

Abstract
STUDY QUESTION
What is the heritability of C-reactive protein (CRP) levels in women with polycystic ovary syndrome (PCOS) and their first-degree relatives?
SUMMARY ANSWER
Women with PCOS and their siblings are more likely to have elevated CRP levels when both of their parents have elevated CRP. This PCOS family-based study indicates that CRP levels are likely a heritable trait.
WHAT IS KNOWN ALREADY
Previous studies have established that an elevated blood level of CRP is variably present in women with PCOS, and may be present independent of metabolic status.
STUDY DESIGN, SIZE AND DURATION
A familial based phenotyping study consisting of 81 families comprised of PCOS patients and their first-degree relatives for 305 subjects.
PARTICIPANTS/MATERIALS, SETTING AND METHODS
Study conducted at an academic health center. An elevated CRP level was defined as >28.6 nmol/l. To account for familial clustering, generalized estimating equations with a logit link were used to model the association between elevated CRP levels in patients with PCOS and their siblings with their parental group (A = neither parent with elevated CRP; B = one parent with elevated CRP; C= both parents with elevated CRP), adjusting for gender, age and BMI of the offspring. We did additional heritability analyses by using a variance component estimation method for CRP levels, adjusting for sex, age and BMI.
MAIN RESULTS AND THE ROLE OF CHANCE
We observed elevated CRP levels in 94% of the offspring in group C, 45% in group B and 10% in group A after adjusting for age, gender and BMI of the offspring. The median BMI of the offspring in group A, B and C were 30.0, 28.7 and 31.2 kg/m2, respectively. Heritability estimates of CRP levels ranged from 0.75 to 0.83 and remained significant after excluding for type 2 diabetes mellitus. Our small sample size increases the possibility of a type 1 error.
LIMITATIONS, REASONS FOR CAUTION
This is a single report in an adequately powered but limited sample size study identifying the strong heritability of CRP levels. Replication in other large family cohorts is necessary.
WIDER IMPLICATION OF THE FINDINGS
These findings support the concept that there is an increased cardiovascular disease risk profile in families of women with PCOS.
STUDY FUNDING/COMPETING INTEREST
This research was supported by National Institutes of Health grants U54HD-034449 and P50 HD044405 (A.D.). Priyathama Vellanki is supported in part by NIH/NIDDK Training Grant T32 DK007169.
Keywords: C-reactive protein, cardiovascular risk, hyperandrogenism, heritability, first-degree relatives
Introduction
Polycystic ovary syndrome (PCOS) is the most common hormonal abnormality occurring in ∼5–8% of female population of reproductive age (Azziz et al., 2004). Previous studies have shown a high incidence of familial clustering of PCOS, suggesting that PCOS has a genetic component (Carey et al., 1993; Legro et al., 1998). The prevalence of metabolic syndrome (MetS) is high in women with PCOS compared with the general US population (Ehrmann et al., 2006), with mixed associations in other populations (Hosseinpanah et al., 2011; Wijeyaratne et al., 2011). First-degree relatives are also affected (Sam et al., 2006; Coviello et al., 2009). The MetS and its components are associated with measures of inflammatory markers, such as C-reactive protein (CRP) levels (Ridker et al., 2003).
Previous studies have shown a positive correlation between parents and their offspring with respect to CRP levels (Pankow et al., 2001; Hersh et al., 2006). CRP is a strong independent predictor of future cardiovascular disease (CVD) and type 2 diabetes mellitus (T2DM) (Ridker et al., 2000). Epidemiological evidence has shown that elevated CRP levels in women with PCOS are associated with other surrogate markers of CVD even in young women with PCOS (Boulman et al., 2004; Cascella et al., 2006; Orio et al., 2006). However, studies have reported comparable CRP levels between women with PCOS and control women who had similar age and BMI distributions (Meyer et al., 2005; Topcu et al., 2006)
Hence, elevated CRP levels in women with PCOS may be confounded by obesity or heritability. As the issue remains controversial whether CRP levels are normal or increased in women with PCOS, our goal was to study the familial aggregation and heritability of CRP in women with PCOS and their first-degree relatives. We hypothesize that CRP levels have a high heritable component in women with PCOS and their families.
Methods
Study population and research design
The study consisted of 81 families comprised of women with PCOS (n = 81) and their first-degree relatives (father (n = 81), mother (n = 81), brothers (n = 21) and sisters (n = 41, of which 34 had PCOS, 5 non-PCOS and 2 unknown PCOS status)) for 305 subjects who participated in our ongoing phenotype and genotype studies of familial PCOS. We have previously reported data on 78 of 81 probands (and family members) (Legro et al., 1998; Sam et al., 2006; Coviello et al., 2009). In this study, the 81 probands were chosen from families recruited from the Central Pennsylvania area who were part of a complete trio (proband, mother and father), and all had available serum for the CRP assay.
