

Abstract
Human papillomaviruses (HPVs) are the causative factor for greater than 90% of cervical cancers and 25% of head and neck cancers. The incidence of HPV positive (+) head and neck squamous cell carcinomas (HNSCCs) has greatly increased in the last 30 years. E6 and E7 are the two key viral oncoproteins that induce and propagate cellular transformation. An immune response generated during cisplatin/radiation therapy improves tumor clearance of HPV(+) cancers. Augmenting this induced response during therapy with an adenoviral HPV16 E6/E7 vaccine improves long term survival in preclinical models. Here we describe the generation of an HPV16 E6/E7 construct, which contains mutations that render E6/E7 non-oncogenic, while preserving antigenicity. These mutations do not allow E6/E7 to degrade p53, pRb, PTPN13, or activate telomerase. Non-oncogenic E6/E7 (E6Δ/E7Δ) expressed as a stable integrant, or in the [E1-, E2b-] adenovirus, lacks the ability to transform human cells while retaining the ability to induce an HPV specific immune response. Moreover, E6Δ/E7Δ plus chemotherapy/radiation statistically enhances clearance of established HPV(+) cancer in vivo.
Keywords: HPV, E6, E7, Adenovirus, immunotherapy, head and neck cancer
INTRODUCTION
The majority of individuals diagnosed with oropharyngeal (base of tongue, tonsil) head and neck squamous cell carcinoma (HNSCCs) are infected with high risk human papillomaviruses (HPVs). Greater than 90% of these individuals with HPV(+) cancers are infected with HPV type 16. While the incidence of HPV negative (−) HNSCC is declining, the incidence of HPV(+) HNSCC is on the rise and has tripled in recent years.1,2 HPV(+) patients often do not have a significant tobacco history and present with a more advanced disease stage compared to their HPV(−) counterparts, suggesting HPV(+) HNSCC is a distinct virally mediated disease.3,4 The development of new treatments, which specifically target HPV(+) disease, and advancements in existing therapies that improve survival are imperative for this cancer type.
Current treatment of HNSCC with non-specific chemotherapy and radiation, which results in significant morbidity, could be improved with addition of immunotherapy. Known viral oncogenes are attractive candidates as targets for immunotherapy. The HPV viral oncoproteins, E6 and E7, are predominantly responsible for transformation in HPV(+) HNSSCs. Like many viral oncoproteins, E6/E7 are expressed early in the course of infection and play multifunctional roles. E6 promotes degradation of p53, indirectly activates telomerase, and disrupts the function of the cellular phosphatase tumor suppressor PTPN13.5–8 E7 inactivates pRb and activates Mi2β.5,9 Together, these oncogenic alterations drive rapid cellular proliferation, suppress or down regulate key tumor suppressor proteins, and lead to cellular immortality. In addition, E6/E7 expression is vital for malignancy and is required to maintain a malignant transformed phenotype.10,11
Currently, no HPV vaccine is effective at treating established disease, despite their success with preventing infection.12 Vaccine prevention of high risk HPV infection will likely fail as HPV infection of the tonsil epithelia occurs early in life, prior to when immunization is currently administered.13 Current HPV16 vaccines utilize viral coat proteins or virus-like particles with HPV16 late gene products; however, many HPV(+) HNSCC express early (E6/E7) rather than late viral genes such as the viral coat proteins. Thus, vaccination with viral coat proteins might be ineffective for treating established cancer14 and an immune based therapeutic vaccine to the viral oncogenes, E6/E7, may prove more effective.
Our pre-clinical model of HPV(+) HNSCC supports the effectiveness of an E6/E7 immunotherapeutic vaccine.15,16 The immune response that develops in animals bearing HPV(+) tumors is required to achieve tumor clearance in conjunction with cisplatin and radiation treatment, a regimen most often clinically used.16 Boosting the immune response with recombinant adenovirus expressing HPV E6/E7 improves long term cures by as much as 30% over standard cisplatin/radiation alone.16 Though encouraging, two obstacles remain before bringing this therapy to trial in humans.
Pre-existing adenoviral immunity severely limits the usefulness of an adenovirus-based vaccine strategy. To negate this problem, we utilized an E1, E2b deleted adenovirus (Ad5 [E1-, E2b-]) that induces effective immune stimulation despite the generation of vector induced Ad5 immunity.17 The second obstacle is the expression of active viral oncogenes presents potential risks despite delivery via a non-replicating recombinant adenovirus. Potential transgene integration of the HPV oncogene(s) into the host genome may initiate cellular proliferation, transformation, and immortalization. To address this risk, we inactivated the E6/E7 coding regions responsible for oncogenic transformation. E6 and E7 have been well studied and their oncogenic coding sites previously described.10,18 In this report, we describe the generation of a full length E6/E7 vaccine in which the known oncogenic transforming regions are inactivated. The E6Δ/E7Δ cassette does not retain cellular oncogenic functions, yet retains antigenicity allowing vaccine treatment of established HPV-related cancer in vivo.
MATERIALS AND METHODS
MUTAGENISIS OF HPV16 E6/E7 ONCOGENES
In vitro site-directed mutagenesis of HPV E6/E7 oncogenes was performed as per manufacturer directions (Aligent Technologies, Santa Clara, CA). Mutated regions: Leucine to Glycine mutation at position 50 of E6 was generated; this region encompasses the p53 binding and telomerase activation site (L50G Forward primer: GACTATTTTGCTTTTCGGGATGGAT-G; L50G Reverse primer: CCCATCTCTATATACTATGCATCCATC). Second, the C-terminal PDZ binding domain of E6 (Glutamic acid, Threonine, Glutamine and Leucine) at residues 146–151 was mutated to four Alanines (PDZ Forward primer: 5’-GAACTCGTAGAGCAGCCGCG-GCGTA-3’; PDZ Reverse primer: 5’-GTGTATCTCCATGCATGATTACGCCG-3’). Third, three single point mutations in HPV16 E7 at the Rb binding sites were generated as follows: Histidine to Proline at residue 2 (H2P Forward primer; 5’-CAGCCGCGGCGTAATCATGCCT-GGA-3’; H2P Reverse primer: 5’-GCAATGTAGGTGTATCTCCAGGCATG-3’), Cysteine to Glycine at residue 24 (C24G Forward primer: 5’-CCAGAGACAACTGATCTCTACGGTTA-3’; C24G Reverse primer: 5’-GCTGTCATTTAATTGCTCATAACCGTA-3’) and Glutamic acid to Alanine at residue 46 (E46A Forward primer: 5’-GGTCCAGCTGGACAAGCAGCACCGG-3’; E46A Reverse primer: 5’-GTAATGGGCTCTGTCCGGTGCTGCTT-3’). A final single point mutation of Leucine to Arginine at residue 67 of E7 was generated to disrupt Mi2β binding (L67R Forward primer: 5’-CGTGTGTGCTTTGTACGCACCTCCGA-3’; L67R Reverse primer: 5’-GTGTGACTCTACGCTTCGGAGGTGC-3’). Fidelity of the final construct was verified by direct DNA sequencing.
