

Abstract
Domestic and nondomestic cats have been shown to be susceptible to feline spongiform encephalopathy (FSE), almost certainly caused by consumption of bovine spongiform encephalopathy (BSE)-contaminated meat. Because domestic and free-ranging nondomestic felids scavenge cervid carcasses, including those in areas affected by chronic wasting disease (CWD), we evaluated the susceptibility of the domestic cat (Felis catus) to CWD infection experimentally. Cohorts of 5 cats each were inoculated intracerebrally (i.c.) or orally (p.o.) with CWD-infected deer brain. At 40 and 42 months postinoculation, two i.c.-inoculated cats developed signs consistent with prion disease, including a stilted gait, weight loss, anorexia, polydipsia, patterned motor behaviors, head and tail tremors, and ataxia, and the cats progressed to terminal disease within 5 months. Brains from these two cats were pooled and inoculated into cohorts of cats by the i.c., p.o., and intraperitoneal and subcutaneous (i.p./s.c.) routes. Upon subpassage, feline CWD was transmitted to all i.c.-inoculated cats with a decreased incubation period of 23 to 27 months. Feline-adapted CWD (FelCWD) was demonstrated in the brains of all of the affected cats by Western blotting and immunohistochemical analysis. Magnetic resonance imaging revealed abnormalities in clinically ill cats, which included multifocal T2 fluid attenuated inversion recovery (FLAIR) signal hyperintensities, ventricular size increases, prominent sulci, and white matter tract cavitation. Currently, 3 of 4 i.p./s.c.- and 2 of 4 p.o. secondary passage-inoculated cats have developed abnormal behavior patterns consistent with the early stage of feline CWD. These results demonstrate that CWD can be transmitted and adapted to the domestic cat, thus raising the issue of potential cervid-to-feline transmission in nature.
INTRODUCTION
Trans-species transmission of prion diseases is unpredictable and remains poorly understood, yet has obvious public health implications. These facts drive continued appraisal of present concepts regarding both transmissible spongiform encephalopathy (TSE) spread and intervention strategies. It is clear that variant Creutzfeldt-Jakob disease (vCJD) (1) and feline spongiform encephalopathy (FSE) (2) can be attributed to the ingestion of meat and bone from bovine spongiform encephalopathy (BSE)-infected cattle. Transmissible mink encephalopathy (TME) in the United States is also attributed to the consumption of scrapie- or BSE-infected meat products (3). It has been hypothesized that sheep scrapie may have crossed the species barrier from scrapie-infected sheep to cattle as BSE, and possibly from sheep to deer as chronic wasting disease (CWD) (4), yet this remains far from certain, and spontaneous generation cannot be excluded. Thus, there is accruing evidence for the trans-species transmission of prions, with potentially grave consequences for animals and humans.
FSE, the fatal TSE of domestic and nondomestic cats, was first described shortly after the BSE epidemic (5) and strain typed to be the result of the consumption of BSE-contaminated beef (1). This provided support for the adaptation and transmission of orally consumed prions, as well as evidence that felids are susceptible to prion infection. Chronic wasting disease has been shown to be readily transmitted via multiple means, including direct contact with a CWD-infected cervid (6–8), exposure to infectious CWD prions in the environment (9, 10), and ingestion of bodily fluids and tissues from a CWD-infected cervid (11–15). Recent research by Miller and colleagues (16) performed in an area of Colorado where CWD is endemic, where nearly one-fourth of the adult mule deer are prion infected, confirmed the assumption that afflicted animals become prey to mountain lions more often than healthy ones. In addition, experimental studies have demonstrated that prions infect and cause disease in several rodent scavenger species (i.e., voles and deer mice) overlapping in distribution with CWD and scrapie outbreaks (17, 18). Such scavenging rodents are known to cannibalize and are also an important food source for many larger predators and scavengers in nature, hence providing a reservoir and a potential bridge for cross-species transmission of prions from the natural host to predator/scavenger species.
Thus, the facile transmission of CWD, coupled with its growing geographical distribution (19) and propensity to persist in the environment (20–22), provide ample opportunity for the bioavailability of CWD prions to many species coexisting with CWD in nature, including felids.
We undertook this study to determine (i) whether felids are susceptible to deer origin CWD prion infection and disease progression and (ii) whether disease course and transmission efficiency are altered upon subsequent passage of feline-adapted CWD in cats.
MATERIALS AND METHODS
Domestic cats, genotyping, and prion protein sequence comparison.
Four-month-old domestic cats were provided by the Andrea D. Lauerman specific-pathogen-free (SPF) colony, Department of Microbiology, Immunology and Pathology (DMIP), College of Veterinary Medicine and Biological Sciences (CVMBS), Colorado State University (CSU).
All animals were handled in strict accordance with good animal practice, as defined by relevant national and/or local animal welfare bodies, and all animal work was approved by Colorado State University Animal Care and Use Committee (ACUC approval no. 02-151A, 08-175A, and 11-2622A). All cats were genotyped to determine the prion protein gene (PRNP) coding sequence in the laboratory of Wilfred Goldmann, The Roslin Institute, University of Edinburgh, Scotland, United Kingdom.
Prion protein amino acid sequences were recovered from GenBank for the domestic cat (ACA50727.1) and 8 other mammalian species, including mountain lions (ACA50739.1), bobcats (ACA50731.1), Canadian lynx (ACA50730.1), white-tailed deer (AAP37447.1), mule deer (AAC33174.2), elk (AAC12860.2), cattle (CAA39368.1), and humans (BAG32276.1). The sequences were aligned and pairwise identities calculated using default parameters for the MUSCLE algorithm (multiple sequence comparison by log expectation) in the Geneious v.5.6 (Biomatters, Ltd.) software.
Biocontainment protocols.
Protocols to preclude extraneous exposure and cross-contamination between cohorts of animals as previously described (12) included protective shower-in requirements, Tyvek clothing, masks, head covers, and footwear, while maintaining stringent husbandry. Tissues were harvested with sample-specific instruments to minimize possibility of cross-contamination. Solid and liquid waste from each suite was either incinerated or collected and denatured by alkaline digestion.
Inocula. (i) Primary passage DeerCWD.
Deer origin CWD (DeerCWD) brain inoculum that was confirmed as CWD positive by immunohistochemistry (IHC) was from a free-ranging mule deer (989-09147) provided by Terry Spraker at the Colorado State Veterinary Diagnostic Laboratory (CSVDL), which was utilized in previous bioassay studies (12). The CWD-negative brain inoculum for sham controls was sourced from two white-tailed deer (UGA 1 and 2) born and raised in a CWD-free area provided by the Warnell School of Forestry and Natural Resources, University of Georgia, Athens (UGA).
(ii) Secondary passage FelCWD.
Inocula for feline-adapted CWD (FelCWD) secondary passage studies consisted of pooled brain sections from two primary passage cats (4152 and 4137) confirmed as CWD positive by Western blotting and IHC or one primary passage CWD-negative sham-inoculated cat (4149).
Inoculation cohorts.
