

Abstract
Nocardicin A is a monocyclic β-lactam isolated from the actinomycete Nocardia uniformis that shows moderate antibiotic activity against a broad spectrum of Gram-negative bacteria. The monobactams are of renewed interest due to emerging Gram-negative strains resistant to clinically available penicillins and cephalosporins. Like isopenicillin N, nocardicin A has a tripeptide core of nonribosomal origin. Paradoxically, the nocardicin A gene cluster encodes two nonribosomal peptide synthetases (NRPSs), NocA and NocB, predicted to encode five modules pointing to a pentapeptide precursor in nocardicin A biosynthesis, unless module skipping or other non-linear reactions are occurring. Previous radiochemical incorporation experiments and bioinformatic analyses predict the incorporation of p-hydroxy-L-phenylglycine (L-pHPG) into positions 1, 3, and 5 and L-serine into position 4. No prediction could be made for position 2. Multi-domain constructs of each module were heterologous expressed in Escherichia coli for determination of the adenylation domain (A-domain) substrate specificity using the ATP/PPi exchange assay. Three of the five A-domains, from modules 1, 2, and 4, required the addition of stoichiometric amounts of MbtH family protein NocI to detect exchange activity. Based on these analyses, the predicted product of the NocA+NocB NRPSs is L-pHPG–L-Arg–D-pHPG–L-Ser–L-pHPG, a pentapeptide. Despite being flanked by nonproteinogenic amino acids, proteolysis of this pentapeptide by trypsin yields two fragments from cleavage at the C-terminus of the L-Arg residue. Thus, a proteolytic step is likely involved in the biosynthesis of nocardicin A, a rare but precedented editing event in the formation of nonribosomal natural products which is supported by the identification of trypsin-encoding genes in N. uniformis.
Introduction
The peptide core of many bioactive natural products, such as the antibiotics penicillin, vancomycin, and daptomycin, is biosynthesized by large modular proteins known as nonribosomal peptide synthetases (NRPS).1 Interactions between NRPSs and other proteins are essential for overall catalytic function. NRPS proteins must interact with 4′-phosphopantethienyl transferases (PPTase) that catalyze the transfer of a phosphopanethienyl side chain from coenzyme A to the active site serine residue of each peptidyl carrier or thiolation (T) domain of the NRPS, converting the apo-NRPS to its holo-form.1,2 In addition, there are increasing numbers of examples of associated biosynthetic proteins known to catalyze reactions, typically oxidations and halogenations, on T-domain tethered substrates.3,4 At the N- and C-termini of paired NRPS proteins, “COM domains”, consisting of α-helical recognition units, are points of mutual interaction linking the product assembly reactions on each.5 The importance of NRPS interaction with one or more auxillary proteins for optimal activity is particularly well exemplified by recent discoveries that identify the crucial role of the MbtH family of proteins. The MbtH family comprise a group of relatively small proteins (~8–9 KDa) often found embedded in biosynthetic clusters for nonribosomal peptide-derived secondary metabolites. They are collectively named for the protein identified in the mycobactin biosynthetic cluster of Mycobacterium tuberculosis.6 In vivo studies of Streptomyces coelicolor A3(2) concluded that MbtH family proteins, CchK associated with the coelichelin gene cluster and CdaX associated with the calcium-dependent antibiotic (CDA) gene cluster, are indispensable for production of these secondary metabolites.7 Another study demonstrated that heterologous expression of clorobiocin in S. coelicolor M512 was barely detectable upon the removal of all MbtH protein coding regions but complementation with mbtH genes cchK, cdaX, cloY, or couY (from the related coumermycin gene cluster) substantially restored production.7,8
Despite the prevalence of MbtH encoding genes in NRPS-containing gene clusters and in vivo studies demonstrating their importance and structure determinations,9,10 the role of these proteins in nonribosomal peptide biosynthesis remained unclear until the ATP/PPi exchange activity of the NRPS protein VbsS was found to be dependent on its co-expression with VbsG, an MbtH protein encoded next to the NRPS in the vicibactin gene cluster.11 A minimal NRPS module is three domains – an adenylation (A) domain, a thiolation (T) domain, and a condensation (C) domain. A-domains activate their cognate substrate, typically an L-α-amino acid, by catalyzing the adenylation of the carboxylate moiety by reaction with ATP. This reaction is usually reversible and can be monitored by an ATP/PPi exchange assay. The activated substrate reacts with the terminal thiol group of the pantethienyl side chain of the T-domain, forming a thioester. C-domains catalyze peptide bond formation between the free amino group of the upstream T-domain tethered aminoacylthioester with the downstream T-domain thioester. NRPSs usually terminate with a thioesterase (TE) domain, which catalyzes the release of the nascent peptide chain by hydrolysis or macrocyclization.