VIRAL CONSTRUCTION
Ad5 [E1-, E2b-] containing E6Δ/E7Δ (Ad5 [E1-, E2b-]-E6Δ/E7Δ) and wild type E6/E7 (Ad5 [E1-, E2b-]-wt-E6/E7) were constructed and produced as previously described.17,19 Briefly, the transgenes were sub-cloned into the E1 region of the Ad5 [E1-, E2b-] vector using a homologous recombination-based approach. The replication deficient virus was propagated in the E.C7 packaging cell line, CsCl2 purified, and titered as previously described.17 Viral infectious titer was determined as plaque forming units (PFU) on an E.C7 cell monolayer. The viral particle (VP) concentration was determined by sodium dodecyl sulfate (SDS) disruption and spectrophotometry at 260 nm and 280 nm.17,20 The ratio of VP to plaque forming units (PFU) was 36.7/1 VP/PFU. The E6Δ/E7Δ insert as well as the wt-E6/E7 were then cut and ligated into the retroviral vector pLXSN using EcoRI and BamHI restriction sites. Retrovirus particles were generated in the Phoenix A cell line according to manufacturer’s recommendations (American Type Culture Collection, Manassas, VA) with polybrene (Sigma St. Louis, MO) added to a final concentration of 8µg/ml.
CELLS
A549 (human adenocarcinoma alveolar basal epithelium cell line) cells were grown in Dulbecco’s Modified Eagle Medium (Thermo Fisher, Logan, UT) supplemented with 10% Fetal Bovine Serum (Thermo Fisher). Primary human tonsil epithelial (HTE) cells were isolated from surgical tonsillectomy of consented patients under institutional IRB approval as previously described.21 HTE cells were maintained in Keratinocyte SFM media (KSFM; Invitrogen, Carlsbad, CA). Mouse tonsil epithelial cells expressing HPV16 E6, E7, Ras and luciferace (mEERL) have been previously described21 and were maintained in DMEM supplemented with 22.5% Hams F-12 medium, 10% heat inactivated FCS, 100 U/mL penicillin, 100 µg/mL streptomycin, 0.5 µg/mL hydrocortisone, 0.0084 µg/mL cholera toxin, 5 µg/mL transferrin, 5 µg/mL insulin, 0.00136 µg/mL tri-iodo-thyronine, and 5 µg/mL EGF (Invitrogen).
RETROVIRAL INFECTION OF CELL LINES
HTE and A549 cells were infected with retroviral supernatant containing wt-E6/E7, E6Δ/E7Δ, or empty vector retrovirus and incubated at 37°C in 5% CO2 overnight. Media was aspirated 24 hours post-infection and fresh media supplemented with neomycin (RPI, Mount Prospect, IL) was added for selection. Individual colonies were ring cloned and put under further selection using 800 µg/ml neomycin. Clones shown in figures are representative of multiple clones tested. Due to their density dependence for cell growth, HTE cell lines were not placed under antibiotic selection but maintained in KSFM until cell death or immortalization.
Standard PCR was performed to analyze mRNA in stable cell lines expressing LXSN, LXSN wt-E6/E7, or LXSN E6Δ/E7Δ to validate that the changes made in E6Δ/E7Δ did not affect its transcription rate. E6/E7 forward primer 5’-CAAACCGTTGTGTGATTTGTTAATTA-3’ and reverse primer 5’-GCTTTTTGTCCAGATGTCTTTGC-3’; GAPDH forward primer 5’-GG-GAAGGTGAAGGTCGGAGT-3’and reverse primer 5’-TGGAAGATGGTGATGGGATTTC-3’ were used. All primer concentrations were at 450 nM. Preincubation was 94°C for 10 min. Cycling conditions were 94°C for 40 sec, 55°C for 40 sec, and 72°C for 1 min for a total of 30 cycles using the Stratagene Mx3000P thermocycler.
The telomere repeat amplification protocol (TRAP) assay to test telomerase activity in the A549 parental, A549 wt-E6/E7 and A549 E6Δ/E7Δ cell lines were measured as described previously.22,23 Cells were grown to 80% confluency, harvested by trypsinization, counted and resuspended to 400 000 cells/ml. Cells were lysed for 30 min on ice in CHAPS lysis buffer (0.5% Chaps; 10 mM Tris, pH 7.5; 1 mM MgCl2; 1 mM EGTA, 5 mM 2-ME; 10% glycerol; 0.1 mM 4-(2-amino-ethyl)benzene-sulfonyl fluoride hydrochloride (AEBSF). Membranes were pelleted by centrifugation (10 000 rpm at 4°C) for 20 min and soluble proteins collected. The primer pair used were TS primer: 5’-AATCCGTCGAGCAGAGTT-3’ and ACX primer: 5’-GCGCGG[CTTACC]3CTAACC- 3 each at a final concentration of 100 ng. The PCR reaction contained SYBERGreen PCR Master Mix (Applied Biosystems, Carlsbad, CA), 1 mM EGTA pH 7.5 and 1 ul Lysate (2 000 cells) in a 50 µl reaction volume and completed in triplicate. The PCR reaction was incubated at room temperature for 30 min. Cycling conditions were 95 °C for 10 min with 40 cycles at 95 °C for 15 sec and 60 °C for 60 sec using the Stratagene Mx3000P thermocycler. The human foreskin keratinocyte (HFK) hTERT cell line was used as a reference gene.
ADENOVIRAL INFECTION OF CELL LINES
HTE cells grown to 80% confluency were infected with Ad5 [E1-, E2b-]-null (empty vector), Ad5 [E1-, E2b-]-wt-E6/E7, Ad5 [E1-, E2b-]-E6Δ/E7Δ, or Ad GFP at an MOI of 100 for 24 hours. DNA was collected at passages 1, 2, 3, 4 and 5 post-infection. Cells were trypsinized, rinsed, and re-suspended in 1X Phosphate Buffered Saline. DNA extraction was performed using a standard animal tissue spin-column protocol from DNeasy DNA Blood and Tissue Kit (Qiagen, Valencia, CA).
Quantitative real-time polymerase chain reaction was performed to assay for HPV16 copy number. Briefly, HPV 16 primer set 520 5’-TTGCAGATCATCAAGAACACGTAGA-3’ and 671 5’-CTTGTCCAGCTGGACCATCTATTT-3’ along with an 18S primer set (Applied Biosystems) were used. The amplification reaction contained SyberGreen Universal Master Mix (Applied Biosystems) 250 nM HPV 16 primers or 100 nM 18S primers and 25 ng template. Cycling conditions were 95 °C for 10 min with 40 cycles at 95 °C for 15 sec and 60 °C for 60 sec using the Stratagene Mx3000P thermocycler.