Cohorts of 2 to 5 4-month-old SPF domestic cats were kept in separate group housing suites throughout the study. Suite-specific and dedicated protective clothing, utensils, and waste disposal were utilized to exclude cross-contamination by fomites, bedding, food, excretions, or contact.
Primary passage.
Cohorts of 5 SPF domestic cats were inoculated with DeerCWD brain either intracerebrally (i.c.; 0.5 g into the left parietal cortex) or orally (p.o.; a total of 1 g administered by syringe over 2 consecutive days at 0.5 g/day). Age-matched sham controls were inoculated in the same manner with brain from CWD-negative deer.
Secondary passage.
FelCWD brain pool was inoculated by multiple routes into cohorts of SPF domestic cats: i.c. (0.5 g into the left parietal cortex; n = 5), p.o. (a total of 1 g syringe administered over 2 consecutive days at 0.5 g/day; n = 4), or intraperitoneally and subcutaneously (i.p./s.c.) (0.25 g each; n = 4). Two cats were cohoused with the i.c.-inoculated cohort to monitor for horizontal/environmental shedding and transmission of disease-causing prions. As lingual abrasions have been shown to facilitate prion transmission following oral exposure (23, 24), two abrasions (2 cm in length) were made on the dorsal and ventral aspects of the tongue by lightly scratching the surfaces with a 20-gauge needle prior to second-passage p.o. inoculations.
Monitoring and sample collection.
Following inoculation, cats were monitored daily for behavioral changes and euthanized at or before onset of late-stage clinical signs. At study termination, bodily secretions (blood and saliva), excreta (urine and feces), and tissues from each cat were harvested, fixed, and/or frozen for the detection of FelCWD.
Western blotting.
Tissue homogenates were prepared from the obex region of the medulla oblongata at 10% (wt/vol) in NP-40 buffer (10 mM Tris-HCl buffer [pH 7.5], 0.5% NP-40, 0.5% sodium deoxycholate) in a FastPrep FP120 cell disrupter (Qbiogene) with one 45-s cycle at a speed setting of 6.5 and then cooled on ice. Homogenates were mixed with proteinase K (PK) (Invitrogen) at a final concentration of 50 μg/ml and incubated at 37°C for 30 min with shaking. Samples were mixed with 10× reducing agent–4× lithium dodecyl sulfate (LDS) sample buffer (Invitrogen) at a final concentration of 1×, heated to 95°C for 5 min, and then run through a NuPAGE 10% Bis-Tris gel at 100 V for 2.5 h. Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane at 100 V for 1 h in transfer buffer (0.025 M Trizma base, 0.2 M glycine, 20% methanol [pH 8.3]). The membrane was loaded into a prewetted SNAP i.d. blot holder (Millipore) and then placed in the SNAP i.d. system (Millipore). Blocking buffer (Blocker casein in Tris-buffered saline [TBS] [Thermo Scientific] with 0.1% Tween 20) was added to the blot holder for 10 min, followed by vacuum removal. Primary antibody BAR224 (Cayman Chemical) conjugated to horseradish peroxidase (HRP) was diluted in blocking buffer to 0.2 μg/ml, added to the blot holder, incubated for 12 min, and then vacuumed through the membrane. The membrane was washed three times with 30 ml of wash buffer (50% Blocker casein in TBS, 50% 1× TBS, 0.1% Tween 20) with continuous vacuum and then developed with ECL Plus enhanced chemiluminescence Western blotting detection reagents (GE) and viewed on a Luminescent image analyzer LAS-3000 (Fujifilm).
IHC.
Tissues were fixed in 10% neutral buffered formalin or paraformaldehyde-lysine-periodate (PLP) for 5 days or 2 days, respectively, and stored in 60% ethanol. Tissues were treated for 1 h with 88% formic acid, rinsed with tap water, and embedded in paraffin, and 6-μm sections were placed on positively charged slides. Deparaffinized and rehydrated sections were PK digested (20 mg/ml) at 37°C in 1× TBS for 15 min, washed for 10 min in TNT buffer (0.1 M Tris-HCl [pH 7.5], 0.15 M NaCl, 0.05% Tween 20), and subjected to heat-induced epitope retrieval in the 2100-Retriever (Prestige Medical) in sodium citrate buffer (0.01 M sodium citrate, 0.05% Tween 20 [pH 6.0]). A tyramide signal amplification (TSA) detection kit (PerkinElmer) was used to enhance the signal, as described in previous work (25), using the anti-prion protein antibody L42 (R-Biopharm) at 0.077 μg/ml and Envision+ anti-mouse HRP-labeled polymer (Dako). Following TSA treatment, slides were incubated with 3-amino-9-ethylcarbazole (AEC) substrate-chromogen (Dako), counterstained with hematoxylin and bluing reagent (0.1% sodium bicarbonate), and coverslipped with an aqueous mounting medium (Vector Laboratories).
RT-QuIC.
Real-time quaking-induced conversion (RT-QuIC), as first described by Atarashi (26), Wilham (27), and Orru (28), was used to assess prion-converting activity in brain tissue from all terminated cats. Serial dilutions (10−2 to 10−9) of tissue homogenates or “seeds” were prepared from the obex region of the medulla oblongata in seed buffer (0.1% SDS, 1× phosphate-buffered saline [PBS], 1× N2 medium supplement [Gibco]). Positive and negative assay controls included serial dilutions (10−2 to 10−9) of naïve and CWD-infected white-tailed deer and cat obex. Reaction mixtures, set up in duplicate in 96-well black bottom optic plates (Nalgene Nunc), consisted of 2 μl seed in 98 μl real-time quaking-induced conversion (RT-QuIC) buffer (final concentrations of 1× PBS, 300 mM NaCl, 1 mM EDTA, 10 μM thioflavin T [ThT], 0.1 mg/ml recombinant hamster PrPC substrate [with glycerol stocks generously provided by Byron Caughey, NIH]). Plates containing reaction mixtures were placed in a BMG Fluostar plate reader at 42°C with cycles of 1 min of shaking and 1 min at rest and with ThT fluorescence measurements taken at 15-min intervals for 50 h.
MRI.
Anatomic brain magnetic resonance imaging (MRI) scans, including proton density, T2-weighted, fluid attenuated inversion recovery (FLAIR), and T1-weighted (pre- and postcontrast) scans were performed on 10 i.c.-inoculated cats: 6 clinically ill (1 primary passage, 5 secondary passage), 2 asymptomatic (primary passage), and 2 age-matched sham controls, as well as 1 uninoculated age-matched control. MRI was done with a Signa LX 1.5 T MR HiSpeed Plus system (General Electric), as previously described (29). Briefly, anesthetized cats were positioned sternally with the head centered within the quadrature head coil. The pituitary fossa was centered in the imaging coil, and laser light localization was used to standardize a consistent longitudinal orientation along the z-axis. The anatomic brain MRI scan included transverse dual fast-spin echo proton density/T2-weighted scan (repetition time [TR] = 4,000 ms, echo time [TE] = 17/115 ms, echo train length = 16, slice thickness = 3 mm, image matrix = 256 × 256), T2 FLAIR (TR = 8,125 ms, TE = 120, inversion time [TI] = 2,200 ms), and T1-weighted scans (TR = 550 ms, TE = minimal) pre- and postcontrast after intravenous diethylenetriaminepentaacetic acid (gd-DTPA; Magnevist) at 0.5 mmol/kg (Bayer Pharmaceuticals). Diffusion-weighted imaging (DW-MRI) was performed with b factor 2000 using TE = 90 to 110 ms, TR = 8,000 ms, field of view (FOV) = 24 mm, slice thickness = 5 mm, image matrix = 124 × 124, and a number of excitations (NEX) of 4. All MRI pulse sequences were reviewed qualitatively for gross lesions.
