional mutations in the CP111/126 coding sequence, respectively. In all of the tests performed, they demonstrated identical characteristics, including the ability to form VLPs and, most importantly, a lack of evolution to a more efficiently replicating phenotype. Passaging of VEEV/CRK2−/GFP and VEEV/CRK4−/GFP variants, but not VEEV/CRK−/GFP, reproducibly led to a gradual decrease of infectious titers to undetectable levels. The results of one representative experiment from a series of repeated experiments are shown in Fig. 4B.
Since VEEV/CRK2−/GFP demonstrated characteristics indistinguishable from those of VEEV/CRK4−/GFP, only the latter construct was used in the next rounds of experiments, represented schematically in Fig. 5. Mutations present in CP111/126 of VEEV/CRK4−/GFP did not affect the virus's ability to form virions (VLPs). The VEEV/CRK4−/GFP-infected cells produced VLPs with essentially the same efficiency as that for cells infected with VEEV/GFP or VEEV/CRK−/GFP (Fig. 5B, C, and D). Cells infected with any of these three viruses released particles containing glycoproteins and capsid at comparable ratios (Fig. 5C), suggesting a similar virion organization. However, most of the released virions containing defective capsid proteins remained genome-free, and samples from VEEV/CRK−/GFP- and VEEV/CRK4−/GFP-infected cells contained viral genomic RNA quantities a few orders of magnitude lower than those of cells infected with VEEV/GFP (Fig. 5E). Interestingly, samples of particles released from VEEV/CRK4−/GFP-infected cells demonstrated a more pronounced difference between the infectious titers and concentrations of packaged viral genomes than that for particles released from cells infected with the VEEV/CRK−/GFP control. This suggests that mutations in CP111/126 might also interfere with proper genome release from the nucleocapsids and/or initiation of genome replication. However, this possibility needs additional experimental support. Other experiments also demonstrated that both replicating VEEV/CRK4−/GFP and VEEV/CRK−/GFP packaged viral subgenomic RNAs more efficiently than viral genomes (Fig. 5F). The most probable explanation for this is that the newly designed mutated capsids are no longer capable of specific RNA packaging, and thus they package viral subgenomic RNAs more efficiently due to the presence of these RNAs in the cells at higher concentrations than that of genomic RNA. However, the defective capsid proteins did not package β-actin mRNA, one the most abundant cellular mRNAs, at concentrations that could reliably be detected (data not shown). Taken together, these data suggest that additional mutations introduced into CP111/126 have a strong negative effect on virus evolution but do not affect formation and release of VLPs.
Fig 5.
Results of comparative analysis of VEEV virions and VLPs formed by replicating PIV genomes. (A) Schematic diagram of the experiments conducted for particle analysis. The data were collected in three independent experiments. In each experiment, the released viral particles were analyzed for their infectivity, stability, and RNA and protein content (see Materials and Methods for details). (B) Representative Coomassie blue-stained gel with particles pelleted from 1 ml of medium harvested from cells infected with VEEV/GFP- or PIV genome-containing particles. (C) Representative Western blot of released viral particles. (D) Results of quantitative analysis of the membrane presented in panel C. (E) Comparative analysis of infectious titers of released viral particles and levels of packaged viral and PIV genomic RNAs (see Materials and Methods for details). Infectious titers and concentrations of the genomic RNAs were normalized to those of VEEV/GFP. (F) Subgenomic/genomic RNA ratios in released viral particles were determined by RT-qPCR. Panels B and C are representative images from one of three highly reproducible experiments. The quantitative data from three independent experiments were used for calculation of means and standard deviations for panels D, E, and F.
Mutations of the positively charged aa in the VEEV capsid protein affect nucleocapsid stability.
Considering that most of the virions released from the PIV-infected cells were genome-free VLPs, it was important to evaluate the effect(s) of the introduced mutations on the stability of the released virions and nucleocapsids. To achieve this, we performed a series of parallel experiments, as shown in Fig. 5A. (i) Particles were prepared in FBS-free medium and then incubated at 56°C for 60 min. After this treatment, they were pelleted by ultracentrifugation through 25% sucrose (see Materials and Methods and Fig. 5A for details) and further analyzed by Western blotting using VEEV-specific antibodies (Fig. 6A, B, and C). In parallel, the temperature-treated samples were analyzed for residual virus infectivity (Fig. 6D). (ii) Another set of samples was treated for 15 min with 1% Triton X-100, and then the particles were also pelleted through 25% sucrose and further analyzed by Western blotting (Fig. 6A and E). (iii) Samples were incubated at 56°C for 60 min, treated with 1% Triton X-100, and subsequently pelleted through a sucrose solution and analyzed by Western blotting (Fig. 6A). The results indicated that the numerous mutations introduced into the capsid coding sequence had a detectable but not deleterious effect on the stability of the released particles (Fig. 6A, B, and C). Upon incubation at 56°C, and following ultracentrifugation, VEEV/CRK−/GFP- and VEEV/CRK4−/GFP-produced virions reproducibly demonstrated a noticeable loss of E2 glycoprotein but apparently retained their structural integrity and could be pelleted. Titers of VEEV/GFP, VEEV/CRK−/GFP, and VEEV/CRK4−/GFP decreased at very similar rates (data not shown), and a 60-min incubation at 56°C caused a drop of infectious titers of ∼3 orders of magnitude for all of the tested viruses (Fig. 6D).