WESTERN BLOT ANALYSIS
The following stable cell lines A549 wt-E6/E7, A549 E6Δ/E7Δ, and parental A549 cells were grown to 80–90% confluency, rinsed with PBS and harvested with lysis solution (50 mM Tris HCl pH 7.5; 150 mM NaCl; 5 mM EDTA; 2 mM Na3VO4; 100 mM NaF; 10 mM NaPPi; 10% glycerol; 1% Triton; 1X Halt Protease Inhibitors; 17.4 µg/µl PMSF). Membranes were pelleted by centrifugation (10 000rpm at 4°C) and soluble proteins collected. Total protein was quantified using the BCA protein assay kit as per the manufacturer’s directions (Pierce, Logan, UT) and equal total protein was analyzed by western blot. Briefly, proteins were separated by SDS-PAGE, transferred to PVDF-membranes (Immobilon-P), blocked with either 5% bovine serum albumin (MP Biomedicals, Solon, OH) or non-fat dry milk and visualized by chemiluminescence on film or via UVP bioimaging system (Upland, CA). Membranes were incubated with the following antibodies: FAP-1 (1:500, Santa Cruz, Santa Cruz, CA), p53 (1:500, Calbiochem, Darmstadt, Germany), pRb (1:250, BD Biosciences, Sparks, MD) and GAPDH (1:5 000, Ambion, Carlsbad, CA)
IMMUNIZATION OF MICE WITH Ad5 [E1-, E2b-]-E6Δ/E7Δ FOR IMMUNE ASSAYS
Male C57Bl/6 mice (N=4) 8 to 10 weeks old were immunized three times intranasally at 7-day intervals. Control mice (N=4) were given with buffer solution or with 1010 VP Ad5 [E1-, E2b-]-null intranasally using the same injection schedule as that for mice immunized with Ad5 [E1, E2b-]-E6Δ/E7Δ. Two weeks after the last injection/immunization, all mice were sacrificed and spleens harvested. Splenocytes from each mouse were isolated for ELISpot testing as described below. Serum from each mouse was collected and stored at −20 °C until testing.
PREPARATION OF mEERL CELL LYSATES FOR ELISpot
HPV(+) (mEERL) cells were grown in a T125 flask until confluent, after which cells were aseptically scraped off the plastic surface, washed three times with sterile PBS, and re-suspended in 1 mL of sterile PBS. Cells were lysed by freeze-thawing 3 times and cellular debris removed by centrifugation. Soluble protein was brought to a final volume of 2 mL with sterile PBS. The presence of HPV-E7 in the lysate was confirmed by western blot analysis performed as described.24
ENZYME-LINKED IMMUNOSPOT (ELISpot) ASSAYS
HPV16 E6 and E7 specific IFN-γ and IL-2 production from splenocytes isolated from individual mice following immunizations was detected by ELISpot as previously described.19 Briefly, cells were stimulated with HPV16 E6 and E7 peptides (15-mer peptide complete sets for each; JPT Peptide Technologies, Berlin, Germany). Peripheral blood mononuclear cells (PBMC) were used at a concentration of 2 × 105 cells/well and reported as the number of spot forming cells (SFC) per 106 cells per well. All E6 peptides were combined and tested as a single pool. Similarly, all E7 peptides were combined and tested as a single pool. Each peptide pool was tested in duplicate. To test for specificity, splenocytes were also exposed to an HIV-gag peptide pool and a cytomegalovirus (CMV) peptide pool. Peptides were utilized at 0.1 µg of each peptide/well. To test for reactivity to mEERL cell lysate, 25 µL of lysate was added to test wells in duplicate. In all ELISpot assays, cells stimulated with concanavalin A (ConA) at a concentration of 1 µg/well served as positive controls. Colored SFC were counted using an Immunospot ELISpot plate reader (Cellular Technology, Shaker Heights, OH) and responses considered positive if, 1) 50 SFC were detected/106 cells after subtraction of the negative control or 2) SFC were at least 2-fold greater than those in the negative control wells and significantly elevated.
ELISpot: MOUSE GRANZYME B ASSAY
ELISpot mouse Granzyme B kit (R&D Bio Systems, Minneapolis, MN) methods were followed as per manufacturer’s instructions with the following changes. Briefly, splenocytes were incubated with mEERL for 4 hrs to stimulate Granzyme B production. 5 × 105 splenocytes were added to wells of ELISpot plate and placed at 37° C with CO2 for 2 hrs.
IN VIVO TUMOR GROWTH AND TREATMENT
Male C57Bl/6 mice were obtained from the Jackson Labs and maintained by Sanford Research LARF in accordance with USDA guidelines. All experiments were approved by Sanford Research IACUC and performed within institutional guidelines. Briefly, 1 × 106 mEERL cells were implanted subcutaneously in the right hind flank of mice using a 23-gauge needle. After palpable tumors were present, on days 7, 14, and 21 mice were given 1010 VP Ad5[E1-, E2B-]-null or Ad5 [E1-, E2b-]-E6Δ/E7Δ intranasally. Cisplatin (CalBiochem) 20mg/m2 administered intraperitoneally and concurrent radiation (8 Gy Xray (Rad Source RS2000 irradiator, Brentwood, TN)) was administered on days 13, 20, 27 days post-tumor implantation. Tumor growth was monitored weekly using caliper measurements and tumor volume calculated using the following formula, volume = (width2)(depth). Mice were euthanized when tumors reached 1.5 cm in any dimension, the animal became emaciated, or demonstrated functional leg impairment. Long-term survival was followed for greater than 70 days.
STATISTICAL ANALYSIS
Statistically significant differences in the mean immune responses between groups of animals were determined by Student’s t-test using GraphPad Prism® (GraphPad Software, Inc., LaJolla, CA). Statistically significant differences for mouse in vivo survival analysis were determined by Kaplan-Meier log rank survival using SigmaPlot® 11 (Systat Software Inc., San Jose, CA).
RESULTS
E6/E7 MUTATIONS
To decrease the risk associated with adenoviral delivery of HPV E6/E7 we mutated six known oncogenic regions within E6 and E710, 25 (Figure 1A). The introduced mutations destroy the ability of E6 to mediate degradation of p53, activate telomerase, and bind at the PDZ motif.6,7 The E6 PDZ binding motif participates in transformation by inactivating the tumor suppressor protein PTPN13 and other proteins with PDZ domain interactions.8,26,27. Mutations introduced in E7 prevent its ability to bind/inactivate pRb and other pocket proteins, and to associate with Mi2β that enhances cellular growth.9 Figure 1B shows an amino acid alignment of wild type E6/E7 (wt-E6/E7) and non-oncogenic E6/E7 (E6Δ/E7Δ) proteins.
Figure 1.
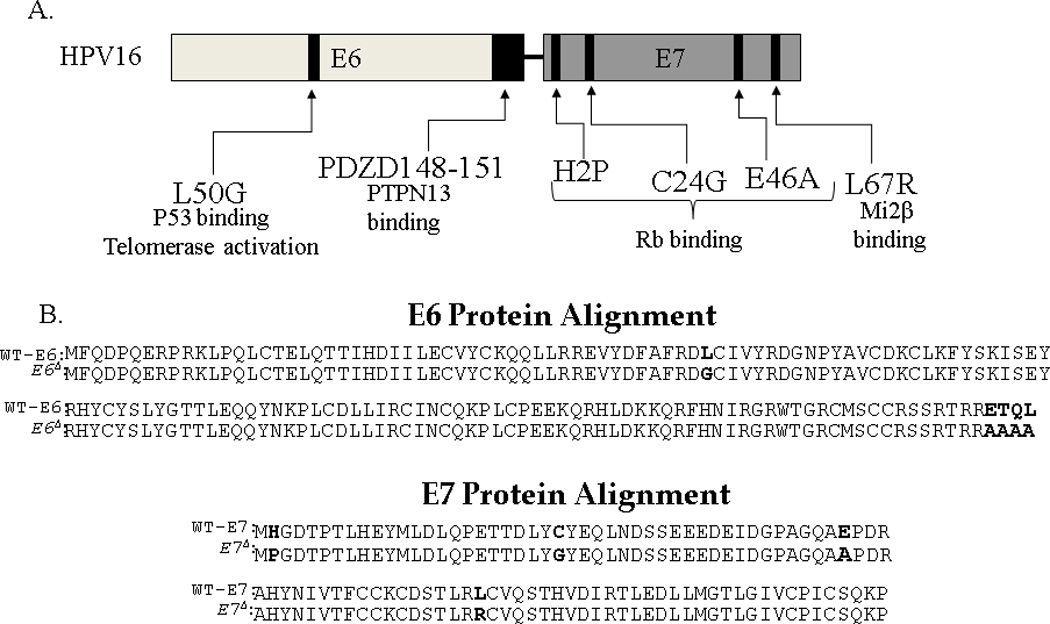
Schema of molecular changes to inactivate oncogene function of HPV16 E6/E7. The specific changes that disrupt E6 and E7 function are outlined in the schema (A) and specific protein changes are shown in bold in the protein alignment (B).
