

Abstract
Current literature fully supports HPV-associated oropharyngeal squamous cell carcinoma (OPSCC) as a unique clinical entity. It affects an unambiguous patient population with defined risk factors, has a genetic expression pattern more similar to cervical squamous cell carcinoma than non-HPV-associated head and neck squamous cell carcinoma (HNSCC), and may warrant divergent clinical management compared to HNSCC associated with traditional risk factors. However, a detailed understanding of the molecular mechanisms driving these differences and the ability to exploit this knowledge to improve clinical management of OPSCC has not yet come to fruition. This review summarizes the etiology of HPV positive (HPV+) OPSCC and provides a detailed overview of HPV virology and molecular pathogenesis relevant to infection of oropharyngeal tissues. Methods of detection and differential gene expression analyses are also summarized. Future research into mechanisms that mediate tropism of HPV to oropharyngeal tissues, improved detection strategies, and the pathophysiologic significance of altered gene and microRNA expression profiles is warranted.
I. INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) is a major global health problem, ranking as the 6th most prevalent cancer worldwide, with >405,000 new cases/year, 2/3 of which are in the developing world [1]. Mortality is thought to be 200,000 deaths annually, and US deaths occur at a rate of 1 death/hour. These tumors occur in the oral cavity, larynx, orophaynx, and other locations throughout the head and neck [Fig. 1A,B]. The major risk factors of HNSCC include the synergistic effects of tobacco and ethanol abuse in addition to physical irritation, malnutrition, and immune defects. Due to public health efforts encouraging smoking cessation, the overall incidence of HNSCC has decreased in recent years. In contrast, the incidence of oropharyngeal squamous cell carcinoma (OPSCC) has increased significantly at a rate of 1.3% and 0.6% yearly, between 1973 and 2004, for base of tongue and tonsillar carcinomas, respectively. In this same period, the incidence of oral cavity cancer decreased 1.9% [2, 3]. Recent statistics show a striking 225% increase in population-level incidence of HPV-positive (HPV+) OPSCC between 1984 and 2004, while HPV-negative (HPV−) OPSCC incidence decreased by 50% in the same time period [4]. Oropharyngeal refers to the anatomic location from the junction of hard and soft palate to the plane of the hyoid bone and includes the base of tongue, palatine arch (including tonsillar foassae and pillars), uvula, vallecula epiglottica, and lateral and posterior oropharyngeal walls [Fig. 1A,B]. Molecular studies of malignancies arising in this area have shown that 40–80% of OPSCC diagnosed in the US contain human papilloma virus (HPV) and the IARC Multicenter Study estimated that 18% of oral and oropharyngeal cancers worldwide are HPV associated [3, 7]. Recent studies support a viral etiology for oral cancer in these sites [2–8]. Thus, the decline in oral cavity cancer has been attributed to a reduction in tobacco use, whereas the increasing incidence of oropharynx cancer appears to reflect a rise in HPV-related malignancy. Similar observations have been made in Europe [10, 11].
Fig. 1.
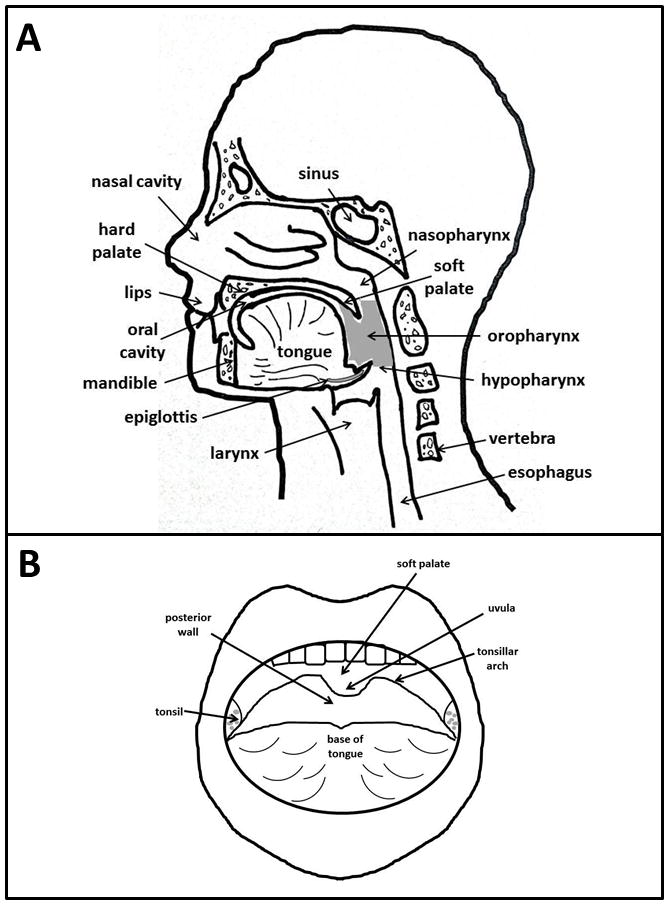
Sites of incidence of (A) head and neck and (B) oropharyngeal squamous cell carcinoma.
HPV-associated cancers have a different biology than HPV− cancers, with distinct phenotypic features distinguishing HPV+ OPSCC including poor differentiation, scant keratinization, and basaloid phenotype compared to the typically keratinizing morphology of HPV-negative tonsillar squamous cell carcinomas [3, 9] [Fig 2]. Indeed, analysis of HPV+ versus HPV− lesions reveal that tumors harboring HPV genomic DNA lack many of the characteristic mutations of disease thought to be attributed to traditional risk factors of tobacco and alcohol abuse. Moreover, viral oncogene expression and variable degrees of genomic integration have been observed in numerous case studies [10, 12–14]. Furthermore, an inverse relationship between p53 mutation and HPV infection has been reported [15], strengthening the evidence in support of a role for HPV in OPSCC etiology. Overall, HPV+ tumors are more likely to have p53 degradation and pRB inactivation with resulting p16 upregulation [16]. In contrast, HPV− tumors tend to have p53 mutation and downregulation of p16 [7, 9].
Fig. 2. Distinct phenotypic features characteristic of (A) HPV+ OPSCC relative to (B) HPV− OPSCC.
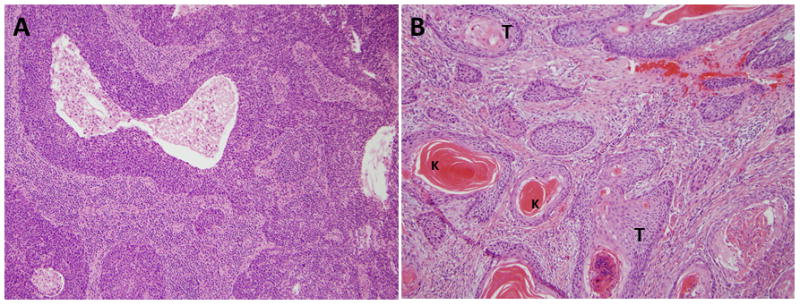
Nonkeratinizing hyperchromatic tumor cells with ill-defined borders, abundant mitoses, and areas of necrosis (A). Keratinizing tumor cells with abundant pink cytoplasm composed in discrete nests (B).
In HPV infection, the life cycle of the virus is linked to the differentiation state of the host cell and requires that the host cell remain active in the cell cycle [21, 22]. The highest level of viral replication occurs in the granular layer of stratified epithelia where keratinocytes are terminally differentiated and are in the process of enucleation and death [Fig. 3] [23]. An infection can lead to unchecked cellular proliferation via overriding the mechanisms that halt the cell cycle in addition to altered cellular differentiation. This pathological state allows for replication of viral DNA in synchrony with chromosomal DNA. The uncontrolled cellular proliferation in the context of manipulated cell cycle checkpoints leads to chromosomal instability and accumulation of genetic mutations and subsequently the initiation of premalignant and malignant lesions. The cellular factors that are co-opted by HPV proteins are involved in the proliferation of cells during their differentiation, including viral E6 mediated ubiquitination of p53 and viral E7 mediated modulation of Rb (section IV.D below).
Fig. 3. HPV infection of oral mucosa.
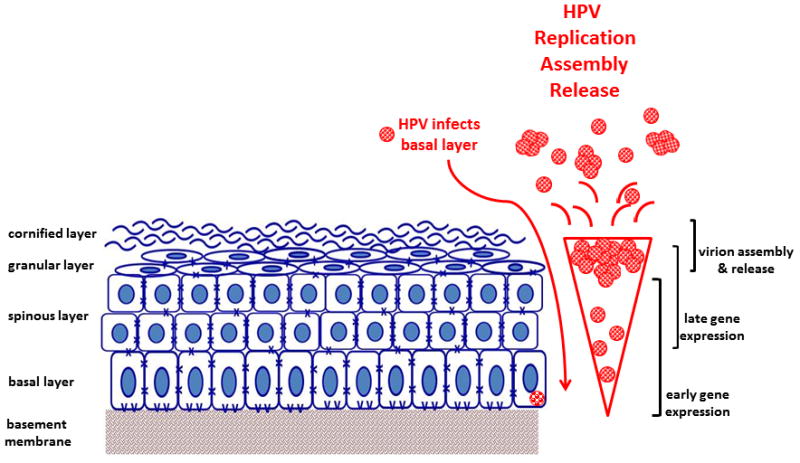
HPV (red) infects proliferating cells in the basal layer that are exposed by wounding. The virus replicates in synchrony with cellular DNA replication. The highest level of viral replication occurs in the granular layer, as expression of HPV E6 and E7 maintain proliferation of these normally terminally differentiated cells. Mature virions are released through the cornified layer.
II. ETIOLOGY AND DETECTION
Certain high-risk subtypes of the human papillomavirus are causative agents for a number of human neoplasia including disease of the cervix, anus, penis, and vulva. Approximately 291 million women worldwide are carriers of HPV DNA, of which 32% are infected with the high risk subtypes HPV16 or HPV18, or both. Type 16 (85–95%) and to a lesser extent 18 are the subtypes most commonly identified among HPV-positive OPSCC [27, 28]. Interestingly, the low risk subtypes HPV6 and HPV11 have been detected in oropharyngeal cancers, suggesting that these types may not in fact be benign at this anatomic location [14, 29]. While detection of HPV genomic DNA was noted in head and neck cancer over 20 years ago [3], an etiological role is now undeniably supported. The involvement of HPV in head and neck squamous cell carcinogenesis is supported by a series of observations. First, HPV is a virus with broad and essential tropism for epithelial tissues. In vitro studies clearly prove that high risk viral oncoproteins immortalize human keratinocytes, including oral keratinocytes [23]. The high risk HPV genotypes can be detected in squamous cell carcinoma with PCR techniques and fluorescent in-situ hybridization [13]. In addition, genotype concordant viral DNA can be found in the lymph nodes of patients with metastatic OPSCC [25]. Moreover, multiple gene signatures detected with DNA microarrays are able to predict HPV-16 prevalence in primary HNSCC with a false discovery rate of <0.2 [26]. Lastly, the established role for high risk HPV in cervical SCC is foundational support for a causative role for HPV in OPSCC because theses distinct anatomical locations share morphological, embryological, and immunological features and can be infected with the same virus [7].
HPV+ OPSCC has identifiable histology, biochemical characteristics and surrogate markers that give it singular identity. As a result of its prognostic value, the American Joint Committee on Cancer Staging Criteria recommends testing HPV status for oral and pharyngeal cancers [30]. Efficient and effective detection of HPV associated tumors is an essential component of disease management as well as research. The optimal method to detect HPV associated tumors, however, is still unresolved. The histopathologic characteristics of HPV+ OPSCC are instructive but not definitive in the detection of HPV. More commonly, polymerase chain reaction, immunohistochemistry, and in-situ hybridization are used as part of a multi-step detection protocol. Histopathologic characteristics of HPV+ OPSCC include poor differentiation, scant keratinization, and basaloid phenotype [Fig. 2]. The usefulness of these findings for diagnostic and prognostic purposes is debated. The non-keratinizing OPSCC phenotype correlates positively with immunohistochemical staining for p16 and favorably associates with prognostic data [31]. Others report a positive association between the non-keratinizing phenotype and HPV+ disease, but a portion of non-keratinizing OPSCC cases are HPV-negative [32, 33]. Further, histologic findings are subject to inter-observer variability. While there is value in completing a histological profile, other methods have largely replaced histologic findings for definitive detection of HPV+ OPSCC.
