

Abstract
MicroRNA (miRNA) function has been studied extensively in the last two decades. These short, non-coding RNAs influence a variety of cellular processes through repression of target genes. With the number of genes that a single miRNA can target, the biological effects of one miRNA alone can be vast. In cancer, aberrant miRNA expression is ubiquitous and consequently it can provoke progression of the disease. Though much is known about the downstream effects of miRNA, the mechanisms that control the level of miRNA expression itself are not well documented. In this review, we will focus on how miRNAs are regulated as well as potential therapeutic targets that can be exploited for cancer therapy.
Keywords: microRNA, miRNA, Cancer, Regulation, Transcriptional, Post-transcriptional, Epigenetic, RNA structure, Single nucleotide polymorphisms, SNPs, Pathobiology
Introduction
Hundreds of miRNAs have been extensively studied since the premier of Lin4 in 1993 [1]. Lin4 was found to regulate several genes associated with development in C. elegans and soon after, the field exploded with studies describing the ability of miRNA to downregulate a variety of genes in several diseases. After nearly twenty years of research, miRNAs are thought to regulate over 50% of the human genome and their role in cancer progression is of considerable interest [2,3,4]. Numerous review articles have nicely summarized miRNA downstream signaling pathways and many miRNA gene targets have been validated mechanistically. Less studied, however, are the modes of miRNA regulation and how these regulatory mechanisms are disrupted in cancer, which will be the focal point of this review.
Transcriptional Regulation of miRNA
The regulation of primary miRNA transcripts is one of the most important aspects of the miRNA regulation and is controlled by polymerase. The first investigation in this area (2002) suggested that polymerase III (Pol III) may be able to terminate miRNA transcription due to the length of miRNA transcripts and the number of uracil repeats[5]. In 2004, polymerase II (Pol II) was shown to transcribe miRNAs as well [6]. To distinguish polymerase II promoters from polymerase III promoters, Zhou’s group employed a bioinformatics-based tool that predicted miRNA promoter regions and the polymerase that transcribed it. The accuracy of their system was validated by its sensitivity and specificity to detect known Pol II and Pol III promoters across the genome [7]. Other promoter-predicting computational methods began to find that Pol III had the ability to transcribe miRNAs between Alu repeats. Finally, Borchert’s group found Alu repeats upstream of predicted miRNA promoter regions and showed that 52 human miRNAs are wedged between Alu repeats. They suggested that miRNA transcription may be controlled by transcription factors during cell stress, similar to Pol III–mediated transcription [8]. Currently, it is thought that miRNAs on the same strand as their host gene (intragenic) are transcribed predominantly by Pol II, while miRNAs between clusters of genes (intergenic) are transcribed either by their own Pol II promoter or from Pol III [9]. Often, the same polycistronic transcript is shared between clusters of 2–19 hairpins that encode miRNAs [10]. Nearly 50% of miRNAs are believed to originate from non-protein coding RNA transcripts and around 40% are located in intronic regions of genes that code proteins [11,12]. It is known that abnormal miRNA alteration is a ubiquitous feature in human cancer as greater than 50% of miRNA genes are found in either fragile sites or cancer-associated genomic regions indicating that many are involved in the initiation or progression of cancer [13]. Though much is known about the transcription of miRNA, the mechanisms underlying global deregulation of miRNA in human cancers remain obscure.
Systems biology and genomics-based approaches, especially in the context of studying human cancers, are increasingly utilized to determine information about miRNA promoters in addition to their target genes. These approaches are powerful in elucidating the mechanisms of miRNA regulation. Marson et al. found computationally that Nanog, Sox2, Pou5f1 and Tcf3 along with other embryonic stem cell transcription factors regulate ~20% of annotated mammalian miRNAs [14]. However, we must keep in mind that not all predicted promoter regions or transcription factor binding sites have been validated. Nevertheless, there is a growing body of literature confirming that various RNA-binding proteins, such as transcription factors and hormones, play a role in directly regulating miRNA transcription. In prostate cancer, for example, miR-21 stands out as one of the most studied miRNAs. Unlike most other miRNAs, the gene locus and promoter region of miR-21 were identified and extensive studies have shown that various factors control miR-21 expression by binding to the putative promoter. Ras, ERK1/2 and EGFR induce miR-21 whereas NF1, C/EBP, and Gfi1 suppress its expression in prostate cancer [15,16,17,18,19].
In hormone responsive cancers, estrogen and androgen receptors have the ability to transcriptionally regulate miRNAs. Ribas et al. identified 16 androgen-induced miRNAs from a microarray screen in two prostate cancer cell lines. The same study found that androgen-induced AR bound directly to a highly conserved androgen response element on the promoter of miR-21 [20]. miR-125b was also found to be androgen responsive when comparing androgen-independent to androgen-dependent cell lines [21]. Ambs et al. found the miR-181b-1, miR-181c, miR-221 clusters along with miR-338 and miR-126 contain AR binding elements in regions flanking their promoter; however, the only miRNA induced by androgen in LNCap cells was miR-338 [22]. As for estrogen receptor regulation, a study by Ferraro et al. used ChIP-Seq to validate 6 miRNAs modulated by ERα [23]. Furthermore, ERβ binds two sites proximal to the miR-30a gene to inhibit synthesis of the primary transcript [24]. In addition to hormone-responsive elements, many other common transcription factors modulate miRNA.
