

Abstract
The hepatitis C virus (HCV) NS3 helicase shares several conserved motifs with other superfamily 2 (SF2) helicases. Besides these sequences, several additional helicase motifs are conserved among the various HCV genotypes and quasispecies. The roles of two such motifs are examined here. The first motif (YRGXDV) forms a loop that connects SF2 helicase motifs 4 and 5, at the tip of which is Arg393. When Arg393 is changed to Ala, the resulting protein (R393A) retains a nucleic acid stimulated ATPase but cannot unwind RNA. R393A also unwinds DNA more slowly than wild type and translocates poorly on single-stranded DNA (ssDNA). DNA and RNA stimulate ATP hydrolysis catalyzed by R393A like the wild type, but the mutant protein binds ssDNA more weakly both in the presence and absence of the non-hydrolyzable ATP analog ADP(BeF3). The second motif (DFSLDPTF) forms a loop that connects two anti-parallel sheets between SF2 motifs 5 and 6. When Phe444 in this Phe-loop is changed to Ala, the resulting protein (F444A) is devoid of all activities. When Phe438 is changed to Ala, the protein (F438A) retains nucleic acid-stimulated ATPase, but does not unwind RNA. F438A unwinds DNA and translocates on ssDNA at about half the rate of the wild type. Equilibrium binding data reveal that this uncoupling of ATP hydrolysis and unwinding is due to the fact that the F438A mutant does not release ssDNA upon ATP binding like the wild type. A model is presented explaining the roles of the Arg-clamp and the Phe-loop in the unwinding reaction.
The hepatitis C virus (HCV)1 has caused a global crisis by infecting more than 2% of the world’s population (1). Combination therapies using interferon and the antiviral drug ribavirin eliminate HCV in a majority of patients but such treatments are costly and cause debilitating side effects. Vaccine and drug development has been difficult because the only natural hosts for HCV are humans and chimpanzees, and the virus cannot be conventionally cultivated in vitro. As a result, the enzymes encoded by the virus have been intensely studied for rational drug design (2). Several groups have reported potent compounds that inhibit the HCV RNA-dependent RNA polymerase (3) and its serine protease (4). However, few inhibitors of the HCV helicase have been explored as antivirals because less is known about the mechanism of action of the HCV helicase and if or how it differs from similar cellular enzymes.
HCV helicase is part of the viral non-structural protein 3 (NS3). The N-terminal one-third of NS3 is a chymotrypsin-like serine protease, and the C-terminal NS3 portion is a helicase that is fueled by the hydrolysis of ATP. Although the HCV helicase only unwinds duplex RNA during viral replication, the purified protein unwinds DNA dramatically more efficiently than RNA (5). In these reactions, HCV helicase requires a 3′ single-stranded (ss) DNA or RNA overhang to initiate unwinding and is therefore classified as a 3′-5′ helicase. The structure of the HCV helicase has been determined four times using x-ray crystallography: twice as an apoenzyme (6, 7), once with a bound ssDNA (8) and once as part of the full-length NS3 protein (9). NMR structures of parts of the HCV helicase have also been reported (10, 11). HCV helicase consists of 3 domains, with ATP likely binding between the structurally similar domains 1 and 2. One strand of the unwound duplex binds in the cleft that separates domain 3 from domains 1 and 2 (8). Gorbalenya and Koonin (12) have classified the HCV helicase as a member of helicase superfamily 2, which contains several large protein families that all share several signature sequences. The SF2 helicase signature sequence motifs line the “ATP-binding” cleft between domains 1 and 2. Numerous studies have elucidated the roles of the SF2 motifs in HCV helicase function, and structure-based mutagenesis has revealed a few other critical conserved residues (reviewed in Refs. 2 and 13). Little is known about the function of residues outside these conserved motifs.
In the present study, sequence alignments and structure-based site-directed mutagenesis were used to identify two additional conserved motifs in domain 2 of the HCV helicase that are critical for helicase action. After introducing individual point mutations into these regions of the enzyme, ATPase, unwinding, translocation rates, and DNA binding were analyzed. The first conserved motif examined is part of a loop connecting SF2 helicase motifs 4 and 5. At the tip of this loop is Arg393, which contacts the backbone of DNA (8). To test the importance of Arg393, the residue was changed to Ala, and the R393A mutant was purified and characterized. Data show how this “Arg-clamp” motif aids the unwinding of the double helix. The second conserved motif spans two conserved phenyalanines and connects a pair of β-sheets extending from domain 2. This Phe-loop motif is far from the known ssDNA binding site and the putative ATP binding site. To examine the role of the Phe-loop, two conserved Phe in this motif were changed to Ala. Analyses of the purified mutants reveal that the Phe-loop might help stabilize the rotation of domain 2 by anchoring it to domain 3. Such a conformational change can be visualized by comparing currently available structures of HCV helicase and could occur upon ATP binding or hydrolysis. A model is presented to explain the role of the Arg-clamp and the Phe-loop in the helicase mechanism that combines the domain rotation model proposed by Yao et al. (6) and the ratcheting inchworm model proposed by Kim et al. (8).
EXPERIMENTAL PROCEDURES
Materials
RNase-free reagents were purchased from Ambion Inc. (Austin, TX), and RNA oligonucleotides were from Dharmacon (Lafayette, CO). DNA oligonucleotides were obtained from either Integrated DNA Technologies (Corralville, IA) or Genelink (Hawthorne, NY). Plasmid pET24a and BL21(DE3) cells were obtained from Novagen (Madison, WI). Beryllium fluoride was from Alfa Aesar (Ward Hill, MA).
Mutagenesis
The QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used to construct each mutant from the plasmid p24Hel-1a (14), which contains the helicase region of NS3 isolated from an infectious clone of HCV genotype 1a isolate H77c (15). The following oligonucleotides were utilized in this PCR-based mu-tagenesis: R393A(+), 5′-CAATGCCGTGGCCTACTACGCCGGTCTTGACGTGTCTGTC-3′; R393A(−), 5′-GACAGACACGTCAAGACCGGCGTAGTAGGCCACGGCATTG-3′; F438A(+), 5′-GTCACTCAGACAGTCGATGCCAGCCTGGATCCTACCTTTACCATTGAG-3′; F438A(−) 5′-CTCAATGGTAAAGGTAGGATCCAGGCTGGCATCGACTGTCTGAGTGAC-3′, and F444A(+) 5′-CAGCCTTGACCCTACCGCTACCATTGAGACAAC CAC-3′; and F444A(−), 5′-GTGGTTGTCTCAATGGTAGCGGTAGGGTCAAGGCTG-3′. Mutations were confirmed by sequencing the plasmid DNA. All constructs place the HCV helicase under control of a T7(lac) promoter, which contains a lac operator between the T7 promoter and the transcription start site (16). Use of this hybrid promoter was necessary to control basal expression of the HCV helicase, which appears to be toxic to Escherichia coli. Each of the four recombinant proteins possesses an additional MAS sequence at their N termini and a C-terminal His tag (PNSSSVDKLAAALEHHHHHH). BL21(DE3) cells harboring plasmids were grown in LB broth, induced at 0.5 OD600 with 1 mm isopropyl-1-thio-β-d-galactopyranoside, and grown for 3 h at 23 °C.
Protein Purification
All proteins were purified as previously described (14). Briefly, cells were harvested and lysed by two passes through a French Press, and the clarified soluble extract was fractionated on three columns: a 10-ml Ni-NTA column, a 100-ml Sephacryl S-200HR gel filtration column, and finally a 0.5-ml DEAE column. All constructs yielded 1–2 mg of pure helicase/liter culture except for those containing the F444A mutation. Only a small amount of the F444A mutant was present in the soluble fraction of a cellular crude extract. The overwhelming majority of F444A was found in inclusion bodies, which were discarded. About 50 μg of F444A was purified from a 2-liter culture.
ATPase Assays
Initial rates of ATP hydrolysis were measured as described previously (14). Reactions were run at 37 °C in 25 mm MOPS (pH 6.5), 5 mm MgCl2 0.1% Tween20, ATP, and 10–100 nm helicase. Data were fit to the Michaelis-Menten equation by non-linear regression to calculate the Km and Vmax in the presence and absence of nucleic acids. To determine the constant KRNA, defined as the concentration of RNA that supports a half maximum rate of catalysis, reactions were performed in the presence of 4 mm ATP with various concentrations of RNA as previously described (14).