RESULTS
PRNP genotypes and sequence comparison.
No single-amino-acid polymorphisms were detected in any of the 39 inoculated cats. Four cats were heterozygous for the nonapeptide repeats in the N terminus: 1 i.c. DeerCWD inoculate (clinical disease at 40 months postinfection [p.i.]; cat 4152) and 2 sham controls from the primary passage cohort (cats 4154 and 4151), as well as 1 s.c./i.p. FelCWD inoculate (no clinical disease at 40 months p.i.; cat 4338) from the secondary passage cohort (Table 1).
Table 1.
Study cohorts, design, and outcome of cats inoculated with CWDa
| Cohort | Animal no. | Inoculation route | Genotype codon 57 | MPI: |
Qualitative MRI analysis result | Age (mo) at MRI | |
|---|---|---|---|---|---|---|---|
| To early/late stage of TSE disease | At euthanasia | ||||||
| Primary passage | |||||||
| DeerCWD brain | 4137 | i.c. | TC | 40/45 | 45 | Abnormal | 56 |
| 4142 | TT | − | 25 | ND | ND | ||
| 4146 | CC | − | 86 | Normal | 81 | ||
| 4152 | CND | 42/47 | 47 | ND | ND | ||
| 4156 | TC | − | 86 | Abnormal | 80 | ||
| 4133 | p.o. | CC | − | 85 | ND | ND | |
| 4143 | TT | − | 25 | ND | ND | ||
| 4148 | TC | − | 85 | ND | ND | ||
| 4150 | TT | − | 85 | ND | ND | ||
| 4155 | CC | − | 19 | ND | ND | ||
| Naïve deer brain | 4136 | i.c. | TC | − | 85 | ND | ND |
| 4144 | TC | − | 49 | Normal | 57 | ||
| 4149 | TT | − | 26 | ND | ND | ||
| 4154 | CND | − | 85 | ND | ND | ||
| 4157 | TC | − | 45 | ND | ND | ||
| 4132 | p.o. | TC | − | 21 | ND | ND | |
| 4141 | CC | − | 85 | ND | ND | ||
| 4145 | CC | − | 26 | ND | ND | ||
| 4147 | CC | − | 85 | ND | ND | ||
| 4151 | CND | − | 85 | ND | ND | ||
| Secondary passage | |||||||
| FelCWD brain | 4332 | i.c. | CC | 19/23 | 23 | Abnormal | 32 |
| 4346 | TT | 21/27 | 27 | Abnormal | 35 | ||
| 4357 | TT | 19/25 | 25 | Abnormal | 29 | ||
| 4360 | TT | 23/25 | 25 | Normalb | 28 | ||
| 4341 | CC | 24/26 | 26 | Abnormal | 34 | ||
| 4329 | p.o. | CC | ° | CTM | ND | ND | |
| 4337 | CC | 42/* | CTM | ND | ND | ||
| 4352 | TT | ° | CTM | ND | ND | ||
| 4344 | CC | 35/* | CTM | ND | ND | ||
| 4361 | i.p./s.c. | TT | 31/* | CTM | ND | ND | |
| 4338 | CND | ° | CTM | ND | ND | ||
| 4345 | CC | 29/* | CTM | ND | ND | ||
| 4348 | CC | 29/* | CTM | ND | ND | ||
| 4340 | Cohoused | CC | ° | CTM | ND | ND | |
| 4356 | TT | ° | CTM | ND | ND | ||
| Primary passage-negative feline brain | 4334 | i.c. | TT | ° | CTM | ND | ND |
| 4359 | CC | − | CTM | Normal | 31.5 | ||
| 4349 | TT | ° | CTM | ND | ND | ||
| 4353 | CC | ° | CTM | ND | ND | ||
| Uninoculated | 4135 | ND | ND | − | 65 | Normal | 65 |
MPI, month postinfection; CND, cannot determine repeat number variation in heterozygous state; −, did not develop TSE clinical disease, nor was PrPCWD detected from terminal brain tissue; *, no late-stage clinical signs at the time of this report (continuing to monitor [CTM]at 42 MPI); °, no clinical signs at the time of this report (continuing to monitor at 42 MPI); ND, not done.
In this cat, MRI revealed a normal brain but mandibular lymph node asymmetry.
The alignment and pairwise analysis of the PRNP amino acid sequences of domestic cats, bobcats, Canadian lynx, white-tailed deer, mule deer, elk, cattle, and humans (Table 2; see Fig. 5) revealed that pairwise genetic identity for these species ranges from 84% (humans and mountain lions) to 100% (white-tailed deer and mule deer). The domestic cat PRNP shares 94.9 to 99.1% homology with nondomestic cats and 92.1 to 92.5% homology with cervid species. While an octarepeat deletion (amino acids [aa] 69 to 77) was identified in the mountain lion sequence, the PRNP amino acid sequences for domestic and nondomestic cats are highly homologous compared to those of the other 5 species analyzed. For comparison, we analyzed the PRNP amino acid sequence for species in which successful cross-species prion transmission has been documented—cattle to humans (87.9% homology) and cattle to domestic cats (87.7% homology).
Table 2.
Pairwise genetic distances between domestic cats and 7 other mammalian species sharing a home range in North America
| Species | Genetic distance (%) froma: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Domestic cats | Mountain lions | Bobcats | Canadian lynx | White-tailed deer | Mule deer | Elk | Humans | Cattle | |
| Domestic cats | 100 | 94.9 | 98.6 | 99.1 | 92.1 | 92.1 | 92.5 | 89.7 | 87.7 |
| Mountain lions | — | 100 | 96.2 | 96.5 | 88 | 88 | 88.4 | 84 | 84.6 |
| Bobcats | — | — | 100 | 99.6 | 90.4 | 90.4 | 90.8 | 86.4 | 86.9 |
| Canadian lynx | — | — | — | 100 | 90.8 | 90.8 | 91.2 | 86.8 | 87.3 |
| White-tailed deer | — | — | — | — | 100 | 100 | 99.6 | 90.2 | 94.7 |
| Mule deer | — | — | — | — | — | 100 | 99.6 | 90.2 | 94.7 |
| Elk | — | — | — | — | — | — | 100 | 89.8 | 94.3 |
| Humans | — | — | — | — | — | — | — | 100 | 87.9 |
| Cattle | 100 | ||||||||
—, repeat comparison.