Polymerase chain reaction (PCR) of HPV sequences requires specimens of fresh frozen or formalin-fixed paraffin-embedded tissue. Tissue sections represent a heterogenous mixture of tumor, stroma, and inflammatory cells, such that laser capture microdissection is necessary to localize HPV signal to the specific cell type of interest. As mentioned previously, the expression of E6 and E7 does not necessarily signal integrated HPV, and whether the virus is integrated or episomal remains unanswered with conventional consensus primer-based PCR. However since the E2 gene is a common point of alteration in integrated HPV, cells with integrated HPV will not amplify a PCR product using primers specific for the E2 gene. The ratio of E2:E6 is then assessed and, if very low, it is taken to reflect integrated HPV [14]. Although this technique is intriguing, it falls short in terms of sensitivity because multiple studies have shown that genes besides E2 can be disrupted secondary to viral integration [36–38] and that viral oncoproteins are expressed regardless of integration status. Quantitative real time RT-PCR (Q-PCR) has provided data that is important clinically in establishing prognosis and providing additional evidence that OPSCC is a unique clinicopathologic entity. The addition of quantification to detection methods has allowed for studies comparing the number of viral genomes present to the disease site and also to patient outcome. The viral load varies widely among OPSCC cases. Recent data shows that it correlates with patient outcome, such that high viral load is associated with improved prognosis. It is thought to occur because most HPV+ tumors with high viral load have high p16 and reduced or low Rb, p53, and cyclin-Dl, while low viral load tumors show reduced p16, increased cyclin-D1 and p53, and normal Rb [8].
As described previously, HPV oncoprotein E7 inactivates the RB gene product leading to sufficient upregulation of p16 for detection by immunohistochemical methods. As a result, p16 serves as a surrogate marker for HPV+ OPSCC, suggesting HPV viral transcription. p16 is a sensitive marker, elevated in cases involving HPV16 as well as less frequently present viral serotypes [40–42]. Immunohistochemical analysis does not, however, differentiate HPV serotype and false positives might be present in the context of HPV-unrelated disturbances in the RB gene product. In-situ hybridization permits identification of specific HPV serotype and direct visualization of the distribution of HPV+ cells within the tissue sample. Signal amplification steps significantly improve the sensitivity of this technique to the point of viral detection with as few as 1 copy per cell [40]. Complete concordance with PCR results was demonstrated in a small sample when unequivocally positive or negative in-situ hybridization findings are present [39]. However, this technique does not provide evidence of viral integration as may be possible with PCR.
III. HPV VIROLOGY
A. Overview
HPVs are small, 50–55nm in diameter, non-enveloped double-stranded DNA viruses, which carry out their life cycle in either mucosal or cutaneous epithelia. Infection may result in an asymptomatic carrier state or a variety of both benign and malignant neoplasia. These viridae have icosahedral capsids composed of 72 capsomeres, surrounding a circular DNA genome of ~7900 base pairs [22]. Hundreds of different genotypes have been identified and the viruses are grouped according to distinctions in type-specific antigens on the virion surface. Each type shares less than 90% DNA sequence homology in the region of their major capsid protein, L1. Specific clinical manifestations are associated with many of the types and for sake of convenience, the more common subtypes are grouped according to high- or low- potential symptoms or the ability to effect cellular transformation. Although there are 15 known high-risk types (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73, and 82) and 12 low-risk types (6, 11, 40, 42, 43, 44, 54, 61, 70, 72, 81, and CP6108), types 16, 18, and 31 are the major types associated with mucosal epithelial cancers, while HPV 16 accounts for 90–95% of HPV positive OPSCC [43].
The genome can be divided into an early (E) region (containing genes E1, E2, E4, E5, E6 and E7), late (L) region (containing genes L1 and L2), and an upstream regulatory region (URR) [Fig. 4]. Two major viral promoters induce transcription of polycistronic mRNAs. The DNA itself associates with cellular histones and is compacted into aggregates similar to chromatin [44]. The conformation of the chromatin and epigenetic modifications of histones are significantly altered upon cellular differentiation, and this effectively divides gene expression into early and late events. During early stages of the viral life cycle, early transcripts are initiated by a promoter referred to as either p97 or p105 in the HPV-16/31 or HPV-18 subtypes, respectively. The p97 promoter is located just upstream of the E6 open reading frame (ORF) [44]. The major promoter of late genes is located further downstream and varies slightly depending on the virus sub-type, but it is generally referred to as p742 [44]. The latter transcriptional events are activated in concert with epithelial differentiation via segregated temporal usage of these promoters. The early genes E1–E7 play a role in regulating, promoting, and supporting viral DNA transcription and replication. The late genes, L1 and L2, are transcribed only in productively infected cells and encode the major and minor capsid proteins required for assembly of progeny virions and eventual accumulation and release into the environment.
Fig. 4. HPV genome organization.
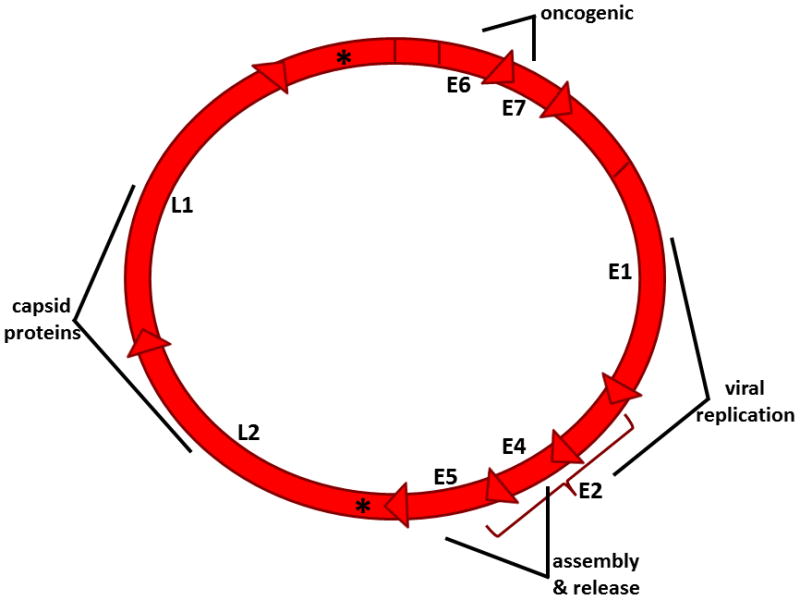
The viral genome of HPV is a double-stranded circular genome of approximately 8 kb transcribed as polycistronic mRNAs with 8 open reading frames. High-risk HPV genomes contain two viral promoters (*) encoding early (E) and late (L) genes.
B. Mode of Viral Entry
The basal cell is fundamental to papillomavirus infection and may be the only cell within epethelia capable of establishing infection. This dependence on terminally differentiated keratinocytes has made the study of viral entry challenging because propogation of virions in cell culture is does not occur [45]. It is thought that infection occurs at sites of injury in the proliferating basal layer of epithelial surfaces. This proliferation due to microtrauma induces basal cell migration and enhanced cell division therefore increasing the probability of a productive infection. A remarkably complicated and still poorly understood process is required for binding to the host cell surface, subsequent internalization, and intracellular trafficking, but the basic evolutionary strategy is dependent upon processes for excluding access to the differentiated cells and fostering access to the basal cell layers. This is achieved by preferential binding to basement membrane proteins specific to the basal layers [45].
Most papillomavirus types utilize heparan sulfate proteoglycans (HSPGs) located within the basement membrane and extracellular matrix, specifically syndecan-1 that is the HSPG isotype expressed predominantly in epithelia, not just for initial attachment but also to promote conformational changes in the viral capsid [49]. These interactions are thought to initiate attachment and be essential for infectious internalization but not directly mediate virion internalization [45]. HPV capsids also bind laminin-5, but the significance of this interaction is not entirely understood [49]. A number of candidate integrins have been studied in vitro and it appears that at least 3 integrins, α6, β4, and β1 may be involved in secondary HPV binding, although α6 is likely paramount [46–48]. Interestingly, there has not been definitive evidence of an amino acid binding motif present on the surface of HPV representative of a known recognition motif for any of these host cell receptors [49]. However, a particular conformational cluster of basic acids is thought to be critical and involved in inducing a conformational change of the capsid upon contact with the host cell surface [49]. Virus internalization may require additional factors such as syndecans and glypicans, two heparan sulfate proteoglycans also thought to be upregulated in response to injury. There is compelling evidence for 3 modes of viral entry: clathrin-mediated endocytosis, caveolar endocytosis, and a clathrin- and caveolae-independent pathway involving tetraspanin-enriched microdomans [49]. HPV-16 is proposed to bind cells and subsequently colocalize with transmembrane tetraspanins CD63 and CD151, although the role of these tetraspanins is not fully elucidated [50]. After binding and endocytosis, HPV is thought to gain access to the microfilament network via an interaction with L2 and the motor protein complex dynein for transport around the cytoplasm and to the nucleus [51].
C. Host Immune Response
Clearly HPV infections that lead to cancer require alterations in host cellular immune response. Prior to viral replication, host immune intervention via tumor necrosis factor a (TNFa) and interleukin-1 (IL-1) production may influence the fate of HPV-infected cells, as a robust response results in antiviral activity and specifically downregulates HPV16 E6/E7 mRNA transcription [52]. Alternatively, some authors have suggested that HPV may mediate resistance to TNF early in its life cycle [53, 54]. From studies in women with cervical HPV infection, it has been demonstrated that the majority of women infected mount an effective immune response and clear the infection, ~10% develop persistent infection, and only 5–10 per 100,000 progress from premalignant to malignant cervical disease in developed countries [23]. Similar comparisons are not yet available for head and neck infections. However, evasion of the innate immune response is a biologic hallmark of HPV infection in both premalignant and malignant disease and results from viral stimulation and/or repression of host signaling pathways [23]. Although the molecular pathways responsible for this evasion are postulated to lie in viral downregulation of cytokine expression, an alternative or perhaps parallel explanation is the evolutionary niche created by infection of keratinocytes, which have a naturally short lifespan and are generally sheltered from large populations of antigen presenting cells [23]. Because of the short keratinocyte lifespan, cell lysis is not necessary for viral escape [Fig. 3]; rather virions are released as cells propogate through the mucosal layers. Nevertheless, the hope of vaccination is to bolster adaptive immunity by introducing highly immunogenic repeat structures based upon L1 termed L1 virus-like particles (VLPs). These VLPs are brought into lymph nodes by antigen presenting cells and are presented to T cells that will drive the production of a B-cell response and capsid specific neutralizing antibodies [56].
Type I interferons are potently produced in response to viral nucleic acids. Under normal conditions, various toll-like receptors such as TLR9 recognize unmethylated CpG DNA, leading to interferon regulatory factor (IRF) activation and expression of IFN-alpha and —beta. IFN subsequently acts in a paracrine fashion to protect neighboring cells that are not yet infected and to induce an antiviral state where replication is hindered, class I MHC molecules are expressed, anti-angiogenic programs stimulated, and immunostimulatory properties induced. E6/7 proteins have been shown to inhibit IFN-alpha signal transduction [55, 56, 58, 59]. E7 binds IRF-1 in vivo and in vitro, inhibiting the IRF-1-mediated activation of IFN-beta promoter [56, 57]. Although the mechanism of this downstream inhibition is not fully elucidated, E7 appears to recruit histone deacetylase to the IRF-1 promoter via the carboxy-terminal zinc finger of E7 [56, 57].
Once the virus escapes the immune system and the infection persists, progression of pathology can ensue. Persistent viral infection with a high-risk subtype is the central etiological factor in the development of anogenital malignancies with 99% of cervical cancers positive for HPV DNA [60] and a sub-set of head and neck cancers. As was mentioned previously, initial infection is in the basal layer where both stem cells and transit-amplifying cells reside. The continuously dividing transit-amplifying cells act as a reservoir of cells for suprabasal cells [44]. It is here that the virus establishes itself as a low copy number extrachromosomal plasmid. The mechanism for extrachromosomal HPV DNA persistence in cycling basal cells is unknown [61] although this copy number is maintained throughout the course of the infection [44]. This process amplifies the genome to around 50–100 copies/cell and is thought to be independent of the cell cycle [55].
IV. PATHOGENESIS- A CONSQUENCE OF VIRAL GENE EXPRESSION
Although HPV has been identified in squamous cell cancers at other head and neck sites, the overwhelming anatomic locations for disease burden associated with HPV are the mucosal lining of the tonsils and the base of tongue [Fig. 1B]. The pharyngeal tonsillar tissue functions as part of the secondary immune system. Their location allows for exposure to both the respiratory and alimentary tracts and therefore allows access to both inspired and ingested antigens. There are four functional immunological structures of the tonsils: the outer endodermally derived reticular crypt epithelium, the mesodermally derived follicular area, the mantle zone, and the germinal center of the lymphoid follicle. Antigen presenting cells at the outer epithelium transport antigen to helper T-cells and may subsequently stimulate B cells to produce IgA antibodies which are transported to the surface. It has been speculated that the immunologic function of the tonsils may contribute to harboring the virus and that transformation of the keratinocytes requires some contribution from the microenvironment created by the lymphocytes and antigen presenting cells populating the tonsils [58, 59, 62]. Some studies have suggested that HPV associated tonsilar SCC originates from the deep invaginations of the cryptic epithelium whereas non-HPV related SCCs develop from the surface epithelium [63–65]. The numerous branching crypts that line the palatine and lingual tonsils are common sites of inflammation due to accumulations of dead lymphocytes and desquamated epithelium [66]. The epithelium of the crypts is reticulated, which refers to a discontinuous epithelium where nonepithelial cells such as lymphocytes and antigen presenting cells pass between squames. This organization of cells facilitates direct transport of antigens, a favorable environment for contact between the effector cells of the immune system, and is thought to help provide a pool of immunoglobulins [67]. However, this reticulated structure also is a compromise in epithelial integrity, leaving the deep layers of the epithelium exposed to deposition of viral particles [68]. The determinants of infectivity and susceptibility are not well established with respect to HPV and oropharyngeal cancers. It has been postulated that the common endodermal orgin of the squamous lining of both the cervix and tonsils, their shared locations at the junction between external and internal environments, and human microbiota at mucosal surfaces at these locations play a role in allowing persistent HPV infection and provide a favorable mileu for cacinogenesis initiation and propogation.