Fig 6.

Mutations in the RNA-binding domain of the VEEV capsid protein affect the stability of nucleocapsids. The schematic representation of these experiments is shown as part of the diagram in Fig. 5A. (A) Western blotting of viral particles treated under different conditions. (B) Analysis of particle protein content after incubation at 56°C for 1 h and pelleting by ultracentrifugation. (C) Analysis of glycoprotein/capsid ratio in viral particles before and after incubation for 1 h at 56°C. The concentration of proteins on a quantitative Western blot was determined in fluorescence units. (D) Comparison of infectious titers of samples before and after 1 h of incubation at 56°C. (E) Analysis of nucleocapsid stability after Triton X-100 treatment (see Materials and Methods for details). Panel A shows a representative image from one of three highly reproducible experiments; the quantitative data from all three experiments were used for calculation of means and standard deviations for the other panels.
Treatment with Triton X-100 removed the lipid envelope and glycoprotein spikes from all of the particles (Fig. 6A and E). Nevertheless, nucleocapsids formed by wt capsid protein and containing viral RNA remained stable and were readily pelleted by ultracentrifugation. However, nucleocapsids composed of mutated capsids and lacking viral RNAs were unstable, and we were unable to isolate them. Interestingly, incubation of viral samples at 56°C apparently produced a significant alteration in the conformation of the spikes in the envelope. This alteration prevented envelope removal from both wt virions and VLPs by Triton X-100 treatment (Fig. 6A).
Thus, the above-described experiments demonstrated that the mutations introduced into the capsid protein made VLP-specific nucleocapsids unstable after removal of the lipid envelope. However, the same mutations had no such deleterious effects on the thermostability of the released VLPs.
VEE PIVs are potent type I IFN inducers.
Recently published data strongly suggest that replication of VEEV genomes lacking all of the structural protein genes (replicons) induces type I IFN, and likely other proinflammatory cytokines, more efficiently than wt virus replication does (29). Therefore, VEEV replicons were suggested for application in vivo as efficient adjuvants (29, 30). This high level of cytokine expression is likely determined at least partially by the inability of replicons to express capsid protein, and thus to interfere with cellular transcription. Therefore, in this study, we compared the levels of IFN-β induction caused by replication of VEE PIV genomes encoding mutated capsid proteins with those caused by replication of (i) VEEV/GFP, (ii) VEEV replicon, and (iii) other modified VEEV genomes encoding different combinations of structural proteins. The results of one representative and highly reproducible experiment are shown in Fig. 7. NIH 3T3 cells, which are competent in both type I IFN expression and signaling, were infected at the same MOI with each of the packaged constructs, and IFN-β levels were measured at 20 h postinfection. As expected, replication of the VEErep/GFP replicon induced a 10-fold higher level of IFN-β than did replication of VEEV/GFP expressing wt VEEV capsid. This higher level of replicon-specific IFN induction was abrogated by wt VEEV capsid protein expression in VEErep/CWT/GFP replicon-infected cells. Mutations in the capsid NLS and supraNES-NLS connecting peptide of VEEV/C1/GFP, VEEV/CRK−/GFP, and VEEV/CRK4−/GFP made these defective viruses even more efficient IFN-β inducers than VEErep/GFP, suggesting that expression of viral structural proteins has an additional positive effect on IFN induction. The detected increase was not a result of either mutated capsid or E1 expression, because VEErep/CRK4−E1/GFP and VEErep/Csin-veevE1/GFP constructs, expressing mutated VEEV capsid and chimeric SINV-VEEV capsid proteins, respectively, induced IFN-β to the same level as that in VEErep/GFP-infected cells. E2 glycoprotein expression by VEErep/CRK4−E2/GFP had some positive effect on IFN induction, but this effect was more profound in the cells infected with the VEErep/Csin-veevE2/GFP construct. The latter construct had a SINV-specific 5′UTR and a SINV-specific translational enhancer in the 5′ terminus of the subgenomic RNA and thus could produce higher levels of E2 (31). To rule out the possibility that incoming viral particles themselves were the primary type I IFN inducers, we infected NIH 3T3 cells with genome-free VLPs harvested after infection of BHK-21 cells with packaged VEEV/CRK4−/GFP. Despite using an MOI of ∼1,000 particles per cell, the VLPs induced IFN-β at barely detectable levels, which likely resulted from the very low residual levels of PIV genome-containing virions in the samples used for infection.