Unwinding Assays
To generate substrates for helicase assays, two synthetic oligonucleotides were annealed by heating them to 95 °C and allowing them to cool slowly to room temperature. Before annealing, the shorter strand was 32P-labeled using polynucleotide kinase. The DNA substrate consisted of a shorter strand DNA oligonucleotide 5′-[32P]GCCTCGCTGCCGTCGCCA-3′ annealed to a longer strand DNA oligonucleotide 5′-TGGCGACGGCAGCGAGGCTTTTTTTTTTTTTTTTTTTT-3′. The resulting duplex has a 3′-ssDNA tail. The duplex RNA substrate consisted of the RNA oligonucleotide 5′-UGGCGACGGCAGCGAGGCUUUUUUUUUUUUUUUUUUUU-3′ annealed to the oligonucleotide 5′-[32P]GCCUCGCUGCCGUCGCCA-3′. The DNA or RNA substrate (1 nm) and HCV helicase (50–200 nm) were incubated in reaction buffer (25 mm MOPS pH 6.5, 3 mm MgCl2, 0.1% Tween 20) for 30 min. 10-μl reactions were initiated by the addition of 5 mm ATP, terminated with 2.5 μl of 5× stopping buffer after 20 s, 40 s, 1 min, 2 min, 3 min, 5 min, 7.5 min, and 10 min. 5× stopping buffer contained 250 mm Tris-Cl, pH 7.5, 20 mm EDTA, 0.5% SDS, 0.1% Nonidet P-40, 0.1% bromphenol blue, 0.1% xylene cynanole FF, 50% glycerol. 250 nm trap DNA was also added before electrophoresis to prevent re-annealing. Products were analyzed by non-denaturing polyacrylamide gel electrophoresis as described previously (14). In some assays, excess trap DNA, which consisted of the unlabeled shorter oligonucleotide, was added when the reactions were initiated. RNA unwinding assays were performed at 37 °C, but DNA unwinding assays were performed at 23 °C because rates of DNA unwinding at 37 °C were too rapid to be accurately measured.
Translocation Assays
Translocation on ssDNA was assessed using an assay, developed by Raney and co-workers (17, 18), that measures the helicase catalyzed displacement of streptavidin from a biotinylated [32P]oligonucleotide. The streptavidin-bound biotinylated DNA substrate 5′-[32P]T(bio)GGCGACGGCAGCGAGGCTTTTTTTTTTTTTTTTTTTT-3′ (5 nm) and various concentrations of HCV helicase were incubated in reaction buffer (25 mm MOPS, pH 6.5, 3 mm MgCl2, 0.1% Tween 20, 37 °C) for 30 min. Reactions were initiated by the addition of 5 mm ATP and 6 μm biotin (to trap released streptavidin) and terminated at various times with the stopping buffer used in the unwinding assays. After electrophoresis on a 15% non-denaturing polyacrylamide gel, reactions were analyzed with a Molecular Dynamics phosphorimager using ImageQuant software (Amersham Biosciences, Piscataway, NJ).
Fluorescence-based Nucleic Acid Binding Assay
The quenching of intrinsic protein fluorescence upon formation of an enzyme-nucleic acid complex was monitored as described previously (14). Aliquots of DNA were added to the helicase in 2 ml of 25 mm MOPS, pH 6.5, 3 mm MgCl2, and 0.1% Tween 20 at 23 °C. Fluorescence was measured by exciting the sample at 280 nm and reading the emission at 340 nm with a Varian Carey Eclipse fluorescence spectrophotometer. After correcting data for dilution and inner filter effects, fractional fluorescence FNA was calculated by dividing the fluorescence values by the initial fluorescence of the protein in the absence of DNA. FNA was fit to the total concentration of added 18-nt long poly(U) DNA oligonucleotide ([NA]T) using Equation 1, which accounts for ligand depletion.
| (Eq. 1) |
In Equation 1, ΔFMAX is the maximum change in fractional fluorescence resulting when all enzyme molecules are bound to nucleic acid. KD is the dissociation constant, and ET is total enzyme concentration. The procedure was repeated at three different enzyme concentrations for each enzyme/oligonucleotide pair. KD was calculated by globally fitting 3 data sets to Equation 1 using Graphpad Prism Version 4.0 (San Diego, CA).
Gel Shift Assays
The same [32P]DNA duplex used in the unwinding assays (1 nm) was incubated with increasing concentrations of helicase (1–1,000 nm) in 25 mm MOPS, pH 6.5, 3 mm MgCl2, 0.1% Tween 20. After electrophoresis in a 15% Tris borate/EDTA polyacrylamide gel, the amounts of bound and free DNA were determined using a phosphorimager.
RESULTS
The helicase encoded by Hepatitis C virus has been extensively studied both as a model helicase and as a potential drug target. One problem with designing drugs that function by inhibiting viral enzymes is that the virus often rapidly evolves resistance by selecting mutations in inhibitor binding sites. Ideal inhibitors should therefore bind to critical invariant residues.
Sequence Alignments
To locate critical invariant residues in the HCV helicase, 163 NS3 sequences were obtained from the Hepatitis C Virus data base (s2as02.genes.nig.ac.jp/) and aligned using the program ClustalX (version 1.83) (19). The results are shown in Fig. 1A. The consensus generated by ClustalX is shown below the sequence of the helicase from genotype 1a (isolate H77c). The NS3 protease occupies the first 180 amino acids that are not shown in Fig 1A. Such an analysis reveals that only about one-third of the residues in the helicase are invariant in all HCV sequences reported. About one-third are conserved, and the rest of the residues are highly variable among the known HCV genotypes and quasispecies.
Fig. 1. Identification of the Arg-clamp and Phe-loop in the HCV helicase.

A, consensus generated from an alignment of HCV NS3 sequences obtained from 163 different isolates is shown below the sequence of the helicase from HCV genotype 1a used in this study (isolate H77c, NCBI accession AF011751). Asterisks designate amino acids that are identical in all sequences, colons mark very conserved residues, and periods indicate conserved residues as defined by the program ClustalX (19). Locations of conserved motifs are designated above the sequence. B, domain 2 of the HCV helicase was aligned with the analogous domain of other SF2 helicases using the Vector Alignment Search Tool (41) available at the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/Structure/VAST/vast.shtml). These structures include eIF4a from Saccharomyces cerevisiae (NCBI accession 1FUU), the DNA repair protein UvrB from Bacillus caldotenax (NCBI accession 1D9Z), and a DEAD-box protein from Methanococcus Jannaschii (NCBI accession 1HV8). Also included are key cellular proteins whose structures are still not known; spliceosome component prp16 (NCBI accession P15938), nuclear DNA helicase II (NCBI accession O70133), the cockayne syndrome-linked protein ERCC-6 (NCBI accession Q03468), and the human DEAD box protein DDX10 (NCBI accession Q13206) Conserved residues are shaded, and the locations of conserved motifs and residues targeted for site-directed mutagenesis are noted above the alignment.
Many of the conserved amino acids cluster around the helicase “signature” motifs described by Gorbalenya and Koonin (12), but it should be noted that not all residues are strictly conserved even within the signature motifs. For example, Lys210 in Motif 1, which helps form the Walker-type phosphate binding “P-loop,” is sometimes replaced by an Arg. Likewise, His293 in the signature “DECH-box” of motif 2 is a Tyr in HCV genotype 4a (20). Also shown on Fig. 1A are a TXGX motif that was shown to interact with bound nucleic acid (21), and Trp501, which intercalates between the nucleic acid bases acting like a bookend to aid unwinding (22-25).
Fig. 1A highlights additional conserved motifs outside the SF2 motifs. This study focuses on two of these motifs, which we have named the Arg-clamp and the Phe-loop. The Arg-clamp spans amino acids 392–397, where 5 of 6 conserved amino acids are invariant. An examination of the structure of HCV helicase bound to a oligonucleotide reveals that the side chain of Arg393 is only 2.3 Å from a bridging phosphate in the DNA backbone (8). To test if this is a functionally important contact, Arg393 was altered to an Ala using site-directed mutagenesis. The R393A mutant protein was purified and compared with the wild type (see below). The second conserved feature examined here is the motif spanning the absolutely conserved amino acids 437–444. In NS3, this region is the tip of an extended β-loop. This Phe-loop links two phenylalanines, one on each end of each β-strand that forms the β-loop. Kim et al. (8) proposed that this β-loop might function like the L45 loop that binds ssDNA in ssDNA-binding proteins. In support of this role, Lam et al. (14) has shown that a nearby residue at position 450 is involved in DNA binding and unwinding. Alternatively, this Phe-loop could play a more structural role. As noted by Yao et al. (6) the two conserved Phe in the Phe-loop pack into a pocket containing Phe531 and Trp532 of domain 3. Thus, the Phe-loop could either be involved in nucleic acid interactions or as an important contact to link domains 2 and 3. To test these ideas, Phe438 and Phe444 in the Phe-loop were changed to Ala, and the recombinant purified proteins were characterized.