Fig 5.

Amino acid alignment of 8 mammalian prion proteins. The prion protein amino acid sequences of mountain lions, bobcats, Canadian lynx, white-tailed deer, mule deer, elk, humans, and cattle were aligned against the domestic cat sequence. All sequences were recovered from GenBank. Periods represent identical residues; dashes represent deletions. Residues differing from those of the domestic cat are shown.
Clinical signs of TSE. (i) Primary passage of DeerCWD into domestic cats.
Project-dedicated caretakers intimately familiar with each animal observed the cats daily. Subtle clinical signs consistent with TSE, including a stilted gait, weight loss, anorexia, polydipsia, patterned motor behaviors, head and tail tremors, and ataxia, were detected 40 to 42 months p.i. in 2/5 primary passage i.c.-inoculated cats, which ultimately mandated euthanasia 45 to 47 months p.i. (Table 1). Of the 3 remaining i.c.-inoculated cats, 1 was euthanized at 25 months p.i. for disease screening, while 2 remained asymptomatic for TSE disease and were euthanized at 86 months p.i. The p.o.-inoculated cohort did not develop signs of TSE disease (0/5); 2/5 were euthanized at 19 and 25 months p.i., and the remaining 3 were euthanized at 85 months p.i. to permit examination for FelCWD. No cats receiving naïve deer brain developed TSE disease.
(ii) Secondary passage of FelCWD into domestic cats.
Five of 5 secondary passage i.c.-inoculated cats developed slowly progressing signs of TSE sooner than the primary passage cats, at 20 to 24 mo p.i., with some individuals displaying additional signs of aggressiveness and hyperreactivity. Symptomatic i.c.-inoculated cats were euthanized 23 to 27 months p.i. (Table 1). Currently (40 months p.i.), 3/4 i.p./s.c.- and 2/4 p.o.-inoculated cats are showing early TSE signs, including a stilted gait, head and tail tremors, and hyperreactivity. All remaining cats (i.p./s.c. or p.o. inoculated and cohoused) will continue to be monitored for further development of TSE disease.
Detection of FelCWD in tissues at necropsy. (i) Primary passage of DeerCWD into domestic cats.
At necropsy, 2/5 i.c.-inoculated symptomatic cats were prion positive, as indicated by Western blotting, IHC, and RT-QuIC detection of FelCWD in the medulla oblongata at the level of the obex. This PK-resistant material had a slightly lower molecular mass than CWD-infected deer brain homogenate (Fig. 1A). IHC revealed small amounts of FelCWD immunostaining, primarily within neurons (Fig. 2A). Neuroanatomically, the FelCWD deposits were confined to nuclei of the caudal brain stem, including the dorsal motor nucleus of the vagus and the nucleus of the solitary tract. Morphologically, the FelCWD deposits were finely granular and intraneuronal. No evidence of FelCWD was found in animals that did not develop clinical disease.
Fig 1.
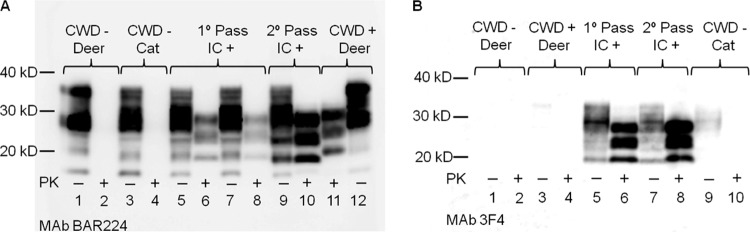
Immunoblot demonstration of FelCWD in brain (medulla) of cats. (A) Lanes 2 and 4 demonstrate complete proteinase K (PK) digestion of PrPC in CWD-negative inoculated cat and deer. Lanes 6 and 8 demonstrate FelCWD detection after PK digestion in 2 cats i.c. inoculated with deer origin CWD (primary passage DeerCWD), while lane 10 demonstrates FelCWD detection in 1 representative cat i.c. inoculated with cat-adapted CWD (secondary passage FelCWD). Lane 11 shows the molecular mass shift upon partial PK digestion of DeerCWD in a CWD+ deer. The MAb used was BAR224. (B) Lanes 1 to 4 demonstrate the absence of detection of PrPC and DeerCWD in deer. Lanes 5 to 8 show detection of PrPC and FelCWD in cats inoculated with DeerCWD or FelCWD. The MAb used was 3F4.
Fig 2.

FelCWD detection by immunohistochemistry in brain (medulla oblongata at obex) of cats. (A and B) Detection (red deposits) demonstrated in cats inoculated with deer origin DeerCWD (A) and cat-adapted FelCWD (B). Panel C demonstrates the lack of FelCWD detection in cats inoculated with age-matched cat-adapted CWD-negative deer obex. Scale bars, 10 μm. The MAb used was L42. Panels D and E represent DeerCWD detection and the lack thereof in CWD-infected and naïve deer brain, respectively.
(ii) Secondary passage of FelCWD into domestic cats.
Western blot analysis of the medulla oblongata at the level of the obex region of all five i.c.-inoculated cats revealed PK-resistant material with a molecular mass signature similar to that of primary passage cats (Fig. 1A). While antibody BAR224 recognized both deer and cat origin PrP, monoclonal antibody (MAb) 3F4 recognized only cat origin PrPs (Fig. 1). Morphologically, in contrast to the initial passage cats, in which mild, intraneuronal, and finely granular FelCWD deposits dominated, the FelCWD deposits in the obex regions of the second passage cats were (i) more extensive, (ii) more heterogenous (finely granular, coarsely granular, and clumped), and (iii) identified in multiple cellular patterns (intraneuronal, perineuronal, and linear) (Fig. 2).
Detection of prion-converting activity by RT-QuIC.
PrPC converting activity, analyzed by RT-QuIC of brain tissue, was detected in 7/7 i.c.-inoculated cats that developed prion disease (Fig. 3), providing 100% specificity and 92% sensitivity compared to Western blotting and IHC analysis. (A total of 13/14 duplicates were positive in cats that were positive by Western blotting and IHC.) RT-QuIC did not detect prions in CWD-inoculated cats that tested Western blot and IHC negative (3 i.c.-inoculated and 5 p.o.-inoculated primary passage cats) or in sham-inoculated cats.
Fig 3.

RT-QuIC detection of PrPC converting activity in the brain (medulla) of DeerCWD- and FelCWD-inoculated cats. Reaction mixtures were seeded, in duplicate, with cat or deer brain homogenate at a dilution of 10−4. Each data point represents the average ThT fluorescence of each duplicate reaction set. (A) Detection of PrPC converting activity in 2 of 5 primary passage DeerCWD i.c.-inoculated cats (cats 1 and 4). (B) Detection of PrPC converting activity in 5 of 5 secondary passage FelCWD i.c.-inoculated cats. Panels A and B demonstrate detection of PrPC converting activity in CWD+ deer (red dotted lines with X) and lack of converting activity in CWD-negative deer (black circles) and cat (black diamonds) controls.