LOSS OF ONCOGENIC FUNCTION IN E6Δ/E7Δ
To confirm the loss of oncogene function, A549 cells were infected with a retrovirus containing wt-E6/E7, E6Δ/E7Δ, or control vector and subsequently ring cloned. Clones were analyzed by western blot. Figure 2A shows that expression of wt-E6/E7 decreases PTPN13, pRb, and p53 protein expression, while the E6Δ/E7Δ rescues this phenotype. PCR analysis of clones confirmed similar levels of viral oncogene expression (Figure 2B) suggesting that the changes evident by western blot were a consequence of altered oncogene function rather than expression and confirm the biochemical loss-of-function in the E6Δ/E7Δ construct. Telomerase activity was also examined in these clones. As predicted, the E6Δ/E7Δ and vector control did not show telomerase activation while the wt-E6/E7 enhanced it (Figure 2C). Previous studies have shown that HPV wt-E6/E7 induces morphological mesenchymal type changes.28 Therefore, the morphological characteristics of clones were also examined. Figure 2D shows that control and E6Δ/E7Δ grow in tight colonies, while wt-E6/E7 expression induces a mesenchymal-like change in morphology and cells grow in a non-adherent manner. Together, these data suggest that, unlike expression of wt-E6/E7, stable expression of E6Δ/E7Δ does not induce the biochemical or morphological changes associated with cellular transformation.
Figure 2.
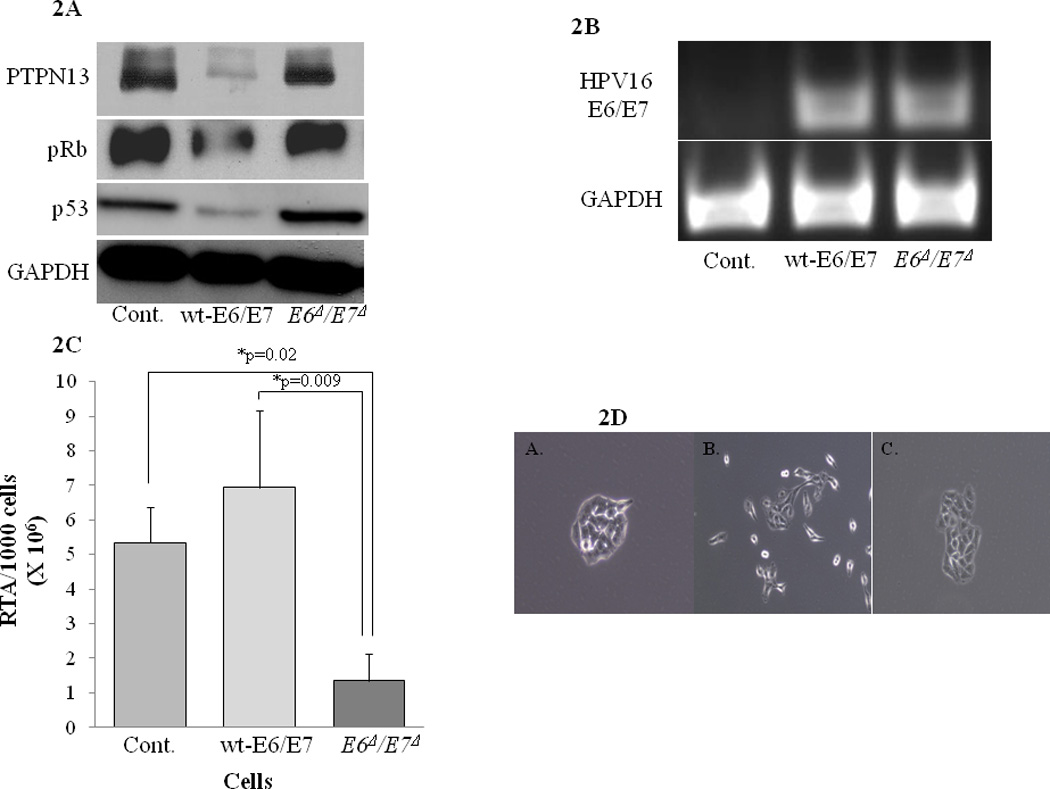
A. Detection of E6/E7 protein targets in cells stably expressing wild type E6/E7 (wt-E6/E7) vs. non-oncogenic HPV16 E6/E7 (E6Δ/E7Δ). Stable A549 cell lines were generated using infection with retrovirus containing either control virus, wt-E6E7, or E6Δ/E7Δ. Western blot analysis from the cell lysates were analyzed for cellular levels of E6 (p53, PTPN13) and E7 (pRB) proteins these oncogenes degrade. GAPDH levels shown as a control for loading
B. Expression of mRNA in cells stably expressing wt-E6/E7 vs. E6Δ/E7Δ. Expression of E6/E7 message was examined in the stably transduced A549 cells, using RT-PCR for E6. RT-PCR for GAPDH was also examined to verify similar amounts of quality mRNA were tested.
C. TRAP assay to measure telomerase activity in wt-E6/E7 vs. E6Δ/E7Δ expressing cell lines. Telomerase activity was analyzed completing TRAP assay on the indicated A549 cells stably expressing E6Δ/E7Δ or wt-E6/E7. E6Δ/E7Δ decline in RTA was statistically lower than wt or control with p=0.009 and p=0.02 respectively. Results shown represent repeat experiments done in triplicate.
D. Morphology of A549 colonies after infected with either (A) control, (B) wt-E6E7, or (C) E6Δ/E7Δ retrovirus. Colony growth in the cells stably expressed the various constructs was examined by plating 50 cells on 150mm tissue culture dishes and photos were taken 48hrs after plating. A549 control and A549 E6Δ/E7Δ grow in tight colonies, while A549 wt-E6E7 exhibit spindle/mesenchymal morphology an do not form tight colonies.
To further confirm that E6Δ/E7Δ cannot transform cells, HTEs were infected with retrovirus containing wt-E6/E7, E6Δ/E7Δ, or empty vector. Expression of wt-E6/E7 results in cellular immortalization, consistent with previous findings.29 However, HTEs expressing E6Δ/E7Δ and uninfected (control) HTEs did not immortalize (Figure 3A). These findings along with the biochemical evidence above demonstrates that stable expression of the E6Δ/E7Δ, even with an integrating retrovirus, does not result in cellular immortalization.
Figure 3.