Although the Arg-clamp and Phe-loop are conserved in HCV and related viruses, structural alignments of the HCV helicase and other SF2 helicases reveal that these motifs are unique to the HCV-like family of enzymes (Fig. 1B). This alignment reveals that neither the Phe-loop nor the Arg-clamp is present in representatives of other families in SF2. Alignments of the HCV helicase with 21 other human DEAD-box proteins also failed to uncover a human equivalent of these motifs (data not shown). However, as pointed out by Caruthers et al. (26), other DEAD-box proteins contain a nearby conserved Arg (Arg298 in eIF4a) that could likewise interact with bound nucleic acids.
Three site-directed mutants were constructed to examine the role of the Arg-clamp and Phe-loop: R393A, F438A, and F444A. Each mutant protein retained the same C-terminal His tag as the wild type (Hel-1a), was expressed in E. coli, and was purified to apparent homogeneity using the same protocol (Fig. 2). Here, the wild-type enzyme is the NS3 protein isolated from infectious clone H77c of HCV genotype 1a (15). The proteins lack the first 166 amino acids of NS3, which comprise the NS3 protease. Both R393A and F438A behaved like the wild type throughout expression and purification. However, F444A was far less soluble in crude extracts than the other proteins. Only a small amount of F444A (50 μg) was obtained from a 1-liter culture, whereas over 2 mg of the other proteins were purified from 1-liter cultures.
Fig. 2. Expression and purification of the HCV helicase domain and site-directed mutants.
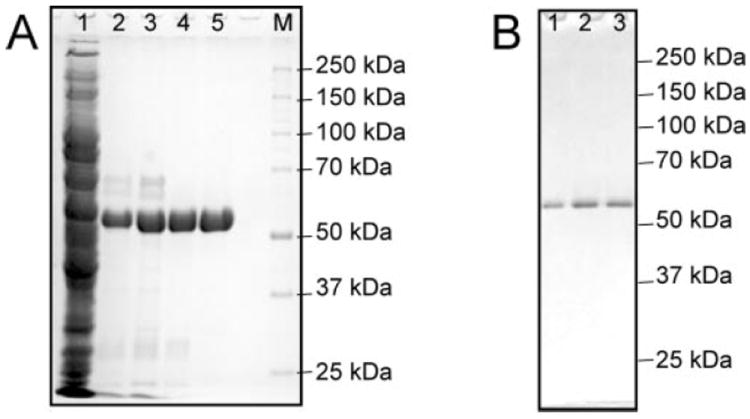
A, 10% polyacrylamide gel containing 1% sodium dodecyl sulfate is shown with the HCV helicase with the R293A at various stages of purification. Lane 1 contains 20 μg of the soluble fraction of the crude cell lysate. Lane 2 contains 10 μg of the protein after NiNTA chromatography. Lane 3 contains 7 μg of the protein following fractionation with ammonium sulfate. Lane 4 contains 5 μg of R393A following gel filtration chromatography, and lane 5 shows 5 μg of R393A after DEAE chromatography. Lane M contains proteins standards with the indicated molecular masses. B, SDS gel with 1 μg of F444A (lane 1), F438A (lane 2), and Hel-1a (lane 3) after each was expressed and purified as shown in panel A.
ATPase Assays
ATP hydrolysis fuels the action of the HCV helicase. Based on structural alignments with similar helicases that have been crystallized in the presence of ATP analogs (27), ATP likely binds and is hydrolyzed in the cleft between domains 1 and 2 of HCV helicase, far from the Arg-clamp and Phe-loop motifs. Thus, it is not surprising that the R393A and F438A substitutions have little effect on ATP hydrolysis (Fig. 3).
Fig. 3. ATP hydrolysis catalyzed by HCV helicase (Hel-1a) and site-directed mutants.
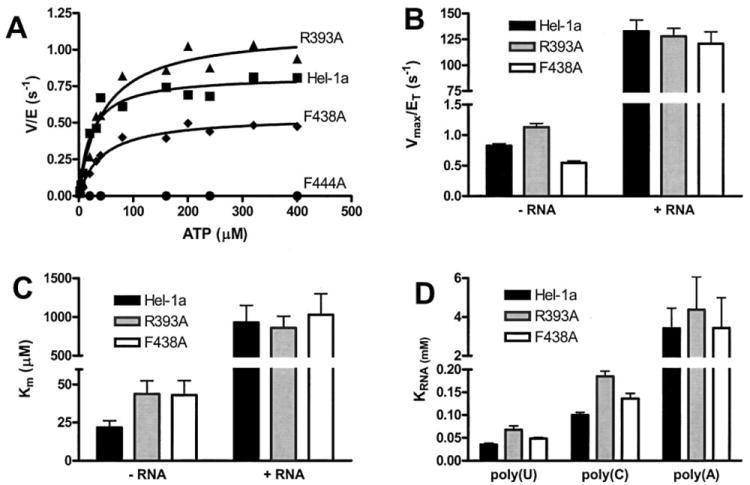
A, initial rates of ATP hydrolysis (v, nmol/s) divided by the amount of total enzyme in the reaction (E, nmol) plotted versus ATP concentration for wild type (squares), R393A (triangles), F438A (diamonds), and F444A (circles). Data are fit to the Michaelis-Menten equation to yield kinetic constants plotted in B and C. B, Vmax (nmol/s) divided by total enzyme (nmol), which is equivalent to the turnover rate constant kcat, for Hel-1a (black), R393A (gray), and F438A (white). Data were obtained from 12 reactions (performed in triplicate) at ATP concentrations ranging from 2 μm to 4 mm. Reactions were repeated in the presence and absence of 2 mg/ml poly(U) RNA. C, Km values for ATP hydrolysis obtained from the same reactions described in B. D, concentration of poly(U), poly(C), or poly(A) RNA necessary to support a half maximum rate of ATP hydrolysis (KRNA) catalyzed by Hel-1a (black), R393A (gray), or F438A (white). KRNA values were obtained by fitting rates obtained in the presence of 9 different RNA concentrations to the equation; v = Vmax [RNA]/KRNA+[RNA]. Error bars in B, C, and D show the S.E. in non-linear regression analyses.
On the other hand, the F444A protein retains no detectable ability to hydrolyze ATP under any condition (Fig. 3A). Furthermore, F444A did not exhibit any detectable helicase activity and did not bind DNA or RNA in any assays described below. Since insufficient amounts of F444A were available for biophysical analysis, the possibility that the F444A protein was not folded properly could not be ruled out. Therefore, because no general conclusions regarding the essential role of F444A can be firmly established, the negative data obtained with this protein are not shown in the assays below.
HCV helicase hydrolyzes ATP fairly rapidly even in the absence of DNA or RNA. In the absence of nucleic acids, Hel-1a hydrolyzes ATP with a turnover rate (kcat) of 0.82 ± 0.36 s−1, assuming the protein functions as a monomer and is 100% active. R393A hydrolyzes ATP faster at 1.1 ± 0.2 s−1, and F438A hydrolyzes somewhat more slowly at 0.54 ± 0.1 s−1. The confidence intervals suggest that these differences are not significant. In the presence of a saturating amount of poly(U) RNA, ATP hydrolysis by Hel-1a is stimulated 160 fold to 132 ± 10 s−1. R393A-catalyzed ATP hydrolysis is stimulated by RNA 115-fold to 127 ± 7 s−1, and the F438A catalyzed reaction is stimulated 222-fold to 120 ± 11 s−1. Again, these rate constants are not significantly different (Fig. 3B).
The Km values for ATP hydrolysis are likewise similar for reactions catalyzed by Hel-1a, R393A, and F439A (Fig. 3C). The only minor differences between the mutants and the wild type occur in the absence of RNA, where the Km with Hel-1a is 21 ± 4 μm, and the Km with R393A is 44 ± 9 μm and the Km with F438A is 43 ± 9 μm. In the presence of RNA, there are no Km differences: Hel-1a, 920 ± 200 μm; R393A, 860 ± 150 μm; F438A, 1000 ± 270 μm.
Although the Phe-loop and the Arg-clamp are far from the putative ATP binding site, interactions with nucleic acids could conceivably influence the activation of ATP hydrolysis by various nucleic acids. For this reason, rates of ATP hydrolysis were measured in the presence of various concentrations of different RNA homopolymers (average lengths 2500 nt), and the parameter KRNA was determined. KRNA is analogous to a Km and is defined as the concentration of RNA that leads to 50% maximum ATP hydrolysis. Fig. 3D shows that the nucleic acid stimulation profiles of the mutants are unchanged. Poly(U) stimulates each protein better (lower KRNA) than poly(C). Poly(A) stimulates each enzyme poorly, whereas poly(G) does not stimulate any of the helicases (not shown). Poly(U) RNA stimulates ATP hydrolysis by R393A with a KRNA (67 ± 8 μm) about 2-fold higher than Hel-1a (35 ± 3 μm). The poly(U)-stimulated KRNA seen with F438A, 48 ± 3 μm, is more similar to that seen with the wild type. Similar differences between Hel-1a, R393A, and F438A are seen when the enzymes are stimulated by poly(C) or poly(A) RNA (Fig. 3D). The experiments summarized in Fig. 3D were also performed using all the DNA and RNA oligonucleotides used below in the helicase and binding assays. No differences in the stimulation of ATP hydrolysis catalyzed by the wild type or mutant proteins were noted (data not shown).