The Institutional Review Board of Pennsylvania State University College of Medicine, Hershey, PA, approved the study protocol. Written informed consent was obtained from all participants. Women with PCOS and their siblings and parents were eligible to participate. All of the PCOS participants included in the study met the NIH PCOS definition criteria (Legro et al., 1998): (i) spontaneous intermenstrual periods of ≥45 days or ≤8 periods per year, and (ii) serum total testosterone >2 nmol/l or bioavailable testosterone >0.52 nmol/l, while not taking oral contraceptives. The quantitative determination of CRP levels was done using Mesoscale Discovery CRP assay kit (Gaithersburg, MD) having an interassay coefficient of variation of 9.7%. Clinical significance of CRP levels based on the American Heart Association (http://www.americanheart.org) are (i) <9.5 nmol/l = low risk for CVD, (ii) 9.5–28.6 nmol/l = intermediate risk for CVD and (iii) >28.6 nmol/l = high risk for CVD. We defined a CRP level >28.6 nmol/l as elevated in our study. T2DM was defined as fasting plasma glucose (FPG) ≥7 mmol/l. MetS as defined by the American Heart Association 2005 guidelines (Grundy, 2005): waist circumference (WC) ≥102 cm in men and ≥88 cm in women, triglyceride levels (TTG) ≥1.7 mmol/l or on drug treatment for elevated TTG, high-density lipoprotein cholesterol levels (HDL-C) <1.03 mmol/l in men or <1.3 mmol/l in women, systolic blood pressure ≥130 mmHg or diastolic blood pressure ≥85 mmHg or on drug treatment to lower blood pressure, FPG ≥5.55 mmol/l. If three of the five criteria were met, subjects were identified as having MetS.
We used two different analyses, logit link model and variance component analysis, to test the effects of parental CRP levels on their offspring. To account for familial clustering, generalized estimating equations (GEE) (Zeger and Liang, 1986) with a logit link were used to model the association between elevated CRP levels in women with PCOS and their siblings (i.e. dependent variable) with their parental CRP group (i.e. group A: neither parent having elevated CRP, group B: only one parent having elevated CRP, or group C: both parents having elevated CRP). Analyses were adjusted for gender, age and BMI of the offspring. GEE with a logit link is an extension of logistic regression that accounts for correlated data, e.g. children from the same family. As a secondary analysis, a mixed-effects model was used to assess CRP levels on a continuous scale to compare female offspring with and without PCOS. As the CRP levels were skewed, the natural logarithm of the CRP levels were taken to meet mixed-effects modeling assumptions and thus comparisons between parental groups are reported as the ratio of geometric means, due to back-transforming, and 95% confidence intervals (CIs). All hypothesis tests were two sided. All baseline characteristics and hypothesis tests were performed using SAS software (version 9.2; SAS Institute Inc., Cary, NC).
We also calculated heritability estimates (h2) of natural log transformed CRP levels for the 81 families included in this study. A heritability estimate (h2) shows the proportion of phenotypic variance that can be explained by genetic variance (Almasy and Blangero, 1998). As CRP levels could be elevated in T2DM, additional analyses excluding participants with T2DM were performed. In these analyses, the whole family was excluded if a proband had T2DM (n = 2 families and 7 people). If a non-proband family member had T2DM (1 non-PCOS sister, 1 brother, 2 mothers, 10 fathers), they were also excluded from the analysis. However, the family was still included because all families had at least one unaffected family member in addition to the proband. FPG was missing from 4 fathers and 2 mothers; they were included in the analysis.
We also investigated if there was a sex-specific parental effect on heritability of CRP levels. For calculation of maternal h2, we did a heritability analysis by making the phenotypic information on fathers unknown. For paternal h2, we made the phenotypic information on mothers unknown. This was done excluding participants with T2DM. All h2 were performed using Sequential Oligogenic Linkage Analysis Routines (SOLAR, version 4.2.7, Southwest Foundation for Biomedical Research). Proband ascertainment was employed and all covariates were adjusted for age, age2, BMI and sex.
Sample size justification
A sample size of 27 families per parental CRP group (81 families in total) would provide 81% power to detect a trend across parental CRP groups in the elevated CRP proportions among offspring using a two-sided test for trend from a logistic regression model with a type I error rate of 5%. We assumed that the proportion of offspring having elevated CRP levels would be 0.03 in parental group A, 0.10 in parental group B and 0.30 in parental group C.
Results
Baseline characteristics (Table I) show that all family members were overweight, except for probands who were obese. Diabetes was present (<5%) in each group except for fathers. MetS was most prevalent in PCOS probands. Among the probands, most met criteria for MetS due to a high WC.
Table I.