A. E1
E1 contains a nuclear localization signal and it has been proposed that maintenance of the viral episome in undifferentiated keratinocytes is a function of a conserved region of E1 unique to anogenital subtypes of the virus [69]. E1 is a 68kDa protein with ATPase and helicase with 3′–5′ activities. As a monomer, E1 weakly binds AT-rich sequences upstream to the start sites of early transcribed HPV genes [70–75]. E1 recognition sequences in origins of HPV replication are adjacent to E2 DNA binding sites and the latter act to load E1 onto the origin and facilitate higher affinity binding, thereby initiating origin-specific viral DNA replication [44]. The recruitment of cellular replication complexes including chaperone proteins and chromatin remodeling complexes leads to E1 binding host DNA polymerase alpha [76–78]. E1 proteins bound to DNA oligomerize into an hexameric ring and act as a 3′–5′ helicase to unwind supercoiled DNA. The regulation of E1 activity is not fully understood, but it is known that the protein interacts with mitogen-activated protein kinases and the cell cycle regulators cyclin E and A, both of which are expressed after cells have progressed through the restriction point [77]. Mutagenesis studies have revealed essential cyclin binding motifs, serine residues that are CDK sites, and a dominant leucine-rich nuclear export sequence (NES) located on E1 which are important for its nuclear localization and the overall ability of the virus to adapt the cellular regulatory mechanisms to support viral replication [79, 80].
B. E2
E2 is a 50kDa transcription factor, which plays an integral role in transcriptional regulation of early genes, DNA replication, and genome maintenance functions. E2 binds and interacts with numerous cellular factors. The C-terminus encodes a dimeric beta-barrel that binds DNA and interacts with E1 while the N-terminus encodes a transactivation alpha-helix domain rich in glutamine [81–83]. The URR (also known as LCR) of mucosal HPV DNA contains four consensus palindromic sequence specific binding sites for E2 (E2BSs), three of which are in close proximity to E1 recognition sequences [44]. These E2SBs are overlapped by recognition sequences for cellular transcription factors such as TATA box-binding associated factor (TFIID) and specificity protein 1 [84, 85]. During early stages of infection, viral gene expression is minimal and is initiated by transcription factor binding to sequences in the LCR [86]. E2 ineracts with these transcription factors as well as C/EBP transcription factors, activation domain-modulating factor-1, p/CAF, p300/CBP, nucleosome assembly protein (NAP-1), and Brd4 [87]. E2 represses only genes from the early promoter and helps maintain virus copy number low in undifferentiated cells [44]. The mechanisms of transcriptional repression are poorly understood but likely involve E2 mediated recruitment and coordination of several distinct cellular pathways. In support of this, genome-wide siRNA screening identified up to 96 cellular genes contributing to repression [88]. Some of these genes were subsequently validated to independently and additively mediate E2 induced repression, including the cellular demethylase JARID1C/SMCX and EP400, a component of the NuA4/TIP60 histone acetyltransferase complex. High-risk HPV E2 has also been demonstrated to transactivate and upregulate SR (serine/arginine-rich) proteins, highly conserved host splicing regulators for alternative splicing that are co-opted to participate in viral RNA processing and can act as oncoproteins [89, 90]. This leads to dysregulation of the tightly controlled process of constitutive and alternative splicing of host genes and elaborates the involvement of E2 in the pathogenesis of HPV induced malignancies. E2 and p16(INK4a) show exclusive immunohistochemistry staining patterns in precursor stages of cervical carcinoma in paraffin-embedded clinical samples. Staining for cytokeratin K13, a marker of squamous cell differentiation, overlapped with E2 while proliferation markers Ki67 and p63 inversely correlated [91]. Studies in HeLa cells show that expression of E2 results in a suppression of transcription of E6 and E7, reflecting a role of E2 in early stages of HPV infection [92].
C. E4 and E5
Many of the cellular pathways co-opted by HPV discussed so far are logically consistent with viral replication. The E4 and E5 proteins are expressed early, but function to modulate the late phase of the viral life cycle. As E4 and E5 are the third and fourth transcripts on polycistronic early mRNA, respectively, very little protein is translated because the ribosome-scanning mechanisms of HPV are relatively inefficient [93]. After epithelial differentiation, E4 becomes the most highly expressed viral gene due to utilization of the late gene promoter, therefore placing these transcripts first and second on polycistronic late mRNA [44]. The E4 ORF lacks a unique AUG codon but is translated with the first 5 amino acids from E1 because of splicing events, allowing for an E1–E4 fusion protein [44]. In vitro functions of E1–E4 include E4 mediated G2/M arrest via a cyclin binding motif [94], association with and reorganization of keratin intermediate filaments [95], and regulation of viral gene expression perhaps by interacting with E4-DBD RNA helicase [96]. In vivo effects remain debated, but it is known that there is minimal sequence homology between low- and high-risk E4 protein and that the high-risk types are more likely to interact with and possibly collapse keratin networks.
E5 is an 83 amino acid hydrophobic protein and therefore localizes to membrane-bound compartments including the golgi, endosomes, endoplasmic reticulum, and the nuclear membrane. The function of this protein is controversial. Some studies suggest that, similar to bovine papillomavirus E5 association with platelet-derived growth factor receptor, that HPV E5 activates epidermal growth factor receptor (EGFR) [97]. However mutational analyses have shown that EGFR is not a direct target of E5 action [98]. Instead, E5 affects EGFR turnover from the plasma membrane by binding an endosomal vacuolar ATPase, the result of which is impaired early endosome acidification, increased recycling of receptors to the cell surface, and therefore amplified receptor signaling [22, 23]. In vitro studies have shown increased phosphoylation of Akt and Erk1/2 associated with E5 mediated enhancement of EGFR signaling, leading to increased expression of VEGF [64]. The contribution of E5 to carcinogenesis or malignant potential in head and neck cancer is unclear; however because EGFR activity is a prognostic indicator and therapeutic target, further investigation of E5 regulation of EGFR in OPSCC is warranted.
D. E6 and E7
It has long been thought that the viral genome is maintained in a non-integrated episomal form in benign warts and integrates into the host genome in most cancers. Increased oncogene expression has been demonstrated subsequent to E2 disruption/deletion and integration and is proposed as a central event in oncogenesis [99, 100]. As with other oncogenic viruses, the integration into the host genome appears to be a random event. However, the pattern of integration is clonal. In cervical cancer, integration of high-risk HPV into host genomic material induces genomic instability in the host and also alters gene expression in the virus. When integrated, the open reading frame of the viral genome appears to be consistently interrupted such that the viral repressor E2 is lost, resulting in overexpression of two oncoproteins, E6 and E7. E6/7, when overexpressed, disrupt the function of wildtype Rb and p53 leading to the development of a malignant phenotype. As a selective mechanism, this favorable gene expression profile allows cells with integrated virus a significant growth advantage. Indeed, in anogenital cancer, a high proportion of HPV tumors with integrated virus are observed. Studies from head and neck cancers have revealed that viral integration is not necessary for initiation of oncogenesis and these cancers vary greatly with respect to the physical state of the HPV genome (episomal versus integrated) [63, 101, 102]. The significance of this discordance is not well understood, nor is the mechanism of overexpression of E6/E7 in tumors of the head and neck. However, in both cases, in a subset of infected patients, viral infection leads to transcriptionally active HPV with high E6/E7 mRNAs levels, and subsequently to cell proliferation and eventually to genomic instability due in part to mitotic spindle defects that result in aneuploidy and damaged chromosome structure [102, 103]. Interestingly, E6/E7 gene products are required for the maintenance of proliferation in HeLa cells [23]. These cells, that have been cultured for 50 years, have been demonstrated to contain integrated sequences of HPV-18 expressing E6 and E7 and repression at the HPV-18 promoter or knockdown with RNAi of E6/7 results in senescence in 10–14days [23]. Repression of E6/7 in HeLa cells also results in diminished telomerase activity, cyclin-dependent kinase activity, and c-myc expression [104].
The upper layers of the mucosal surface, predominantly the stratum spinosum and granulosum, are sites of viral assembly and high-level viral gene expression [Fig. 3]. This is a key point in understanding HPV infection, as the life cycle of the virus is linked to the differentiation state of the host cell. Under normal conditions, cell division and cell cycle entry are confined to the basal layer. HPV requires that the host cell remain active in the cell cycle as the cell progresses through the strata because the virus is dependent on the enzymatic machinery of the cell [22]. There appears to be temporal and spatial regulation of viral gene expression. In normal epithelia, the cell cycle is halted after the divisions taking place in the basal layer. An important molecular correlate to this is the activity of p63, an essential regulator of keratinocyte differentiation. An infection may initiate carcinogenesis when the regulation of differentiation is disrupted, the cell undergoes resistance to growth inhibition, evades the immune response, subverts apoptosis, and is effectively immortalized. This pathological state allows for replication of viral DNA in synchrony with chromosomal DNA. Importantly, as the cell enters the upper, differentiating compartment of the epithelium, viral gene expression transitions from minimal to very high, such that the virus is able to utilize the enzymatic machinery of the host cell to produce multiple copies per cell. This replication is due to entry of the host cell into S phase, a phenomena largely attributed to the viral protein E7 since knockdown of this protein fails to initiate cellular proliferation and viral genome amplification [105].
The uncontrolled cellular proliferation in the context of manipulated cell cycle checkpoints leads to chromosomal instability and accumulation of genetic mutations [Fig. 5]. The cellular factors that are co-opted by HPV proteins are involved in the proliferation of cells during their differentiation. The cooperative activities of the viral proteins E6 and E7 are fundamental in the development of genomic instability because they synergize to effect myriad cellular factors involved in cell cycle progression, signaling pathways involved in cell survival, differentiation, proliferation (independent of direct effects on the cell cycle), and inhibition of apoptosis. The centrality of E6 and E7 in HPV pathogenesis is reflected by the observation that expression in organotypic raft cultures results in identical phenotypic changes as those seen high-grade cervical squamous neoplasia in vivo [106]. When E6 and E7 are present, it has been observed that these cells have a higher percentage of polyploidy in addition to inappropriate centrosomal duplication [107]. The result of disrupted mitotic events is chromosomal instability. Because cells of stratified squamous epithelia eventually desquamate, this instability of the structures required for coordinated gene expression is likely to cause malignancy only when there is long-term viral presence, possibly in the time frame of years to decades [23].
Fig. 5. HPV E6 and E7 promote cellular transformation and development of malignant phenotype.
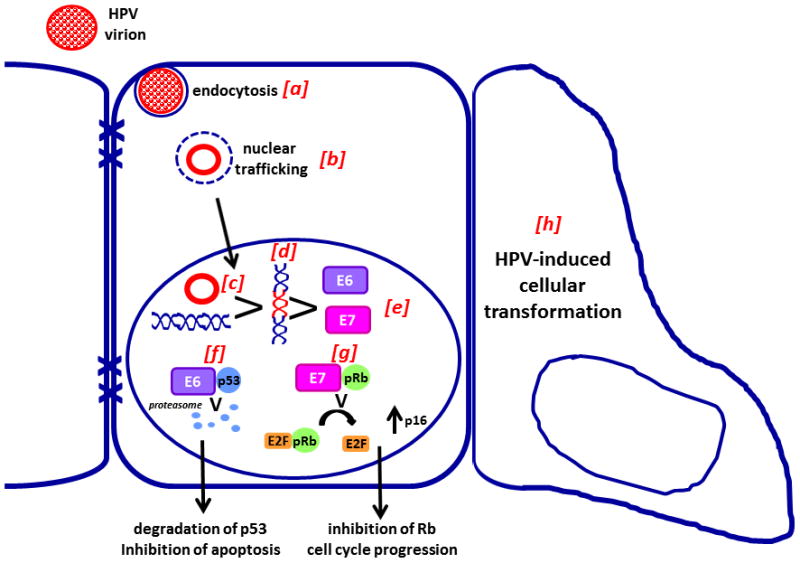
Virions enter the cell via endocytosis (a) and are trafficked to the nucleus (b) where they persist in episomal form (c) or are integrated into the host genome (d). Both episomal and integrated viral DNA produce E6 and E7 (e). Interaction of E6 with p53 and the ubiquitin ligase E6-associated protein target p53 for proteasomal degradation (f) and prevents apoptosis. Retinoblastoma family tumor suppressor proteins including pRb, p130, and p107 interact with E7 (g) and are inactivated, resulting in release of E2F and promoting cell cucle progression. Together these functions of E6 and E7 promote cellular transformation (h). Adapted with permission from [13].