The results of these experiments suggested that VEE PIVs are more potent type I IFN inducers than previously described VEEV replicons. This additional increase in IFN induction could result from E2 glycoprotein expression but was more prominent for the constructs capable of producing E2-E1 glycoprotein spikes. Their expression appears to induce additional cell stress (32), which leads to an increase in type I IFN secretion. However, this phenomenon needs additional investigation.
PIV genomes can be packaged to high titers in vitro by use of strongly modified helper RNAs.
Although the H/C helper proved to be effective in PIV genome packaging, its application could potentially result in recombination events between the PIV genome and helper RNA (33, 34). These events are in all likelihood very rare, but they are still possible and could potentially lead to formation of viral genomes encoding an RNA packaging-competent capsid protein under the control of an additional subgenomic promoter. Therefore, to additionally decrease the possibility of recombination and appearance of spreading-competent virus, we designed a helper RNA lacking the subgenomic promoter and expressing a noncytopathic capsid protein (C1) with mutations in the NLS and supraNES-NLS connecting peptide (18). In this helper RNA (H/2A-C1), the mutated noncytopathic capsid protein was fused with the amino-terminal fragment of the nsP1 gene, and the FMDV 2A protease gene was placed between the nsP1 and capsid coding sequences to mediate proper protein processing (Fig. 8A). To visualize helper RNA replication in the cells, the helper construct was followed by the codons for the first 4 aa of E3 fused with either the GFP or Cherry gene. After coelectroporation of the in vitro-synthesized H/2A-C1 helper and PIV RNAs, PIV genomes were reproducibly packaged into infectious virions to titers exceeding 1 × 109 infectious units/ml. Passaging of the PIV genome-containing viral particles led to a rapid reduction in titers, indicating that helper RNAs were packaged very inefficiently.
Preparation of large samples of packaged PIV genomes by using H/C and H/2A-C1 helpers requires repeated electroporations. Therefore, we made an attempt to design a packaging system that would require electroporation only at the first step, which could be followed by in vitro passaging of samples on an escalating scale. Previously, this approach was successfully employed for propagation of a flavivirus PIV as a virus with a two-component genome (14, 16, 17).
For this system, we tested numerous helper RNA designs and eventually constructed a passaging-competent helper RNA which demonstrated no recombination events with the PIV genome during passaging. This helper encoded all of the nonstructural proteins and carried a noncytopathic capsid protein gene in the strongly modified subgenomic RNA, which was identical to the H/2A-C1 helper (Fig. 8A). While this passaging helper, H1234/2A-C1-Cherry, was self-replicating, it lacked viral glycoprotein-encoding genes and produced no spreading virus. Thus, cells released infectious particles only upon containing both replicating PIV and helper RNAs, which together expressed a complete set of RNA packaging-competent structural proteins. In the experiments presented below, we used a variant of these helper RNAs which encoded a fluorescent Cherry protein in addition to nsPs and capsid. This allowed us to distinguish between the cells containing replicating PIV and helper genomes and to easily evaluate the titers.
Coelectroporation of the in vitro-synthesized VEEV/CRK4−/GFP PIV and H1234/2A-C1-Cherry RNAs led to efficient packaging of both genomes into infectious virions (Fig. 8B). Titers of particles containing either genome were close to 109 infectious units/ml. Titration of harvested stocks confirmed that only simultaneous infection with both viral genomes could lead to plaque formation; therefore, there was a lack of proportionality between the dilutions and the numbers of plaques formed. This was an indication that the PIV and helper genomes were packaged into separate virions and thus the virus population existed as a two-component genome virus (Fig. 8C). During the next passages, performed at MOIs of >0.02 infectious units/cell, this virus was able to develop a spreading infection, with a high percentage of cells having both PIV- and helper-specific markers, i.e., GFP and Cherry, respectively. Both genomes were again packaged to similar titers, approaching 109 infectious units/ml (Fig. 8B).
To efficiently produce VLPs, VEE PIV genomes can be delivered into cells in DNA form.
DNA vaccines represent one of the promising directions in development of safe and efficient vaccine candidates. Therefore, in the next experiments, we tested whether it is possible to combine the advantages of a DNA-based delivery system and the efficiency of PIV replication and antigen production.