Perhaps it is unsurprising that the DNA binding elements that regulate miRNA transcription are considerably equivalent to those that control protein-coding genes. For instance, c-Myc is thought to regulate 10–15% of protein-coding genes and has been found to control the expression of a number of miRNAs [25,26]. The miR-17-92 cluster was the first set of miRNAs reported as having oncogenic potential due to transcriptional activation by c-Myc. O’Donnell et al. show that c-Myc is capable of binding to an E-box in the first intron of miR-17-92 cluster’s host gene. Not long after, c-Myc was reported to influence metabolism, cell cycle, angiogenesis, metastasis and apoptosis through direct transcriptional regulation of more than ten miRNAs and that number is still rising [27]. Moreover, Ju et al. found that more that 46% of 326 miRNA promoter regions identified from miRbase contained potential p53-binding sites and may be regulated by the protein [28]. For example, p53 directly modulates miRNA-34 expression through a binding site on miRNA-34’s host gene [29]. Upon miR-34 inactivation, p53-dependent apoptosis was effectively inhibited, suggesting that miR-34 acts as a tumor suppressor. Given that p53 is the most frequently mutated gene in human cancer, it is not surprising that miRNA deregulation is also a common occurrence. These results are consistent with numerous studies that find decreased miR-34 expression in tumors [30]. It is apparent from the studies described above that transcription factors have the ability to activate or suppress miRNA expression allowing miRNAs to play either oncogenic or tumor suppressive roles in cancer.
Cell type specific transcription factors also play a role in miRNA transcription (Figure 1). miR-223 is one example, activated by myeloid transcription factors C/EBP and PU.1, and highly expressed in myeloid cells [31]. The miRNA-181 family is another example. All four family members were found to be directly induced by the Wnt/β-catenin signaling pathway in hepatocellular carcinoma [32]. During development, the synchronized expression of tissue-specific miRNAs and mRNAs leads to proper cell differentiation. In cancer, aberrant miRNA expression may change cell fate and lead to progression of the disease.
Fig. 1.
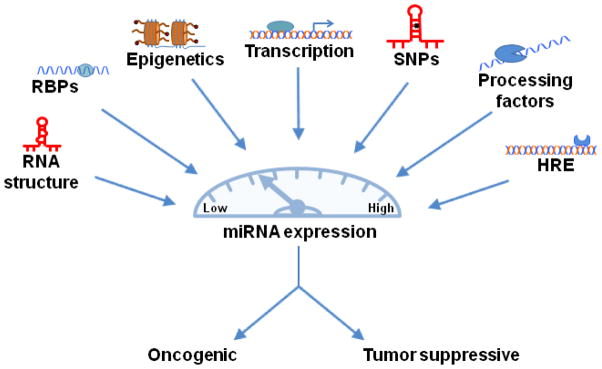
Many factors influence miRNA expression. Mutations, aberrant protein expression, or epigenetic alterations are only a few examples of defects in regulatory mechanisms that cause abnormal miRNA expression and lead to cancer. Abbreviations: RBPs, RNA-binding proteins; SNPs, Single nucleotide polymorphisms; HRE, Hormone-responsive element
Though several transcription factors bind to and regulate miRNA transcripts, many miRNAs themselves participate in self-regulating feedback loops by targeting the transcription factors or other proteins that modulate their expression. E2F and c-Myc, for example, induce the expression of the miR-17-92 cluster while the cluster itself works to inhibit both E2F and c-Myc, forming a negative feedback loop [33]. It is through this loop that miR-17-92 operates as both a tumor suppressor and oncogene in cancer. Another, more complex, inflammatory feedback loop involves HNF4α, miR-124, IL6R, STAT3, miR-24 and miR-629, in the control of hepatocellular transformation and liver cancer [34]. Also in hepatocellular carcinoma, miR-21 promotes invasion and migration through a feedback loop involving programmed cell death 4 (PDCD4) and activating protein 1 (AP-1) [35]. Evidence of a feedback loop between LKB1, a serine/threonine-protein kinase, and miR-451 plays a role in cell metabolism, apoptosis, and DNA damage response in glioma cells [36]. Understanding the intricate networks between transcription factors and miRNAs is critical to advance our knowledge of cancer development and improve clinical strategies to stop cancer progression.
Post-transcriptional Regulation of miRNA
In addition to the transcriptional regulation of miRNA, there are numerous opportunities for miRNAs to be regulated post-transcriptionally (Figure 1). The biogenesis of miRNAs has been studied in detail and is thoroughly described in other articles [37,38]. In brief, after transcription, the primary transcript of miRNA (pri-miRNA, ~3000 nucleotides) is recognized by the Drosha complex and cropped into a shorter precursor (pre-miRNA, 60–100 nucleotide stem-loop). Exportin 5 binds to the precursor and translocates it into the cytoplasm where the Dicer complex cleaves the pre-miRNA into a driver and passenger (miRNA:miRNA*) strand complex (20–22 nucleotides). Typically the RNA-induced Silencing Complex (RISC) will bind only to the stable driver (guide) strand and recruit Ago2 protein. This final step allows the seed sequence of the miRNA (typically 6–8 nucleotides in length) to interact with its target 3′UTR on mRNA. If the binding complementarity is complete between the miRNA and mRNA, Ago2 protein is thought to cause degradation of the mRNA. However, if the binding sites between the miRNA and mRNA are not completely complementary, translation is prevented but the mRNA remains intact.