Several bioinformatic algorithms have been developed for substrate prediction of A-domains. The first algorithms were based on the crystal structure of PheA, the L-phenylalanine activating domain of the gramicidin NRPSs.12 Structure studies of PheA show the substrate binding pocket and ATP binding region at the interface of the N-terminal and C-terminal domains of the enzyme. A-domain substrate prediction algorithms were derived by correlating the residues directly lining the substrate binding pocket to its cognate substrate.13,14 More recent A-domain prediction algorithms have used transductive support vector machines (TSVMs) or hidden Markov Model (HMM) methodologies to improve prediction capabilities.15–17
The activation of ATP/PPi exchange activity in VbsS by VbsG suggests the MbtH protein interacts with the A domain of VbsS. The contribution of MbtH homologues VioN, CmnN, and PacJ to the activity of NRPSs involved in the biosynthesis of viomycin, capreomycin and pacidamycin, respectively, were subsequently reported.18,19 CmnN and VioN were found to be necessary to activate the ATP/PPi exchange reactions in the β-lysine activating modules of CmnO and VioO and modules 1 and 2 from CmnA, but are not required for ATP/PPi exchange in the A-domains of NRPSs CmnF and CmnG. PacJ is required for activation of ATP/PPi exchange in PacL. The mechanism for A-domain activation by MbtH proteins remains unclear. Kinetic measurements of the NovH tyrosine activating domain in novobiocin biosynthesis indicated that the Km and the turnover number measured for NovH were dissimilar when paired with different but complementary MbtH proteins CloY and YbdZ.20
Nocardicin A is the most potent antibiotic of the monobactam nocardicins isolated from N. uniformis subsp. tsuyamanensis ATCC 21806, with activity against gram-negative bacteria. Nocardicin A has also been isolated from other actinomycetes, including Actinosynnema mirum.21 The genus Actinosynnema is characterized by the formation of synnemata from the substrate mycelium, at the tip of which, zoospores are produced.22 Classical morphological comparison of N. uniformis ATCC 21806 to A. mirum strain NR 0364 revealed a high degree of similarity in the formation of synnemata and zoospores suggesting that it should be reclassified into the Actinosynnema genus.21
While both A. mirum and N. uniformis produce nocardicin A and have nearly identical gene clusters, the biosynthetic pathway has only been studied in N. uniformis. The structures of all the nocardicins isolated from N. uniformis contain a tripeptide core known to be derived from two units of p-hydroxy-L-phenylglycine (L-pHPG) and one unit of L-Ser.23 While a three module NPRS would be expected in the nocardicin A gene cluster, two NRPSs, NocA and NocB, together containing five modules were found.24 Recent in vivo mutagenesis experiments indicate that each of these five modules is essential, demonstrating that module skipping is not likely occurring and implying the formation of pentapeptide precursor in nocardicin A biosynthesis by the conventional linear paradigm.24 Determination of each A-domain substrate specificity would provide the identity of this putative pentapeptide and may suggest mechanisms by which subsequent truncation occurs to form the tripeptide backbone of the nocardicins. Bioinformatic algorithms have predicted L-pHPG to be the preferred substrate for A1, A3, and A5 and L-Ser to be the preferred substrate for A4.13 The substrate specificity for A2 has been more problematic with low confidence predictions ranging from L-ornithine or L-δ-N-hydroxyornithine,24 “a large amino acid such as L-Orn, L-Lys or L-Arg”, a hydrophilic amino acid, L-Asp, L-Asn, L-Glu, or L-Gln, or L-Arg.15,25 The inability to obtain clear experimental data for A2 has prevented identification of the presumed pentapeptide and left unknown whether the tripeptide backbone of nocardicin A originates from modules 1–3 or 3–5.
Among the proteins encoded by the nocardicin A biosynthetic cluster, NocI, a small 74 amino acid protein, shows clear homology to the MbtH family of proteins, and 2.3 kbp upstream of the cluster, a 73 amino acid paralog encoded by nocP is found. In this study, we report MbtH protein NocI is required for ATP/PPi exchange to be observed in three of the five A-domains of NocA and NocB. Surprisingly, the MbtH protein encoded just upstream of the cluster, NocP, was found to only partially complement NocI. The A-domain•NocI/NocP protein interaction was further characterized by co-expression studies. HPLC and binding analyses of dependent modules co-expressed with NocI indicate a 1:1 stoichiometry. Determining the function of NocI was requisite to experimentally demonstrating the identities of the amino acids recognized and activated by each module of NocA and NocB. As a result, a pentapeptide precursor can now be proposed for nocardicin A biosynthesis, as well as possible mechanisms for proteolysis to a tripeptide later in the biosynthetic pathway.