MRI detects abnormalities in clinically ill cats.
Abnormalities were detected in 4/6 i.c.-inoculated clinically ill cats (1/1 primary passage and 3/5 secondary passage) (Fig. 4). The abnormalities in 1 DeerCWD i.c.-inoculated cat (4137) consisted of small sparse T2 and T2 FLAIR signal hyperintensities associated with the white matter of the internal capsule and minimal lateral ventricular enlargement. The multifocal T2 hyperintensities in this cat lacked mass effect or contrast enhancement. Two FelCWD i.c.-inoculated cats (4346 and 4357) had asymmetric lateral ventricle enlargement, one (4346) presenting with a focal dilation of the ventricular extension into the olfactory bulb. These changes are consistent with ex vacuo ventricular enlargement and probable loss of brain volume, since there was otherwise a lack of mass effect or signs of ventricular obstruction. One cat (4341) had a small linear cerebrospinal fluid (CSF)-filled cavitation involving a portion of the corona radiata of the parietal cerebrum, consistent with previous brain degeneration or other form of insult. Three cats (4137, 4156, and 4341) had mild subjective ventricular and sulcal prominences that could accompany early brain atrophy. Scans of three age-matched controls were normal.
Fig 4.

Abnormalities recorded in magnetic resonance imaging brain scans of cats i.c. inoculated with CWD. (A to D) CWD+ inoculated cats; (E) CWD-negative inoculated cat. White arrows indicate abnormalities. (A) Inflammatory lesions; (B) focal dilation of the olfactory extension/horn; (C) distended lateral ventricle; (D) focal cavitation. Panel E demonstrates the lack of abnormalities in an age-matched cat i.c. inoculated with CWD-naïve deer obex.
DISCUSSION
Transmission of deer origin CWD to domestic cats.
CWD, the only prion disease in a wildlife population, exhibits high transmission rates among cervids in their natural environment. Because of the facile transmission between cervids, questions arise regarding interspecies transmission and alternate host reservoirs in scavenger/predator species sympatric with CWD in nature. In this study, we have demonstrated transmission of deer origin CWD (DeerCWD) to one such scavenger species, the domestic cat, via the classical i.c. route of inoculation.
Transmission of cat-adapted CWD to domestic cats.
The successful transmission of prions to alternate hosts is determined by the species barrier, the strength of which, for prions, is dependent upon interactions between host PrPC and the infecting prion strain (30). Transmission across a species barrier requires adaptation by both the host and the infectious agent, as the original host PrPRES templates its conformation onto the new host PrPC (31). The new host PrPRES thus formed takes on strain-specific properties in the tertiary structure. Therefore, subsequent passage into this new host requires less time for efficient templating, conversion, rogue prion deposition, and progression to terminal clinical disease (31, 32). The results of this study support this host adaptation hypothesis, as upon secondary passage of cat-adapted CWD (FelCWD), 100% of FelCWD i.c.-inoculated cats developed clinical disease in about half the time required for DeerCWD-inoculated cats. In conjunction with an appreciably shortened course of clinical disease, FelCWD deposition and distribution were more extensive and diffuse (Fig. 2), suggesting a broadening of host cell tropism, i.e., the ability of feline-adapted CWD prions to infect and amplify in or on a greater variety of feline cell types.
Clinical disease in peripherally inoculated domestic cats.
A plausible route for interspecies transmission of prion diseases is via breach of mucosal surfaces during ingestion of infectious prions found in carcasses, their by-products, and in the environment. Precedence for prion trans-species transmission has been well documented with vCJD, FSE, and TME (4, 33). Studies have also demonstrated that prion diseases can be orally transmitted to many species: i.e., CWD to voles (34), Peromyscus mice (34), and ferrets (35), scrapie to squirrel monkeys (36) and hamsters (37–40), BSE to sheep (41–43), goats (41), cynomolgus macaques (44, 45), and lemurs (46), and CJD and Kuru to squirrel monkeys (36), with some requiring prior in vivo or in vitro adaptation.
In our studies, p.o. inoculation of DeerCWD was not sufficient to induce TSE. Western blotting, IHC, and RT-QuIC failed to show the presence of FelCWD in these animals, demonstrating the existence of a considerable species barrier. However, early signs of disease are currently observed in second passage p.o.- and i.p./s.c FelCWD-inoculated cats, suggesting that the ingestion of and/or open wound exposure to CWD-contaminated material could cause feline TSE disease. Naïve cats cohoused with the i.c.-inoculated FelCWD-positive cohort continue to serve as horizontal/environmental shedding test subjects and are currently housed with the i.p./s.c.-inoculated cohort. These remaining cats will continue to be monitored for further development of clinical disease, and terminally harvested tissues will be analyzed for the presence of FelCWD.
Detection of MRI abnormalities in clinically ill cats.
Cerebral MRI has been used to detect specific abnormalities between sporadic CJD (sCJD), vCJD, and nonprion rapidly progressive dementias (47–52). In sCJD patients, T2 hyperintensities are observed in the cortex and deep gray matter, in particular the striatum, whereas in vCJD, T2 hyperintensities are most commonly seen in posterior pulvinar and medial thalamus (47, 52–56). The histological basis of the T2 hyperintensities is not certain, but for at least certain subtypes of CJD, it may be related to vacuolar fluid accumulation (57). Cerebral atrophy can also become evident on MRI in CJD patients (58). DW-MRI is physiologically based and is a highly sensitive and specific marker of restricted brain diffusion in CJD patients (59, 60) and has improved the imaging diagnosis over conventional MRI. Restricted diffusion and DW-MRI signal hyperintensity have been found in the basal ganglia, thalamus, and cortex of CJD patients (60). Again, the proposed explanations for restricted diffusion vary with the CJD subtype but may be related to intraneuronal microvacuolation (57), prion protein deposition, and spongiform change (61). The combination of DW-MRI with FLAIR imaging has sensitivity and specificity reported at >91 to 98% (59, 62). Those sequences are important for early detection to inform appropriate diagnostic testing (61) and to enable timely intervention as new therapies are developed (59). Although only a small number of studies have used MRI to investigate animal models of prion disease, it has been shown that focal blood-brain barrier disruption occurs in a hamster scrapie model (63), that there are increases in T2 measurements in the septum, hippocampus, thalamus, and cortex of mice inoculated with 139A scrapie (64), and that the diffusion of tissue water was significantly reduced in the late preclinical period in mice inoculated with ME7 scrapie (65). Furthermore, a recent MRI study in sheep naturally exposed to scrapie demonstrates a generalized cerebral atrophy (66).
In this study, MRIs were performed on cats inoculated with deer or cat origin CWD. Abnormalities resembling those found in aforementioned MRI studies in CJD patients (47, 52–56, 58), including nonspecific T2 and T2 FLAIR hyperintense signal changes and ventricular enlargement consistent with cerebral atrophy, were present in the symptomatic cat inoculated with DeerCWD, whereas only ventricular enlargement was found in symptomatic cats inoculated with FelCWD. Based on the limited MRI data in other prion animal models, it appears that FelCWD most closely resembles the atrophy demonstrated in sheep scrapie. How or whether these signal abnormalities correlate with histopathological data is currently being assessed.