Post-transcriptional regulation of miRNA can occur at each step during the biogenesis and processing of miRNAs. In cancer, expression of the mature form of miRNA often deviates from the expression of its transcript. Thomson et al. analyzed 68 tumors versus 21 normal samples with both miRNA and mRNA expression data and found no correlation between the pri-miRNA and mature expression in the tumor samples though there was significant correlation in normal tissue. Decreased mature expression compared to pri-miRNA suggests that alterations occur in processing steps rather than during transcription [39]. A study in 111 invasive epithelial ovarian cancer patients revealed that 60% of patients had decreased Dicer expression and 51% of patients had decreased Drosha levels. Patients with high levels of both Dicer and Drosha were associated with >11 years increased median survival [40]. In addition, global repression of miRNA processing by knockdown of Drosha, Dicer, or DGCR8 (which encodes a cofactor of Drosha) in lung adenocarcinoma cells leads to more aggressive tumors. A conditional deletion of Dicer1 alone enhanced tumorigenesis in the same cells [41]. The same group went on to compare hemizygous versus homozygous knockdown of Dicer and found that one copy was enough to increase the tumor burden whereas loss of both copies inhibited tumorigenesis. This suggests that Dicer functions as a haploinsufficient tumor suppressor and other miRNA processing factors may work similarly [42]. A second group confirmed these results in retinoblastoma [43]. On the contrary, Ambs et al. found increased Dicer and DGCR8 expression in tumor tissue of prostate cancer patients compared to nontumor tissue. Dicer was also found to be increased in a prostate adenocarcinoma and a Burkitt lymphoma–derived cell line [44,45]. It is possible that miRNA processing factors play varied roles in cancer dependent on miRNA abundance in the tumor tissue and varying degrees of cellular context. If less miRNA expression in a particular tumor leads to progressive disease then less Dicer is warranted, while the opposite might be true for tumors with an overabundance of miRNA.
Both Dicer and Drosha have been shown to be associated with other cellular proteins that modulate their function in a way that is necessary for the correct orientation and cleavage of mature miRNA. DGCR8, for example, is thought to direct Drosha cleavage by recognizing the region between the single-stranded RNA arms of the primary transcript and the stem-loop. Since cleavage by Dicer occurs at a specific distance from the initial Drosha cleavage site, Drosha effectively determines both the 3′ and 5′ ends of the mature miRNA sequence [46]. Improper cleavage may result in a seed sequence unable to bind targets or in instability of the driver strand making it unrecognizable by the RISC complex. One study used mass spectrometry and found that Drosha interacted with at least 20 different proteins [47]. Two of those proteins, p68 and p70, form a complex that interacts with both Drosha and the precursor sequence to regulate miRNA, but their precise role is not yet clarified [48]. It is thought that many of the proteins in the Drosha complex speed up RNA cleavage activity, enhance recruitment of Drosha, or act as a scaffold to assist with processing.
TAR RNA binding protein (TRBP) and protein kinase R–activating protein (PACT) are the two most studied proteins that interact with Dicer and help to enhance both stability and processing activity along with RISC assembly [49,50]. Any disruption in the normal activity of these processing assistants may result in slower cleavage of miRNAs or halt the biogenesis of some miRNAs altogether. One study in colorectal neoplasia showed that miR-143 and miR-145 exhibited decreased expression in tumors compared to normal tissue. However, the precursor levels of both miRNAs were equal in tumor and non-tumor tissue suggesting that abnormal Dicer processing might be the culprit [51].
Though many miRNAs are expressed only in specific tissues, a recent study found that the precursor form might be ubiquitous. For instance, pre-miR-138 is ubiquitously expressed in all tissues however, the mature form is only found in the fetal liver and adult mouse brain [52]. The precursor, but not the mature, form was also found in the cytoplasm suggesting functional nuclear export but no Dicer cleavage. Interestingly, other mature miRNAs are found in the cytoplasm as well suggesting Dicer’s ability to cleave some, but not all, miRNAs. The cause of obstruction between the interaction of Dicer and miR-138, however, remains unknown. A similar situation occurs for let-7 family members and Dicer, but in this case Lin28 mediates the interaction [53,54]. Lin28 not only has the ability to compete with Dicer for Let-7 binding, it promotes 3′-polyuridylation of pre-let-7 causing its degradation [55,56,57]. Notably, 15 additional miRNAs contain the same motif that allows Lin28 to bind and they also experience uridylation. The examples above indicate that Dicer cleavage may work with co-factors to specifically interact with particular miRNAs. The expression of these cofactors and the Dicer method of cleavage may be obstructed in cancer due to variation of the cell microenvironment.