E6 is an 18kDa 150 amino acid protein that contains two zinc binding domains with two Cys-X-X-Cys motifs [Fig. 6] [44, 108]. It is expressed predominantly in the nucleus but can also be found in the cytoplasm. There are over 12 binding proteins for E6 all of which have a permissive effect on the actions of E7. The combined effects of these proteins result in cells which do not effectively segregate and coordinate the events of the cell cycle leading to the accumulation of mutations because cell cycle checkpoints are attenuated at the achilles heel: p53. E6 is exquisitely evolved to affect cell immortalization via both direct and indirect mechanisms. First, E6 forms a trimeric complex with p53 and a cellular E3 ubiquitin ligase, termed E6AP [109]. This interaction results in ubiquinylation of p53 and subsequent degradation by the 26S proteosome, drastically reducing the half-life of p53 [110]. Furthermore, E6 can inhibit p53 mediated gene transcription by direct binding and inhibition of its transcriptional activities [111]. P53 is known to heterodimerize with various isoforms of the p63 and p73 family. Because the steady-state level of p53 is reduced as a consequence of E6 expression, it is thought that the differentiation process is altered because the relative balance between p53 and p63 is disrupted, possibly leading to poor coordination of the genes necessary for normal squamous differentiation [23]. Indirectly, E6 associates with and inactivates three histone acetyltransferases; p300, Creb binding protein, and ADA3. These increase the stability of p53 via acetylation [112–114]. Degradation of ADA3 has also been proposed to underlie the inhibition of p14/ARF dependent activation of p53 [115, 116]. E6 contributes additional mechanisms to allow for immortalization of cells by activating transcription of telomerase reverse transcriptase (TERT) [117, 118]. This is the catalytic sub-unit of telomerase, an enzyme usually not expressed in somatic cells. Activation is dependent on E6AP, as knockdown fails to recapitulate the phenotype [108]. All high-risk E6 proteins contain a C-terminal motif termed XT/SXV that mediates binding to cellular proteins with PDZ domains [22, 119]. The PDZ domain proteins are also implicated in the oncogenic activity of HPV because mutant E6 proteins that lose their ability to bind p53 can still immortalize cells [119–121], while transgenic mice expressing E6 mutant XT/SXV domains do not develop tumors [122]. PDZ proteins are often found in epithelial tight junctions and it is thought that E6 binding modulates signal transduction pathways downstream of these cell-cell contact proteins [44, 119].
Fig. 6. Representation of HPV16 E6 structure and binding partners.
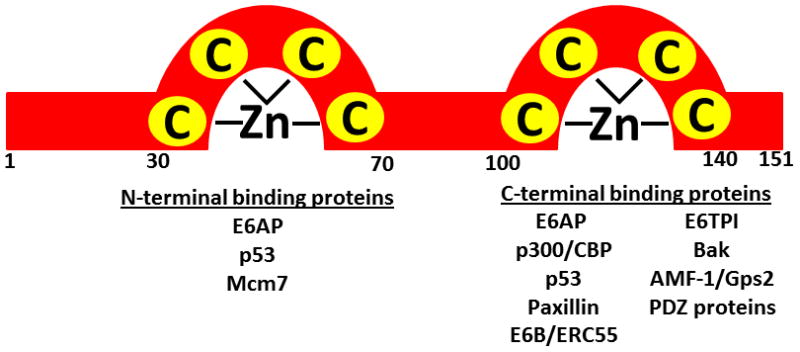
HPV E6 proteins are approximately 150 amino acids and contain two zinc finger domains. Identified binding partners of the N- and C-terminal domains are indicated below. Adapted with permission from [118].
Cell cycle and S-phase dependent amplification of the HPV DNA in the differentiated upper levels of the epithelium is most prominently set into motion by HPV E7. Many coalescing signaling pathways can induce the cellular transition from G1→S phase, but the regulating factors essential for binding and subsequently inhibiting transcription factors controlling S-phase DNA replication machinery are the retinoblastoma family members p105 (Rb-tumor suppressor), p107, and p130 [23]. Rb protein appears to be the predominant tumor suppressor, supported by observations obtained using knockout mouse models as well as the general observation that Rb inactivation (but not p107 or p130) is a hallmark of many sporadic human cancers [123]. These small so called nuclear phospho-‘pocket proteins’ regulate the activity of the many members of the E2F family of transcription factors [23]. The cell cycle can progress when coordinated signaling pathways provide biological feedback to the Rb family members via cyclin-dependent kinase complexes, resulting in their hyperphosphoylation and release from the transactivation domain of E2F family transcription factors and the dissociation of the chromatin remodeling proteins, the HDACs. E2F family members bind DNA in the promoter region of many genes involved in DNA synthesis, cell cycle progression (cyclin A and cyclin E), and mitosis. All E7 proteins from HPV virions have evolved to bind cellular Rb family members but only high risk E7 can efficiently target them for degradation thereby disrupting Rb-E2F-HDAC complexes. [22, 118]. The difference between low risk E7 and high risk E7 appears to be attributed to an Asp vs Gly at positions 21 and 22 for the high risk and low risk, respectively [118]. Upon binding, E7 targets Rb family members for ubiquitin-dependent proteasomal degradation [118]. The result of degraded Rb is constitutive expression of E2F responsive genes and the hallmark event in a cell transitioning from resting or G1 phase to S-phase and subsequent preparation for DNA replication and mitosis.
E7 is a 13 kDa protein ~100 amino acids in length with conserved sequence homologies with Adenovirus E1A and SV40 large T antigen [Fig. 7] [118]. Expression is predominantly nuclear but some studies report a cytoplasmic presence [22, 124]. Interestingly, E7 has no intrinsic enzymatic or DNA-binding activities [22]. Instead, it is known to bind an almost overwhelming multitude of cellular factors, the biological significance of which is not fully understood. However, E7 represents an exquisitely evolved viral protein that adapts many host signaling pathways to the benefit of viral replication. It is well known that E7 has extremely low immunogenicity, and the solution structure provides some explanation as to why this may be; the highly conserved N-terminal region is intrinsically unstructured [124]. The N-terminus contains strong interaction domains with the Rb family members within an LXCXE motif with the conserved region 2, in addition to binding domains for p300 and p600 [118, 124]. The C-terminus contains a zinc-binding domain composed of two CXXC motifs and subsequently creates a more rigid structure [118, 124]. There are multiple binding motifs within the C-terminus, including residues that interact with E2F directly, a weaker Rb interaction domain thought to be important for Rb destabilization in HR-infections, a binding site for class I HDACs, and domains that directly interact with and effectively titrate cyclin/cdk kinases p21 and p27, both activators of Rb via CDK2 [118]. The binding of E7 to E2F6 has fascinating consequences, as the interaction is thought to maintain a conducive environment for viral replication by recruiting polycomb group complexes to E2F-responsive promoters [22]. This recruitment is thought to inhibit the physiologic repression induced by polycomb complexes at promoter regions and allow for constitutive E2F gene transcription [22]. This function of E7 is similar to the function of binding class I HDACs. E7 facilitates HDAC removal at other non-E2F promoters allowing transcriptional machinery access to promoters [22]. Furthermore, the E7-HDAC interaction is essential for episomal maintenance [22].
Fig. 7. Representation of HPV16 E7 structure and binding partners.
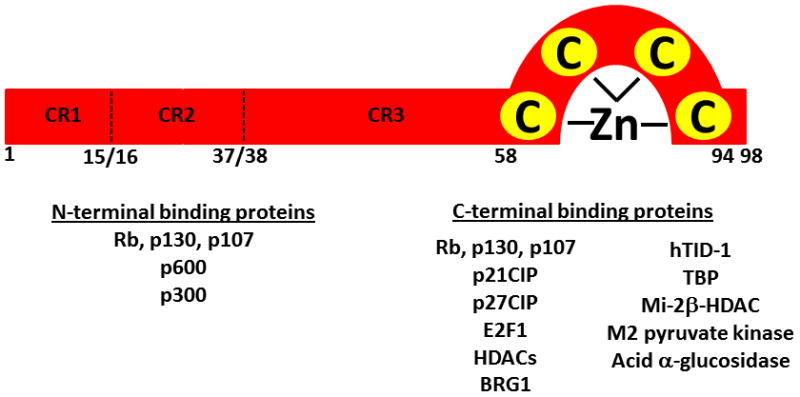
HPV E7 proteins are approximately 100 amino acids and contain a single zinc finger domain. Three conserved regions designated CR1, CR2 and CR3 are homologous to Adenovirus E1A and SV40 large T antigen and are essential for viral oncogenesis. Identified binding partners of the N- and C-terminal domains are indicated below. Adapted with permission from [118].
Unlike E6, the expression of high risk HPV E7 proteins alone can immortalize human keratinocytes at a low rate. In a transgenic mouse model, E7 expression along with low-dose estrogen exposure induced high-grade cervical dysplasia [22]. E7 also may inhibit differentiation by binding and inhibiting the activity of p300/CAF (CREB-binding protein associated factor), the activity of which has been shown to acetylate RB1 at a coordinated time point so that keratinocytes exit the cell cycle and permanently differentiate [23]. Many of the mechanisms promoting anti-apoptotic activities and driving continual cell cycle progression are indirect biochemical effects in the sense that viral proteins are not directly interacting with some of the targets that promote these driving forces of malignant progression. In fact, p53 levels increased downstream of constitutive E2F gene transcription, but once again the synergistic relationship between E6 and E7 acts to neutralize the cell-cycle stalling and pro-apoptotic effects of p53. Signaling pathways associated with keratinocyte survival, such as cell-to-cell contact mediated E-cadherin signaling are also altered to allow for successful viral replication. For example, E7 upregulates the Akt kinases, which are well characterized downstream effectors of E-cadherin and normally encourage pro-survival signals as keratinocytes detach from the basement membrane and stratify upwards [23].
Recent literature has suggested alterations in host miRNA and other epigenetic markers that are induced by HPV in cervical cancer [125–131]. For example, experiments in normal human keratinocytes have shown that the E7 viral oncoprotein blocks normal upregulation of miR-203, a factor that negatively regulates proliferative capacity upon epithelial cell differentiation [125]. E5 expression has also been shown to repress miR-203 and result in increased p63 levels via an unknown mechanism [129]. Furthermore, E5 expression led to significant upregulation of miR-146a and repression of miR-324-5p [133]. miR-146a upregulation by E5 may play significant roles in promoting cell cycle proteins and attenuating immune response to HPV while miR-324-p5 alteration may lead to events associated with epithelial-mesenchymal transition such as N-cadherin upregulation and E-cadherin downregulation [129]. In cervical cancer tissue and in cell lines, the high-risk HPV oncoprotein E6 inhibits tumor suppressive miR-34a by destabilizing p53 [127, 130]. An analysis of miRNA expression profiles in HPV-positive cervical cancer cell lines, lesions, and tumor tissues revealed aberrant expression of miR-218 and targeting of the epithelial cell-specific marker LAMB3 [128]. An additional miRNA expression profile on cervical cancer cell lines provided data important for understanding the mechanisms by which particular miRNAs are involved in carcinogenesis by correlating miRNA expression and tissue differentiation [126]. The relationship between HPV status and miRNA profiles in OPSCC has not been evaluated.
V. DIFFERENTIAL GENE EXPRESSION ANALYSIS
Detected HPV+ OPSCC has increasingly been subjected to molecular study to identify key aberrations present in affected cells that may improve understanding and inform discrepancies in response to therapy and prognosis among tumor populations. Researchers have compared HPV+ OPSCC to HPV+ disease arising in different anatomical sites, HPV− disease, and normal oral mucosa as well as HPV+ tumors in smokers and non-smokers. Key areas of genetic aberrations include DNA replication, DNA repair, cell cycling, and chemotherapy/radiotherapy sensitivity. A summary of genomic and proteomic profile data is presented.
When genome-wide chromosomal profiles compare HPV+ cervical SCCs, HPV+ OPSCCs, and HPV− OPSCCs, hierarchical clusters demonstrate two main groups: one mainly HPV+ and the other HPV− (Fig. 8). However chromosomal alterations were also identified specific for the two tissue types as well as pan-SCC aberrations [132]. A second study corroborates enhanced similarity between HPV+ OPSCC and HPV+ cervical SCC relative to HPV-negative OPSCC, in particular a distinct and larger subset of cell cycle genes and some testis-specific genes normally expressed in meiotic cells are upregulated in concordance with HPV infection [133]. These common changes in transcriptional profiles identified between HPV+ HNSCC and cervical carcinoma, include down regulation of genes involved in immune response (interleukins and interferon-induced proteins) and upregulation of genes within the 20q region along with downregulation of genes on 13q [134]. Upregulation of 20q appears to be caused by E7 expression and is related to inactivation of Rb [134].