Results
Adenylation Domain Specificities of NocA and NocB
Heterologous expression of a multidomain construct from each module of NocA and NocB in Escherichia coli (E. coli) was pursued so that the substrate specificity could be experimentally determined for each A-domain using the standard ATP/PPi exchange assay. For all modules, except module 2, each construct was cloned into pET28b(+) for expression of C-terminal His6 fusion proteins in E. coli BL21(DE3) Rosetta 2 cells and purification by NTA affinity chromatography and Q-Sepharose ion exchange chromatography. Yields typically ranged from 0.5–1 mg protein/L culture. Because this expression protocol failed for several constructs of module 2, the A2T2-His6 coding sequence was cloned into pMALc2x for expression of an N-terminal maltose binding protein (MBP) fusion protein. Heterologous expression of MBP-A2T2-His6 in E. coli BL21(DE3) Rosetta 2 and isolation by NTA affinity chromatography yielded 2–3 mg protein/L culture. Protein products were confirmed by MALDI analysis of trypsin digests.
Bioinformatic analysis of the A-domains of modules 1, 3 and 5 predicted L-pHPG to be their preferred substrate although protein sequence alignment of these three A-domains highlights a long linker region between motifs 2 and 3 in the A1 domain that is not found in the A3 or A5 domains.24 The ATP/PPi exchange assay results for A1T1-His6, A3T3 E3C4-His6 and C5A5T5TE-His6 are shown in Figure 1A. Very strong ATP/PPi exchange was observed in the presence of L-pHPG for these three modules with a marked lack of activity for any other amino acid substrate. Although some exchange above background was observed with D-pHPG as substrate (ca. 6% relative to L-pHPG), this observed activity is likely the result of a small L-isomer impurity. It should be noted that no exchange activity was observed for closely related analogues of pHPG such as L-phenylglycine (PG), D-PG, D/L-4-fluorophenylglycine (FPG) or D/L-4-chlorophenylglycine (ClPG), a further indication of the high specificity of these A-domains.
Figure 1.

Representative results of the ATP/PPi exchange assay for L-pHPG activating modules A1T1-His6, A3T3 E3C4-His6 and C5A5T5TE-His6 and L-serine activating module, A4T4-His6 at 0.5uM. For modules A1T1-His6 and A4T4-His6 which require addition of a MbtH protein for activity, His6-NocI was added at 2 uM final concentration. All assays were performed at room temperature.
For the present discussion, however, it is important to note that despite the similarity of the amino acid binding pockets of A1, A3 and A5, ATP/PPi exchange in A1T1-His6 was only observed in the presence of the MbtH proteins NocI or NocP. Of the three L-pHPG activating modules, ATP/PPi exchange was dependent on stoichiometric amounts of MbtH protein only in A1T1-His6. The addition of NocI to A3T3 E3C4-His6 and C5A5T5TE-His6 had no effect on the ATP/PPi exchange reaction under identical experimental conditions.
The substrate binding pocket of the A4 domain in NocB was predicted to activate L-serine by several algorithms and was consistent with past experiments that established the β-lactam ring of nocardicin A is derived from L-serine.23 Initial ATP/PPi exchange assays of A4T4-His6 failed to demonstrate any significant activity in the presence of any amino acid. As observed in experiments with A1T1-His6, A4T4-His6 was also dependent on the presence of stoichiometric amounts of NocI for the observation of ATP/PPi exchange with its preferred substrate, L-serine as shown in Figure 1B.
Substrate determination for the A2 domain has been the most difficult to achieve of the five modules, as noted earlier. Recently, the A6 domain of the lysobactin NRPS was characterized to activate L-Arg, independent of the presence of a MbtH protein.26 A2 shares the 8 residue L-Arg binding pocket signature with this lysobactin A-domain, as well as showing good overall similarity to it (40.2% identical, 55.8% similar). Upon expression and purification of MBP-A2T2-His6, initial ATP/PPi exchange experiments conducted under conditions used to characterize A1T1-His6, A3T3 E3C4-His6, A4T4-His6 and C5A5T5TE-His6 demonstrated only weak exchange activity and poor selectivity in the presence of small hydrophobic amino acids such as L-Val, L-Leu, and L-Ile and no exchange activity for L-Arg. As depicted in Figure 2A, the observation of L-Arg dependent ATP/PPi exchange was only observed under altered assay conditions26 involving a 10-fold higher concentration of protein, conducting experiments at 30 °C instead of room temperature (~23 °C), and the addition of a 2-fold excess of NocI to the assay. Of note, under these conditions significant ATP/PPi exchange was also observed for the hydrophobic amino acids L-Val, L-Leu, L-Ile, and L-Phe, but this activity was not dependent on the addition of the MbtH protein, NocI. The dramatic activation of ATP/PPi exchange by NocI in the presence of L-Arg compared to the small hydrophobic amino acids is seen in a difference plot (Figure 2B). Also, unlike the lysobactin A6 domain, ATP/PPi exchange activity by MBP-A2T2-His6 was not observed for D-Arg nor was ATP/PPi exchange activity observed for similar amino acids predicted from bioinformatics such as L-ornithine, L-δ-N-hydroxyornithine, L-δ-N-acetyl-δ-N-hydroxyornithine, or L-Lys.