The newly designed plasmids pVEEV/CRK4−/GFP and pVEEV/CRK4− (Fig. 9A) carried VEE PIV genomes under the control of the CMV promoter. At 24 h post-plasmid transfection (see Materials and Methods for details), cells released essentially the same amount of VLPs as did cells infected with packaged PIV genomes (Fig. 9B). To compare the efficiencies of this PIV DNA-based approach and standard CMV-driven protein expression, we cloned the cDNA of a VEEV subgenomic RNA encoding all of the viral structural proteins and a capsid protein lacking the NLS under the control of the CMV promoter into p26S (Fig. 9A). Cells transfected with this plasmid compared to those transfected with pVEEV/CRK4−/GFP produced 8-fold lower levels of VEEV capsid protein and levels of glycoproteins that were barely detectable (data not shown). Additional sequencing of p26S did not detect any mutations in the 26S RNA-specific genes. Thus, the low levels of E2 and E1 expression were likely the result of additional RNA splicing after transcription from the CMV promoter. The PIV genome-expressing cassettes require only single copies of unspliced RNA to reach the cytoplasm for further amplification, and therefore this approach might be less dependent on splicing. Thus, on one hand, the DNA-based approach eliminates the requirement for in vitro PIV genome synthesis and electroporation, and on the other hand, it might strongly improve the expression efficiency of standard DNA-based vaccines and add the above-described self-adjuvant characteristics. However, inefficient delivery of plasmid DNA into cells in vivo certainly remains a problem.
Fig 9.
PIV genomes efficiently replicate and produce VLPs upon delivery into cells in DNA form. (A) Schematic representations of plasmids encoding PIV genomes and VEEV structural proteins under the control of the CMV promoter. (B) BHK-21 cells were either infected with VEEV/GFP or particles containing the packaged VEEV/CRK4−/GFP genome at an MOI of 20 infectious units/cell or transfected with plasmids carrying the indicated PIV genomes under the control of the CMV promoter. Media and cells were harvested at 24 h postinfection or -transfection, and viruses or VLPs were pelleted from equal volumes of medium and analyzed by SDS-PAGE followed by Coomassie blue staining or Western blotting using VEEV-specific antibodies.
DISCUSSION
Previously, development of efficient vaccines against viral infections was focused mostly on either selection of attenuated virus mutants incapable of inducing disease or the use of inactivated viruses. Live attenuated virus vaccines offer many advantages over the use of inactivated and subunit immunogens. Live vaccines induce strong, long-lived protective immunity characterized by the presence of neutralizing antibodies and induction of the cell-mediated component of the immune response. Thus, they closely mimic the immune response induced by wt virus infections. However, attenuation usually results from serial virus passaging in tissue culture, chicken embryos, or mouse brains. Thus, the resulting attenuated phenotype relies on very few point mutations, which can potentially revert to the wt sequence during virus replication in vivo. Development of arbovirus vaccine candidates is even more complicated because these attenuated viruses can be transmitted by mosquito vectors, in which they cause persistent infection. Infected mosquitoes represent another system that can increase the chances for reversion of attenuated viruses to the natural, more pathogenic phenotype. Inactivated, subunit, or SVP/VLP-based vaccines demonstrate high safety but typically require frequent boosters, which make the vaccination process lengthy and expensive. They also induce a different immune response from that found after natural viral infection. Inactivated vaccines might also require large-scale production of biohazardous viruses under strict biocontainment conditions.
The approach described in this study was aimed at designing pseudoinfectious VEEV, a defective virus which combines the safety of inactivated vaccines and the efficiency of live attenuated viruses. This PIV was expected (i) to demonstrate the same RNA replication and protein expression levels as wt VEEV, (ii) to mimic wt virus replication in terms of the levels of expression of structural proteins and their compartmentalization in infected cells, (iii) to efficiently release VLPs having the same structure as wt virions but lacking viral genetic material (which also renders them incapable of transmission to mosquito vectors), and (iv) to induce an abortive or single round of infection. Our data demonstrate that the achievement of these goals is possible by introducing clustered mutations into the VEEV capsid protein. It should be noted that our and other laboratories have previously designed SINV and Semliki Forest virus (SFV) variants containing extended deletions in the capsid coding sequence (35–37). However, these smaller capsid proteins were dramatically less efficient at virion formation. In this study, the introduction of 30 point mutations eliminated almost all of the positively charged capsid-specific aa, which mediate protein binding to viral genomic RNA. However, they caused only minor changes in the size of the VEEV capsid protein and did not affect the capsid's ability to form nucleocapsid and, ultimately, VLPs. Without a viral genome, the nucleocapsids of released VLPs were less stable than those of the wt, RNA-containing virus, and no preformed nucleocapsids were found in the cytoplasm of infected cells (data not shown). However, as indicated above, VLP formation was very efficient, and this strongly suggested that at least for VEEV infection, preformation of nucleocapsids in the cytoplasm is not an absolutely essential prerequisite of virus budding.