Argonaut (Ago) proteins play a key role during miRNA biogenesis as they coordinate with the RISC complex to direct mature miRNAs to their targets. These proteins also represent important regulators of miRNA that are affected by cellular context. One study found that ectopic expression of the protein boosted mature miRNA expression dramatically while silencing Ago2 expression resulted in reduced levels of mature miRNAs [58]. Another study found that Lin-41 can bind Ago2 directly to cause ubiquitination and degradation of the protein. Since Lin-41 is normally expressed in stem cells and undifferentiated cell types, it is reasonable to consider that Ago2 may play a role in maintaining a differentiated cell phenotype [59]. Lin-41 is one example of a factor that modulates key processing-assistant proteins during miRNA biogenesis, but other factors also influence the biogenesis pathway.
Numerous RNA-binding proteins (RBPs) are also capable of interacting with miRNA to modify their expression. A recent study analyzed RBP and transcription factor binding to precursor miRNAs and found the 3′ end of the 5′ arm to be enriched for the binding of both [60]. A study by Guil et al. showed that hnRNP A1, a ribonucleoprotein, bound to the stem-loop of miR-18a to assist in context-dependent processing. The investigators suggest hnRNP A1 may bind the stem-loop to recruit Drosha or DGCR8 or to prevent other factors from inhibiting further miRNA processing [61].
Extracellular stimuli may affect miRNA expression. One study found that SMAD signaling, through transforming growth factor-β (TGF-β) and bone morphogenic protein (BMP) stimulation, promotes the Drosha-mediated maturation process of miR-21 by binding to the stem-loop region of the precursor [62]. In addition, extracellular growth factor (EGF) expression was capable of increasing Ago2 expression in breast cancer cells [63]. Furthermore, during DNA damage, p53 interacts with Drosha processing to upregulate the expression of miRNAs that suppress growth such as miR-143 and miR-145 [64]. These examples represent only a few of the many miRNA binding proteins that influence the localization, processing, or stability of miRNA expression (Figure 1).
RNA Organization and Single Nucleotide Polymorphisms
Another major influence in the regulation of miRNAs is the RNA structure itself. At each step in the biogenesis process, the structure of RNA changes and may affect how the RNA interacts with itself, other ligands and, perhaps most importantly, RNA-binding proteins (RBPs). For instance, importin 8 (Imp 8), a protein that interacts with the Ago protein complex is thought to modify the structure of the complex and allow for efficient mRNA targeting [65].
The biological state of a cell may provoke structural changes to RNA as well. One prominent example is Pumilio-1 (PUM1), an RNA-binding protein that interacts with the 3′UTR of p27, a cyclin-dependent kinase. Upon growth factor stimulation, PUM1 is upregulated and binds to the 3′UTR of p27 instigating a conformational change to the RNA secondary structure that creates an accessible target site for miR-221 and miR-222 [66]. miR-221 and miR-222 in turn regulate cell cycle progression by repressing p27. However, without proper regulation, p27 can accumulate in the cytoplasm leading to increased cellular motility and tumor metastasis [67].
The level of mature miRNAs in the cytoplasm may remain high, such as the case with miR-221 and miR-222 in quiescent cells, but have no consequence due to steric hindrance of their target site. From this study, it is reasonable to imagine that other secondary mRNA structure modifications may alter the accessibility, or even prevent the binding of, miRNA, the RISC complex, and Argonaut proteins to their target 3′UTR site. Aberrant gene, and consequently protein, expression follows leaving the cell susceptible to disease states such as cancer.
Single nucleotide polymorphisms (SNPs) also modify mRNA structure and alter the binding site for several RBPs and miRNAs. In fact, a single nucleotide change in the 3′UTR of mRNA can either disrupt the ability of a miRNA to bind its target or generate a new target site altogether [68]. Likewise, a single nucleotide modification in the seed region of miRNA, the section of the miRNA about 7 nucleotides long that targets mRNA, may significantly change the targeting profile or inhibit targeting capabilities of the miRNA completely. Both situations result in altered gene expression associated with cancer. For example, Landi et al. found that variant alleles of CD86 are significantly associated with increased risk of CRC [69]. Another study confirmed that twelve unique SNPs in miRNA binding sites have an abnormal allele frequency in cancer [70]. In addition, a genome-wide SNP identification study found 48 SNPs occur in the seed regions of miRNA and greatly influence the number of putative targets [71]. This group went on to experimentally validate eight SNPs in various miRNA seed regions and found that five were consistent with their target-binding predictions.
Above are two examples of SNP locations that have an effect in regulating miRNAs. However, there are a myriad of other possibilities. SNPs in the precursor sequence may prevent accessory proteins from binding and slow the processing rate of miRNAs; a SNP in the a miRNA promoter may impede transcription by preventing Pol II, Pol III or transcription factors from binding; or a SNP in a mature miRNA may affect its stability and cause the passenger strand to be mistaken as the driver by the RISC complex. Interestingly, some SNPs may even confer a protective effect. One SNP, rs895819, is located in the terminal loop of miR-27a and is thought to decrease the size of the loop and reduce mature miRNA production by either limiting the binding of Drosha or increasing the binding affinity of a Drosha inhibitor such as Lin28 [72,73,74]. This SNP in miR-27a’s terminal loop is actually associated with a reduced risk of breast cancer and may present miRNA terminal loops as a new target for therapeutic intervention. For more information on the role of SNPs in miRNA regulation, please refer to a comprehensive review written by Ryan et al. published in 2010.