Fig. 8. Common and distinct chromosomal profiles of HPV(+) vs HPV(−) HNSCC relative to HPV(+) cervical SCC (CxSCC).
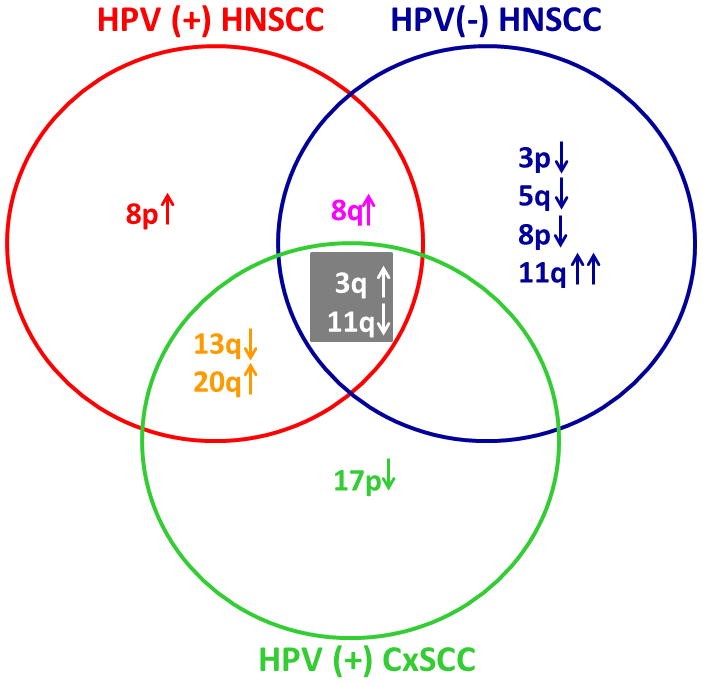
Array-based comparative genomic hybridization identified specific chromosomal alterations in HPV(+) and HPV(−) SCC from head and neck or cervical tissue. Gain at 3q and losses at 11q were common in all sample groups while loss at 13q and gain at 20q were representative of HPV(+) disease, regardless of the tissue of origin. Adapted with permission from [134].
An interesting recent study compared gene expression profiles of HPV+ versus HPV−oropharyngeal cancer and oral cavity cancer [132]. In oral cavity tumors, no significant difference in gene expression was noted when comparing HPV+ to HPV− specimens. However, analysis of oropharyngeal tumors shows significant differences (347 differentially expressed genes) in HPV+ versus HPV− lesions. Differences were particularly common among genes involved in DNA regulation and repair, cell cycle, and chemo- or radio-therapy sensitivity. These results underscore the observation that HPV+ oropharyngeal disease represents a divergent biologic entity from HPV− disease. An additional study compared gene expression profiles of HPV+ HNSCC with normal oral mucosa. A larger number of genes were differentially expressed in HPV+ HNSCC versus cancer free oral mucosa (397) and in comparison of HPV− HNSCC to normal oral tissue [137]. A subgroup of 59 genes was present only in HPV+ HNSCC and not in HPV− HNSCC or normal oral mucosa. A summary table of key genomic regions altered in HPV+ disease is presented (Table 1). These genes are predominantly involved in mitosis, DNA repair and transcriptional regulation.
Table 1.
Key genes upregulated in HPV+ tumors.
| Gene Upregulated in HPV + tumors | Function | Reference |
|---|---|---|
| CDKN2A | Cell cyle inhibitor, product of E2f transcription | 147, 151 |
| PCNA | Cell cycle-dependent auxillary protein for DNA polymerase (pol) delta (also required for SV40 DNA replication in vitro). | 147, 151 |
| RFC4 | Accessory protein (with PCNA) for DNA pol delta and epsilon. Part of a large multisubunit protein complex of DNA damage sensors called BASC. | 147, 151 |
| MCM2 | Early S phase protein involved in DNA replication, likely regulated by E2F motifs. | 147, 151 |
| MCM3 | Involved in DNA replication, likely serves as helicase that links a histone chaperone to a histone H3–H4 bridge | 147, 151 |
| CDC7 (cell- division cycle-7)- | Protein kinase essential for the G1/S transition and initiation of DNA replication. May regulate DNA replication by modulating MCM functions. | 147, 151 |
| TYMS (thymidylate synthetase)- | Maintains the dTMP (thymidine-5-prime monophosphate) pool critical for DNA replication and repair. | 147, 151 |
| CCNE2 (cyclin E2 | Activates CDK2 and is involved in DNA synthesis. | 147, 151 |
| USP1 (Ubiquitin- specific protease 1)- | Negatively regulates PCNA polyubiquitination, a process normally induced by DNA damaging agents. | 147, 151 |
| BRG1 | Involved in a switch in ATP-dependent chromatin- remodeling mechanisms that coincide with the final mitotic division. Note that this essential transition is mediated by repression of BAF53a by miR-9* and miR-124. | 147, 151 |
As tobacco use is a known risk factor for HNSCC, smoking status was considered in a recent gene expression profiling study. Comparison of gene expression patterns from never smokers that were HPV+ to those that were HPV− revealed that genes involved in viral defense and immune response were downregulated in HPV+ tumors (IFIT1, IFITM1-3, IFI6-16, IFI44L, and OAS2), consistent with the immuno-modulating nature of HPV infections, predisposing to development into malignancies [136, 137]. Upregulated genes include those involved in CDK inhibition (cyclin-dependent kinase inhibitor-2C or CDKN2C), G1/S transition (CDC7), replication and DNA repair (RFC4-5), and dimerization with E2F (TFDP2). For HPV+ smokers versus nonsmokers, differential expression was observed in several genes regulated by p53 or E2F-transcription factors. Replication factor-C4 (RFC4) was consistently upregulated in HPV− samples from never-smokers. Other differentially expressed genes based on smoking status include: CDC7, MCM2, cytochrome P450 (CYP4V2), insulin-like growth factor (IGF), and keratin associated proteins (KRT6B, 10, 11). Significant upregulation of a key component of the mammalian synaptonemal complex, a structure regulating the arrangement of homologous chromosomes in meiosis (SYCP2) and normally transiently expressed in gametes, was also observed.
In addition to modified gene expression profiles, chromosomal aberrations and amplifications are significantly less frequent in HPV+ compared to HPV− OPSCC [41, 135]. A group of chromosomal aberrations in OPSCC clearly occur more frequently amongst HPV-cases, including gains at 11q13 and losses of 3p, 9p, and 18q. Based on these data, it has been proposed that the number of required genetic alterations for progression to malignancy is lower in HPV+ oncogenesis [135]. These findings are interesting considering that HPV+ disease is associated with a favorable prognosis, as these specific chromosomal aberrations also appear to be significant indicators of clinical outcome. A notable exception is 16q loss, which predominates in but is not exclusive to HPV+ OPSCC and may be an independent positive prognostic variable [135]. The 11q13 amplification, however, portends a poorer prognosis regardless of HPV status but tends to be rare in HPV related disease [135]. Thus, these genetic differences may have dramatic implications for differing clinical management for the two disease phenotypes.
VI. UNANSWERED QUESTIONS AND FUTURE DIRECTIONS
Like other HPV-associated malignancies, HPV+ oropharyngeal cancers appear to be a sexually transmitted disease; however there is evidence that non-sexual transmission can occur [7, 138]. HPV+ tumors are more likely to affect white young men (<50 years old) with no history of ethanol or tobacco abuse and are associated with distinct risk factors including > 6 lifetime oral-sex partners (odds ratio 3.4), > 26 lifetime vaginal-sex partners (odds ratio 3.1), a younger age at first sexual intercourse, and seropositivity for HPV type 16 L1 capsid protein (odds ratio 32.2) [139, 140]. Similarly, an odds ratio of 230 was reported for the development of oropharyngeal cancer with high-risk HPV DNA detected in the oral cavity when adjusted for alcohol and tobacco use [139–141], while a similar landmark study reported an odds ratio of 14.4 for the association of HPV-16 seropositivity with the risk of OPSCC [141]. In individuals with OPSCC, seropositivity for HPV-16, was strongly associated with no history of tobacco or alcohol abuse [139]. The mechanism predisposing white young men to this type of cancer remains obscure, but is currently supported in the literature both in the US and in Europe where high rates of HPV+ OPSCC have been reported in Scandinavian countries [10, 11, 132]. This is in stark contrast to cervical cancer, where trends show that overall HPV prevalence and HPV16 prevalence is highest in sub-Saharan Africa [142]. However, the same study showed HPV+ women in Europe were significantly more likely to be infected with HPV16 than were those in sub-Saharan Africa (OR 2.64, p=0.0002). Since type 16 is the most commonly identified virus in OPSCC, widespread vaccination with a HPV vaccine in women may lead to a decline in the incidence of male HPV+ OPSCC [143].
Patients with HPV+ OPSCC more often present with stage III–IV disease and paradoxically, data show the disease responds better to chemotherapy and radiation, such that HPV status is a strong positive prognostic factor for survival [17]. A review of the literature suggests that patients with HPV+ HNSCC had a lower risk of death (meta HR:.85, 95% CI 0.7–1.0) and less chance of recurrence (meta HR: 0.62, 95% CI: 0.5–0.8) [6]. When anatomic site is taken into consideration, patients with HPV+ OPSCC have a 28% reduced risk of death (meta HR: 0.72, 95%CI: 0.5–1.0) compared to HPV− OPSCC [7]. The biologic mechanism that underlies this increased responsiveness to treatment and improved overall survival is currently unknown [15, 16–19]. Further questions remain regarding the details of why HPV has a tropism for oropharyngeal tissues, how virus enters keratinocytes preferentially at this specific anatomic site, and how it is able to induce malignant transformation. The cell- and tissue-level events associated with the distinct histology observed in HPV+ OPSCC specimens raises the question of whether HPV+ OPSCC arise from a cell type distinct from that of HPV− OPSCC, or whether the viral life cycle alters keratin expression and deposition by infected cells. Another area in need of further exploration involves determining the prevalence of HPV16 in non-cancerous tissues. Interestingly one survey identified that 13 of 206 cases of tonsillitis and 1 of 174 normal tonsils were positive for HPV 16 DNA by PCR in the absence of L1 antibody production [144]. Indeed, even in HPV+ OPSCC, the question of whether the virus is integrated or episomal remains unanswered. Further, it is currently unknown whether a specific viral load threshold is associated with carcinogenesis and whether viral load is a significant prognostic factor.
In the last few years, HPV status has been linked to racial survival discrepancies in the United States [145–148]. A recent study using National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) data reflected lower overall survival rates for black patients with HNSCC and particularly OPSCC [147, 148]. It is important to note that black patients tend to present at more advanced stages of disease, with 38.6% of patients presenting with stage IV disease compared with 34.0% and 30.2% of Hispanic and white patients, respectively [148]. Further, of all head and neck cancer sites considered, OPSCC was associated with the greatest difference in survival between blacks and whites. This finding is supported by results of an additional study showing that only 4% of HNSCC were HPV+ in black patients, while 34% were HPV+ in white patients [146]. Related to this, a critical area for examination is the issue of screening for oral and oropharngeal infection, dysplastic changes, and malignancies in a cost-effective and wide-spread manner. Research into microRNAs, because of their relative stability in serum and possibly saliva, holds particular interest in this regard.
VIII. Conclusion
Current clinical literature fully supports HPV-associated OPSCC as a unique entity: it is histologically distinct, affects an unambiguous patient population with defined risk factors, and may warrant divergent clinical management compared to HNSCC associated with traditional risk factors. Although epidemiological and clinical studies support HPV+ OPSCC as a distinct clinical entity, the pathobiology of HPV-associated OPSCC initiation and progression is poorly understood. A more detailed understanding of molecular events associated with HPV+ OPSCC progression is therefore a prerequisite for improved staging and diagnostic/prognostic stratification, as well as for the design of novel therapeutic strategies specific for this population.
Acknowledgments
The authors are partially supported by a National Institutes of Health (NIH)/National Institute of Dental and Craniofacial Research Ruth L. Kirschstein National Research Service Award Individual Fellowship F31DE021926 (D.L.M.) and a NIH/National Cancer Institute Research Grant RO1CA085870 (M.S.S.).