An additional benefit of this extensive mutagenesis was inactivation of the VEEV capsid's ability to interfere with cellular transcription. The redundant mutations destroyed the capsid-specific NLS and modified the peptide located between the supraNES and the NLS, which is also critically involved in capsid binding to the karyopherin receptors CRM1 (an exportin) and importin-α/β (26). Thus, the designed capsid mutants were incapable of inhibition of nucleocytoplasmic trafficking and activation of the antiviral response. PIV replication induced significantly more efficient secretion of type I IFN, and most likely other cytokines, than that induced by replication of wt VEEV. Moreover, VEE PIVs were better IFN-β inducers than VEEV replicons that had been proposed for application as virus-derived adjuvants (29, 30).
An important area of vaccine development that we also attempted to examine in this study was the method of delivery of defective PIV genomes into cells. In theory, the simplest way would be a direct, transfection-mediated RNA delivery, but so far this is not efficiently achievable, particularly in in vivo experiments. Another possibility is to package PIV genomes into infectious viral particles. Alphavirus packaging systems have been developed by numerous research teams and are widely applied (38–42). Usually packaging is achieved by cotransfecting cells with an in vitro-synthesized defective viral genome and helper RNAs that lack some or all of the nonstructural genes but encode one or more structural proteins under the control of the subgenomic promoter or internal ribosome entry sequence (IRES) (36, 43). Such packaging systems appear to be very efficient, but helper RNA can potentially recombine with a defective genome, leading to production of an infectious virus (34). To avoid recombination, we developed helpers that lack the subgenomic promoter and all of the viral genes except that encoding the capsid protein. In the numerous experiments performed in this study, we never detected any appearance of a recombinant infectious virus, indicating that formation of such a virus from the VEE PIV genome and a subgenomic promoter-free helper appears to require multiple, rare recombination events. Standard packaging with helper RNA resulted in stocks of packaged VEE PIV genome-containing infectious particles with titers of 2 × 109 to 5 × 109 infectious units/ml.
In addition to development of a replication-deficient helper, we also designed a self-replicating helper system for PIV genome packaging. This approach has already been tested for flavivirus vaccine production in our previous studies (16, 17). During testing of 16 different, potentially passaging-competent helpers, we stumbled upon an incredible ability of VEEV genomes to recombine and evolve. The designed constructs contained the capsid protein coding sequence under the control of either the subgenomic promoter or IRESs, and most of them were efficient in PIV genome packaging and passaging. However, nonhomologous recombination events between PIV and helper genomes resulted in formation of infectious viruses encoding packaging-competent capsid protein under the control of additional subgenomic promoters. The latter event was greatly beneficial for virus replication and spread.
Because the newly designed subgenomic, promoterless helper H/2A-C1 (Fig. 8) did not exhibit any recombination activity with the PIV genome, we developed a passaging helper expressing H/2A-C1 RNA from the subgenomic promoter. This unusual helper packaged PIV genomes very efficiently and, most importantly, did not engage in any recombination events during passaging. This was likely a result of the ability of transcribed subgenomic RNA to self-replicate (data not shown) and to serve as a defective interfering (DI) RNA. In case of possible recombination events, expression of DI RNA from the viral genome appears to be greatly disadvantageous for virus replication and thus serves as a negative selective pressure.
DNA transfection provides another potential method for VEE PIV genome delivery into the cell. Currently, DNA immunization is a rapidly developing area. Plasmid DNA used for vaccination usually carries the gene of interest under the control of DNA-dependent RNA polymerase II (Pol II) (44). Thus, gene expression depends mostly on the number of plasmids delivered into the nucleus and the efficiency of the promoter. However, placement of the VEE PIV genome under the control of the RNA Pol II promoter in the plasmid DNA makes antigen expression mostly independent of these conditions. Regardless of the number of PIV genome RNA molecules synthesized by cellular RNA polymerase, they are rapidly amplified by viral RNA-dependent RNA polymerase (RdRp) and express viral structural proteins and VLPs independent of the cellular transcription machinery. Our data demonstrate that a plasmid carrying the VEE PIV genome under the control of the CMV promoter was more productive in terms of expression of viral structural proteins than a control construct in which viral structural proteins were cloned under the control of the CMV promoter. Importantly, the structural proteins expressed from the DNA-delivered PIV genome were also efficiently released in the VLP form.
The observation of more efficient production of immunogens from plasmids encoding self-replicating PIVs supports previous findings that alphavirus replicon-based DNA vaccination requires ∼100- to 1,000-fold less plasmid DNA for induction of a comparable protective immune response to that induced by a conventional DNA vaccine (45, 46). An additional benefit of self-replicating PIV genome delivery instead of the use of standard RNA polymerase II-dependent cassettes is the ability of PIV RNA replication to efficiently induce proinflammatory cytokines, and thus to serve as an adjuvant (30, 47).