Baseline characteristics of study participants.
| PCOS, probands median (Q1,Q3) [n] | Fathers median (Q1,Q3) [n] | Mothers median (Q1,Q3) [n] | Brothers median (Q1,Q3) [n] | Sisters median (Q1,Q3) [n] | |
|---|---|---|---|---|---|
| Age (years) | 28 (25, 32) [81] | 57 (52, 62) [81] | 54 (50, 57) [81] | 28 (24, 35) [21] | 29 (23, 35) [41] |
| BMI (kg/m2) | 37.1 (30.3, 41.3) [81] | 29.8 (26.4, 32.3) [79] | 29.1 (25.4, 34.4) [81] | 26.0 (24.8, 29.6) [21] | 26.7 (22.3, 29.2) [41] |
| CRP (nmol/l) | 114 (29, 524) [81] | 76 (29, 362) [81] | 86 (38, 324) [81] | 38 (19, 162) [21] | 48 (19, 181) [41] |
| Percentage with diabetes, MetS and MetS components meeting MetS criteria within each group | |||||
| Diabetes % (n/total n) | 2.5 (2/81) | 13.0 (10/77) | 2.5 (2/79) | 4.8 (1/21) | 2.4 (1/41) |
| MetS % (n/total n) | 57.7 (45/78) | 48.6 (36/74) | 50.7 (38/75) | 15.8 (3/19) | 13.7 (5/37) |
| WC % (n/total n) | 80.2 (65/81) | 51.4 (37/72) | 58.4 (45/77) | 11.8 (2/17) | 45.0 (18/40) |
| TTG % (n/total n) | 51.2 (41/80) | 47.5 (41/79) | 51.9 (41/79) | 23.8 (5/21) | 17.1 (7/41) |
| HDL-C % (n/total n) | 81.2 (65/80) | 42.5 (34/80) | 49.4 (39/79) | 28.6 (6/21) | 56.1 ( 23/41) |
| HTN % (n/total n) | 35.5 (27/76) | 64.1 (43/67) | 54.8 (40/73) | 60.0 (9/15) | 11.1 (3/27) |
| FPG % (n/total n) | 17.3 (14/81) | 49.3 (38/77) | 29.11 (23/79) | 9.5 (2/21) | 4.9 (2/41) |
n, number of people with metabolic syndrome or diabetes; total n, number of people who have information to determine if they have the disease status. All percentages were calculated for each group using the total number of people for whom information was available.
CRP, C-reactive protein; Diabetes, fasting plasma glucose ≥7 mmol/l; FPG, fasting plasma glucose, ≥5.55 mmol/l; HDL-C, high-density lipoprotein cholesterol, <1.04 mmol/l in men, <1.30 mmol/l in women; HTN, hypertension, systolic blood pressure ≥130 mmHg or diastolic blood pressure ≥85 mmHg; MetS, metabolic syndrome; TTG, triglyceride levels, ≥1.7 mmol/l; WC, waist circumference, ≥102 cm in men, ≥88 cm in women.
Sixty-two percent of the families (n = 50) were classified into parental group C where both parents had elevated CRP levels, 27% (n = 22) in parental group B where one parent had an elevated CRP level, and 11% (n = 9) in parental group A where neither parent had an elevated CRP level (88, 37 and 18 offspring per group, respectively). As hypothesized, and not adjusting for covariates, a greater percentage of offspring had elevated CRP levels in parental group C (84%), followed by parental group B (43%), and then parental group A (17%). Adjusting for offspring gender, age and BMI using GEE with a logit link, 94% of the offspring had elevated CRP levels in parental group C, 45% in parental group B and 10% in parental group A (linear trend test P < 0.0001; Fig. 1). The median (25th, 75th percentile) for age, CRP and BMI of the offspring were as follows: parental group A [27 (23, 34) years, 10 (5, 29) nmol/l, 30.0 (24.3, 37.1) kg/m²], parental group B [30 (25, 33) years, 29 (14, 48) nmol/l, 28.7 (25.8, 37.6) kg/m²] and parental group C [28 (24, 33) years, 190 (76, 548) nmol/l, 31.2 (25.9, 39.5) kg/m²]. Offspring in parental group C and the offspring in parental group A were both borderline obese with no differences between parental groups.
Figure 1.
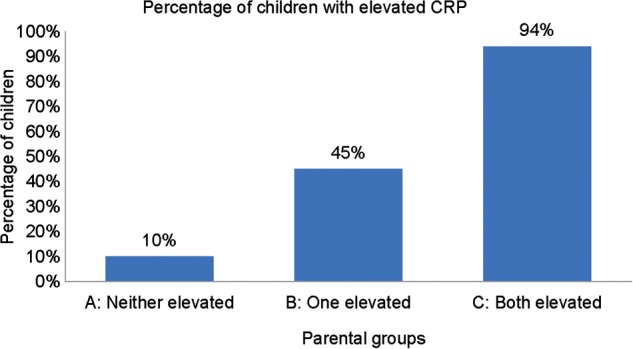
Primary analysis: percentage of elevated CRP levels defined as >28.6 nmol/l in offspring adjusted for their gender, age, and BMI using GEE with a logit link to account for familial clustering. Linear trend test P < 0.0001.