In Fig. 3, ATP hydrolysis was measured under optimal conditions for RNA unwinding (MOPS buffer, pH 6.5). It should be noted that assays performed with the wild type enzyme at physiological pH yield somewhat different results (14). At pH 7.5 (Tris-Cl buffer), all values for KRNA are about 100-fold higher, although the enzyme retains the same nucleic acid stimulation profile (poly(U) > (poly(C) > (poly(A)). No additional differences between the mutants and wild type were noted when rates of ATP hydrolysis were measured at pH 7.5 or in other buffer systems (data not shown).
Unwinding Assays
Considering that both R393A and F438A interact with RNA (Fig. 3D) and hydrolyze ATP as rapidly as wild type (Fig. 3B), one might reasonably expect that the mutant proteins would also unwind DNA and RNA like the wild type. However, although many point mutants in HCV helicase affect both ATPase and helicase, some disrupt the helicase function but leave the ATPase intact (21-24). Rigorous analyses of mutations that uncouple ATPase from helicase have yielded valuable insights into the mechanism of helicase action (28).
Unwinding assays were based on those described by Pang et al. (5). RNA and DNA duplexes were made from two complementary oligonucleotides. The shorter oligonucleotide is labeled with 32P, and the longer strand forms a 3′- single stranded tail necessary for helicase action. The helicase and substrate were combined, and the unwinding reaction was initiated with ATP. After termination, unwound products were detected using non-denaturing polyacrylamide gel electrophoresis. No unwinding was observed in the absence of ATP, although such a reaction has been previously reported for HCV helicase (29). Also, when a boiled substrate was incubated under these conditions in the absence of protein, no re-annealing was observed.
When RNA is used as a helicase substrate, a minimum of 50 nm of Hel-1a is required to detect unwinding at 37 °C (Fig. 4A). The total amount of RNA unwound reaches a maximum between 100 and 250 nm enzyme. 500 nm Hel-1a unwinds RNA initially more rapidly but unwinds less total RNA possibly because of ATP depletion. Even under optimal conditions (250 nm enzyme) no RNA unwinding by either R393A or F438A can be detected (Fig. 4B). Likewise, neither mutant unwinds RNA when present in higher or lower concentrations (Fig 4C).
Fig. 4. RNA unwinding catalyzed by Hel-1a, R393A, and F438A.
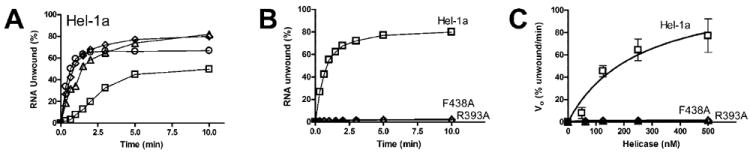
A, duplex [32P]RNA substrate (1 nm) was incubated with 50 nm (□), 125 nm (Δ), 250 nm (◇), 500 nm (○) of Hel-1a. Reactions were initiated by adding ATP, incubated at 37 °C, and terminated at various times. Percentage of unwound RNA was determined by separating reaction products on non-denaturing polyacrylamide gels. B, reactions containing 250 nm Hel-1a (▫) were repeated substituting either F438A (◇) or R393A (Δ). C, initial rates of RNA unwinding for reactions containing various amounts of Hel-1a (□), F438A (◇), or R393A (Δ). Data were fit to Equation 2 to yield the following constants for Hel-1a: Vmax = 124 ± 39, KHel = 274 ± 170.
As has been previously established, the HCV helicase unwinds DNA better than RNA (5, 30). DNA unwinding assays were therefore used to detect and analyze residual helicase activity in these mutants that lack RNA helicase activity. DNA unwinding assays were performed under the same conditions as the RNA unwinding assays except that the reactions were incubated at 23 °C. HCV helicase unwinds DNA so rapidly at 37 °C that rates could not be accurately measured. Under these conditions at 23 °C, both R393A and F438A retain some ability to unwind DNA (Fig. 5). Only 25 nm of the wild-type enzyme is necessary to detect DNA unwinding, which reaches a maximum at 100 nm enzyme (Fig. 5A). More than 50 nm of R393A is necessary to detect unwinding, and even in the presence of 200 nm R393A, more than half of the DNA remains annealed. A more linear response is seen in the presence of various amounts of F438A, but that protein also unwinds DNA more slowly than wild type (Fig. 5C). The initial rates (0–120 s) of DNA unwinding can be fit to protein concentration using Equation 2,
| (Eq. 2) |
for both Hel-1a and F438A (the data for R393A do not describe a hyperbola). Since protein exceeds substrate, the value, KHel, represents an apparent dissociation constant for the enzyme and DNA substrate. The Vmax obtained for F438A (19 ± 4) is about one-third that with Hel-1a (56 ± 20), and the KHel for F438A (151 ± 20) is 3-fold higher than Hel-1a (48 ± 20).
Fig. 5. DNA unwinding catalyzed by Hel-1a, R393A, and F438A.
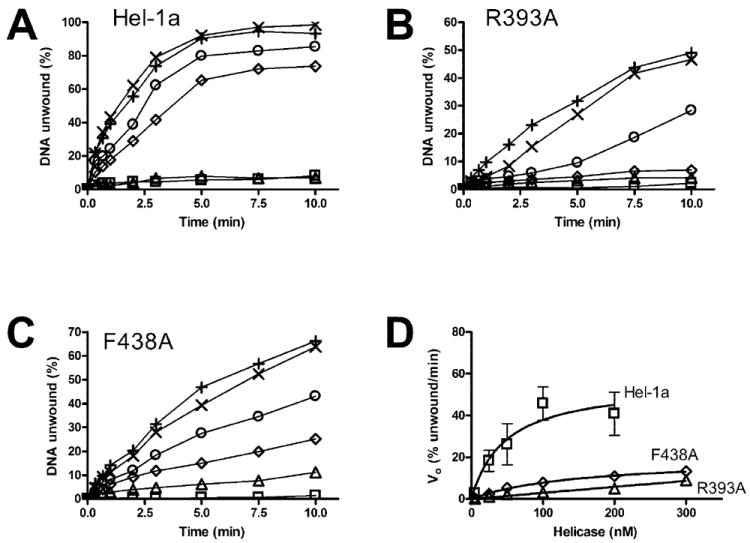
In all reactions, 1 nm of duplex [32P]DNA substrate was preincubated with indicated amounts of HCV helicase. Reactions were initiated with ATP, incubated at 23 °C for various times, terminated, and analyzed using native polyacrylamide gels. A, reactions containing Hel-1a at 1 nm (□), 5 nm (Δ), 25 nm (◇), 50 nm (○), 100 nm (×), or 200 nm (+). B, reactions containing R393A at 5 nm (□), 25 nm (Δ), 50 nm (◇), 100 nm (○), 200 nm (×), or 300 nm (+). C, reactions containing F438A at 5 nm (□), 25 nm (Δ), 50 nm (◇), 100 nm (○), 200 nm (×), or 300 nm (+). D, initial rates of DNA unwinding for reactions containing various amounts of Hel-1a (□), F438A (◇), or R393A (Δ). Data were fit to Equation 2.
The above analysis suggests that the mutant proteins have a lower affinity for the DNA duplex than the wild type. Although all three proteins are fully associated with the DNA before the reaction is initiated, as shown below in direct binding studies, the mutants might fall from the substrate before unwinding can be detected. To test this hypothesis, a DNA trap was added to the unwinding reactions when they were initiated. The trap should act to sequester helicase that falls from the substrate as the reaction proceeds. The trap used here consists of the shorter of the two oligonucleotides that form the duplex substrate. The reactions were initiated with ATP and various trap concentrations and contained 100 nm of either Hel-1a (Fig. 6A), R393A (Fig. 6B), or F438A (Fig. 6C). Although even the highest concentrations of trap only slightly inhibited the reaction catalyzed by the wild type, as little as 500 nm trap (5× the enzyme concentration), significantly affected the unwinding rates of the two mutants (Fig. 6D).
Fig. 6. Effect of a DNA trap on DNA unwinding catalyzed by Hel-1a, R393A, and F438A.