Comparison of CWD and BSE transmission to domestic cats.
Striking similarities are seen in the clinical presentation and duration of TSEs (4, 55). In cats, both BSE and CWD are marked by changes in behavior and gait, hyperaesthesia, ataxia, and tremors, and both progress to terminal-phase disease 3 weeks to 5 months postobservation of initial TSE signs (67–69).
BSE appears to be more efficiently transmitted to domestic cats by oral exposure than CWD. In this study, while we have shown that cats are susceptible to CWD infection by the classical intracranial route, oral transmission was seen only after subpassage into a second cohort of cats that had received lingual abrasions known to enhance transmission potential (23, 24). While the presence or absence of oral lesions was not reported in FSE cases observed by Wyatt (68, 69) and Legget (67), it is hypothesized that the affected cats consumed BSE-contaminated meat and bone in commercial cat foods.
Implications associated with trans-species transmission of CWD prions.
It has been determined that, once a prion strain has been adapted to a new host species, the prions from this new host species propagate more efficiently in a third host. Specifically, the passage of cow BSE prions in sheep or goats markedly increased the transmission efficiency into human transgenic mice (70). Although a substantial species barrier appears to exist between deer and cats, barring an invasive route of inoculation, we must consider the epidemiologic and ecologic implications associated with CWD transmission to felids, which could potentially result in the generation of prion strains adapted for natural (mucosal) routes of transmission to felids and/or other noncervid species. The high genetic identity homology observed between domestic and nondomestic cats compared to 5 other mammalian species sharing a home range with CWD-infected cervids, while warranting further analysis of protein structure and protein-protein interactions, suggests that nondomestic cats may be a susceptible reservoir species in nature (Fig. 5). If CWD has the ability to infect and establish alternate host reservoirs in nature or to initiate the adaptations necessary for trans-species transmission, this will impact not only wildlife, but also domestic species, which can lead to serious consequences for human health. Here, we have demonstrated that one scavenger/predator species (the domestic cat) that cohabitates with the natural hosts of CWD could become an intermediate host for this prion disease in nature.
ACKNOWLEDGMENTS
We thank Sallie Dahmes and David Osborn for providing the naïve white-tailed deer used in our studies, Terry Spraker for CWD-positive mule deer inoculum, Gary Mason and Nathaniel Denkers for contributions to inoculations, Nicholas Haley, Clare Hoover, Craig Miller, and Martha MacMillan for necropsy expertise, Kelly Walton, Tracy Webb, Cristina Weiner, Matthew Rosenbaum, Winona Burgess, and Elizabeth Magden for animal evaluations, Alan Elder and Amber Mayfield for laboratory support, Anca Selariu for editing, Wilfred Goldmann and Paula Stewart for generously genotyping the cats, and Justin Lee for sequence analysis.
This work was supported by contract N01-AI25491 and grant R01-NS061902 from the National Institutes of Health (NIH) and by the College Research Counsel (CRC) at Colorado State University, College of Veterinary Medicine and Biological Sciences (CSU-CVMBS), and the Department of Microbiology, Immunology and Pathology (DMIP).
Footnotes
Published ahead of print 12 December 2012
REFERENCES
- 1. Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. 1997. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature 389:498–501 [DOI] [PubMed] [Google Scholar]
- 2. Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. 1996. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 383:685–690 [DOI] [PubMed] [Google Scholar]
- 3. Marsh RF, Bessen RA. 1993. Epidemiologic and experimental studies on transmissible mink encephalopathy. Dev. Biol. Stand. 80:111–118 [PubMed] [Google Scholar]
- 4. Imran M, Mahmood S. 2011. An overview of animal prion diseases. Virol. J. 8:493 doi:10.1186/1743-422X-8-493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aldhous P. 1990. Spongiform encephalopathy found in a cat. Nature 345:194 doi:10.1038/345194c0 [DOI] [PubMed] [Google Scholar]
- 6. Miller MW, Williams ES. 2003. Prion disease: horizontal prion transmission in mule deer. Nature 425:35–36 [DOI] [PubMed] [Google Scholar]
- 7. Miller MW, Wild MA, Williams ES. 1998. Epidemiology of chronic wasting disease in captive Rocky Mountain elk. J. Wildl. Dis. 34:532–538 [DOI] [PubMed] [Google Scholar]
- 8. Williams ES, Young S. 1992. Spongiform encephalopathies in Cervidae. Rev. Sci. Tech. 11:551–567 [DOI] [PubMed] [Google Scholar]
- 9. Mathiason CK, Hays SA, Powers J, Hayes-Klug J, Langenberg J, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hoover EA. 2009. Infectious prions in pre-clinical deer and transmission of chronic wasting disease solely by environmental exposure. PLoS One 4:e5916 doi:10.1371/journal.pone.0005916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miller MW, Williams ES, Hobbs NT, Wolfe LL. 2004. Environmental sources of prion transmission in mule deer. Emerg. Infect. Dis. 10:1003–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haley NJ, Mathiason CK, Zabel MD, Telling GC, Hoover EA. 2009. Detection of sub-clinical CWD infection in conventional test-negative deer long after oral exposure to urine and feces from CWD+ deer. PLoS One 4:e7990 doi:10.1371/journal.pone.0007990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hays SA, Hayes-Klug J, Seelig DM, Wild MA, Wolfe LL, Spraker TR, Miller MW, Sigurdson CJ, Telling GC, Hoover EA. 2006. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 314:133–136 [DOI] [PubMed] [Google Scholar]
- 13. Sigurdson CJ, Williams ES, Miller MW, Spraker TR, O'Rourke KI, Hoover EA. 1999. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J. Gen. Virol. 80:2757–2764 [DOI] [PubMed] [Google Scholar]