Significant differences were observed when examining all pairwise comparisons between the three parental groups with the effect size quantified using odds ratios (OR) and 95% CIs. The odds of having offspring with elevated CRP levels were 138 times greater in parental group C compared with parental group A [OR = 138, 95% CI (19, 998), P < 0.0001]. Similarly, it was 18 times greater in parental group C compared with parental group B [OR = 17.6, 95% CI (4.5, 68.1), P < 0.0001] and eight times in parental group B compared with parental group A [OR = 7.8, 95% CI (1.6, 38.6), P < 0.01]. We noted no significant difference between history of confounding medication use and elevated CRP levels using a chi-squared test (P = 0.39). Of the parents with elevated CRP, 41% (51 of 121) were on medications that may affect CRP levels (i.e. insulin sensitizing agents, antihypertensives, lipid lowering agents etc.) versus 50% (20 of 40) parents without an elevated CRP level (with one parent missing a full medication history).
In our secondary analysis, we compared female siblings with PCOS (n = 86) and those without PCOS (n = 34). Not adjusting for covariates, the percentage of female offspring having elevated CRP levels increases from parental group A to parental group C. This increasing trend in female siblings in both the women with and without PCOS is similar to our observed results when using all offspring (males and females) regardless of female PCOS status. Adjusting for age, BMI and family cluster, significant differences were observed when examining all pairwise comparisons among the three parental groups (Table II). A significant dose–response is found with significantly increased odds of an elevated CRP level in female siblings with increasing parental load of elevated CRP levels, regardless of their PCOS status. We examined the prevalence of individual components of the MetS in the children of PCOS parents according to their parental load of CRP. We noted no association between increasing parental CRP load and affected status for the individual MetS components (Table III).
Table II.
Mixed-effects models for comparing CRP levels among parental groups (elevated CRP defined as >28.6 nmol/l) for PCOS and non-PCOS daughters adjusted for age, BMI and family clusters.
| Pairwise comparisons of parental groups | |||
|---|---|---|---|
| Proband and sisters | Parental group in relation to CRP level status | Ratio of means (95% CI) | P |
| PCOS | Both versus neither elevated | 11.8 (5.5, 25.1) | <0.0001 |
| Both versus one elevated | 6.0 (3.5, 10.5) | <0.0001 | |
| One versus neither elevated | 2.0 (0.8, 4.5) | 0.11 | |
| Non-PCOS | Both versus neither elevated | 21.7 (5.8, 80.7) | <0.0001 |
| Both versus one elevated | 5.6 (2.4, 13.3) | 0.0001 | |
| One versus either elevated | 3.9 (1.0, 15.8) | 0.06 | |
CRP, C-reactive protein; PCOS, polycystic ovary syndrome.
Table III.
Prevalence of components of the metabolic syndrome among probands with PCOS and their siblings based on parental CRP status.
| A: Neither parent elevated |
B: One parent elevated |
C: Both parents elevated |
||||
|---|---|---|---|---|---|---|
| Raw proportion (%) | Adjusted proportiona (95% CI) | Raw proportion (%) | Adjusted proportiona (95% CI) | Raw proportion (%) | Adjusted proportiona (95% CI) | |
| Abnormal blood pressure | 2/13 (15%) | 0.24 (0.08, 0.55) | 12/31 (39%) | 0.65 (0.40,0.84) | 29/74 (39%) | 0.54 (0.36,0.71) |
| Abnormal glucose | 1/18 (6%) | 0.03 (0.00,0.28) | 4/37 (11%) | 0.06 (0.02,0.16) | 7/88 (8%) | 0.02 (0.01,0.07) |
| Abnormal triglycerides | 5/18 (28%) | 0.23 (0.09,0.46) | 19/37 (51%) | 0.47 (0.24,0.71) | 29/87 (33%) | 0.25 (0.14,0.42) |
| Abnormal HDL | 10/18 (56%) | 0.49 (0.26,0.72) | 22/37 (59%) | 0.52 (0.28,0.75) | 62/87 (71%) | 0.65 (0.44,0.82) |
| Abnormal waist circumference | 13/18 (72%) | 0.96 (0.82,0.99) | 21/34 (62%) | 0.79 (0.31,0.97) | 50/86 (58%) | 0.51 (0.11,0.90) |
| Metabolic syndrome | 5/15 (33%) | 0.25 (0.07,0.58) | 16/34 (47%) | 0.41 (0.15,0.73) | 33/82 (40%) | 0.24 (0.08,0.52) |
CRP, C-reactive protein; HDL, high-density lipoprotein; PCOS, polycystic ovary syndrome.
aAdjusted Adjusted for gender, age, BMI and familial cluster.