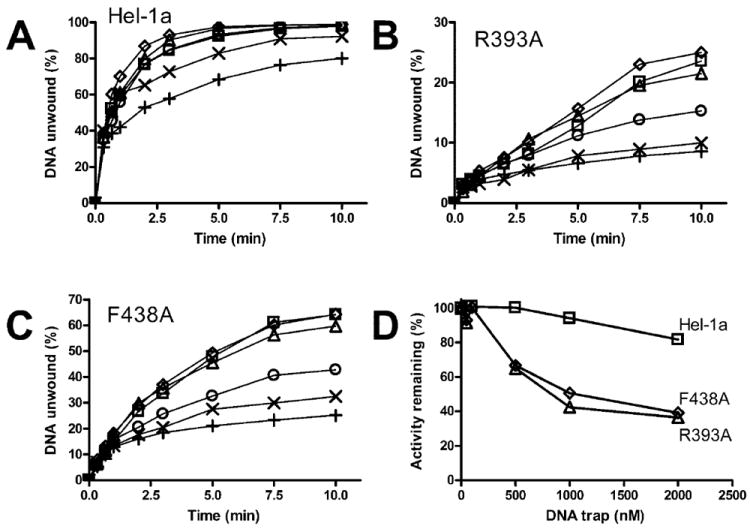
Unwinding assays were performed at 23 °C using a [32P]DNA substrate in which the shorter strand is labeled. Reactions were initiated by adding ATP and various amounts of the shorter DNA strand, lacking a radiolabel, which acts as an enzyme trap. Reactions contained no trap (□), 50 nm (Δ), 100 nm (◇), 200 nm (○), 1000 nm (×), or 2000 nm (+) trap oligonucleotide and 100 nm of either Hel-1a (A), R393A (B), or F438A (C). D, amount of DNA unwound after 10 min in the presence of trap was divided by the amount unwound in the presence of trap. This activity remaining is plotted against trap concentration for Hel-1a (□), R393A (Δ), and F428A (◇).
Translocation Assays
The mechanism by which a helicase unwinds a double helix is complex and still not completely understood. Most current models suggest that a helicase moves along one strand while actively displacing the complementary strand. These models are primarily based on the fact that most helicases require a single-stranded region in order to unwind a double helix, and it is widely assumed that this single-stranded region is required for helicase loading. The polarity of the required ssDNA tail is usually indicative of the directionality of helicase movement. The HCV helicase requires a 3′ ssDNA tail and such a tail is provided on the longer strand of the helicase substrate utilized above. The results of the unwinding assays suggest that the mutant helicases might be defective in their ability to move along DNA (or RNA) in a 3′-5′ direction. To more directly analyze the movement of the helicases on ssDNA, the translocation of each protein on ssDNA was measured using an assay developed by Raney and co-workers (17, 18). This assay measures helicase catalyzed displacement of streptavidin from a biotinylated oligonucleotide. An oligonucleotide with the same sequence as the longer strand used in the unwinding assay, which contains a biotin at its 5′-end annealed to streptavidin, is used here to investigate apparent movements of the HCV helicase on ssDNA.
Fig. 7 shows that both F438A and R393A translocate poorly along ssDNA. In these assays, biotinylated [32P]oligonucleotide bound to streptavidin is incubated with the HCV helicase. Reactions are initiated with ATP, terminated at various times, and analyzed on native polyacrylamide gels. In such gels, the streptavidin annealed substrates migrate more slowly than the displaced products. The wild type (Fig. 7A) more rapidly displaces streptavidin from the [32P]oligonucleotide than either R393A (Fig. 7B) or F438A (Fig. 7C). None of the proteins displace streptavidin bound to the same oligonucleotide with a biotin-labeled 3′-end, confirming the 3′-5′ directionality for the helicase.
Fig. 7. Displacement of streptavidin from a 5′-biotinylated oligonucleotide by Hel-1a, R393A, and F438A.
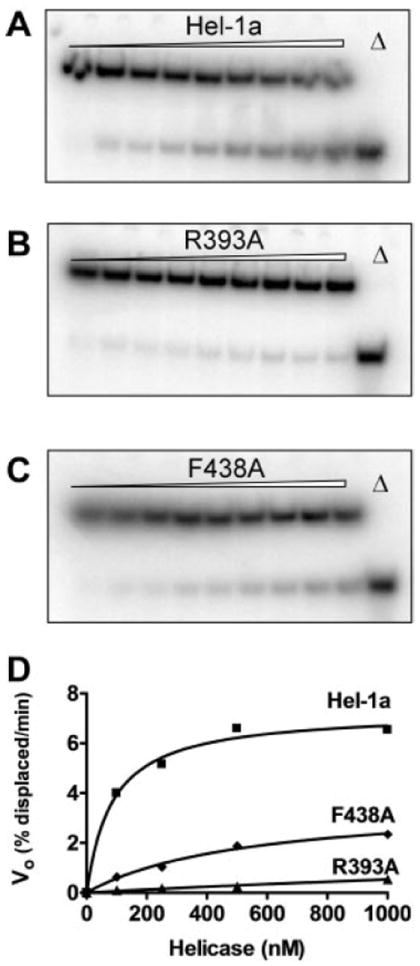
HCV helicase was incubated with a [32P]oligonucleotide with biotin at its 5′-end bound to streptavidin. When ATP is added to the reaction, streptavidin is displaced from the oligonucleotide leading to a shift in the electrophoretic mobility of the [32P]DNA. Autoradiographs of non-denaturing gels are shown of reactions containing 100 nm of Hel-1a (A) R393A (B) or F438A (C). Reactions were analyzed before ATP addition (lane 1) and after 1, 3, 6, 9, 12, 15, 20, and 30 min (lanes 2–9). Also shown are the products after boiling (Δ, lane 10). D, initial rates of streptavidin displacement that were measured in reactions containing various amounts of Hel-1a (squares), R393A (triangles), and F438A (diamonds). Data were fit to Equation 3.
When these translocation assays are performed at different helicase concentrations, it becomes apparent that the streptavidin displacement rates are dependent on protein concentration (Fig. 7D). R393A displaces streptavidin only at high enzyme concentrations (>500 nm). However, even at the highest concentration tested, R393A functions at less than one-tenth the rate of Hel-1a. For Hel-1a and F438A, initial rates of streptavidin displacement can be fit to the general rate Equation 3.
| (Eq. 3) |
In these assays, enzyme is in excess of substrate, and thus total enzyme approximately equals free enzyme. Such an analysis suggests that F438A interacts with the ssDNA with a Kapp (530 ± 120 nm) that is 6-fold higher than that seen with Hel-1a (85 ± 17 nm). It should be noted, however, that these apparent dissociation constants, like the KRNA and KHel values determined above, are not direct measurements of the affinity of the proteins for nucleic acids. They are instead complex functions dependent on translocation rates, dissociation rates, association rates, and processivity. The Vmax for the F438A catalyzed reaction (3 ± 1.3) is less than half of Hel-1a (7.2 ± 2).
Binding Assays
Because DNA binds HCV helicase near Trp501 (8), nucleic acid binding significantly affects intrinsic protein fluorescence. Preugschat et al. (31) first used intrinsic protein fluorescence quenching to measure the affinity of HCV helicase for nucleic acids (31). Their data were later confirmed using the same techniques by Levin and Patel (32), who subsequently showed that the affinity of HCV helicase for DNA decreases substantially in the presence of the non-hydrolysable ATP analog ADP[BeF3] (33). Thus, by monitoring changes in protein fluorescence upon titration with an oligonucleotide in the presence and absence of ADP[BeF3], one can determine the thermodynamic parameters describing the interaction of HCV helicase at different points in the reaction cycle. This was done with Hel-1a, R393A, and F438A.
Before examining DNA binding, an interaction between AD-P[BeF3] and the HCV helicase mutants was confirmed by examining the ability of ADP[BeF3] to inhibit ATP hydrolysis catalyzed by the mutants (Fig. 8A). BeF3 has been proposed to coordinate ADP in a position analogous to the γ-phosphate of ATP (34). The formation of this complex requires excess F−, which is supplied in the form of NaF. Although NaF and ADP alone inhibit ATP hydrolysis (35), the addition of beryllium fluoride leads to very potent inhibition (33). This effect is seen not only with the wild type, but also with R393A and F438A (Fig. 8A).
Fig. 8. Interactions between Hel-1a, R393A, and F438A and a DNA oligonucleotide in the presence and absence of ADP(BeF3).