Similar to the databases used to find the location of miRNA promoters and obtain information about miRNA target genes, there are web-based resources to estimate both RNA structure conformations and the location of SNPs. Although there are algorithms that can predict RNA structure from the primary sequence, they are unable to determine the various conformational changes that may occur upon interaction with other RNAs or RBPs and do not take into account secondary structures that RNA may form in solution [75]. SNPs however are more easily predicted using programs such as Patrocles and PolymiRTS in conjunction with dbSNP, but their functional consequence must be validated experimentally.
Epigenetic Regulation of miRNA
Epigenetic alterations such as DNA methylation and other histone modifications manipulate miRNA expression in much the same way as they control gene expression [76]. Both hyper- and hypo-methylation are frequent events in cancer and induce functional changes to gene expression resembling that of a genetic mutation. To date, there have been many studies investigating how epigenetic changes affect miRNA expression. Roman-Gomez et al. used chromatin immunoprecipitation (ChIP) in acute lymphoblastic leukemia (ALL)-derived cell lines to identify 13 miRNAs with high levels of repressive H3K9me2 marks in their CpG islands. The histone modifications correlated with CpG hypermethylation and low miRNA expression in each ALL cell line [77]. Methylation of the promoter region of miR-145 caused decreased expression in nearly 50 different cancer cell lines (five cancer types) and treatment of seven cells lines with 5-aza-2′-deoxycytidine, a DNA methyltransferase inhibitor, led to the re-expression of miR-145 [78].
In 2009, Weber et al. reported that CpG islands are associated with 155 out of 332 miRNA genes (47%) [79]. Many of those miRNAs have been reported in multiple studies as silenced in a variety of cancer types [80]. The miR-127 gene in particular is one of many embedded in a CpG island and was found to be significantly upregulated upon the combination treatment of T24 bladder cancer cells with a DNA methyltransferase inhibitor (DNMTi) and a histone deacetylase inhibitor (HDACi) [81]. The same study showed that 17 of the 313 human miRNAs analyzed were increased greater than 3 fold after treatment. Other miRNA genes surrounded by CpG islands are also methylated in cancer such as miR-124a in colorectal cancer [82] and Let-7a-3 in lung cancer. Similar to miR-127, a combination treatment with HDAC and DNMT inhibitors led to the demethylation and transcription of the Let-7a-3 gene in A549 cells [83]. However, the Let-7a-3 gene is better left methylated as increased expression promotes human lung carcinogenesis. Indeed, the methylation of miRNA greatly affects the expression of many target genes. In the above examples, miR-127 targets BCL6, a protoncogene that upregulates p53 expression; miR-124 targets CDK6 which triggers the inactivation of tumor suppressor Rb; and finally Let-7a-3 has nearly 200 targets, many with roles in tumorigenesis.
In addition to associations between methylated CpG islands and decreased miRNA expression, one study has shown that the AML1/ETO fusion protein in leukemia recruits DNMTs, MeCp2, and HDAC1 to an AML1 site on the promoter of miR-223 [84]. As mentioned above, miR-223 expression leads to proper myeloid cell differentiation. Once the fusion protein binds to the promoter, miR-223 gene expression is silenced and halts cell differentiation.
Studies such as those described above, convey the importance of using DNMT and HDAC inhibitors to encourage re-expression of miRNAs in cancers where they are markedly downregulated. In the future, it will be interesting to witness reactivated miRNA expression by means of even more specific modes of therapy.
Conclusion
The current estimate indicates that the human miRNA family comprises 1,733 mature miRNAs derived from 1,424 precusors, each of which is capable of targeting hundreds of protein-coding genes [85]. From this review it is apparent that there are many avenues by which miRNAs can be regulated and that abnormal regulation results in cancer development (Figure 1). Cancer arises as a consequence of an imbalance in cellular homeostasis and compelling evidence indicates that aberrant alterations of miRNA signaling pathways are a ubiquitous feature in human cancers. The elaborate networks between miRNA regulation and downstream signaling pathways are perfect examples of how a cell develops intricate processes to maintain homeostasis. Understanding this process is critical for our ability to define effective targets for cancer management and to improve outcome. As described above, there is a great deal of exciting research being accomplished in this field; however, it should also be clear that this is the tip of a very large iceberg. The efforts to date have opened the door to many additional questions and a considerable amount of carefully validated research lies ahead. For example, it is now known that the same miRNA has the ability to act as either a tumor suppressor or an oncogene depending on its cellular context and microenvironment. But how exactly does that cellular context affect the miRNA? Are there changes at the transcriptional level or are miRNAs influenced predominantly during processing? Further, the manner in which miRNAs are regulated directly affects their mRNA targets, which may lead to aberrant protein expression and disease states. Deciphering how miRNAs are regulated is crucial to understand their role both physiologically and in cancer. It is only through this additional knowledge that we will be able to develop targeted therapeutics that extend the survival of those suffering from the “emperor of all maladies”.
Acknowledgments
This work was supported by the intramural Research Program of the Center for Cancer Research, National Cancer Institute (Z01 BC 010876).