LITERATUE CITED
- 1.Duvvuri U, Myers JN. Cancer of the head and neck is the sixth most common cancer worldwide. Curr Probl Surg. 2009;46:114–7. doi: 10.1067/j.cpsurg.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Chaturvedi AK, Engels EA, Anderson WF, Gillison ML. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J Clin Oncol. 2008;26:612–9. doi: 10.1200/JCO.2007.14.1713. [DOI] [PubMed] [Google Scholar]
- 3.Wilczynski SP, Lin BT, Xie Y, Paz IB. Detection of human papillomavirus DNA and oncoprotein overexpression are associated with distinct morphological patterns of tonsillar squamous cell carcinoma. Am J Pathol. 1998;152:145–56. [PMC free article] [PubMed] [Google Scholar]
- 4.Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, Jiang B, Goodman MT, Sibug-Saber M, Cozen W, Liu L, Lynch CF, Wentzensen N, Jordan RC, Altekruse S, Anderson WF, Resenberg PS, Gillison ML. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29:4294–301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strati K, Lambert PF. Human Paillomavirus association with head and neck cancers: Understanding virus biology and using it in the development of cancer diagnostics. Expert Opin Med Diagn. 2008;2:11–20. doi: 10.1517/17530059.2.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petersen PE. Oral cancer prevention and control--the approach of the World Health Organization. Oral Oncol. 2009;45:454–60. doi: 10.1016/j.oraloncology.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 7.Pannone G, Santoro A, Papagerakis S, Lo Muzio L, De Rosa G, Bufo P. The role of human papillomavirus in the pathogenesis of head & neck squamous cell carcinoma: an overview. Infect Agent Cancer. 2011;6:4. doi: 10.1186/1750-9378-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Syrjanen S. The role of human papillomavirus infection in head and neck cancers. Ann Oncol. 2010;21(Suppl 7):vii243–vii245. doi: 10.1093/annonc/mdq454. [DOI] [PubMed] [Google Scholar]
- 9.Marur S, D’Souza G, Westra WH, Forastiere AA. HPV-associated head and neck cancer: a virus-related cancer epidemic. Lancet Oncol. 2010;11:781–9. doi: 10.1016/S1470-2045(10)70017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dayyani F, Etzel CJ, Liu M, Ho CH, Lippman SM, Tsao AS. Meta-analysis of the impact of human papillomavirus (HPV) on cancer risk and overall survival in head and neck squamous cell carcinomas (HNSCC) Head Neck Oncol. 2010;2:15. doi: 10.1186/1758-3284-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Licitra L, Zigon G, Gatta G, Sanchez MJ, Berrino F. EUROCARE Working Group. Human papillomavirus in HNSCC: a European epidemiologic perspective. Hematol Oncol Clin North Am. 2008;22:1143–53. vii–viii. doi: 10.1016/j.hoc.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Snijders PJ, Heideman DA, Meijer CJ. Methods for HPV detection in exfoliated cell and tissue specimens. APMIS. 2010;118:520–8. doi: 10.1111/j.1600-0463.2010.02621.x. [DOI] [PubMed] [Google Scholar]
- 13.Allen CT, Lewis JS, Jr, El-Mofty SK, Haughey BH, Nussenbaum B. Human papillomavirus and oropharynx cancer: biology, detection and clinical implications. Laryngoscope. 2010;120:1756–72. doi: 10.1002/lary.20936. [DOI] [PubMed] [Google Scholar]
- 14.Syrjanen S. Human papillomavirus (HPV) in head and neck cancer. J Clin Virol. 2005;32:S59–66. doi: 10.1016/j.jcv.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 15.Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, Shah KV, Sidransky D. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92:709–20. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 16.Psyrri A, DiMaio D. Human papillomavirus in cervical and head-and-neck cancer. Nat Clin Pract Oncol. 2008;5:24–31. doi: 10.1038/ncponc0984. [DOI] [PubMed] [Google Scholar]
- 17.Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, Forastiere A, Gillison ML. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;100:261–9. doi: 10.1093/jnci/djn011. [DOI] [PubMed] [Google Scholar]
- 18.D’Souza G, Sugar E, Ruby W, Gravitt P, Gillison ML. Analysis of the effect of DNA purification on detection of human papillomavirus in oral rinse samples by PCR. J Clin Microbiol. 2005;43:5526–35. doi: 10.1128/JCM.43.11.5526-5535.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, Westra WH, Chung CH, Jordan RC, Lu C, Kim H, Axelrod R, Silverman CC, Redmond KP, Gillison ML. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer CA, Kampmann M, Zlobec I, Green E, Tornillo L, Lugli A, Wolfensberger M, Terracciano LM. p16 expression in oropharyngeal cancer: its impact on staging and prognosis compared with the conventional clinical staging parameters. Ann Oncol. 2010;10:1961–6. doi: 10.1093/annonc/mdq210. [DOI] [PubMed] [Google Scholar]
- 21.Melar-New M, Laimins LA. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J Virol. 2010;10:5212–21. doi: 10.1128/JVI.00078-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–60. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 23.McCance DJ. Human papillomaviruses and cell signaling. Sci STKE. 2005;288:pe29. doi: 10.1126/stke.2882005pe29. [DOI] [PubMed] [Google Scholar]
- 24.Shin KH, Min BM, Cherrick HM, Park NH. Combined effects of human papillomavirus-18 and N-methyl-N′-nitro-N-nitrosoguanidine on the transformation of normal human oral keratinocytes. Mol Carcinog. 1994;2:76–86. doi: 10.1002/mc.2940090205. [DOI] [PubMed] [Google Scholar]
- 25.Joo YH, Jung CK, Sun DI, Park JO, Cho KJ, Kim MS. High-risk human papillomavirus and cervical lymph node metastasis in patients with oropharyngeal cancer. Head Neck. 2011 doi: 10.1002/hed.21697. [DOI] [PubMed] [Google Scholar]
- 26.Schlecht NF, Burk RD, Adrien L, Dunne A, Kawachi N, Sarta C, Chen Q, Brandwein-Gensler M, Prystowsky MB, Childs G, Smith RV, Belbin TJ. Gene expression profiles in HPV-infected head and neck cancer. J Pathol. 2007;213:283–93. doi: 10.1002/path.2227. [DOI] [PubMed] [Google Scholar]
- 27.Kreimer AR, Clifford GM, Boyle P, Franceschi S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomarkers Prev. 2005;14:467–75. doi: 10.1158/1055-9965.EPI-04-0551. [DOI] [PubMed] [Google Scholar]
- 28.Gillison ML, Lowy DR. A causal role for human papillomavirus in head and neck cancer. Lancet. 2004;363:1488–9. doi: 10.1016/S0140-6736(04)16194-1. [DOI] [PubMed] [Google Scholar]
- 29.Kahn T, Turazza E, Ojeda R, Bercovich A, Stremlau A, Lichter P, Poustka A, Grinstein S, zur Hausen H. Integration of human papillomavirus type 6a DNA in a tonsillar carcinoma: chromosomal localization and nucleotide sequence of the genomic target region. Cancer Res. 1994;54:1305–12. [PubMed] [Google Scholar]
- 30.Brandwein-Gensler M, Smith RV. Prognostic indicators in head and neck oncology including the new 7th edition of the AJCC staging system. Head Neck Pathol. 2010;4:53–61. doi: 10.1007/s12105-010-0161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chernock RD, El-Mofty SK, Thorstad WL, Parvin CA, Lewis JS., Jr HPV-related nonkeratinizing squamous cell carcinoma of the oropharynx: utility of microscopic features in predicting patient outcome. Head Neck Pathol. 2009;3:186–94. doi: 10.1007/s12105-009-0126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El-Mofty SK, Patil S. Human papillomavirus (HPV)-related oropharyngeal nonkeratinizing squamous cell carcinoma: characterization of a distinct phenotype. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:339–45. doi: 10.1016/j.tripleo.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 33.Lee WT, Tubbs RR, Teker AM, Scharpf J, Strome M, Wood B, Lorenz RR, Hunt J. Use of in situ hybridization to detect human papillomavirus in head and neck squamous cell carcinoma patients without a history of alcohol or tobacco use. Arch Pathol Lab Med. 2008;132:1653–6. doi: 10.5858/2008-132-1653-UOISHT. [DOI] [PubMed] [Google Scholar]
- 34.Pim D, Collins M, Banks L. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene. 1992;7:27–32. [PubMed] [Google Scholar]
- 35.Maufort JP, Shai A, Pitot HC, Lambert PF. A role for HPV E5 in cervical carcinogenesis. Cancer Res. 2010;70:2924–31. doi: 10.1158/0008-5472.CAN-09-3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wentzensen N, Wilson LE, Wheeler CM, Carreon JD, Gravitt PE, Schiffman M, Castle PE. Hierarchical clustering of human papilloma virus genotype patterns in the ASCUS-LSIL triage study. Cancer Res. 2010;70:8578–86. doi: 10.1158/0008-5472.CAN-10-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hafner N, Driesch C, Gajda M, Jansen L, Kirchmayr R, Runnebaum IB, Durst M. Integration of the HPV16 genome does not invariably result in high levels of viral oncogene transcripts. Oncogene. 2008;27:1610–7. doi: 10.1038/sj.onc.1210791. [DOI] [PubMed] [Google Scholar]
- 38.Ragin CC, Taioli E. Survival of squamous cell carcinoma of the head and neck in relation to human papillomavirus infection: review and meta-analysis. Int J Cancer. 2007;121:1813–20. doi: 10.1002/ijc.22851. [DOI] [PubMed] [Google Scholar]
- 39.Agoston ES, Robinson SJ, Mehra KK, Birch C, Semmel D, Mirkovic J, Haddad RI, Posner MR, Kindelberger D, Krane JF, Brodsky J, Crum CP. Polymerase chain reaction detection of HPV in squamous carcinoma of the oropharynx. Am J Clin Pathol. 2010;134:36–41. doi: 10.1309/AJCP1AAWXE5JJCLZ. [DOI] [PubMed] [Google Scholar]
- 40.Singhi AD, Westra WH. Comparison of human papillomavirus in situ hybridization and p16 immunohistochemistry in the detection of human papillomavirus-associated head and neck cancer based on a prospective clinical experience. Cancer. 2010;116:2166–73. doi: 10.1002/cncr.25033. [DOI] [PubMed] [Google Scholar]
- 41.Smeets SJ, Hesselink AT, Speel EJ, Haesevoets A, Snijders PJ, Pawlita M, Meijer CJ, Braakhuis BJ, Leemans CR, Brakenhoff RH. A novel algorithm for reliable detection of human papillomavirus in paraffin embedded head and neck cancer specimen. Int J Cancer. 2007;121:2465–72. doi: 10.1002/ijc.22980. [DOI] [PubMed] [Google Scholar]
- 42.Klingenberg B, Hafkamp HC, Haesevoets A, Manni JJ, Slootweg PJ, Weissenborn SJ, Klussman JP, Speel EJ. p16 INK4A overexpression is frequently detected in tumour-free tonsil tissue without association with HPV. Histopathology. 2010;56:957–67. doi: 10.1111/j.1365-2559.2010.03576.x. [DOI] [PubMed] [Google Scholar]
- 43.Kreimer AR, Clifford GM, Boyle P, Franceschi S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systemic review. Cancer Epidemiol Biomarkers Prev. 2005;14:467–475. doi: 10.1158/1055-9965.EPI-04-0551. [DOI] [PubMed] [Google Scholar]
- 44.Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004;68:362–72. doi: 10.1128/MMBR.68.2.362-372.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bienkowska-Haba M, Sapp M. The cytoskeleton in Paillomavirus infection. Viruses. 2011;3:260–271. doi: 10.3390/v3030260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Letian T, Tianyu Z. Cellular receptor binding and entry of human papillomavirus. Virol J. 2010;7:2. doi: 10.1186/1743-422X-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bretscher MS. Circulating integrins: alpha 5 beta 1, alpha 6 beta 4 and Mac-1, but not alpha 3 beta 1, alpha 4 beta 1 or LFA-1. EMBO J. 1992;11:405–10. doi: 10.1002/j.1460-2075.1992.tb05068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurpakus MA, Quaranta V, Jones JC. Surface relocation of alpha 6 beta 4 integrins and assembly of hemidesmosomes in an in vitro model of wound healing. J Cell Biol. 1991;115:1737–50. doi: 10.1083/jcb.115.6.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horvath CA, Boulet GA, Renoux VM, Delvenne PO, Bogers JP. Mechanisms of cell entry by human papillomaviruses: an overview. Virol J. 2010;7:11. doi: 10.1186/1743-422X-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, Florin L. Clathrin- and caveolin-independent entry of human papillomavirus type 16--involvement of tetraspanin-enriched microdomains (TEMs) PLoS One. 2008;3:e3313. doi: 10.1371/journal.pone.0003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schneider MA, Spoden GA, Florin L, Lambert C. Identification of the dynein light chains required for human papillomavirus infection. Cell Microbiol. 2011;13:32–46. doi: 10.1111/j.1462-5822.2010.01515.x. [DOI] [PubMed] [Google Scholar]
- 52.Kyo S, Inoue M, Hayasaka N, Inoue T, Yutsudo M, Tanizawa O, Hakura A. Regulation of early gene expression of human papillomavirus type 16 by inflammatory cytokines. Virology. 1994;200:130–9. doi: 10.1006/viro.1994.1171. [DOI] [PubMed] [Google Scholar]