- 14. Tamgüney G, Miller MW, Wolfe LL, Sirochman TM, Glidden DV, Palmer C, Lemus A, DeArmond SJ, Prusiner SB. 2009. Asymptomatic deer excrete infectious prions in faeces. Nature 461:529–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tamgüney G, Richt JA, Hamir AN, Greenlee JJ, Miller MW, Wolfe LL, Sirochman TM, Young AJ, Glidden DV, Johnson NL, Giles K, DeArmond SJ, Prusiner SB. 2012. Salivary prions in sheep and deer. Prion 6:52–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miller MW, Swanson HM, Wolfe LL, Quartarone FG, Huwer SL, Southwick CH, Lukacs PM. 2008. Lions and prions and deer demise. PLoS One 3:e4019 doi:10.1371/journal.pone.0004019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heisey DM, Mickelsen NA, Schneider JR, Johnson CJ, Johnson CJ, Langenberg JA, Bochsler PN, Keane DP, Barr DJ. 2010. Chronic wasting disease (CWD) susceptibility of several North American rodents that are sympatric with cervid CWD epidemics. J. Virol. 84:210–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kurt TD, Telling GC, Zabel MD, Hoover EA. 2009. Trans-species amplification of PrP(CWD) and correlation with rigid loop 170N. Virology 387:235–243 [DOI] [PubMed] [Google Scholar]
- 19. USGS, National Wildlife Health Center October 2012. Map of chronic wasting disease in North America. US Geological Survey, Reston, VA: http://www.nwhc.usgs.gov/disease_information/chro [Google Scholar]
- 20. Saunders SE, Bartelt-Hunt SL, Bartz JC. 2012. Occurrence, transmission, and zoonotic potential of chronic wasting disease. Emerg. Infect. Dis. 18:369–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saunders SE, Shikiya RA, Langenfeld K, Bartelt-Hunt SL, Bartz JC. 2011. Replication efficiency of soil-bound prions varies with soil type. J. Virol. 85:5476–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sharp A, Pastor J. 2011. Stable limit cycles and the paradox of enrichment in a model of chronic wasting disease. Ecol. Appl. 21:1024–1030 [DOI] [PubMed] [Google Scholar]
- 23. Bartz JC, Kincaid AE, Bessen RA. 2003. Rapid prion neuroinvasion following tongue infection. J. Virol. 77:583–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Denkers ND, Telling GC, Hoover EA. 2011. Minor oral lesions facilitate transmission of chronic wasting disease. J. Virol. 85:1396–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seelig DM, Mason GL, Telling GC, Hoover EA. 2011. Chronic wasting disease prion trafficking via the autonomic nervous system. Am. J. Pathol. 179:1319–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, Onwubiko HA, Priola SA, Caughey B. 2008. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat. Methods 5:211–212 [DOI] [PubMed] [Google Scholar]
- 27. Wilham JM, Orrú CD, Bessen RA, Atarashi R, Sano K, Race B, Meade-White KD, Taubner LM, Timmes A, Caughey B. 2010. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 6:e1001217 doi:10.1371/journal.ppat.1001217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Orrù CD, Wilham JM, Vascellari S, Hughson AG, Caughey B. 2012. New generation QuIC assays for prion seeding activity. Prion 6:147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bucy DS, Brown MS, Bielefeldt-Ohmann H, Thompson J, Bachand AM, Morges M, Elder JH, Vandewoude S, Kraft SL. 2011. Early detection of neuropathophysiology using diffusion-weighted magnetic resonance imaging in asymptomatic cats with feline immunodeficiency viral infection. J. Neurovirol. 17:341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Collinge J, Clarke AR. 2007. A general model of prion strains and their pathogenicity. Science 318:930–936 [DOI] [PubMed] [Google Scholar]
- 31. Prusiner SB. 1998. Prions. Proc. Natl. Acad. Sci. U. S. A. 95:13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Plinston C, Hart P, Chong A, Hunter N, Foster J, Piccardo P, Manson JC, Barron RM. 2011. Increased susceptibility of human-PrP transgenic mice to bovine spongiform encephalopathy infection following passage in sheep. J. Virol. 85:1174–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Imran M, Mahmood S. 2011. An overview of human prion diseases. Virol. J. 8:559 doi:10.1186/1743-422X-8-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kurt TD, Seelig DM, Schneider JR, Johnson CJ, Telling GC, Heisey DM, Hoover EA. 2011. Alteration of the chronic wasting disease species barrier by in vitro prion amplification. J. Virol. 85:8528–8537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perrott MR, Sigurdson CJ, Mason GL, Hoover EA. 2012. Evidence for distinct chronic wasting disease (CWD) strains in experimental CWD in ferrets. J. Gen. Virol. 93:212–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gibbs CJ, Amyx HL, Bacote A, Masters CL, Gajdusek DC. 1980. Oral transmission of kuru, Creutzfeldt-Jakob Disease, and scrapie to nonhuman primates. J. Infect. Dis. 142:205–208 [DOI] [PubMed] [Google Scholar]
- 37. Bergström A-L, Jensen TK, Heegaard PMH, Cordes H, Hansen VB, Laursen H, Lind P. 2006. Short-term study of the uptake of PrP(Sc) by the Peyer's patches in hamsters after oral exposure to scrapie. J. Comp. Pathol. 134:126–133 [DOI] [PubMed] [Google Scholar]
- 38. Murayama Y, Yoshioka M, Okada H, Takata M, Yokoyama T, Mohri S. 2007. Urinary excretion and blood level of prions in scrapie-infected hamsters. J. Gen. Virol. 88:2890–2898 [DOI] [PubMed] [Google Scholar]
- 39. Prusiner SB, Cochran SP, Alpers MP. 1985. Transmission of scrapie in hamsters. J. Infect. Dis. 152:971–978 [DOI] [PubMed] [Google Scholar]
- 40. Thomzig A, Kratzel C, Lenz G, Krüger D, Beekes M. 2003. Widespread PrPSc accumulation in muscles of hamsters orally infected with scrapie. EMBO Rep. 4:530–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Foster JD, Hope J, Fraser H. 1993. Transmission of bovine spongiform encephalopathy to sheep and goats. Vet. Rec. 133:339–341 [DOI] [PubMed] [Google Scholar]
- 42. Jeffrey M, Martin S, González L, Ryder SJ, Bellworthy SJ, Jackman R. 2001. Differential diagnosis of infections with the bovine spongiform encephalopathy (BSE) and scrapie agents in sheep. J. Comp. Pathol. 125:271–284 [DOI] [PubMed] [Google Scholar]
- 43. McCutcheon S, Alejo Blanco AR, Houston EF, de Wolf C, Tan BC, Smith A, Groschup MH, Hunter N, Hornsey VS, MacGregor IR, Prowse CV, Turner M, Manson JC. 2011. All clinically-relevant blood components transmit prion disease following a single blood transfusion: a sheep model of vCJD. PLoS One 6:e23169 doi:10.1371/journal.pone.0023169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Herzog C, Salès N, Etchegaray N, Charbonnier A, Freire S, Dormont D, Deslys J-P, Lasmézas CI. 2004. Tissue distribution of bovine spongiform encephalopathy agent in primates after intravenous or oral infection. Lancet 363:422–428 [DOI] [PubMed] [Google Scholar]