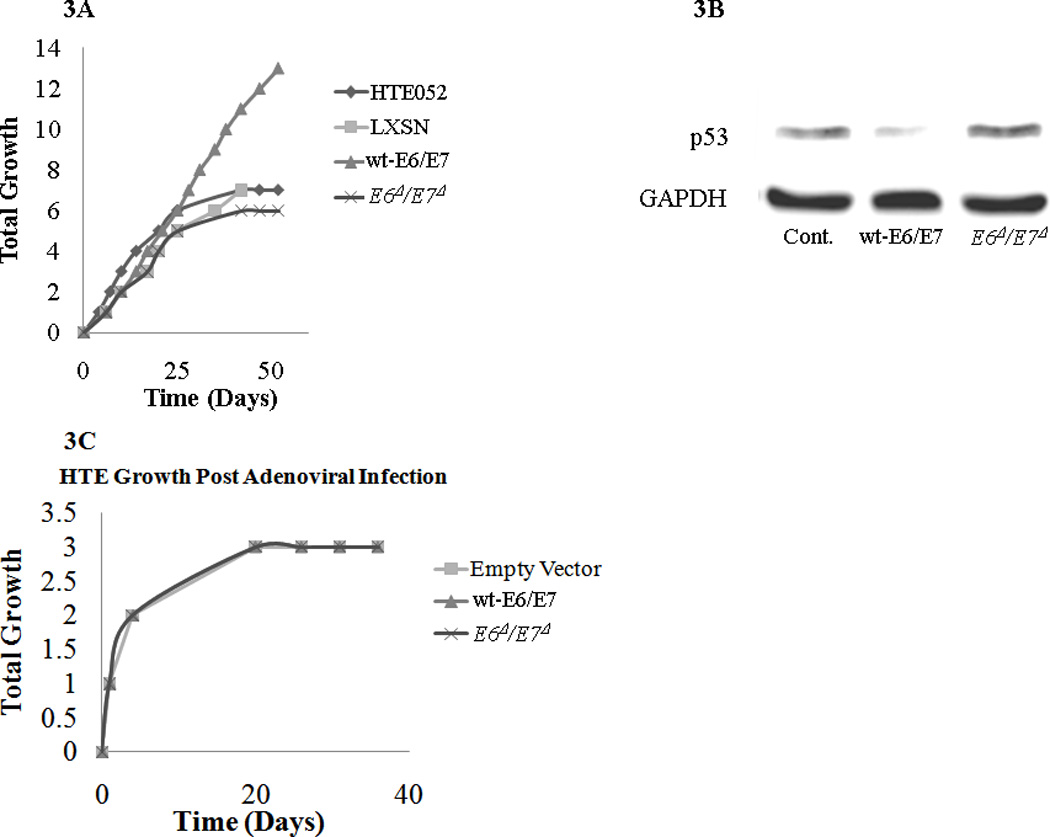
A. Immortalization of primary human tonsil epithelia cells with retrovirus expressing wt-E6/E7 vs. E6Δ/E7Δ. Primary human tonsil epithelia (HTEs) infected with LXSN control vector, wt-E6/E7, E6Δ/E7Δ, and uninfected HTEs served as an additional control and passaged in culture until immortalization or senescence. Wt-E6/E7 had the ability to immortalize HTEs while the E6Δ/E7Δ and controls all were senescent by passage 8.
B. P53 degradation after infection of primary human tonsil epithelia cells with replication defective Ad5 [E1-, E2b-]-wt-E6/E7 vs. Ad5 [E1-, E2b-]-E6Δ/E7Δ. Primary human tonsil epithelial cells (HTE) were infected with Ad5GFP control Ad5 [E1-, E2b-]wt-E6/E7or Ad5 [E1-, E2b-]-E6Δ/E7Δ at an MOI of 100. Approximately 40 % of the cells showed GFP expression in the control(data not shown). After 48 hours cells were lysed and p53 levels were examined by western blot to determine function of E6 expression. GAPDH served as a protein loading control.
C. Growth of primary human tonsil epithelia cells after infection with replication defective Ad5 [E1-, E2b-]-wt-E6/E7 vs. E6Δ/E7Δ. Infection using an adenovirus containing wt-E6E7, E6Δ/E7Δ, or control vector with an MOI of 100 was completed and cells were kept in culture to evaluate for immortalization. In all conditions HTEs were underwent senescence and did not immortalize.
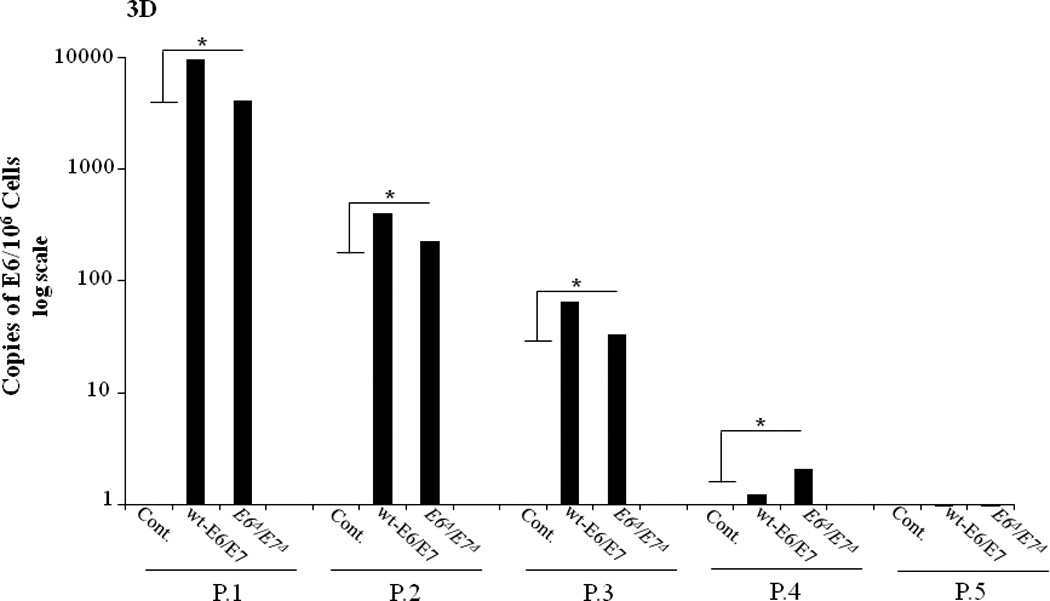
D. Loss of HPV status of primary HTEs infected with adenovirus with passage. HTEs were infected with either Empty Vector (control), Ad5 [E1-, E2b-]wt-E6/E7, or Ad5 [E1-, E2b-]-E6Δ/E7Δ and were passaged. QPCR analysis shows that as the passage number increases the number of Copies of E6 decreases. Results shown represent repeat experiments done in triplicate. (* indicates p-value of <0.001 for both HPV infection vectors compared to controls for passage 1–4)
NEITHER WT-E6/E7 NOR E6Δ/E7Δ GENES IN Ad5 [E1-, E2b-] TRANSFORM PRIMARY HUMAN TONSIL EPITHELIAL CELLS
To determine the function of wt-E6/E7 or E6Δ/E7Δ in a non-replicative adenoviral vector, transfection of HTE was examined and compared to a control expressing GFP. Previous studies have demonstrated that the Ad5 [E1-, E2b-] vector transgenes do not replicate in transfected cells30,31. However, the effect(s) of HPV E6/E7 in the context of this vector have not been previously examined. Wt-E6/E7 was able to induce loss of p53, while E6Δ/E7Δ was able to rescue p53 levels compared to control (Figure 3B). However, neither wt-E6/E7 nor E6Δ/E7Δ were able to immortalize primary tonsil epithelial cells after infection (Figure 3C). To determine if this was due to viral loss with replication, we examined the persistence of viral DNA in association with cell growth. Q-PCR demonstrated that as cells replicated, viral DNA was lost at a similar rate with all inserts, suggesting the viral genes did not integrate into the host cell DNA (Figure 3D). Therefore, HPV genes from the Ad5 [E1-, E2b-] vector do not persist as cellular division continues. Moreover, the transient expression of wt-E6/E7 in this setting was not sufficient to transform primary cells.