- 53.Boccardo E, Manzini Baldi CV, Carvalho AF, Rabachini T, Torres C, Barreta LA, Brentani H, Villa LL. Expression of human papillomavirus type 16 E7 oncoprotein alters keratinocytes expression profile in response to tumor necrosis factor-alpha. Carcinogenesis. 2010;31:521–31. doi: 10.1093/carcin/bgp333. [DOI] [PubMed] [Google Scholar]
- 54.Boccardo E, Lepique AP, Villa LL. The role of inflammation in HPV carcinogenesis. Carcinogenesis. 2010;31:1905–12. doi: 10.1093/carcin/bgq176. [DOI] [PubMed] [Google Scholar]
- 55.Stanley M. Immune response to human papillomavirus. Vaccine. 2006;24:S16–22. doi: 10.1016/j.vaccine.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 56.Um SJ, Rhyu JW, Kim EJ, Jeon KC, Hwang ES, Park JS. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett. 2002;179:205–12. doi: 10.1016/s0304-3835(01)00871-0. [DOI] [PubMed] [Google Scholar]
- 57.Park JS, Kim EJ, Kwon HJ, Hwang ES, Namkoong SE, Um SJ. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem. 2000;275:6764–9. doi: 10.1074/jbc.275.10.6764. [DOI] [PubMed] [Google Scholar]
- 58.Albers A, Abe K, Hunt J, Wang J, Lopez-Albaitero A, Schaefer C, Gooding W, Whiteside TL, Ferrone S, DeLeo A, Ferris RL. Antitumor activity of human papillomavirus type 16 E7-specific T cells against virally infected squamous cell carcinoma of the head and neck. Cancer Res. 2005;65:11146–55. doi: 10.1158/0008-5472.CAN-05-0772. [DOI] [PubMed] [Google Scholar]
- 59.Hoffmann TK, Arsov C, Schirlau K, Bas M, Friebe-Hoffmann U, Klussmann JP, Scheckenbach K, Balz V, Bier H, Whiteside TL. T cells specific for HPV16 E7 epitopes in patients with squamous cell carcinoma of the oropharynx. Int J Cancer. 2006;118:1984–91. doi: 10.1002/ijc.21565. [DOI] [PubMed] [Google Scholar]
- 60.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–9. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 61.Van Tine BA, Dao LD, Wu SY, Sonbuchner TM, Lin BY, Zou N, Chiang CM, Broker TR, Chow LT. Human papillomavirus (HPV) origin-binding protein associates with mitotic spindles to enable viral DNA partitioning. Proc Natl Acad Sci U S A. 2004;101:4030–5. doi: 10.1073/pnas.0306848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams R, Lee DW, Elzey BD, Anderson ME, Hostager BS, Lee JH. Preclinical models of HPV+ and HPV− HNSCC in mice: an immune clearance of HPV+ HNSCC. Head Neck. 2009;31:911–8. doi: 10.1002/hed.21040. [DOI] [PubMed] [Google Scholar]
- 63.Syrjanen S. HPV infections and tonsillar carcinoma. J Clin Pathol. 2004;57:449–55. doi: 10.1136/jcp.2003.008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim SH, Juhn YS, Kang S, Park SW, Sung MW, Bang YJ, Song YS. Human papillomavirus 16 E5 up-regulates the expression of vascular endothelial growth factor through the activation of epidermal growth factor receptor, MEK/ ERK1,2 and PI3K/Akt. Cell Mol Life Sci. 2006;63:930–8. doi: 10.1007/s00018-005-5561-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim SH, Koo BS, Kang S, Park K, Kim H, Lee KR, Lee MJ, Kim JM, Choi EC, Cho NH. HPV integration begins in the tonsillar crypt and leads to the alteration of p16, EGFR and c-myc during tumor formation. Int J Cancer. 2007;120:1418–25. doi: 10.1002/ijc.22464. [DOI] [PubMed] [Google Scholar]
- 66.Avery JK, Steele PF, Avery N. Oral development and histology. 3. Stuttgart: 2002. [Google Scholar]
- 67.Perry ME. The specialised structure of crypt epithelium in the human palatine tonsil and its functional significance. J Anat. 1994;185:111–27. [PMC free article] [PubMed] [Google Scholar]
- 68.Pai SI, Westra WH. Molecular pathology of head and neck cancer: implications for diagnosis, prognosis, and treatment. Annu Rev Pathol. 2009;4:49–70. doi: 10.1146/annurev.pathol.4.110807.092158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cote-Martin A, Moody C, Fradet-Turcotte A, D’Abramo CM, Lehoux M, Joubert S, Poirier GG, Coulombe B, Laimins LA, Archambault J. Human papillomavirus E1 helicase interacts with the WD repeat protein p80 to promote maintenance of the viral genome in keratinocytes. J Virol. 2008;82:1271–83. doi: 10.1128/JVI.01405-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng S, Schmidt-Grimminger DC, Murant T, Broker TR, Chow LT. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 1995;9:2335–2349. doi: 10.1101/gad.9.19.2335. [DOI] [PubMed] [Google Scholar]
- 71.Frattini MG, Laimins LA. The role of the E1 and E2 proteins in the replication of human papillomavirus type 31b. Virology. 1994;204:799–804. doi: 10.1006/viro.1994.1596. [DOI] [PubMed] [Google Scholar]
- 72.Frattini MG, Laimins LA. Binding of the human papillomavirus E1 origin-recognition protein is regulated through complex formation with the E2 enhancer-binding protein. Proc Natl Acad Sci. 1994;91:12398–12402. doi: 10.1073/pnas.91.26.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hughes FJ, Romanos MA. E1 protein of human papillomavirus is a DNA helicase/ATPase. Nucleic Acids Res. 1993;21:5817–5823. doi: 10.1093/nar/21.25.5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Muller F, Giroglou T, Sapp M. Characterization of the DNA- binding activity of the E1 and E2 proteins and the E1/E2 complex of human papillomavirus type 33. J Gen Virol. 1997;78:911–915. doi: 10.1099/0022-1317-78-4-911. [DOI] [PubMed] [Google Scholar]
- 75.Seo YS, Muller F, Lusky M, Hurwitz J. Bovine papilloma virus (BPV)-encoded E1 protein contains multiple activities required for BPV DNA replication. Proc Natl Acad Sci. 1993;90:702–706. doi: 10.1073/pnas.90.2.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Amin AA, Titolo S, Pelletier A, Fink D, Cordingley MG, Archambault J. Identification of domains of the HPV11 E1 protein required for DNA replication in vitro. Virology. 2000;272:137–150. doi: 10.1006/viro.2000.0328. [DOI] [PubMed] [Google Scholar]
- 77.Conger KL, Liu JS, Kuo SR, Chow LT, Wang TS. Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human dna polymerase alpha/primase. J Biol Chem. 1999;274:2696–2705. doi: 10.1074/jbc.274.5.2696. [DOI] [PubMed] [Google Scholar]
- 78.Masterson PJ, Stanley MA, Lewis AP, Romanos MA. A C-terminal helicase domain of the human papillomavirus E1 protein binds E2 and the DNA polymerase alpha-primase p68 subunit. J Virol. 1998;72:7407–7419. doi: 10.1128/jvi.72.9.7407-7419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma T, Zou N, Lin BY, Chow LT, Harper JW. Interaction between cyclin-dependent kinases and human papillomavirus replication- initiation protein E1 is required for efficient viral replication. Proc Natl Acad Sci. 1999;96:382–387. doi: 10.1073/pnas.96.2.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sim J, Ozgur S, Lin BY, Yu JH, Broker TR, Chow LT, Griffith J. Remodeling of the human papillomavirus type 11 replication origin into discrete nucleoprotein particles and looped structures by the E2 protein. J Mol Biol. 2007;375:1165–77. doi: 10.1016/j.jmb.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen G, Stenlund A. Two patches of amino acids on the E2 DNA binding domain define the surface for interaction with E1. J Virol. 2000;74:1506–1512. doi: 10.1128/jvi.74.3.1506-1512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hegde RS, Grossman SR, Laimins LA, Sigler PB. Crystal structure at 1.7 A of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature. 1992;359:505–512. doi: 10.1038/359505a0. [DOI] [PubMed] [Google Scholar]
- 83.Harris SF, Botchan MR. Crystal structure of the human papillomavirus type 18 E2 activation domain. Science. 1999;284:1673–1677. doi: 10.1126/science.284.5420.1673. [DOI] [PubMed] [Google Scholar]
- 84.Demeret C, Desaintes C, Yaniv M, Thierry F. Different mechanisms contribute to the E2-mediated transcriptional repression of human papillomavirus type 18 viral oncogenes. J Virol. 1997;71:9343–9349. doi: 10.1128/jvi.71.12.9343-9349.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dostatni N, Lambert PF, Sousa R, Ham J, Howley PM, Yaniv M. The functional BPV-1 E2 trans-activating protein can act as a repressor by preventing formation of the initiation complex. Genes Dev. 1991;5:1657–1671. doi: 10.1101/gad.5.9.1657. [DOI] [PubMed] [Google Scholar]
- 86.Steger G, Corbach S. Dose-dependent regulation of the early promoter of HPV 18 by the viral E2 protein. J Virol. 1997;71:50–58. doi: 10.1128/jvi.71.1.50-58.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schweiger MR, Ottinger M, You J, Howley PM. Brd4-independent transcriptional repression function of the papillomavirus e2 proteins. J Virol. 2007;81:9612–22. doi: 10.1128/JVI.00447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smith JA, White EA, Sowa ME, Powell ML, Ottinger M, Harper JW, Howley PM. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc Natl Acad Sci U S A. 2010;107:3752–7. doi: 10.1073/pnas.0914818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mole S, Veerapraditsin T, McPhillips MG, Graham SV. Regulation of splicing-associated SR proteins by HPV-16. Biochem Soc Trans. 2006;34:1145–7. doi: 10.1042/BST0341145. [DOI] [PubMed] [Google Scholar]
- 90.McFarlane M, Graham SV. Human papillomavirus regulation of SR proteins. Biochem Soc Trans. 2010;38:1116–21. doi: 10.1042/BST0381116. [DOI] [PubMed] [Google Scholar]
- 91.Xue Y, Bellanger S, Zhang W, Lim D, Low J, Lunny D, Thierry F. HPV16 E2 is an immediate early marker of viral infection, preceding E7 expression in precursor structures of cervical carcinoma. Cancer Res. 2010;70:5316–25. doi: 10.1158/0008-5472.CAN-09-3789. [DOI] [PubMed] [Google Scholar]
- 92.Wu MH, Chan JY, Liu PY, Liu ST, Huang SM. Human papillomavirus E2 protein associates with nuclear receptors to stimulate nuclear receptor-and E2-dependent transcriptional activations in human cervical carcinoma cells. Int J Biochem Cell Biol. 2007;39:413–25. doi: 10.1016/j.biocel.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 93.Remm M, Remm A, Ustav M. Human papillomavirus type 18 E1 protein is translated from polycistronic mRNA by a discontinuous scanning mechanism. J Virol. 1999;73:3062–3070. doi: 10.1128/jvi.73.4.3062-3070.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Knight GL, Pugh AG, Yates E, Bell I, Wilson R, Moody CA, Laimins LA, Roberts S. A cyclin-binding motif in human papillomavirus type 18 (HPV18) E1^E4 is necessary for association with CDK-cyclin complexes and G2/M cell cycle arrest of keratinocytes, but is not required for differentiation-dependent viral genome amplification or L1 capsid protein expression. Virology. 2011;412:196–210. doi: 10.1016/j.virol.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McIntosh PB, Laskey P, Sullivan K, Davy C, Wang Q, Jackson DJ, Griffin HM, Doorbar J. E1--E4-mediated keratin phosphorylation and ubiquitylation: a mechanism for keratin depletion in HPV16-infected epithelium. J Cell Sci. 2010;123:2810–22. doi: 10.1242/jcs.061978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wilson R, Ryan GB, Knight GL, Laimins LA, Roberts S. The full-length E1E4 protein of human papillomavirus type 18 modulates differentiation-dependent viral DNA amplification and late gene expression. Virology. 2007;362:453–60. doi: 10.1016/j.virol.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 97.Tsai TC, Chen SL. The biochemical and biological functions of human papillomavirus type 16 E5 protein. Arch Virol. 2003;148:1445–53. doi: 10.1007/s00705-003-0111-z. [DOI] [PubMed] [Google Scholar]
- 98.Fehrmann F, Klumpp DJ, Laimins LA. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J Virol. 2003;77:2819–31. doi: 10.1128/JVI.77.5.2819-2831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alazawi W, Pett M, Strauss S, Moseley R, Gray J, Stanley M, Coleman N. Genomic imbalances in 70 snap-frozen cervical squamous intraepithelial lesions: associations with lesion grade, state of the HPV16 E2 gene and clinical outcome. Br J Cancer. 2004;91:2063–70. doi: 10.1038/sj.bjc.6602237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jeon S, Allen-Hoffmann BL, Lambert PF. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol. 1995;69:2989–97. doi: 10.1128/jvi.69.5.2989-2997.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dahlgren L, Mellin H, Wangsa D, Heselmeyer-Haddad K, Björnestål L, Lindholm J, Munck-Wikland E, Auer G, Ried T, Dalianis T. Comparative genomic hybridization analysis of tonsillar cancer reveals a different pattern of genomic imbalances in human papillomavirus-positive and -negative tumors. Int J Cancer. 2003;107:244–9. doi: 10.1002/ijc.11371. [DOI] [PubMed] [Google Scholar]