For our heritability analyses (Table IV), we found high h2 ranging from 0.75–0.83 for CRP levels within families of women with PCOS. These remained high after adjustments of age, age2, BMI and sex. Even after exclusion of people with T2DM, h2 did not change significantly and remained high. We also found slightly higher maternal than paternal heritability estimates for CRP levels (Table V).
Table IV.
Heritability (h2) estimates for CRP levels in PCOS families.
| All participants |
Excluding participants with T2DM |
||||
|---|---|---|---|---|---|
| n | h2 ± SE | P | n | h2 ± SE | P |
| 305 | 0.75 ± 0.07 | 1.59 × 10−19 | 284 | 0.73 ± 0.08 | 2.20 × 10−17 |
| Model–no covariates | |||||
| 305 | 0.79 ± 0.07 | 7.94 × 10−22 | 284 | 0.77 ± 0.07 | 1.72 × 10−19 |
| Model–age | |||||
| 305 | 0.76 ± 0.07 | 2.27 × 10−20 | 284 | 0.75 ± 0.08 | 5.18 × 10−18 |
| Model–age2 | |||||
| 303 | 0.80 ± 0.07 | 4.30 × 10−21 | 282 | 0.79 ± 0.07 | 3.68 × 10−19 |
| Model–BMI | |||||
| 303 | 0.83 ± 0.07 | 5.14 × 10−23 | 282 | 0.81 ± 0.07 | 1.11 × 10−20 |
| Model–age and BMI | |||||
| 305 | 0.75 ± 0.07 | 1.65 × 10−19 | 284 | 0.73 ± 0.08 | 2.21 × 10−17 |
| Model–sex | |||||
| 303 | 0.83 ± 0.07 | 3.87 × 10−23 | 282 | 0.80 ± 0.07 | 9.66 × 10−21 |
| Model–age, sex and BMI | |||||
CRP, C-reactive protein; PCOS, polycystic ovary syndrome; T2DM, type 2 diabetes mellitus; SE, standard error.
Table V.
Parental effects on heritability (h2) estimates of CRP levels in PCOS families.
| All participants |
Excluding T2DM |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Maternal h2 |
Paternal h2 |
Maternal h2 |
Paternal h2 |
||||||||
| N | h2 ± SE | P | n | h2 ± SE | P | N | h2 ± SE | P | n | h2 ± SE | P |
| 224 | 0.94 ± 0.13 | 9.35 × 10−15 | 224 | 0.85 ± 0.13 | 5.36 × 10−13 | 215 | 0.94 ± 0.14 | 4.12 × 10−14 | 202 | 0.83 ± 0.14 | 2.89 × 10−11 |
| Model–no covariates | |||||||||||
| 224 | 1.00 | 9.54 × 10−18 | 224 | 0.88 ± 0.13 | 7.36 × 10−14 | 215 | 1.00 | 4.67 × 10−17 | 202 | 0.84 ± 0.14 | 6.34 × 10−12 |
| Model–age | |||||||||||
| 224 | 0.95 ± 0.13 | 7.73 × 10−15 | 224 | 0.85 ± 0.13 | 4.71 × 10−13 | 215 | 0.95 ± 0.14 | 3.62 × 10−14 | 202 | 0.83 ± 0.14 | 2.93 × 10−11 |
| Model–age2 | |||||||||||
| 224 | 1.00 | 3.16 × 10−17 | 221 | 1.00 | 5.90 × 10−15 | 215 | 1.00 | 8.46 × 10−17 | 199 | 1.00 | 3.18 × 10−14 |
| Model–BMI | |||||||||||
| 224 | 1.00 | 6.56 × 10−20 | 221 | 1.00 | 1.14 × 10−15 | 215 | 1.00 | 6.23 × 10−19 | 199 | 1.00 | 1.16 × 10−14 |
| Model–age and BMI | |||||||||||
| 224 | 0.97 ± 0.13 | 1.37 × 10−15 | 224 | 0.84 ± 0.13 | 5.97 × 10−13 | 215 | 1.00 | 5.51 × 10−15 | 202 | 0.81 ± 0.14 | 4.40 × 10−11 |
| Model–sex | |||||||||||
| 224 | 1.00 | 5.68 × 10−20 | 221 | 1.00 | 7.16 × 10−16 | 215 | 1.00 | 6.03 × 10−19 | 199 | 1.00 | 8.82 × 10−15 |
| Model–age, sex and BMI | |||||||||||
CRP, C-reactive protein; PCOS, polycystic ovary syndrome; T2DM, type 2 diabetes mellitus; SE, standard error.