E6Δ/E7Δ INDUCES HPV SPECIFIC CELL-MEDIATED IMMUNE RESPONSES
To determine whether the changes induced in the non-oncogenic E6Δ/E7Δ alter the ability to mount an HPV-specific immune response, ELISpot assays were performed to assess cell mediated immune (CMI) responses in mice immunized with Ad5 [E1-, E2b-]-E6Δ/E7Δ. CMI responses were determined in control and vaccinated mice by assessing the numbers of IFN-γ, IL-2, and granzyme B secreting cells in splenocytes harvested from individual mice. As shown in Figures 4A and 4B, CMI responses were detected in mice immunized with Ad5 [E1-, E2b-]-E6Δ/E7Δ. This was demonstrated by the induction of significantly elevated levels of IFN-γ, or IL-2 spot forming cells (SFC) in immunized mice but not control mice injected with buffer solution or Ad5 [E1-, E2b-]-null. The specificity of the CMI responses was demonstrated by a lack of reactivity when splenocytes were exposed to the irrelevant antigens HIV-gag or CMV. Functionally active splenocytes in all groups was verified by positive responses to ConA. Granzyme B assay was also examined because it reflects immune related tumor cell toxicity assays 40. Splenocytes from mice vaccinated with both E6/E7 vaccines showed significant increase in cell mediated tumor cell toxicity compared to non vaccinated mice splenocytes (Figure 4C). These results indicate that the non-oncogenic E6Δ/E7Δ is immunogenic and induces an HPV specific E6/E7 CMI response.
Figure 4.
Cellular immunity in Ad5 naïve mice after immunizations with Ad5 [E1-, E2b-]-E6Δ/E7Δ. Naïve C57BL/6 mice were immunized three times with either injection buffer (N=4), 1010 VP of Ad5 [E1-, E2b-]-null (N=5), or 1010 Ad5 [E1-, E2b-]-E6Δ/E7Δ (N=5) at 7 day intervals. A fourth immunization was administered two weeks after the third. Splenocytes were assessed for IFN-g (A) or IL-2 (B) secreting cells by ELISpot assay four weeks after the final (fourth) immunization. CMI responses to Ad5-null (Ad5), HPV-E6 (E6), HPV-E7 (E7), HPV(+) cells (mEERL), HIV-1 Gag (gag), or CMV (CMV) were determined. Note: mEERL cells expressing HPV-E6E7 antigens were suspended in PBS and lysed by freeze-thawing.
C. ELISpot Granzyme B Assay to test for cellular immunity after immunizations with Ad5 [E1-, E2b-]-wt-E6/E7, Ad5 [E1-, E2b-]-E6Δ/E7Δ, or Ad5 [E1-, E2b-]-null. Naïve mice were vaccinated with either Ad5 [E1-, E2b-]-wt-E6/E7, Ad5 [E1-, E2b-]-E6Δ/E7Δ, or Ad5 [E1-, E2b-]-null (107 VP). 7 days post primary vaccination mice were boosted with the appropriate vaccine, and splenocytes were evaluated for reactivity towards mEERL cells through secretion of Granzyme B. Both Ad5 [E1-, E2b-]-wt-E6/E7 and Ad5 [E1-, E2b-]-E6Δ/E7Δ show significantly elevated Granzyme B production compared to Ad5 [E1-, E2b-]-null.
E6Δ/E7Δ IMMUNOTHERAPY INCREASES SURVIVAL DURING CONCURRENT CHEMOTHERAPY/RADIATION
We have previously shown that mice intranasally vaccinated with and Adenovirus expressing wild-type E6/E7 improved long term survival in mice receiving standard cisplatin/radiation16. While the in vitro tests suggested that mice receiving the Ad5 [E1-, E2b-]-E6Δ/E7Δ vector had a similar anti-HPV immune response we wanted to test if it would enhance clearance of HPV+ tumors similar to the wild-type oncogenes. To test the efficacy of the non-oncogenic E6Δ/E7Δ we established HPV(+) tumors in wild type mice identically as previously shown16. Once tumors had established for one week Ad5 [E1-, E2b-]-E6Δ/E7Δ or Ad5 [E1-, E2b-]-null was delivered intranasally weekly for 3 weeks. Mice receiving only cisplatin/xrt + Ad5 [E1-, E2b-]-null (vector control) had similar and long term clearance as we had previously reported16. However, mice receiving Ad5 [E1-, E2b-]-E6Δ/E7Δ had significantly improved long term survival (Figure 5) mimicking the response we had seen in the past studies when we had delivered wild-type E6/E716. These data show that in immune competent mice vaccination with Ad5 [E1-, E2b-]-E6Δ/E7Δ enhances immune related clearance in vivo during standard therapy for HPV related cancer.
Figure 5.
Survival after Ad5 [E1-, E2b-]-null vs. Ad5 [E1-, E2b-]-E6Δ/E7Δ vaccination during therapy with cisplatin and radiation. Treatment consisted of standard cisplatin/radiation and vaccinated with Ad5 [E1-, E2b-]-null(solid black line), or Ad5 [E1-, E2b-]-E6Δ/E7Δ (dotted black line) as described in methods. Tumors were measured weekly. Gray arrows represent vaccinations, while black arrows represent chemo/radiation treatment. Long-term survival of mice from these mice vaccinated with Ad5 [E1-, E2b-]-null or Ad5 [E1-, E2b-]-E6Δ/E7Δ during treatment with cisplatin/radiation. Ad5 [E1-, E2b-]-E6Δ/E7Δ showed significantly improved survival compared to Ad5 [E1-, E2b-]-null control (p=0.029).
DISCUSSION
We describe the generation of a novel HPV16 E6/E7 construct containing a combination of modifications that render these viral oncogenes non-oncogenic while retaining the antigenicity necessary to produce an immunotherapeutic vaccine against HPV(+) tumors. We demonstrated biochemically that these mutations lack the capacity to degrade p53, pRb, and PTPN13. In addition we found that whether E6Δ/E7Δ is expressed as a stable integrant or in the Ad5 [E1-, E2b-] vector, it lacks the ability to transform human cells in vitro. Despite this, E6Δ/E7Δ retains the ability to induce an HPV specific CMI response and synergizes with standard therapy, enhancing immune-mediated clearance of an HPV associated cancer in vivo.