A, inhibition of HCV helicase catalyzed ATP hydrolysis by a complex comprised of ADP, Be3+, and F−. Rates of ATP hydrolysis catalyzed by Hel-1a, R393A, or F438A were measured in the presence of poly(U) RNA as described in Fig. 3. Reactions contained no additional components (white), 0.1 mm ADP (black), 0.1 mm ADP, and 5 mm NaF (dark gray), or 0.1 mm ADP, 5 mm NaF, and 0.5 mm BeF2 (light gray). B, titration of 17 nm (squares), 34 nm (triangles), and 68 nm Hel-1a (diamonds) with an 18 nucleotide long polyuridylate DNA oligonucleotide. Fractional fluorescence was fit to Equation 1 to obtain a KD. C, titrations in B were repeated using Hel-1a, R393A, and F438A in the presence and absence of 0.1 mm ADP, 5 mm NaF, and 0.5 mm BeF2.
Fig. 8B shows a typical set of titrations that were performed using three amounts of Hel-1a. Because the enzyme concentration was comparable to the DNA concentration, the data were fit to an equation accounting for ligand depletion (Equation 1). This process was repeated for each enzyme in the presence and absence of ADP[BeF3]. The resulting KD values are compared in Fig. 8C. As shown in Fig. 8C, the apparent KD for the wild-type Hel-1a is 2.4 ± 0.6 nm. This value is similar to that previously reported using HCV helicase derived from different HCV isolates (31, 32). In the absence of ADP[BeF3], both mutants bind DNA somewhat weaker. The KD for F438A is 4.1 ± 1.1 and that for R393A is 5.3 ± 0.9 nm. In the presence of ADP[BeF3], HCV helicase releases ssDNA, and the KD obtained with the wild type increases 7-fold to 17 ± 2 nm. R393A also releases some of its grip on DNA and its KD increases 5-fold to 27 ± 3 nm. On the other hand the KD obtained with the Phe-loop mutant F438A does not increase significantly. The KD describing the interaction of F438A with ssDNA in the presence of ADP[BeF3] is 5.7 ± 1.3 nm. This value is similar to that obtained with F438 in the absence of ADP[BeF3] (4.1 ± 1.1 nm), although both values represent a significantly lower affinity for ssDNA than that seen with the wild type in the absence of the ATP analogue. Similar effects were seen when an RNA oligonucleotide was used instead of a DNA oligonucleotide (data not shown). However, as seen by others (32), RNA bound the enzyme about 5-fold weaker than DNA.
The dissociation constants determined for the HCV helicase and DNA in the presence of ADP(BeF3) in this study are lower than those reported by Levin et al. (33). Whereas in some cases Levin et al. found a 100-fold reduction in affinity, only about a 5-fold reduction was seen here. The simplest explanation for this difference is that Levin et al. used a recombinant HCV helicase with an N-terminal fusion peptide (derived from the T7 gene 10 protein) in addition to a C-terminal His tag (see Refs. 36 and 37). The proteins here contained only a C-terminal His tag. It is possible that N-terminal fusion peptides, and likewise the N-terminal NS3 protease domain, influence helicase activity in a manner that affects these dissociation constants.
The above dissociation constants indicate that under the conditions which the unwinding assays were performed, almost all protein is associated with the DNA substrates. Thus, both mutant proteins bind nucleic acids and hydrolyze ATP similar to the wild type. These observations suggest that the mutant proteins fall from the duplex DNA substrate before unwinding occurs or can be detected. In other words, the mutants appear less stably bound to nucleic acids. Electrophoretic mobility shift analyses (gel-shift assays) were used to examine the stability of the helicase-DNA complexes. In these assays, helicase and the substrate used to measure DNA unwinding were incubated in the absence of ATP, and the complex was separated using non-denaturing polyacrylamide gel electrophoresis. The protein remaining bound during electrophoresis caused the DNA to be shifted. As seen in Fig. 9, Hel-1a, R393A, and F438A all caused a gel-shift, but dramatically different amounts of the three proteins were necessary to shift the DNA. Whereas 60 nm of Hel-1a was required to shift 50% of the DNA, 200 nm of F438A and 800 nm of R393A were necessary to achieve the same effect.
Fig. 9. DNA binding by Hel-1a, R393A, and F438A as measured by gel-shift analysis.
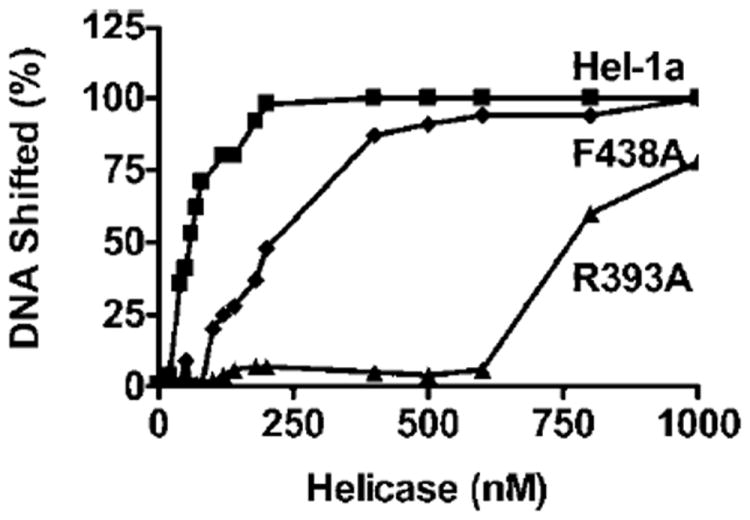
The [32P]DNA duplex used for unwinding assays was incubated with various concentrations of the helicases. Free and bound DNA was separated using non-denaturing polyacrylamide gel electrophoresis. Fraction of DNA shifted at each protein concentration was determined using phosphorimage analysis for Hel-1a (squares), R393A (triangles), and F438A (diamonds).
DISCUSSION
We have demonstrated that two sequence motifs in domain 2 of the HCV helicase are critical for DNA/RNA binding and strand separation. These regions, designated here as the Arg-clamp and Phe-loop, are conserved among all known HCV genotypes and quasispecies (Fig. 1A). Unlike other SF2 motifs, the Arg-clamp and Phe-loop are not shared with other protein families in SF2 (Fig. 1B) and thus may represent better targets for rational drug design. The data support a mechanism for the HCV helicase outlined in Fig. 10 that is based on the rigid domain rotation model proposed by Yao et al. (6), the ratcheting inchworm model proposed by Kim et al. (8) and the Brownian motor mechanism proposed by Levin et al. (33).
Fig. 10. Structure and function of the Arg-clamp and the Phe-loop in the HCV helicase.
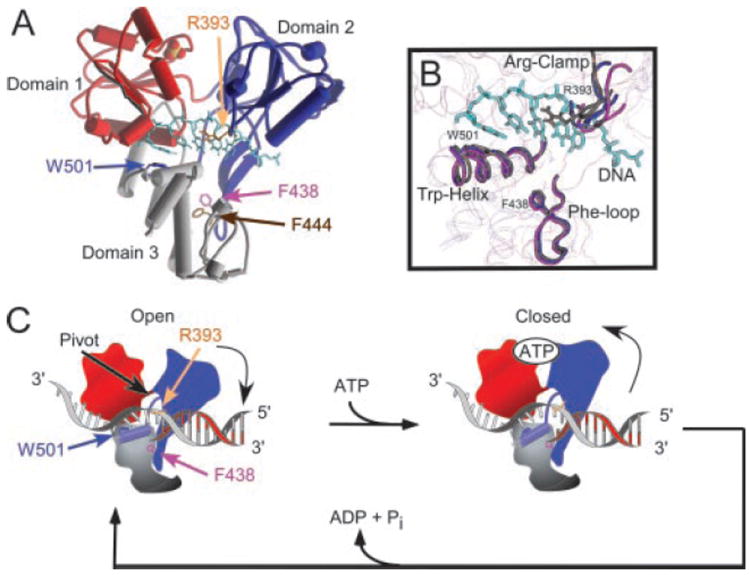
A, structure of HCV helicase bound to DNA as determined by Kim et al. (PDB 1A1V, Ref. 8). Domain 1 is colored red, domain 2 is blue, and domain 3 is gray. The bound DNA oligonucleotide is shown as light blue sticks and the orange/red spheres (sulfate ion) marks the putative ATP binding site. Key amino acid residues are also shown as sticks: Arg393 (orange), Phe438 (purple), Phe444 (brown), and Trp501 (blue). B, structural alignment of three HCV helicase structures from PDB 1HEI (6) (chain A (blue) and chain B (purple)) and PDB 1A1V (8) (brown). The Arg-clamp, Trp-helix, and Phe-loop are highlighted as tubes. C, cartoon depicting DNA unwinding by HCV helicase. In the absence of ATP, the helicase is tightly bound to DNA in an open conformation. Trp501 is intercalated between the bases like a bookend. Upon ATP binding, the cleft between domains 1 and 2 closes leading to a rotation of domain 2. The rotation pulls the Arg-clamp, which remains associated with one strand of double helix and separates that strand from the complementary strand. The Phe-loop remains fixed and provides a pivot point for the rotation of the Trp-helix. Movements of the bookend allow the protein to slide on DNA until ATP is hydrolyzed, and the protein returns to the closed conformation.