Taken together, the results of this study demonstrate that the alphavirus capsid protein can be modified strongly to become almost incapable of packaging viral RNA without a negative effect on its ability to function in viral particle assembly. Thus, VEEV can be transformed into a pseudoinfectious virus. VEE PIV demonstrates high levels of RNA replication and structural protein production, which result in efficient release of virions bearing wt virus architecture. However, most of these virions are genome-free and incapable of developing spreading infection.
Replication of VEE PIV genomes can be initiated by at least three methods of delivery into cells (Fig. 10): (i) direct delivery of in vitro-synthesized RNA into cells; (ii) infection by viral particles containing the PIV genome, generated by helper-based packaging; and (iii) transfection of cells with plasmid DNA containing the PIV genome under the control of the RNA polymerase II promoter. PIV genome replication leads to efficient induction of the cellular antiviral response, characterized by a high level of type I IFN release and expression of viral structural proteins followed by the release of noninfectious VLPs. Most importantly, PIV replication does not lead to infection spread. Thus, PIVs mimic viral infection by complete reproduction of all of the steps of intracellular wt virus replication. However, PIVs' inability to efficiently package their viral genomes makes their safety characteristics similar to those of inactivated or subunit vaccines. Currently, we are comparing the immunogenic and safety characteristics of the designed VEE PIV with those of the standard VEEV TC-83 and other attenuated VEEV variants recently developed in our research.
Fig 10.
PIV genomes can be delivered into cells in several ways: as in vitro-synthesized RNA, as a plasmid carrying the PIV genome under the control of the RNA polymerase II promoter, or packaged into infectious viral particles, which also mediate efficient genome delivery. Regardless of the means of delivery, replication of the PIV genome leads to efficient intracellular expression of viral structural proteins, their presentation at the plasma membrane, and their release in VLP form. Intracellular replication of defective genomes and protein expression also trigger the induction of cytokines, which promotes the development of the adaptive immune response.
In this study, we applied the strategy of PIV design to VEEV only. However, other alphaviruses demonstrate important similarities in RNA replication, protein expression, and the mechanisms of viral particle formation. Therefore, similar approaches in viral genome modification are likely to be applicable to other alphavirus pathogens, such as CHIKV, EEEV, and WEEV.
ACKNOWLEDGMENTS
We thank Niall J. Foy for helpful discussions and for critical reading and editing of the manuscript.
This work was supported by Public Health Service grants AI070207 (S.A., D.Y.K., and I.F.) and AI095449 (M.A. and E.I.F.).
Footnotes
Published ahead of print 5 December 2012
REFERENCES
- 1. Brown DT, Condreay LD. 1986. Replication of alphaviruses in mosquito cells, p 171–207 In Schlesinger S, Schlesinger MJ. (ed), The Togaviridae and Flaviviridae. Plenum Press, New York, NY [Google Scholar]
- 2. Paessler S, Fayzulin RZ, Anishchenko M, Greene IP, Weaver SC, Frolov I. 2003. Recombinant Sindbis/Venezuelan equine encephalitis virus is highly attenuated and immunogenic. J. Virol. 77:9278–9286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dal Canto MC, Rabinowitz SG. 1981. Central nervous system demyelination in Venezuelan equine encephalomyelitis infection. J. Neurol. Sci. 49:397–418 [DOI] [PubMed] [Google Scholar]
- 5. Alevizatos AC, McKinney RW, Feigin RD. 1967. Live, attenuated Venezuelan equine encephalomyelitis virus vaccine. I. Clinical effects in man. Am. J. Trop. Med. Hyg. 16:762–768 [PubMed] [Google Scholar]
- 6. Burke DS, Ramsburg HH, Edelman R. 1977. Persistence in humans of antibody to subtypes of Venezuelan equine encephalomyelitis (VEE) virus after immunization with attenuated (TC-83) VEE virus vaccine. J. Infect. Dis. 136:354–359 [DOI] [PubMed] [Google Scholar]
- 7. Pittman PR, Makuch RS, Mangiafico JA, Cannon TL, Gibbs PH, Peters CJ. 1996. Long-term duration of detectable neutralizing antibodies after administration of live-attenuated VEE vaccine and following booster vaccination with inactivated VEE vaccine. Vaccine 14:337–343 [DOI] [PubMed] [Google Scholar]
- 8. Ludwig GV, Turell MJ, Vogel P, Kondig JP, Kell WK, Smith JF, Pratt WD. 2001. Comparative neurovirulence of attenuated and non-attenuated strains of Venezuelan equine encephalitis virus in mice. Am. J. Trop. Med. Hyg. 64:49–55 [DOI] [PubMed] [Google Scholar]