Footnotes
Disclosure No potential conflicts of interest relevant to this article were reported.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
- 1.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 2.Baek D, Villen J, Shin C, et al. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 4.Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee Y, Jeon K, Lee JT, et al. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee Y, Kim M, Han J, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou X, Wong ST. Computational Systems Bioinformatics and Bioimaging for Pathway Analysis and Drug Screening. Proc IEEE Inst Electr Electron Eng. 2008;96:1310–1331. doi: 10.1109/JPROC.2008.925440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006;13:1097–1101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 9.Schanen BC, Li X. Transcriptional regulation of mammalian miRNA genes. Genomics. 2011;97:1–6. doi: 10.1016/j.ygeno.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.vis-Dusenbery BN, Hata A. MicroRNA in Cancer: The Involvement of Aberrant MicroRNA Biogenesis Regulatory Pathways. Genes Cancer. 2010;1:1100–1114. doi: 10.1177/1947601910396213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saini HK, Griffiths-Jones S, Enright AJ. Genomic analysis of human microRNA transcripts. Proc Natl Acad Sci U S A. 2007;104:17719–17724. doi: 10.1073/pnas.0703890104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marson A, Levine SS, Cole MF, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujita S, Ito T, Mizutani T, et al. miR-21 Gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J Mol Biol. 2008;378:492–504. doi: 10.1016/j.jmb.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 16.Seike M, Goto A, Okano T, et al. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc Natl Acad Sci U S A. 2009;106:12085–12090. doi: 10.1073/pnas.0905234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Talotta F, Cimmino A, Matarazzo MR, et al. An autoregulatory loop mediated by miR-21 and PDCD4 controls the AP-1 activity in RAS transformation. Oncogene. 2009;28:73–84. doi: 10.1038/onc.2008.370. [DOI] [PubMed] [Google Scholar]
- 18.Huang GL, Zhang XH, Guo GL, et al. Expression of microRNA-21 in invasive ductal carcinoma of the breast and its association with phosphatase and tensin homolog deleted from chromosome expression and clinicopathologic features. Zhonghua Yi Xue Za Zhi. 2008;88:2833–2837. [PubMed] [Google Scholar]
- 19.Velu CS, Baktula AM, Grimes HL. Gfi1 regulates miR-21 and miR-196b to control myelopoiesis. Blood. 2009;113:4720–4728. doi: 10.1182/blood-2008-11-190215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ribas J, Ni X, Haffner M, et al. miR-21: an androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res. 2009;69:7165–7169. doi: 10.1158/0008-5472.CAN-09-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi XB, Xue L, Yang J, et al. An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0706641104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ambs S, Prueitt RL, Yi M, et al. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 2008;68:6162–6170. doi: 10.1158/0008-5472.CAN-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferraro L, Ravo M, Nassa G, et al. Effects of oestrogen on microRNA expression in hormone-responsive breast cancer cells. Horm Cancer. 2012;3:65–78. doi: 10.1007/s12672-012-0102-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24•.Paris O, Ferraro L, Grober OM, et al. Direct regulation of microRNA biogenesis and expression by estrogen receptor beta in hormone-responsive breast cancer. Oncogene. 2012;31:4196–4206. doi: 10.1038/onc.2011.583. This study demonstrates the importance of ERb in the regulation of many miRNAs, either independently or by impeding ERa activity, in breast cancer. The results suggest that the expression of this nuclear receptor may contribute to a less aggressive tumor phenotype. [DOI] [PubMed] [Google Scholar]
- 25.O’donnell KA, Wentzel EA, Zeller KI, et al. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 26.Chang TC, Yu D, Lee YS, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27•.Bui TV, Mendell JT. Myc: Maestro of MicroRNAs. Genes Cancer. 2010;1:568–575. doi: 10.1177/1947601910377491. This is an excellent review discussing the role of Myc in modulating the expression of multiple miRNAs in cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xi Y, Shalgi R, Fodstad O, et al. Differentially regulated micro-RNAs and actively translated messenger RNA transcripts by tumor suppressor p53 in colon cancer. Clin Cancer Res. 2006;12:2014–2024. doi: 10.1158/1078-0432.CCR-05-1853. [DOI] [PubMed] [Google Scholar]
- 29.Raver-Shapira N, Marciano E, Meiri E, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 30.He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–822. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukao T, Fukuda Y, Kiga K, et al. An Evolutionarily Conserved Mechanism for MicroRNA-223 Expression Revealed by MicroRNA Gene Profiling. Cell. 2007;129:617–631. doi: 10.1016/j.cell.2007.02.048. [DOI] [PubMed] [Google Scholar]