Fig. 10A shows the locations of the Arg-clamp and the Phe-loop in the structure of HCV helicase with a bound DNA oligonucleotide (determined by Kim et al., Ref. 8). In Fig. 10A, domain 1 is colored red, domain 2 is blue, and domain 3 is gray. Also shown is a sulfate ion that binds domain 1 at a location spatially equivalent to where the β-phosphate of ATP binds similar helicases (38, 39). The bound DNA oligonucleotide is represented in Fig. 10A as light blue sticks. The side chains of Arg393 (orange), Phe438 (purple), Phe444 (brown), and Trp501 (blue) are also shown as sticks in Fig. 10A. Arg393 is less than 2.5 Å from the phosphate backbone of the DNA. Phe438 and Phe444, on the other hand, are about 15 Å from the bound oligonucleotide. Since Trp501 was first observed to intercalate between the DNA bases like a bookend (8), several subsequent studies have confirmed its critical role in the unwinding reaction (22-25).
It was previously unclear whether the DNA bound in the structure shown in Fig. 10A represents the strand on which the helicase is translocating in a 3′ to 5′ direction or the complementary strand. The data obtained here demonstrate that the interaction between Arg393 and the strand, which is seen in the structure, is critical for binding (Figs. 8 and 9) unwinding (Figs. 4-6) and translocation on ssDNA (Fig. 7). Combined, these findings imply that the DNA (Fig. 10A) is the strand on which the helicase is translocating (the longer strand in the helicase substrates). Such an arrangement is depicted in Fig. 10C.
The structure in Fig. 10A provides a snapshot along the helicase reaction pathway. It likely represents a ground state where DNA is tightly bound to the enzyme. To function as a molecular motor, HCV helicase must exit this ground state and temporarily release DNA. The binding and hydrolysis of ATP provides this energy. Levin et al. (33) recently showed using the non-hydrolyzable ATP analog ADP(BeF3) that when HCV helicase binds ATP, its affinity for ssDNA decreases. While ATP is bound, the protein can slide along DNA until hydrolysis occurs and the protein again clamps more tightly in a new position. Kim et al. (8) proposed that a unidirectional movement along ssDNA (or RNA) results from the closing of the cleft between domains 1 and 2 upon DNA binding. Unidirectional translocation occurs because the DNA slides through one end of the protein when the cleft opens and the other end when the cleft closes. When the cleft is open, Trp501 acts like a bookend on the 3′-end of the ssDNA preventing DNA from sliding out of that end of the protein.
Closure of the cleft between domains 1 and 2 of HCV helicase upon ATP binding has not yet been directly visualized. Such conformational changes have, however, been seen in the superfamily 1 helicases Rep (39) and PcrA (38), which are structurally similar to the HCV helicase (27). What might be the molecular impact of an ATP-binding induced cleft closure in HCV helicase? Yao et al. (6) have suggested that since domain 1 and 3 share a more extensive interface, domain 2 could pivot relative to the rest of the protein along flexible linkers. As evidence for such a motion, Yao et al. compared two crystallographically independent HCV helicase structures to show significant variability in the position of domain 2 (6). A similar analysis is presented in Fig. 10B, where the two structures reported by Yao et al. (6) (Protein Data Bank 1HEI chains A and B) are compared with the structure reported by Kim et al. (8) (Protein Data Bank 1A1V). The three structures, which utilize HCV helicase derived from the same viral genotype, were aligned based on structural similarities in domain 1 using NCBI VAST. Fig. 10B highlights variations in the relative positions of the Arg-clamp, the Phe-loop and the helix flanking Trp501, here called the Trp-helix. The brown structure is the one containing the bound oligonucleotide and the purple and blue structures lack a bound ligand. Arg393, Phe438, and Trp501 are displayed as sticks in Fig. 10B. It is clear that the flexibility of domain 2 allows for relatively large movements of the Arg-clamp, whereas the position of the Phe-loop only slightly varies. The position of Trp501 also varies, and interestingly, Trp501 is furthest from the oligonucleotide in the conformation (purple) where Arg393 is also furthest from its position when DNA is bound (brown).
A scheme depicting how conformational changes could lead to duplex unwinding is presented in Fig. 10C. ATP binding leads to domain closure and a rotation of domain 2. The rotation is accomplished by pivoting around the linkage between domains 1 and 2 and simultaneously altering the twist of the anti-parallel β-sheets connecting the Phe-loop to the rest of domain 2, as proposed by Yao et al. (6). Because one end of the Trp-helix is connected to the C terminus of domain 2, a domain 2 rotation would lead to movements of the Trp-helix. A slight movement of Trp501 would remove the bookend and allow DNA to slip past Trp501, as suggested by Kim et al. (8).
The properties of R393A demonstrate that Arg393 clamps ssDNA (or RNA) into the known ssDNA binding site both in the presence and absence of ATP. The model in Fig. 10C proposes that these contacts allow the enzyme to actively disrupt the double helix. Contacts between Arg393 and the backbone of DNA occur both when ATP is absent (open conformation) and present (closed conformation). In the absence of ATP, Hel-1a tightly binds leading to a change in free energy of −49 kJ/mol. R393A binds more weakly with a smaller ΔG of −47 kJ/mol. In the presence of a non-hydrolyzable ATP analog, DNA binds more weakly to both enzymes. The ΔG for binding of the wild type to DNA in the presence of ADP[BeF3] is −44 kJ/mol, whereas that for R393A is −42 kJ/mol. Under both conditions R393 contributes equally to binding (about 2 kJ/mol). The gel-shift experiments (Fig. 9) reveal that these subtle changes in binding energy cause R393A to bind DNA more than 10-fold less stably. Thus, although DNA and RNA still bind R393A, and ATP hydrolysis is stimulated, the mutant enzyme probably falls from the nucleic acid during translocation before unwinding can be detected.
The model in Fig. 10C implies that interactions between Arg393 and DNA in the presence and absence of ATP lead to the active disruption of the double helix. If ATP binding leads to a domain closure, and rotation of domain 2, then one would expect that Arg393 would move away from the DNA backbone. If such a rotation removes Arg393 from contacts with DNA, then there would be no difference between the affinities of Hel-1a and R393A for DNA in the presence of ADP[BeF3]. Because there is such a difference, Arg393 likely interacts with DNA when the protein is both in the open and in the closed conformation. If the DNA remains associated with Arg393, as suggested by the equilibrium binding data, then this would lead to a twisting and disruption of the double helix. Such a disruption would occur much less frequently in the R393A mutant that lacks the appropriate contacts between domain 2 and the nucleic acid backbone.
The F438A substitution also uncouples ATP hydrolysis from unwinding activity but its role in the reaction cycle is more difficult to interpret. The F438A protein retains an ATPase activity indistinguishable from the wild-type protein (Fig. 3), but translocates poorly (Fig. 7) and this leads to reduced DNA unwinding (Figs. 5 and 6) and no RNA unwinding (Fig. 4). This poor translocation correlates with weak DNA binding (Fig. 9) and the curious fact that F438A binds DNA more tightly than wild type in the presence of ADP(BeF3) (Fig. 8). We propose that the characteristics of the F438A mutant result from the fact that the hydrophobic pocket that includes Phe438 provides a pivot point for domain 2 rotations. Phe438 contacts Phe444 and the domain 3 residues His528, Phe531, Trp532, and Phe536. The importance of these contacts in protein folding is clearly evidenced by the complete inactivity of the F444A mutant. If some slippage occurs at this pivot point when domain 2 rotates, then the Trp-helix will remain tightly associated with ssDNA. This model would explain the increased affinity of F438A for ssDNA in the presence of ADP(BeF3). There also may be direct interactions between Phe438 and the Trp-helix because Phe438 is only 4.8 Å from the end of the Trp501 helix (residue Ser488).
Besides Arg393, Phe438, and Phe444, there are clearly other key residues in the Arg-clamp and Phe-loop that deserve further study. For example, Arg393 is next to a conserved tyrosine (Tyr392) that was proposed to play a similar role as Trp501 (23). However, substitution of Tyr392 with Ala leads to a protein that retains some ability to unwind RNA, unlike R393A (23). The Phe-loop is a stretch of 8 invariant amino acids. Of particular interest in the Phe-loop is Asp441, which might provide the major grip on domain 3 by forming hydrogen bonds with Arg601.