The improved HPV(+) tumor clearance obtained using the Ad5 [E1-, E2b-]-E6Δ/E7Δ vaccine in addition to conventional chemotherapy/radiation suggests this vaccine may have clinical usefulness. E6Δ/E7Δ expression as a vaccine in the new Ad5 [E1-, E2b-] vector has several advantages for immune based therapy. The foreign viral E6Δ/E7Δ antigens are “non-self” with less potential to induce autoimmunity than with other tumor associated antigens.
The delivery of the modified viral antigens is also of critical importance to the success of the vaccine. We chose an adenoviral vector as it has been repeatedly used in humans to induce robust CMI responses. Adenovirus can be mass-produced, is stable, and has an extensive safety profile. The new Ad5 [E1-, E2b-] vector has been successfully studied in other pre-clinical animal models of cancer.31,32 Additionally, infectious disease models utilizing the Ad5 [E1-, E2b-] vector suggest efficacy in non-human primates (NHP).33 The CMI responses induced by Ad5 [E1-, E2b-] in Ad5 naïve and Ad5 immune NHP increased over a course of multiple immunizations and the response profiles observed in Ad5 naïve and Ad5 immune NHP were similar. These previous findings suggest that the new Ad5 [E1-, E2b-] based vaccine could be used for homologous vaccination regimes to induce robust CMI responses in the presence of Ad5 vector immunity.
Several pre-clinical studies suggest a therapeutic immune based vaccine strategy using E6 and/or E7 may be of value in the treatment of established disease.34–36 However, these published studies include vaccines utilizing specific regions of either E6 and/or E7.37-39 By expressing slightly varied non-oncogenic full-length forms of both E6 and E7, our strategy has the advantage of maintaining native antigenic processing and thus theoretically increasing the chance of an immune response in vivo. Given the heterogeneity of immune response in humans, which is much less predictable than inbred strains of mice, expression of full-length proteins allows for antigen processing and development of a CMI response that may produce a broad and effective anti-tumor response.
HPV16 is the most common cause of cervical, oropharynx, and anal cancers and common therapies (cisplatin and radiation) are used for treatment in the advanced stages of these cancers. Even though we examined a pre-clinical model of HPV(+) oropharynx cancer, it is likely that similar principles of therapy and vaccination can be used for other HPV(+) associated cancers. Thus, use of the non-oncogenic Ad5 [E1-, E2b-]-E6Δ/E7Δ vaccine in conjunction with chemoradiation represents an important advancement in the therapy of HPV(+) associated cancers. The preclinical and in vitro data show loss of transformation and the equivalent augmentation of immune mediated clearance in mice suggest that further clinical trials in patients in a phase 1 setting would be safe and efficacious. We believe that this new Ad5 [E1-, E2b-]-E6Δ/E7Δ vector vaccine should be investigated further as a new immune based therapy for HPV16 associated cancers.
Supplementary Material
Naïve C57BL/6 (n=6) were injected with 1×106 mEERL cells and followed for growth. One week after implantation tumors enter into log phase growth with none of the mice surviving without treatment, and all mice needed to be euthanized after 30 days. This illustrates the necessity for chemoradiation in order for the mice to survive, and the benefit of the Ad5 [E1-, E2b-]-E6Δ/E7Δ vaccine when given in conjunction with standard treatment.
Acknowledgements
Susan Puumala PhD-Biostatistics, Satoshi Nagat PhD- Immunology consultant, Cathy Christopherson- manuscript preparation.
Sources of Support: JHL supported by NIDCR 7R01DE018386-03 grant and subaward 3SB161 State of South Dakota-2010 Initiative
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Shiboski CH, Schmidt BL, Jordan RC. Tongue and tonsil carcinoma: increasing trends in the U.S. population ages 20–44 years. Cancer. 2005;103:1843–1849. doi: 10.1002/cncr.20998. [DOI] [PubMed] [Google Scholar]
- 2.Hammarstedt L, Dahlstrand H, Lindquist D, Onelov L, Ryott M, Luo J, et al. The incidence of tonsillar cancer in Sweden is increasing. Acta oto-laryngologica. 2007;127:988–992. doi: 10.1080/00016480601110170. [DOI] [PubMed] [Google Scholar]
- 3.Smith EM, Ritchie JM, Summersgill KF, Klussmann JP, Lee JH, Wang D, et al. Age, sexual behavior and human papillomavirus infection in oral cavity and oropharyngeal cancers. International journal of cancer. 2004;108:766–772. doi: 10.1002/ijc.11633. [DOI] [PubMed] [Google Scholar]
- 4.Ritchie JM, Smith EM, Summersgill KF, Hoffman HT, Wang D, Klussmann JP, et al. Human papillomavirus infection as a prognostic factor in carcinomas of the oral cavity and oropharynx. International journal of cancer. 2003;104:336–344. doi: 10.1002/ijc.10960. [DOI] [PubMed] [Google Scholar]
- 5.Smeets SJ, van der Plas M, Schaaij-Visser TB, van Veen EA, van Meerloo J, Braakhuis BJ, et al. Immortalization of oral keratinocytes by functional inactivation of the p53 and pRb pathways. International journal of cancer. 128:1596–1605. doi: 10.1002/ijc.25474. [DOI] [PubMed] [Google Scholar]
- 6.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 7.Veldman T, Liu X, Yuan H, Schlegel R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8211–8216. doi: 10.1073/pnas.1435900100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spanos WC, Hoover A, Harris GF, Wu S, Strand GL, Anderson ME, et al. The PDZ binding motif of human papillomavirus type 16 E6 induces PTPN13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth. Journal of virology. 2008;82:2493–2500. doi: 10.1128/JVI.02188-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brehm A, Nielsen SJ, Miska EA, McCance DJ, Reid JL, Bannister AJ, et al. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. The EMBO journal. 1999;18:2449–2458. doi: 10.1093/emboj/18.9.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, et al. Mechanisms of human papillomavirus-induced oncogenesis. Journal of virology. 2004;78:11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D'Abramo CM, Archambault J. Small molecule inhibitors of human papillomavirus protein - protein interactions. The open virology journal. 2011;5:80–95. doi: 10.2174/1874357901105010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berry JM, Palefsky JM. A review of human papillomavirus vaccines: from basic science to clinical trials. Front Biosci. 2003;8:s333–s345. doi: 10.2741/1003. [DOI] [PubMed] [Google Scholar]
- 13.Duray A, Descamps G, Bettonville M, Sirtaine N, Ernoux-Neufcoeur P, Guenin S, et al. High prevalence of high-risk human papillomavirus in palatine tonsils from healthy children and adults. Otolaryngol Head Neck Surg. 2011;145:230–235. doi: 10.1177/0194599811402944. [DOI] [PubMed] [Google Scholar]
- 14.FUTUREIIStudyGroup. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. The New England journal of medicine. 2007;356:1915–1927. doi: 10.1056/NEJMoa061741. [DOI] [PubMed] [Google Scholar]