- 32.Ji J, Yamashita T, Wang XW. Wnt/beta-catenin signaling activates microRNA-181 expression in hepatocellular carcinoma. Cell Biosci. 2011;1:4. doi: 10.1186/2045-3701-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coller HA, Forman JJ, Legesse-Miller A. “Myc’ed messages”. myc induces transcription of E2F1 while inhibiting its translation via a microRNA polycistron. PLoS Genet. 2007;3:e146. doi: 10.1371/journal.pgen.0030146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34•.Hatziapostolou M, Polytarchou C, Aggelidou E, et al. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell. 2011;147:1233–1247. doi: 10.1016/j.cell.2011.10.043. This study shows that transient expression of hepatocyte nuclear factor 4α (HNF4α) induces an inflammatory feedback loop involving miRNA-24, miRNA-124, and miRNA-629. Upon activation, the circuit suppresses HNF4α expression and promotes hepatocarcinogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Q, Wang Z, Hu Y, et al. miR-21 promotes migration and invasion by the miR-21-PDCD4-AP-1 feedback loop in human hepatocellular carcinoma. Oncol Rep. 2012;27:1660–1668. doi: 10.3892/or.2012.1682. [DOI] [PubMed] [Google Scholar]
- 36.Godlewski J, Nowicki MO, Bronisz A, et al. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol Cell. 2010;37:620–632. doi: 10.1016/j.molcel.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartel B. MicroRNAs directing siRNA biogenesis. Nat Struct Mol Biol. 2005;12:569–571. doi: 10.1038/nsmb0705-569. [DOI] [PubMed] [Google Scholar]
- 38.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 39.Thomson JM, Newman M, Parker JS, et al. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–2207. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merritt WM, Lin YG, Han LY, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med. 2008;359:2641–2650. doi: 10.1056/NEJMoa0803785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar MS, Lu J, Mercer KL, et al. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 42.Kumar MS, Pester RE, Chen CY, et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 2009;23:2700–2704. doi: 10.1101/gad.1848209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43•.Lambertz I, Nittner D, Mestdagh P, et al. Monoallelic but not biallelic loss of Dicer1 promotes tumorigenesis in vivo. Cell Death Differ. 2010;17:633–641. doi: 10.1038/cdd.2009.202. This study reveals the importance of Dicer1 expression and how it effects miRNA expression and cancer progression. Results show that monoallelic loss of Dicer1 leads to a decrease in miRNA expression and promotes tumor growth. Interestingly, biallelic loss of Dicer1 leads to an inhibition of tumor formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiosea S, Jelezcova E, Chandran U, et al. Up-regulation of dicer, a component of the MicroRNA machinery, in prostate adenocarcinoma. Am J Pathol. 2006;169:1812–1820. doi: 10.2353/ajpath.2006.060480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaul D, Sikand K. Defective RNA-mediated c-myc gene silencing pathway in Burkitt’s lymphoma. Biochem Biophys Res Commun. 2004;313:552–554. doi: 10.1016/j.bbrc.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 46.Davis BN, Hata A. Regulation of MicroRNA Biogenesis: A miRiad of mechanisms. Cell Commun Signal. 2009;7:18. doi: 10.1186/1478-811X-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gregory RI, Yan KP, Amuthan G, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 48.Fukuda T, Yamagata K, Fujiyama S, et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol. 2007;9:604–611. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- 49.Chendrimada TP, Gregory RI, Kumaraswamy E, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haase AD, Jaskiewicz L, Zhang H, et al. TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep. 2005;6:961–967. doi: 10.1038/sj.embor.7400509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Michael MZ, O’ Connor SM, Holst Pellekaan NG, et al. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–891. [PubMed] [Google Scholar]
- 52.Obernosterer G, Leuschner PJ, Alenius M, Martinez J. Post-transcriptional regulation of microRNA expression. RNA. 2006;12:1161–1167. doi: 10.1261/rna.2322506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lehrbach NJ, Armisen J, Lightfoot HL, et al. LIN-28 and the poly(U) polymerase PUP-2 regulate let-7 microRNA processing in Caenorhabditis elegans. Nat Struct Mol Biol. 2009;16:1016–1020. doi: 10.1038/nsmb.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wulczyn FG, Smirnova L, Rybak A, et al. Post-transcriptional regulation of the let-7 microRNA during neural cell specification. FASEB J. 2007;21:415–426. doi: 10.1096/fj.06-6130com. [DOI] [PubMed] [Google Scholar]
- 55.Heo I, Joo C, Kim YK, et al. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 56.Hagan JP, Piskounova E, Gregory RI. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat Struct Mol Biol. 2009;16:1021–1025. doi: 10.1038/nsmb.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57•.Michlewski G, Caceres JF. Antagonistic role of hnRNP A1 and KSRP in the regulation of let-7a biogenesis. Nat Struct Mol Biol. 2010;17:1011–1018. doi: 10.1038/nsmb.1874. This study shows that ribonuclear protein A1 interferes with KSRP binding to let-7a and prevents Drosha processing of the miRNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Diederichs S, Haber DA. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell. 2007;131:1097–1108. doi: 10.1016/j.cell.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 59.Rybak A, Fuchs H, Hadian K, et al. The let-7 target gene mouse lin-41 is a stem cell specific E3 ubiquitin ligase for the miRNA pathway protein Ago2. Nat Cell Biol. 2009;11:1411–1420. doi: 10.1038/ncb1987. [DOI] [PubMed] [Google Scholar]
- 60.Jha A, Mehra M, Shankar R. The regulatory epicenter of miRNAs. J Biosci. 2011;36:621–638. doi: 10.1007/s12038-011-9109-y. [DOI] [PubMed] [Google Scholar]