- 9. Kinney RM, Johnson BJB, Welch JB, Tsuchiya KR, Trent DW. 1989. The full-length nucleotide sequences of the virulent Trinidad donkey strain of Venezuelan equine encephalitis virus and its attenuated vaccine derivative, strain TC-83. Virology 170:19–30 [DOI] [PubMed] [Google Scholar]
- 10. Pedersen CE, Jr, Robinson DM, Cole FE., Jr 1972. Isolation of the vaccine strain of Venezuelan equine encephalomyelitis virus from mosquitoes in Louisiana. Am. J. Epidemiol. 95:490–496 [DOI] [PubMed] [Google Scholar]
- 11. Davis NL, Powell N, Greenwald GF, Willis LV, Johnson BJ, Smith JF, Johnston RE. 1991. Attenuating mutations in the E2 glycoprotein gene of Venezuelan equine encephalitis virus: construction of single and multiple mutants in a full-length cDNA clone. Virology 183:20–31 [DOI] [PubMed] [Google Scholar]
- 12. Wang E, Petrakova O, Adams AP, Aguilar PV, Kang W, Paessler S, Volk SM, Frolov I, Weaver SC. 2007. Chimeric Sindbis/eastern equine encephalitis vaccine candidates are highly attenuated and immunogenic in mice. Vaccine 25:7573–7581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akahata W, Yang ZY, Andersen H, Sun S, Holdaway HA, Kong WP, Lewis MG, Higgs S, Rossmann MG, Rao S, Nabel GJ. 2010. A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat. Med. 16:334–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mason PW, Shustov AV, Frolov I. 2006. Production and characterization of vaccines based on flaviviruses defective in replication. Virology 351:432–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rumyantsev AA, Giel-Moloney M, Liu Y, Gao QS, Zhang ZX, Catalan J, Frolov I, Almond J, Kleanthous H, Pugachev KV. 2011. Characterization of the RepliVax platform for replication-defective flavivirus vaccines. Vaccine 29:5184–5194 [DOI] [PubMed] [Google Scholar]
- 16. Shustov AV, Frolov I. 2010. Efficient, trans-complementing packaging systems for chimeric, pseudoinfectious dengue 2/yellow fever viruses. Virology 400:8–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shustov AV, Mason PW, Frolov I. 2007. Production of pseudoinfectious yellow fever virus with a two-component genome. J. Virol. 81:11737–11748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Atasheva S, Krendelchtchikova V, Liopo A, Frolova E, Frolov I. 2010. Interplay of acute and persistent infections caused by Venezuelan equine encephalitis virus encoding mutated capsid protein. J. Virol. 84:10004–10015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Volkova E, Gorchakov R, Frolov I. 2006. The efficient packaging of Venezuelan equine encephalitis virus-specific RNAs into viral particles is determined by nsP1-3 synthesis. Virology 344:315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Atasheva S, Garmashova N, Frolov I, Frolova E. 2008. Venezuelan equine encephalitis virus capsid protein inhibits nuclear import in mammalian but not in mosquito cells. J. Virol. 82:4028–4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garmashova N, Atasheva S, Kang W, Weaver SC, Frolova E, Frolov I. 2007. Analysis of Venezuelan equine encephalitis virus capsid protein function in the inhibition of cellular transcription. J. Virol. 81:13552–13565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liljeström P, Lusa S, Huylebroeck D, Garoff H. 1991. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J. Virol. 65:4107–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lemm JA, Durbin RK, Stollar V, Rice CM. 1990. Mutations which alter the level or structure of nsP4 can affect the efficiency of Sindbis virus replication in a host-dependent manner. J. Virol. 64:3001–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perera R, Navaratnarajah C, Kuhn RJ. 2003. A heterologous coiled coil can substitute for helix I of the Sindbis virus capsid protein. J. Virol. 77:8345–8353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perera R, Owen KE, Tellinghuisen TL, Gorbalenya AE, Kuhn RJ. 2001. Alphavirus nucleocapsid protein contains a putative coiled coil alpha-helix important for core assembly. J. Virol. 75:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Atasheva S, Fish A, Fornerod M, Frolova EI. 2010. Venezuelan equine encephalitis virus capsid protein forms a tetrameric complex with CRM1 and importin alpha/beta that obstructs nuclear pore complex function. J. Virol. 84:4158–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weiss B, Nitschko H, Ghattas I, Wright R, Schlesinger S. 1989. Evidence for specificity in the encapsidation of Sindbis RNAs. J. Virol. 