- 102.Jung AC, Briolat J, Millon R, de Reynies A, Rickman D, Thomas E, Abecassis J, Clavel C, Wasylyk B. Biological and clinical relevance of transcriptionally active human papillomavirus (HPV) infection in oropharynx squamous cell carcinoma. Int J Cancer. 2010;126:1882–94. doi: 10.1002/ijc.24911. [DOI] [PubMed] [Google Scholar]
- 103.Duensing A, Spardy N, Chatterjee P, Zheng L, Parry J, Cuevas R, Korzeniewski N, Duensing S. Centrosome overduplication, chromosomal instability, and human papillomavirus oncoproteins. Environ Mol Mutagen. 2009;50:741–7. doi: 10.1002/em.20478. [DOI] [PubMed] [Google Scholar]
- 104.DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol. 2003;77:1551–63. doi: 10.1128/JVI.77.2.1551-1563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang HK, Duffy AA, Broker TR, Chow LT. Robust production and passaging of infectious HPV in squamous epithelium of primary human keratinocytes. Genes Dev. 2009;23:181–94. doi: 10.1101/gad.1735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McCance DJ, Kopan R, Fuchs E, Laimins LA. Human papillomavirus type 16 alters human epithelial cell differentiation in vitro. Proc Natl Acad Sci. 1988;85:7169–73. doi: 10.1073/pnas.85.19.7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Duensing S, Munger K. Human papillomaviruses and centrosome duplication errors: modeling the origins of genomic instability. Oncogene. 2002;21:6241–8. doi: 10.1038/sj.onc.1205709. [DOI] [PubMed] [Google Scholar]
- 108.Bedard KM, Underbrink MP, Howie HL, Galloway DA. The E6 oncoproteins from human betapapillomaviruses differentially activate telomerase through an E6AP-dependent mechanism and prolong the lifespan of primary keratinocytes. J Virol. 2008;82:3894–902. doi: 10.1128/JVI.01818-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10:4129–35. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brimer N, Lyons C, Vande Pol SB. Association of E6AP (UBE3A) with human papillomavirus type 11 E6 protein. Virology. 2007;358:303–10. doi: 10.1016/j.virol.2006.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lechner MS, Laimins LA. Inhibition of p53 DNA binding by human papillomavirus E6 proteins. J Virol. 1994;68:4262–73. doi: 10.1128/jvi.68.7.4262-4273.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Muench P, Probst S, Schuetz J, Leiprecht N, Busch M, Wesselborg S, Stubenrauch F, Iftner T. Cutaneous papillomavirus E6 proteins must interact with p300 and block p53-mediated apoptosis for cellular immortalization and tumorigenesis. Cancer Res. 2010;70:6913–24. doi: 10.1158/0008-5472.CAN-10-1307. [DOI] [PubMed] [Google Scholar]
- 113.Kumar A, Zhao Y, Meng G, Zeng M, Srinivasan S, Delmolino LM, Gao Q, Dimri G, Weber GF, Wazer DE, Band H, Band V. Human papillomavirus oncoprotein E6 inactivates the transcriptional coactivator human ADA3. Mol Cell Biol. 2002;22:5801–12. doi: 10.1128/MCB.22.16.5801-5812.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Patel D, Huang SM, Baglia LA, McCance DJ. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999;18:5061–72. doi: 10.1093/emboj/18.18.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shamanin VA, Androphy EJ. Immortalization of human mammary epithelial cells is associated with inactivation of the p14ARF-p53 pathway. Mol Cell Biol. 2004;24:2144–52. doi: 10.1128/MCB.24.5.2144-2152.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shamanin VA, Sekaric P, Androphy EJ. hAda3 degradation by papillomavirus type 16 E6 correlates with abrogation of the p14ARF-p53 pathway and efficient immortalization of human mammary epithelial cells. J Virol. 2008;82:3912–20. doi: 10.1128/JVI.02466-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996;380:79–82. doi: 10.1038/380079a0. [DOI] [PubMed] [Google Scholar]
- 118.Wise-Draper TM, Wells SI. Papillomavirus E6 and E7 proteins and their cellular targets. Front Biosci. 2008;13:1003–17. doi: 10.2741/2739. [DOI] [PubMed] [Google Scholar]
- 119.Howie HL, Katzenellenbogen RA, Galloway DA. Papillomavirus E6 proteins. Virology. 2009;384:324–34. doi: 10.1016/j.virol.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu Y, Cherry JJ, Dineen JV, Androphy EJ, Baleja JD. Determinants of stability for the E6 protein of papillomavirus type 16. J Mol Biol. 2009;386:1123–37. doi: 10.1016/j.jmb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–8. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- 122.Nguyen ML, Nguyen MM, Lee D, Griep AE, Lambert PF. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6’s induction of epithelial hyperplasia in vivo. J Virol. 2003;77:6957–64. doi: 10.1128/JVI.77.12.6957-6964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 124.Dreier K, Scheiden R, Lener B, Ehehalt D, Pircher H, Muller-Holzner E, Rostek U, Kaiser A, Fiedler M, Ressler S, Lechner S, Widschwendter A, Even J, Capesius C, Jansen-Durr P, Zwerschke W. Subcellular localization of the human papillomavirus 16 E7 oncoprotein in CaSki cells and its detection in cervical adenocarcinoma and adenocarcinoma in situ. Virology. 2011;409:54–68. doi: 10.1016/j.virol.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Melar-New M, Laimins LA. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J Virol. 2010;84:5212–5221. doi: 10.1128/JVI.00078-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang X, Tang S, Le SY, Lu R, Rader JS, Meyers C, Zheng ZM. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS One. 2008;3:e2557. doi: 10.1371/journal.pone.0002557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang X, Wang HK, McCoy JP, Banerjee NS, Rader JS, Broker TR, Meyers C, Chow LT, Zheng ZM. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA. 2009;15:637–647. doi: 10.1261/rna.1442309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Martinez I, Gardiner AS, Board KF, Monzon FA, Edwards RP, Khan SA. Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene. 2008;27:2575–2582. doi: 10.1038/sj.onc.1210919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Greco D, Kivi N, Qian K, Leivonen SK, Auvinen P, Auvinen E. Human Papillomavirus 16 E5 Modulates the Expression of Host MicroRNAs. PLoS One. 2011;6:e21646. doi: 10.1371/journal.pone.0021646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang X, Meyers C, Guo M, Zheng ZM. Up-regulation of p18Ink4c expression by oncogenic HPV E6 via p53-miR34a pathway. Int J Cancer. 2010;129:1362–72. doi: 10.1002/ijc.25800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zheng ZM, Wang X. Regulation of cellular miRNA expression by human papillomaviruses. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbagrm.2011.05.005. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lohavanichbutr P, Houck J, Fan W, Yueh B, Mendez E, Futran N, Doody DR, Upton MP, Farwell DG, Schwartz SM, Zhao LP, Chen C. Genomewide gene expression profiles of HPV-positive and HPV-negative oropharyngeal cancer: potential implications for treatment choices. Arch Otolaryngol Head Neck Surg. 2009;135:180–8. doi: 10.1001/archoto.2008.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, Woodworth CD, Connor JP, Haugen TH, Smith EM, Kelsey KT, Turek LP, Ahlquist P. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007;67:4605–19. doi: 10.1158/0008-5472.CAN-06-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wilting SM, Smeets SJ, Snijders PJ, van Wieringen WN, van de Wiel MA, Meijer GA, Ylstra B, Leemans CR, Meijer CJ, Brakenhoff RH, Braakhuis BJ, Steenbergen RD. Genomic profiling identifies common HPV-associated chromosomal alterations in squamous cell carcinomas of cervix and head and neck. BMC Med Genomics. 2009;2:32. doi: 10.1186/1755-8794-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Klussmann JP, Mooren JJ, Lehnen M, Claessen SM, Stenner M, Huebbers CU, Weissenborn SJ, Wedemeyer I, Preuss SF, Straetmans JM, Manni JJ, Hopman AH, Speel EJ. Genetic signatures of HPV-related and unrelated oropharyngeal carcinoma and their prognostic implications. Clin Cancer Res. 2009;15:1779–86. doi: 10.1158/1078-0432.CCR-08-1463. [DOI] [PubMed] [Google Scholar]
- 136.Schlecht NF, Burk RD, Adrien L, Dunne A, Kawachi N, Sarta C, Chen Q, Brandwein-Gensler M, Prystowsky MB, Childs G, Smith RV, Belbin TJ. Gene expression profiles in HPV-infected head and neck cancer. J Pathol. 2007;213:283–93. doi: 10.1002/path.2227. [DOI] [PubMed] [Google Scholar]
- 137.Martinez I, Wang J, Hobson KF, Ferris RL, Khan SA. Identification of differentially expressed genes in HPV-positive and HPV-negative oropharyngeal squamous cell carcinomas. Eur J Cancer. 2007;43:415–32. doi: 10.1016/j.ejca.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Smith EM, Ritchie JM, Summersgill KF, Hoffman HT, Wang DH, Haugen TH, Turek LP. Human papillomavirus in oral exfoliated cells and risk of head and neck cancer. J Natl Cancer Inst. 2004;96:449–55. doi: 10.1093/jnci/djh074. [DOI] [PubMed] [Google Scholar]
- 139.D’Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, Westra WH, Gillison ML. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. 2007;356:1944–56. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 140.D’Souza G, Agrawal Y, Halpern J, Bodison S, Gillison ML. Oral sexual behaviors associated with prevalent oral human papillomavirus infection. J Infect Dis. 2009;199:1263–9. doi: 10.1086/597755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hansson BG, Rosenquist K, Antonsson A, Wennerberg J, Schildt EB, Bladstrom A, Andersson G. Strong association between infection with human papillomavirus and oral and oropharyngeal squamous cell carcinoma: a population-based case-control study in southern Sweden. Acta Otolaryngol. 2005;125:1337–44. doi: 10.1080/00016480510043945. [DOI] [PubMed] [Google Scholar]
- 142.Clifford GM, Gallus S, Herrero R, Munoz N, Snijders PJ, Vaccarella S, Anh PT, Ferreccio C, Hieu NT, Matos E, Molano M, Rajkumar R, Ronco G, de Sanjose S, Shin HR, Sukvirach S, Thomas JO, Tunsakul S, Meijer CJ, Franceschi S. Worldwide distribution of human papillomavirus types in cytologically normal women in the International Agency for Research on Cancer HPV prevalence surveys: a pooled analysis. Lancet. 2005;366:991–8. doi: 10.1016/S0140-6736(05)67069-9. [DOI] [PubMed] [Google Scholar]
- 143.de Sanjose S, Diaz M, Castellsague X, Clifford G, Bruni L, Munoz N, Bosch FX. Worldwide prevalence and genotype distribution of cervical human papillomavirus DNA in women with normal cytology: a meta-analysis. Lancet Infect Dis. 2007;7:453–9. doi: 10.1016/S1473-3099(07)70158-5. [DOI] [PubMed] [Google Scholar]
- 144.Chen R, Sehr P, Waterboer T, Leivo I, Pawlita M, Vaheri A, Aaltonen LM. Presence of DNA of human papillomavirus 16 but no other types in tumor-free tonsillar tissue. J Clin Microbiol. 2005;43:1408–10. doi: 10.1128/JCM.43.3.1408-1410.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chaturvedi AK, Engels EA, Anderson WF, Gillison ML. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J Clin Oncol. 2008;26:612–9. doi: 10.1200/JCO.2007.14.1713. [DOI] [PubMed] [Google Scholar]
- 146.Settle K, Posner MR, Schumaker LM, Tan M, Suntharalingam M, Goloubeva O, Strome SE, Haddad RI, Patel SS, Cambell EVr, Sarlis N, Lorch J, Cullen KJ. Racial survival disparity in head and neck cancer results from low prevalence of human papillomavirus infection in black oropharyngeal cancer patients. Cancer Prev Res. 2009;2:776–781. doi: 10.1158/1940-6207.CAPR-09-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen LM, Li G, Reitzel LR, Pytynia KB, Zafereo ME, Wei Q, Sturgis EM. Matched-pair analysis of race or ethnicity in outcomes of head and neck cancer patients receiving similar multidisciplinary care. Cancer Prev Res. 2009;2:782–91. doi: 10.1158/1940-6207.CAPR-09-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schrank TP, Han Y, Weiss H, Resto VA. Case-matching analysis of head and neck squamous cell carcinoma in racial and ethnic minorities in the United States--possible role for human papillomavirus in survival disparities. Head Neck. 2011;33:45–53. doi: 10.1002/hed.21398. [DOI] [PMC free article] [PubMed] [Google Scholar]