- 61.Guil S, Gattoni R, Carrascal M, et al. Roles of hnRNP A1, SR proteins, and p68 helicase in c-H-ras alternative splicing regulation. Mol Cell Biol. 2003;23:2927–2941. doi: 10.1128/MCB.23.8.2927-2941.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adams BD, Claffey KP, White BA. Argonaute-2 expression is regulated by epidermal growth factor receptor and mitogen-activated protein kinase signaling and correlates with a transformed phenotype in breast cancer cells. Endocrinology. 2009;150:14–23. doi: 10.1210/en.2008-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suzuki HI, Yamagata K, Sugimoto K, et al. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 65.Weinmann L, Hock J, Ivacevic T, et al. Importin 8 is a gene silencing factor that targets argonaute proteins to distinct mRNAs. Cell. 2009;136:496–507. doi: 10.1016/j.cell.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 66•.Kedde M, van KM, Zwart W, et al. A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol. 2010;12:1014–1020. doi: 10.1038/ncb2105. The first study to show that RNA binding protein PUM1 induces an RNA structural change that regulates miRNA expression. [DOI] [PubMed] [Google Scholar]
- 67.Larrea MD, Wander SA, Slingerland JM. p27 as Jekyll and Hyde: regulation of cell cycle and cell motility. Cell Cycle. 2009;8:3455–3461. doi: 10.4161/cc.8.21.9789. [DOI] [PubMed] [Google Scholar]
- 68•.Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10:389–402. doi: 10.1038/nrc2867. An outstanding review describing how SNPs in miRNA genes, seed regions, or processing machinery effect miRNA expression in cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Landi D, Gemignani F, Naccarati A, et al. Polymorphisms within micro-RNA-binding sites and risk of sporadic colorectal cancer. Carcinogenesis. 2008;29:579–584. doi: 10.1093/carcin/bgm304. [DOI] [PubMed] [Google Scholar]
- 70.Yu Z, Li Z, Jolicoeur N, et al. Aberrant allele frequencies of the SNPs located in microRNA target sites are potentially associated with human cancers. Nucleic Acids Res. 2007;35:4535–4541. doi: 10.1093/nar/gkm480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71•.Gong J, Tong Y, Zhang HM, et al. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum Mutat. 2012;33:254–263. doi: 10.1002/humu.21641. This study showed that a SNP in the seed region of a miRNA caused it to bind to a new target gene. This is also the first experimental validation of SNP induced changes to miRNA function. [DOI] [PubMed] [Google Scholar]
- 72.Zeng Y, Cullen BR. Efficient processing of primary microRNA hairpins by Drosha requires flanking nonstructured RNA sequences. J Biol Chem. 2005;280:27595–27603. doi: 10.1074/jbc.M504714200. [DOI] [PubMed] [Google Scholar]
- 73.Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA. 2008;14:1539–1549. doi: 10.1261/rna.1155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang J, Zhou F, Xu T, et al. Analysis of sequence variations in 59 microRNAs in hepatocellular carcinomas. Mutat Res. 2008;638:205–209. doi: 10.1016/j.mrfmmm.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 75•.Wan Y, Kertesz M, Spitale RC, et al. Understanding the transcriptome through RNA structure. Nat Rev Genet. 2011;12:641–655. doi: 10.1038/nrg3049. This is an excellent review discussing the importance of RNA structure in understanding RNA function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barski A, Jothi R, Cuddapah S, et al. Chromatin poises miRNA- and protein-coding genes for expression. Genome Res. 2009;19:1742–1751. doi: 10.1101/gr.090951.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roman-Gomez J, Agirre X, Jimenez-Velasco A, et al. Epigenetic regulation of microRNAs in acute lymphoblastic leukemia. J Clin Oncol. 2009;27:1316–1322. doi: 10.1200/JCO.2008.19.3441. [DOI] [PubMed] [Google Scholar]
- 78•.Suh SO, Chen Y, Zaman MS, et al. MicroRNA-145 is regulated by DNA methylation and p53 gene mutation in prostate cancer. Carcinogenesis. 2011;32:772–778. doi: 10.1093/carcin/bgr036. This is the first study to show that DNA methylation and a mutation in p53 cause reduced miR-145 expression in prostate cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weber B, Stresemann C, Brueckner B, Lyko F. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle. 2007;6:1001–1005. doi: 10.4161/cc.6.9.4209. [DOI] [PubMed] [Google Scholar]
- 80•.Wiklund ED, Kjems J, Clark SJ. Epigenetic architecture and miRNA: reciprocal regulators. Epigenomics. 2010;2:823–840. doi: 10.2217/epi.10.51. This review offers a great summary of epigenetic miRNA regulation in cancer. [DOI] [PubMed] [Google Scholar]
- 81.Saito Y, Liang G, Egger G, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–443. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 82.Lujambio A, Ropero S, Ballestar E, et al. Genetic Unmasking of an Epigenetically Silenced microRNA in Human Cancer Cells. Cancer Res. 2007;67:1424–1429. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 83.Brueckner B, Stresemann C, Kuner R, et al. The human let-7a-3 locus contains an epigenetically regulated microRNA gene with oncogenic function. Cancer Res. 2007;67:1419–1423. doi: 10.1158/0008-5472.CAN-06-4074. [DOI] [PubMed] [Google Scholar]
- 84.Fazi F, Racanicchi S, Zardo G, et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell. 2007;12:457–466. doi: 10.1016/j.ccr.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 85.Wang XW, Heegaard NH, Orum H. MicroRNAs in liver disease. Gastroenterology. 2012;142:1431–1443. doi: 10.1053/j.gastro.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]