Discussion
Elevated circulating CRP levels suggest a proinflammatory condition observed in several disorders linked to insulin resistance including obesity, T2DM, CVD, and cancer. In this family-based study, we found a significant positive correlation of CRP levels between the parental groups and their offspring independent of PCOS status or BMI. We noted that the parents of women with PCOS tend to have elevated CRP levels, and in fact in the majority of families (62%) both parents are affected. The risk of having an elevated CRP level in offspring is eight times greater when at least one parent has an elevated CRP when compared with neither parent having elevated CRP. Fecundity in the proband generation declines with an abnormal parental CRP load, but no effect is noted among the mothers of women with PCOS. We also found high heritability estimates of CRP levels in families of women with PCOS. This remained significantly high even after adjustment for covariates known to affect CRP levels, suggesting that CRP levels are influenced by genetic variance within these families.
The present investigation expands on our previous family-based studies showing clustering of insulin resistance and its sequelae in first-degree relatives of women with PCOS (Legro et al., 2002; Sam et al., 2006; Coviello et al., 2009). This study also implicates that CRP levels in PCOS are predominantly heritable on the parent's CRP levels. Our study shows a higher heritability of CRP than other studies (Pankow et al., 2001; Lange et al., 2006; Wessel et al., 2007). These findings are supported from previous studies which have demonstrated a significant heritability of CRP levels in the general population (Pankow et al., 2001; Fox et al., 2008).
There are several limitations to our study. First, we have previously reported a high prevalence of clustering of metabolic abnormalities in these families; therefore, it is likely that we would also find a high prevalence and clustering of CRP levels in these same families. Our study did not utilize control families identified on the basis of a proband without PCOS as we were specifically studying CVD risk within PCOS families. Thus, we cannot answer the question whether CRP levels are elevated in women with PCOS and their first-degree relatives compared with control families. However, our family-based study does include sisters without PCOS allowing for controls within the family. Second, we used a research assay for CRP levels, instead of a clinical assay. However, we corrected for interassay variability by running families on the same plate and any variation between our assay and a clinical assay would have been uniform across families.
Other limitations are a relatively small sample size, lack of a control population and undiagnosed T2DM. We have results with a few wide CIs in relation to some population-based studies of CRP levels. However, our sample size was large in relation to previously reported studies in women with PCOS. We lacked a control population, but our aim was not to compare relative risk of PCOS families, but to examine the heritability of CRP levels within PCOS families. The high heritability estimates in our study could also be accounted for by a small sample size. These families were previously reported to have clustering of metabolic abnormalities; therefore, they could have shared genes and lifestyle habits contributing to high CRP levels. It is difficult to determine how much of the heritability is due to genes or a shared environment. In addition, we defined presence of T2DM as presence of FPG of ≥7 mmol/l. In PCOS, impaired glucose tolerance is more prevalent than impaired fasting glucose (Ehrmann et al., 1999). Participants in our study may have undiagnosed T2DM based on the criteria of a 2-h glucose level ≥11.1 mmol/l after an oral glucose tolerance test. We did not have results for 2-h oral glucose tolerance tests for all the participants in our study; therefore, we may not have excluded all participants with T2DM.
Ultimately, CRP levels are useful as a marker of CVD risk. These events are rare in women with PCOS of reproductive age and in their parents. We recently reported a higher prevalence of stroke and myocardial infarctions in fathers of women with PCOS compared with population-based control men; however, the absolute rate was low (and without increased events noted in mothers) (Taylor et al., 2011). In conclusion, our study showed that circulating CRP levels in women with PCOS are not due to PCOS, but rather are likely inherited from their parents (Pankow et al., 2001; Hersh et al., 2006). Further studies should examine the predictive nature of CRP on CVD events in these families. Recently, studies looking at CRP in CVD did not show a causal link in other populations (Zacho et al., 2008; Wensley et al., 2011). However, given such a high contribution of genetic variance in CRP in our PCOS population, further studies need to be done to examine the causal nature of CRP in CVD in PCOS.
Authors’ roles
A.S., P.V., A.R.K., N.R.-K., A.D. and R.S.L. had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: R.S.L., A.S., A.D.
Acquisition of data: A.S., A.R.K., R.S.L., A.D.
Analysis and interpretation of data: A.S., P.V., A.R.K., N.R.-K., A.D., R.S.L..
Drafting of the manuscript: A.S., P.V., A.R.K., N.R.-K., A.D., R.S.L..
Critical revision of the manuscript for important intellectual content: A.S., P.V., A.R.K., N.R.-K., A.D., R.S.L..
Statistical analysis: A.R.K., P.V.
Study supervision: R.S.L., A.D.
Funding
National Institutes of Health (U54HD-034449 and P50 HD044405 to A.D.); NIH/NIDDK Training (T32 DK007169 to P.V.).