- 15.Lee DW, Anderson ME, Wu S, Lee JH. Development of an adenoviral vaccine against E6 and E7 oncoproteins to prevent growth of human papillomavirus-positive cancer. Archives of otolaryngology--head & neck surgery. 2008;134:1316–1323. doi: 10.1001/archoto.2008.507. [DOI] [PubMed] [Google Scholar]
- 16.Spanos WC, Nowicki P, Lee DW, Hoover A, Hostager B, Gupta A, et al. Immune response during therapy with cisplatin or radiation for human papillomavirus-related head and neck cancer. Archives of otolaryngology--head & neck surgery. 2009;135:1137–1146. doi: 10.1001/archoto.2009.159. [DOI] [PubMed] [Google Scholar]
- 17.Amalfitano A, Hauser MA, Hu H, Serra D, Begy CR, Chamberlain JS. Production and characterization of improved adenovirus vectors with the E1, E2b, and E3 genes deleted. Journal of virology. 1998;72:926–933. doi: 10.1128/jvi.72.2.926-933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen DX, Westbrook TF, McCance DJ. Human papillomavirus type 16 E7 maintains elevated levels of the cdc25A tyrosine phosphatase during deregulation of cell cycle arrest. Journal of virology. 2002;76:619–632. doi: 10.1128/JVI.76.2.619-632.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabitzsch ES, Xu Y, Yoshida LH, Balint J, Amalfitano A, Jones FR. Novel Adenovirus type 5 vaccine platform induces cellular immunity against HIV-1 Gag, Pol, Nef despite the presence of Ad5 immunity. Vaccine. 2009;27:6394–6398. doi: 10.1016/j.vaccine.2009.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mittereder N, March KL, Trapnell BC. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. Journal of virology. 1996;70:7498–7509. doi: 10.1128/jvi.70.11.7498-7509.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams R, Lee DW, Elzey BD, Anderson ME, Hostager BS, Lee JH. Preclinical models of HPV+ and HPV− HNSCC in mice: an immune clearance of HPV+ HNSCC. Head & neck. 2009;31:911–918. doi: 10.1002/hed.21040. [DOI] [PubMed] [Google Scholar]
- 22.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science (New York, NY. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 23.Kim NW, Wu F. Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP) Nucleic acids research. 1997;25:2595–2597. doi: 10.1093/nar/25.13.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabitzsch ES, Xu Y, Yoshida LH, Balint J, Gayle RB, Amalfitano A, et al. A preliminary and comparative evaluation of a novel Ad5 [E1-, E2b-] recombinant-based vaccine used to induce cell mediated immune responses. Immunology letters. 2009;122:44–51. doi: 10.1016/j.imlet.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 26.Lee SS, Weiss RS, Javier RT. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6670–6675. doi: 10.1073/pnas.94.13.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Ham M, Hendriks W. PDZ domains-glue and guide. Molecular biology reports. 2003;30:69–82. doi: 10.1023/a:1023941703493. [DOI] [PubMed] [Google Scholar]
- 28.Peters DM, Dowd N, Brandt C, Compton T. Human papilloma virus E6/E7 genes can expand the lifespan of human corneal fibroblasts. In vitro cellular & developmental biology. 1996;32:279–284. doi: 10.1007/BF02723060. [DOI] [PubMed] [Google Scholar]
- 29.Hoover AC, Spanos WC, Harris GF, Anderson ME, Klingelhutz AJ, Lee JH. The role of human papillomavirus 16 E6 in anchorage-independent and invasive growth of mouse tonsil epithelium. Archives of otolaryngology--head & neck surgery. 2007;133:495–502. doi: 10.1001/archotol.133.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gabitzsch ES, Xu Y, Balcaitis S, Balint JP, Jr, Jones FR. An Ad5[E1-, E2b-]-HER2/neu vector induces immune responses and inhibits HER2/neu expressing tumor progression in Ad5 immune mice. Cancer gene therapy. 2011;18:326–335. doi: 10.1038/cgt.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabitzsch ES, Xu Y, Balint JP, Jr, Hartman ZC, Lyerly HK, Jones FR. Anti-tumor immunotherapy despite immunity to adenovirus using a novel adenoviral vector Ad5 [E1-, E2b-]-CEA. Cancer Immunol Immunother. 2010;59:1131–1135. doi: 10.1007/s00262-010-0847-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osada T, Yang XY, Hartman ZC, Glass O, Hodges BL, Niedzwiecki D, et al. Optimization of vaccine responses with an E1, E2b and E3-deleted Ad5 vector circumvents pre-existing anti-vector immunity. Cancer gene therapy. 2009;16:673–682. doi: 10.1038/cgt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gabitzsch E, Xu Y, JP Balint J, Balcaitis S, Sanders-Beer B, Jones F. Induction and comparison of SIV immunity in Ad5 naive and Ad5 immune non-human primates using an AD5 [E1-, E2b-] based vaccine. Vaccine. 2011 doi: 10.1016/j.vaccine.2011.08.038. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borysiewicz LK, Fiander A, Nimako M, Man S, Wilkinson GW, Westmoreland D, et al. A recombinant vaccinia virus encoding human papillomavirus types 16 and 18, E6 and E7 proteins as immunotherapy for cervical cancer. Lancet. 1996;347:1523–1527. doi: 10.1016/s0140-6736(96)90674-1. [DOI] [PubMed] [Google Scholar]
- 35.Baldwin PJ, van der Burg SH, Boswell CM, Offringa R, Hickling JK, Dobson J, et al. Vaccinia-expressed human papillomavirus 16 and 18 e6 and e7 as a therapeutic vaccination for vulval and vaginal intraepithelial neoplasia. Clin Cancer Res. 2003;9:5205–5213. [PubMed] [Google Scholar]
- 36.Davidson EJ, Boswell CM, Sehr P, Pawlita M, Tomlinson AE, McVey RJ, et al. Immunological and clinical responses in women with vulval intraepithelial neoplasia vaccinated with a vaccinia virus encoding human papillomavirus 16/18 oncoproteins. Cancer research. 2003;63:6032–6041. [PubMed] [Google Scholar]
- 37.Lin CT, Tsai YC, He L, Calizo R, Chou HH, Chang TC, et al. A DNA vaccine encoding a codon-optimized human papillomavirus type 16 E6 gene enhances CTL response and anti-tumor activity. Journal of biomedical science. 2006;13:481–488. doi: 10.1007/s11373-006-9086-6. [DOI] [PubMed] [Google Scholar]
- 38.Peng S, Ji H, Trimble C, He L, Tsai YC, Yeatermeyer J, et al. Development of a DNA vaccine targeting human papillomavirus type 16 oncoprotein E6. Journal of virology. 2004;78:8468–8476. doi: 10.1128/JVI.78.16.8468-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karanam B, Gambhira R, Peng S, Jagu S, Kim DJ, Ketner GW, et al. Vaccination with HPV16 L2E6E7 fusion protein in GPI-0100 adjuvant elicits protective humoral and cell-mediated immunity. Vaccine. 2009;27:1040–1049. doi: 10.1016/j.vaccine.2008.11.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shafer-Weaver K, Sayers T, Strobl S, Derby E, Ulderich T, et al. The Granzyme B ELISPOT assay: an alternative to the 51Cr-release assay for monitoring cell-mediated cytotoxicity. J Transl Med. 2003;1:1479–5876. doi: 10.1186/1479-5876-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Naïve C57BL/6 (n=6) were injected with 1×106 mEERL cells and followed for growth. One week after implantation tumors enter into log phase growth with none of the mice surviving without treatment, and all mice needed to be euthanized after 30 days. This illustrates the necessity for chemoradiation in order for the mice to survive, and the benefit of the Ad5 [E1-, E2b-]-E6Δ/E7Δ vaccine when given in conjunction with standard treatment.