HCV and its viral relatives have evolved a very efficient helicase capable of unwinding both RNA and DNA. Besides providing insights into the mechanism of action of this enzyme, this study highlights two key differences between the HCV helicase and cellular proteins that were likely derived from a single common ancestor (Fig. 1B). The Phe-loop and its connecting β-sheets are absent from cellular DEAD-box proteins and residues that could act in a manner similar to the Arg-clamp are in a somewhat different position. It is noteworthy that HCV helicase and cellular DEAD-box proteins also have numerous different properties. For example eIF4A unwinds RNA in a 3′-5′ or a 5′-3′ direction and does not unwind a DNA double helix (40). Whether or not residues in the Arg-clamp and the Phe-loop account for these differences is unclear, and this question warrants further investigation.
Acknowledgments
We thank Fred Jaffe, Ruth Gallagher, and Ryan Rypma for valuable discussions and technical assistance.
Footnotes
This work was supported by the AASLD Liver Scholar Award from the American Liver Foundation and National Institutes of Health Grant AI052395. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: HCV, hepatitis C virus; NS3, nonstructural protein 3; NS5B, nonstructural protein 5B; eIF-4a, eukaryotic translation initiation factor 4a; ss, single-stranded; VAST, Vector Alignment Search Tool; NCBI, National Center for Biotechnology Information; PDB, Protein Data Bank; MOPS, 4-morpholinepropanesulfonic acid.
References
- 1.Alter MJ, Kruszon-Moran D, Nainan OV, McQuillan GM, Gao F, Moyer LA, Kaslow RA, Margolis HS. N Engl J Med. 1999;341:556–562. doi: 10.1056/NEJM199908193410802. [DOI] [PubMed] [Google Scholar]
- 2.Frick DN. Curr Org Chem. 2003;7 in press. [Google Scholar]
- 3.Leveque VJ, Wang QM. Cell Mol Life Sci. 2002;59:909–919. doi: 10.1007/s00018-002-8478-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narjes F, Koch U, Steinkuhler C. Exp Opin Investig Drugs. 2003;12:153–163. doi: 10.1517/13543784.12.2.153. [DOI] [PubMed] [Google Scholar]
- 5.Pang PS, Jankowsky E, Planet PJ, Pyle AM. EMBO J. 2002;21:1168–1176. doi: 10.1093/emboj/21.5.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao N, Hesson T, Cable M, Hong Z, Kwong AD, Le HV, Weber PC. Nat Struct Biol. 1997;4:463–467. doi: 10.1038/nsb0697-463. [DOI] [PubMed] [Google Scholar]
- 7.Cho HS, Ha NC, Kang LW, Chung KM, Back SH, Jang SK, Oh BH. J Biol Chem. 1998;273:15045–15052. doi: 10.1074/jbc.273.24.15045. [DOI] [PubMed] [Google Scholar]
- 8.Kim JL, Morgenstern KA, Griffith JP, Dwyer MD, Thomson JA, Murcko MA, Lin C, Caron PR. Structure. 1998;6:89–100. doi: 10.1016/s0969-2126(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 9.Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. Structure Fold Des. 1999;7:1353–1363. doi: 10.1016/s0969-2126(00)80025-8. [DOI] [PubMed] [Google Scholar]
- 10.Liu D, Wang YS, Gesell JJ, Wyss DF. J Mol Biol. 2001;314:543–561. doi: 10.1006/jmbi.2001.5146. [DOI] [PubMed] [Google Scholar]
- 11.Gesell JJ, Liu D, Madison VS, Hesson T, Wang YS, Weber PC, Wyss DF. Protein Eng. 2001;14:573–582. doi: 10.1093/protein/14.8.573. [DOI] [PubMed] [Google Scholar]
- 12.Gorbalenya AE, Koonin EV. Curr Opin Struct Biol. 1993;3:419–429. [Google Scholar]
- 13.Kwong AD, Kim JL, Lin C. Curr Top Microbiol Immunol. 2000;242:171–196. doi: 10.1007/978-3-642-59605-6_9. [DOI] [PubMed] [Google Scholar]
- 14.Lam AMI, Keeney D, Eckert PQ, Frick DN. J Virol. 2003;77:3950–3961. doi: 10.1128/JVI.77.7.3950-3961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yanagi M, Purcell RH, Emerson SU, Bukh J. Proc Natl Acad Sci U S A. 1997;94:8738–8743. doi: 10.1073/pnas.94.16.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dubendorff JW, Studier FW. J Mol Biol. 1991;219:45–59. doi: 10.1016/0022-2836(91)90856-2. [DOI] [PubMed] [Google Scholar]
- 17.Morris PD, Raney KD. Biochemistry. 1999;38:5164–5171. doi: 10.1021/bi9822269. [DOI] [PubMed] [Google Scholar]
- 18.Morris PD, Byrd AK, Tackett AJ, Cameron CE, Tanega P, Ott R, Fanning E, Raney KD. Biochemistry. 2002;41:2372–2378. doi: 10.1021/bi012058b. [DOI] [PubMed] [Google Scholar]
- 19.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chamberlain RW, Adams N, Saeed AA, Simmonds P, Elliott RM. J Gen Virol. 1997;78:1341–1347. doi: 10.1099/0022-1317-78-6-1341. [DOI] [PubMed] [Google Scholar]
- 21.Lin C, Kim JL. J Virol. 1999;73:8798–8807. doi: 10.1128/jvi.73.10.8798-8807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Preugschat F, Danger DP, Carter LH, 3rd, Davis RG, Porter DJ. Biochemistry. 2000;39:5174–5183. doi: 10.1021/bi9923860. [DOI] [PubMed] [Google Scholar]
- 23.Paolini C, Lahm A, De Francesco R, Gallinari P. J Gen Virol. 2000;81:1649–1658. doi: 10.1099/0022-1317-81-7-1649. [DOI] [PubMed] [Google Scholar]
- 24.Tai CL, Pan WC, Liaw SH, Yang UC, Hwang LH, Chen DS. J Virol. 2001;75:8289–8297. doi: 10.1128/JVI.75.17.8289-8297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JW, Seo MY, Shelat A, Kim CS, Kwon TW, Lu HH, Moustakas DT, Sun J, Han JH. J Virol. 2003;77:571–582. doi: 10.1128/JVI.77.1.571-582.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caruthers JM, Johnson ER, McKay DB. Proc Natl Acad Sci U S A. 2000;97:13080–13085. doi: 10.1073/pnas.97.24.13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korolev S, Yao N, Lohman TM, Weber PC, Waksman G. Protein Sci. 1998;7:605–610. doi: 10.1002/pro.5560070309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soultanas P, Dillingham MS, Wiley P, Webb MR, Wigley DB. EMBO J. 2000;19:3799–3810. doi: 10.1093/emboj/19.14.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porter DJ, Preugschat F. Biochemistry. 2000;39:5166–5173. doi: 10.1021/bi992384f. [DOI] [PubMed] [Google Scholar]
- 30.Gwack Y, Kim DW, Han JH, Choe J. Eur J Biochem. 1997;250:47–54. doi: 10.1111/j.1432-1033.1997.00047.x. [DOI] [PubMed] [Google Scholar]
- 31.Preugschat F, Averett DR, Clarke BE, Porter DJT. J Biol Chem. 1996;271:24449–24457. doi: 10.1074/jbc.271.40.24449. [DOI] [PubMed] [Google Scholar]
- 32.Levin MK, Patel SS. J Biol Chem. 2002;277:29377–29385. doi: 10.1074/jbc.M112315200. [DOI] [PubMed] [Google Scholar]
- 33.Levin MK, Gurjar MM, Patel SS. J Biol Chem. 2003;278:23311–23316. doi: 10.1074/jbc.M301283200. [DOI] [PubMed] [Google Scholar]
- 34.Ponomarev MA, Timofeev VP, Levitsky DI. FEBS Lett. 1995;371:261–263. doi: 10.1016/0014-5793(95)00898-j. [DOI] [PubMed] [Google Scholar]
- 35.Porter DJ. J Biol Chem. 1998;273:7390–7396. doi: 10.1074/jbc.273.13.7390. [DOI] [PubMed] [Google Scholar]
- 36.Kim DW, Gwack Y, Han JH, Choe J. Biochem Biophys Res Commun. 1995;215:160–166. doi: 10.1006/bbrc.1995.2447. [DOI] [PubMed] [Google Scholar]
- 37.Levin MK, Patel SS. J Biol Chem. 1999;274:31839–31846. doi: 10.1074/jbc.274.45.31839. [DOI] [PubMed] [Google Scholar]
- 38.Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 39.Korolev S, Hsieh J, Gauss GH, Lohman TM, Waksman G. Cell. 1997;90:635–647. doi: 10.1016/s0092-8674(00)80525-5. [DOI] [PubMed] [Google Scholar]
- 40.Du MX, Johnson RB, Sun XL, Staschke KA, Colacino J, Wang QM. Biochem J. 2002;363:147–155. doi: 10.1042/0264-6021:3630147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibrat JF, Madej T, Bryant SH. Curr Opin Struct Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]