63:5310–5318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang R, Hryc CF, Cong Y, Liu X, Jakana J, Gorchakov R, Baker ML, Weaver SC, Chiu W. 2011. A cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J. 30:3854–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thompson JM, Whitmore AC, Konopka JL, Collier ML, Richmond EM, Davis NL, Staats HF, Johnston RE. 2006. Mucosal and systemic adjuvant activity of alphavirus replicon particles. Proc. Natl. Acad. Sci. U. S. A. 103:3722–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Konopka JL, Thompson JM, Whitmore AC, Webb DL, Johnston RE. 2009. Acute infection with Venezuelan equine encephalitis virus replicon particles catalyzes a systemic antiviral state and protects from lethal virus challenge. J. Virol. 83:12432–12442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frolov I, Schlesinger S. 1996. Translation of Sindbis virus mRNA: analysis of sequences downstream of the initiating AUG codon that enhance translation. J. Virol. 70:1182–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barry G, Fragkoudis R, Ferguson MC, Lulla A, Merits A, Kohl A, Fazakerley JK. 2010. Semliki Forest virus-induced endoplasmic reticulum stress accelerates apoptotic death of mammalian cells. J. Virol. 84:7369–7377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kamrud KI, Alterson K, Custer M, Dudek J, Goodman C, Owens G, Smith JF. 2010. Development and characterization of promoterless helper RNAs for the production of alphavirus replicon particle. J. Gen. Virol. 91:1723–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weiss BG, Schlesinger S. 1991. Recombination between Sindbis virus RNAs. J. Virol. 65:4017–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Forsell K, Xing L, Kozlovska T, Cheng RH, Garoff H. 2000. Membrane proteins organize a symmetrical virus. EMBO J. 19:5081–5091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Frolov I, Frolova E, Schlesinger S. 1997. Sindbis virus replicons and Sindbis virus: assembly of chimeras and of particles deficient in virus RNA. J. Virol. 71:2819–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smerdou C, Liljestrom P. 1999. Two-helper RNA system for production of recombinant Semliki Forest virus particles. J. Virol. 73:1092–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Elliott MB, Chen T, Terio NB, Chong SY, Abdullah R, Luckay A, Egan MA, Boutilier LA, Melville K, Lerch RA, Long D, Eldridge JH, Parks CL, Udem SA, Hancock GE. 2007. Alphavirus replicon particles encoding the fusion or attachment glycoproteins of respiratory syncytial virus elicit protective immune responses in BALB/c mice and functional serum antibodies in rhesus macaques. Vaccine 25:7132–7144 [DOI] [PubMed] [Google Scholar]
- 39. Foy BD, Olson KE. 2008. Alphavirus transducing systems. Adv. Exp. Med. Biol. 627:19–34 [DOI] [PubMed] [Google Scholar]
- 40. Gorchakov R, Volkova E, Yun N, Petrakova O, Linde NS, Paessler S, Frolova E, Frolov I. 2007. Comparative analysis of the alphavirus-based vectors expressing Rift Valley fever virus glycoproteins. Virology 366:212–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liljestrom P. 1994. Alphavirus expression systems. Curr. Opin. Biotechnol. 5:495–500 [DOI] [PubMed] [Google Scholar]
- 42. Phillips A, Mossel E, Sanchez-Vargas I, Foy B, Olson K. 2010. Alphavirus transducing system: tools for visualizing infection in mosquito vectors. J. Vis. Exp. 2010:2363 doi:10.3791/2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frolov I, Hoffman TA, Prágai BM, Dryga SA, Huang HV, Schlesinger S, Rice CM. 1996. Alphavirus-based expression systems: strategies and applications. Proc. Natl. Acad. Sci. U. S. A. 93:11371–11377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Donnelly JJ, Wahren B, Liu MA. 2005. DNA vaccines: progress and challenges. J. Immunol. 175:633–639 [DOI] [PubMed] [Google Scholar]
- 45. Hariharan MJ, Driver DA, Townsend K, Brumm D, Polo JM, Belli BA, Catton DJ, Hsu D, Mittelstaedt D, McCormack JE, Karavodin L, Dubensky TW, Jr, Chang SM, Banks TA. 1998. DNA immunization against herpes simplex virus: enhanced efficacy using a Sindbis virus-based vector. J. Virol. 72:950–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leitner WW, Ying H, Driver DA, Dubensky TW, Restifo NP. 2000. Enhancement of tumor-specific immune response with plasmid DNA replicon vectors. Cancer Res. 60:51–55 [PMC free article] [PubMed] [Google Scholar]
- 47. Thompson JM, Whitmore AC, Staats HF, Johnston RE. 2008. Alphavirus replicon particles acting as adjuvants promote CD8+ T cell responses to co-delivered antigen. Vaccine 26:4267–4275 [DOI] [PMC free article] [PubMed] [Google Scholar]