Conflict of interest
The authors have none to declare.
Acknowledgements
We thank Rick Ball from University Park for running the CRP assay, Barbara Scheetz for her longstanding coordination of this study, and Christy Stetter, Sandra Eyer and Ji Young Lee for their valuable help and suggestions.
References
- Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. doi:10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azziz R, Woods KS, Reyna R, Key TJ, Knochenhauer ES, Yildiz BO. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab. 2004;89:2745–2749. doi: 10.1210/jc.2003-032046. doi:10.1210/jc.2003-032046. [DOI] [PubMed] [Google Scholar]
- Boulman N, Levy Y, Leiba R, Shachar S, Linn R, Zinder O, Blumenfeld Z. Increased C-reactive protein levels in the polycystic ovary syndrome: a marker of cardiovascular disease. J Clin Endocrinol Metab. 2004;89:2160–2165. doi: 10.1210/jc.2003-031096. doi:10.1210/jc.2003-031096. [DOI] [PubMed] [Google Scholar]
- Carey AH, Chan KL, Short F, White D, Williamson R, Franks S. Evidence for a single gene effect causing polycystic ovaries and male pattern baldness. Clin Endocrinol. 1993;38:653–658. doi: 10.1111/j.1365-2265.1993.tb02150.x. doi:10.1111/j.1365-2265.1993.tb02150.x. [DOI] [PubMed] [Google Scholar]
- Cascella T, Palomba S, Tauchmanova L, Manguso F, Di Biase S, Labella D, Giallauria F, Vigorito C, Colao A, Lombardi G, et al. Serum aldosterone concentration and cardiovascular risk in women with polycystic ovarian syndrome. J Clin Endocrinol Metab. 2006;91:4395–4400. doi: 10.1210/jc.2006-0399. doi:10.1210/jc.2006-0399. [DOI] [PubMed] [Google Scholar]
- Coviello AD, Sam S, Legro RS, Dunaif A. High prevalence of metabolic syndrome in first-degree male relatives of women with polycystic ovary syndrome is related to high rates of obesity. J Clin Endocrinol Metab. 2009;94:4361–4366. doi: 10.1210/jc.2009-1333. doi:10.1210/jc.2009-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrmann DA, Barnes RB, Rosenfield RL, Cavaghan MK, Imperial J. Prevalence of impaired glucose tolerance and diabetes in women with polycystic ovary syndrome. Diabetes Care. 1999;22:141–146. doi: 10.2337/diacare.22.1.141. doi:10.2337/diacare.22.1.141. [DOI] [PubMed] [Google Scholar]
- Ehrmann DA, Liljenquist DR, Kasza K, Azziz R, Legro RS, Ghazzi MN. Prevalence and predictors of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91:48–53. doi: 10.1210/jc.2005-1329. doi:10.1210/jc.2005-1329. [DOI] [PubMed] [Google Scholar]
- Fox ER, Benjamin EJ, Sarpong DF, Rotimi CN, Wilson JG, Steffes MW, Chen G, Adeyemo A, Taylor JK, Samdarshi TE, et al. Epidemiology, heritability, and genetic linkage of C-reactive protein in African Americans (from the Jackson Heart Study) Am J Cardiol. 2008;102:835–841. doi: 10.1016/j.amjcard.2008.05.049. doi:10.1016/j.amjcard.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM. Metabolic syndrome scientific statement by the American Heart Association and the National Heart, Lung, and Blood Institute. Arterioscler Thromb Vasc Biol. 2005;25:2243–2244. doi: 10.1161/01.ATV.0000189155.75833.c7. doi:10.1161/01.ATV.0000189155.75833.c7. [DOI] [PubMed] [Google Scholar]
- Hersh CP, Miller DT, Kwiatkowski DJ, Silverman EK. Genetic determinants of C-reactive protein in COPD. Eur Respir J. 2006;28:1156–1162. doi: 10.1183/09031936.00147805. doi:10.1183/09031936.00147805. [DOI] [PubMed] [Google Scholar]
- Hosseinpanah F, Barzin M, Tehrani FR, Azizi F. The lack of association between polycystic ovary syndrome and metabolic syndrome: Iranian PCOS prevalence study. Clin Endocrinol. 2011;75:692–7. doi: 10.1111/j.1365-2265.2011.04113.x. doi:10.1111/j.1365-2265.2011.04113.x. [DOI] [PubMed] [Google Scholar]
- Lange LA, Burdon K, Langefeld CD, Liu Y, Beck SR, Rich SS, Freedman BI, Brosnihan KB, Herrington DM, Wagenknecht LE, et al. Heritability and expression of C-reactive protein in type 2 diabetes in the Diabetes Heart Study. Ann Hum Genet. 2006;70:717–725. doi: 10.1111/j.1469-1809.2006.00280.x. doi:10.1111/j.1469-1809.2006.00280.x. [DOI] [PubMed] [Google Scholar]