- 45. Lasmézas CI, Comoy E, Hawkins S, Herzog C, Mouthon F, Konold T, AuvrÉ F, Correia E, Lescoutra-Etchegaray N, Salès N, Wells G, Brown P, Deslys J-P. 2005. Risk of oral infection with bovine spongiform encephalopathy agent in primates. Lancet 365:781–783 [DOI] [PubMed] [Google Scholar]
- 46. Bons N, Mestre-Frances N, Belli P, Cathala F, Gajdusek DC, Brown P. 1999. Natural and experimental oral infection of nonhuman primates by bovine spongiform encephalopathy agents. Proc. Natl. Acad. Sci. U. S. A. 96:4046–4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Collie DA, Summers DM, Sellar RJ, Ironside JW, Cooper S, Zeidler M, Knight R, Will RG. 2003. Diagnosing variant Creutzfeldt-Jakob disease with the pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases. Am. J. Neuroradiol. 24:1560–1569 [PMC free article] [PubMed] [Google Scholar]
- 48. Finkenstaedt M, Szudra A, Zerr I, Poser S, Hise JH, Stoebner JM, Weber T. 1996. MR imaging of Creutzfeldt-Jakob disease. Radiology 199:793–798 [DOI] [PubMed] [Google Scholar]
- 49. Gertz HJ, Henkes H, Cervos-Navarro J. 1988. Creutzfeldt-Jakob disease: correlation of MRI and neuropathologic findings. Neurology 38:1481–1482 [DOI] [PubMed] [Google Scholar]
- 50. Schröter A, Zerr I, Henkel K, Tschampa HJ, Finkenstaedt M, Poser S. 2000. Magnetic resonance imaging in the clinical diagnosis of Creutzfeldt-Jakob disease. Arch. Neurol. 57:1751–1757 [DOI] [PubMed] [Google Scholar]
- 51. Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres-Chae C, Johnson DY, Miller BL, Geschwind MD. 2011. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology 76:1711–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Young GS, Geschwind MD, Fischbein NJ, Martindale JL, Henry RG, Liu S, Lu Y, Wong S, Liu H, Miller BL, Dillon WP. 2005. Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am. J. Neuroradiol. 26:1551–1562 [PMC free article] [PubMed] [Google Scholar]
- 53. Linguraru MG, Ayache N, González Ballester MA, Bardinet E, Galanaud D, Haïk S, Faucheux B, Cozzone P, Dormont D, Brandel J-P. 2005. New ratios for the detection and classification of CJD in multisequence MRI of the brain. Med. Image Comput. Comput. Assist. Interv. 8:492–499 [DOI] [PubMed] [Google Scholar]
- 54. Moreno F, Arriola Larrarte L. 2006. Neuroimaging in the diagnosis of human transmissible spongiform encephalopathies. Neurología 21:428–436 [PubMed] [Google Scholar]
- 55. Sikorska B, Knight R, Ironside JW, Liberski PP. 2012. Creutzfeldt-Jakob disease. Adv. Exp. Med. Biol. 724:76–90 [DOI] [PubMed] [Google Scholar]
- 56. Zeidler M, Sellar RJ, Collie DA, Knight R, Stewart G, Macleod MA, Ironside JW, Cousens S, Colchester AC, Hadley DM, Will RG. 2000. The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt-Jakob disease. Lancet 355:1412–1418 [DOI] [PubMed] [Google Scholar]
- 57. Collie DA, Sellar RJ, Zeidler M, Colchester ACF, Knight R, Will RG. 2001. MRI of Creutzfeldt-Jakob disease: imaging features and recommended MRI protocol. Clin. Radiol. 56:726–739 [DOI] [PubMed] [Google Scholar]
- 58. Lou X, Ma L, An N, Cai YQ, Liang Y, Guo XG. 2006. Diffusion-weighted magnetic resonance imaging in diagnosis of Creutzfeldt-Jakob disease. Chin. Med. J. 119:1242–1247 [PubMed] [Google Scholar]
- 59. Hyare H, Thornton J, Stevens J, Mead S, Rudge P, Collinge J, Yousry TA, Jäger HR. 2010. High-b-value diffusion MR imaging and basal nuclei apparent diffusion coefficient measurements in variant and sporadic Creutzfeldt-Jakob disease. Am. J. Neuroradiol. 31:521–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lee H, Hoffman C, Kingsley PB, Degnan A, Cohen O, Prohovnik I. 2010. Enhanced detection of diffusion reductions in Creutzfeldt-Jakob disease at a higher B factor. Am. J. Neuroradiol. 31:49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kandiah N, Tan K, Pan AB, Au WL, Venketasubramanian N, Tchoyoson Lim CC, Tan NC. 2008. Creutzfeldt-Jakob disease: which diffusion-weighted imaging abnormality is associated with periodic EEG complexes? J. Neurol. 255:1411–1414 [DOI] [PubMed] [Google Scholar]
- 62. Riva-Amarante E, Jiménez-Huete A, Toledano R, Calero M, Alvarez-Linera J, Escribano J, Sánchez Migallón MJ, Franch O. 2011. Usefulness of high b-value diffusion-weighted MRI in the diagnosis of Creutzfeldt-Jakob disease. Neurología 26:331–336 [DOI] [PubMed] [Google Scholar]
- 63. Chung YL, Williams A, Beech JS, Williams SC, Bell JD, Cox IJ, Hope J. 1995. MRI assessment of the blood-brain barrier in a hamster model of scrapie. Neurodegeneration 4:203–207 [DOI] [PubMed] [Google Scholar]
- 64. Sadowski M, Tang CY, Aguinaldo JG, Carp R, Meeker HC, Wisniewski T. 2003. In vivo micro magnetic resonance imaging signal changes in scrapie infected mice. Neurosci. Lett. 345:1–4 [DOI] [PubMed] [Google Scholar]
- 65. Broom KA, Anthony DC, Lowe JP, Griffin JL, Scott H, Blamire AM, Styles P, Perry VH, Sibson NR. 2007. MRI and MRS alterations in the preclinical phase of murine prion disease: association with neuropathological and behavioural changes. Neurobiol. Dis. 26:707–717 [DOI] [PubMed] [Google Scholar]
- 66. McKnight AL, Minkoff LA, Sutton DL, Thomsen BV, Habecker PL, Sweeney RW, Smith G, Dasanu CA, Ichim TE, Alexandrescu DT, Stutman JM. 2010. Generalized cerebral atrophy seen on MRI in a naturally exposed animal model for Creutzfeldt-Jakob disease. J. Transl. Med. 8:125 doi:10.1186/1479-5876-8-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Leggett MM, Dukes J, Pirie HM. 1990. A spongiform encephalopathy in a cat. Vet. Rec. 127:586–588 [PubMed] [Google Scholar]
- 68. Wyatt JM, Pearson GR, Smerdon TN, Gruffydd-Jones TJ, Wells GAH. 1990. Spongiform encephalopathy in a cat. Vet. Rec. 126:513. [DOI] [PubMed] [Google Scholar]
- 69. Wyatt JM, Pearson GR, Smerdon TN, Gruffydd-Jones TJ, Wells GAH, Wilesmith JW. 1991. Naturally occurring scrapie-like spongiform encephalopathy in five domestic cats. Vet. Rec. 129:233–236 [DOI] [PubMed] [Google Scholar]
- 70. Padilla D, Béringue V, Espinosa JC, Andreoletti O, Jaumain E, Reine F, Herzog L, Gutierrez-Adan A, Pintado B, Laude H, Torres JM. 2011. Sheep and goat BSE propagate more efficiently than cattle BSE in human PrP transgenic mice. PLoS Pathog. 7:e1001319 doi:10.1371/journal.ppat.1001319 [DOI] [PMC free article] [PubMed] [Google Scholar]