- Legro RS, Driscoll D, Strauss JF, III, Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci USA. 1998;95:14956–14960. doi: 10.1073/pnas.95.25.14956. doi:10.1073/pnas.95.25.14956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legro RS, Bentley-Lewis R, Driscoll D, Wang SC, Dunaif A. Insulin resistance in the sisters of women with polycystic ovary syndrome: association with hyperandrogenemia rather than menstrual irregularity. J Clin Endocrinol Metab. 2002;87:2128–2133. doi: 10.1210/jcem.87.5.8513. doi:10.1210/jc.87.5.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C, McGrath BP, Cameron J, Kotsopoulos D, Teede HJ. Vascular dysfunction and metabolic parameters in polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90:4630–4635. doi: 10.1210/jc.2004-1487. doi:10.1210/jc.2004-1487. [DOI] [PubMed] [Google Scholar]
- Orio F, Jr, Giallauria F, Palomba S, Cascella T, Manguso F, Vuolo L, Russo T, Tolino A, Lombardi G, Colao A, et al. Cardiopulmonary impairment in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91:2967–2971. doi: 10.1210/jc.2006-0216. doi:10.1210/jc.2006-0216. [DOI] [PubMed] [Google Scholar]
- Pankow JS, Folsom AR, Cushman M, Borecki IB, Hopkins PN, Eckfeldt JH, Tracy RP. Familial and genetic determinants of systemic markers of inflammation: the NHLBI family heart study. Atherosclerosis. 2001;154:681–689. doi: 10.1016/s0021-9150(00)00586-4. doi:10.1016/S0021-9150(00)00586-4. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843. doi: 10.1056/NEJM200003233421202. doi:10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation. 2003;107:391–397. doi: 10.1161/01.cir.0000055014.62083.05. doi:10.1161/01.CIR.0000055014.62083.05. [DOI] [PubMed] [Google Scholar]
- Sam S, Legro RS, Essah PA, Apridonidze T, Dunaif A. Evidence for metabolic and reproductive phenotypes in mothers of women with polycystic ovary syndrome. Proc Natl Acad Sci USA. 2006;103:7030–7035. doi: 10.1073/pnas.0602025103. doi:10.1073/pnas.0602025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MC, Reema Kar A, Kunselman AR, Stetter CM, Dunaif A, Legro RS. Evidence for increased cardiovascular events in the fathers but not mothers of women with polycystic ovary syndrome. Hum Reprod. 2011;26:2226–2231. doi: 10.1093/humrep/der101. doi:10.1093/humrep/der101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topcu S, Caliskan M, Gullu H, Ozcimen EE, Erdogan D, Uckuyu A, Zeyneloglu H, Muderrisoglu H. Do women with polycystic ovary syndrome really have predisposition to atherosclerosis? Aust N Z J Obstet Gynaecol. 2006;46:164–167. doi: 10.1111/j.1479-828X.2006.00541.x. doi:10.1111/j.1479-828X.2006.00541.x. [DOI] [PubMed] [Google Scholar]
- Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, Shah T, Engert JC, Clarke R, Davey-Smith G, Nordestgaard BG, et al. Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ. 2011;342:d548. doi: 10.1136/bmj.d548. doi:10.1136/bmj.d548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel J, Moratorio G, Rao F, Mahata M, Zhang L, Greene W, Rana BK, Kennedy BP, Khandrika S, Huang P, et al. C-reactive protein, an ‘intermediate phenotype’ for inflammation: human twin studies reveal heritability, association with blood pressure and the metabolic syndrome, and the influence of common polymorphism at catecholaminergic/beta-adrenergic pathway loci. J Hypertens. 2007;25:329–343. doi: 10.1097/HJH.0b013e328011753e. doi:10.1097/HJH.0b013e328011753e. [DOI] [PubMed] [Google Scholar]
- Wijeyaratne CN, Seneviratne Rde A, Dahanayake S, Kumarapeli V, Palipane E, Kuruppu N, Yapa C, Seneviratne Rde A, Balen AH. Phenotype and metabolic profile of South Asian women with polycystic ovary syndrome (PCOS): results of a large database from a specialist Endocrine Clinic. Hum Reprod. 2011;26:202–213. doi: 10.1093/humrep/deq310. doi:10.1093/humrep/deq310. [DOI] [PubMed] [Google Scholar]
- Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Willesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med. 2008;359:1897–1908. doi: 10.1056/NEJMoa0707402. doi:10.1056/NEJMoa0707402. [DOI] [PubMed] [Google Scholar]
- Zeger SK, Liang KY. Longitudinal data analysis for discrete and continuous outcomes. Biometrics. 1986;42:121–130. doi:10.2307/2531248. [PubMed] [Google Scholar]