Figure 2.

Representative results of the ATP/PPi exchange assay for 5 uM of the L-Arg activating MBP-A2T2-His6 performed at 30 °C. A. Data plot for activity with and without addition of 10 uM His6-NocI. B. Difference plot – the activity of MBP-A2T2-His6 with His6-NocI with the background activity seen in the absence His6-NocI subtracted.
Interaction of MbtH Proteins NocI and NocP with Nocardicin A-domains
To understand further its interaction with each nocardicin module, NocI was cloned into pCDFDuet for heterologous expression as an N-terminal His6 fusion protein as well as an untagged protein in E. coli. In addition, untagged NocI for in vitro experiments was prepared by heterologous expression of NocI fused to an intein attached to a chitin binding domain (CBD) tag followed by binding to chitin-binding affinity resin and cleavage of the CBD tag from NocI with DTT. The rate of ATP/PPi exchange as a function of NocI (untagged) concentration for 0.5 μM concentrations of A1T1-His6 and A4T4-His6 is shown in Figure 3. Although the overall exchange rate is much higher for A1T1-His6 compared to A4T4-His6, both hyperbolic curves plateau at ~ 2 μM NocI and have Km values of 0.41 μM and 0.59 μM, respectively. In a similar experiment with A3T3 E3C4-His6 and C5A5T5TE-His6, NocI had no effect on the rate of ATP/PPi exchange of their preferred substrate L-pHPG. This observation was supported by analysis of the ATP/PPi exchange reaction for proteins isolated from an A3T3 E3C4-His6 – A4T4-His6 co-expression experiment. Addition of His6-NocI to the assay greatly enhanced ATP/PPi exchange activity observed in the presence of L-Ser but showed little effect on the activity observed in the presence of L-pHPG.
Figure 3.

ATP/PPi exchange rate for modules A1T1-His6 (A) and A4T4-His6 (B) as a function of increasing NocI (untagged) concentration. The fitted line was generated by curve fitting to the Box Lucas model equation: y = a(1 − bx) using OriginLab software version 8.6.
To characterize further the interaction between each module and NocI, untagged NocI was co-expressed with each C-terminally labeled His6 module protein. Following expression, the individual modules were isolated using the standard NTA affinity isolation protocol. SDS-PAGE analysis suggested that untagged NocI co-eluted (or was pulled down) by A1T1-His6, MBP-A2T2-His6 and A4T4-His6, indicating strong interaction. This interaction was not observed during the purifications of A3T3 E3C4-His6 or C5A5T5TE-His6 co-expressed with NocI. For a more quantitative evaluation, the final eluant for each co-expression reaction was denatured by the addition of urea and analyzed by HPLC to confirm the presence of NocI and estimate the module: NocI stoichiometry. The chromatogram and SDS-Page gel for the A4T4-His6 – NocI co-expression are shown in Figure 4. As expected owing to its smaller size, NocI elutes first followed much later by A4T4-His6. Based on the area of each peak and the molar absorptivity of each protein calculated by Vector NTI, the A4T4-His6:NocI and A1T1-His6:NocI stoichiometric ratios were each calculated to be 1:1. The observation of a 1:1 stoichiometic relationship is consistent with the shape of the activity curves shown in Figure 3. Due to the complexity of the HPLC chromatogram, the stoichiometry of the MBP-A2T2-His6 –NocI co-expression could not be confidently determined. Chromatograms for the co-expression of A3T3 E3C4-His6 – NocI and C5A5T5TE-His6 – NocI failed to show any peak in the NocI region, consistent with SDS-PAGE analysis and in keeping with the absence of a strong binding between A3 or A5 with NocI.
Figure 4.

HPLC chromatogram and SDS-Page gel (inset) of A4T4-His6–NocI (untagged) coexpression, purified by NTA affinity chromatography.
The ability to reconstitute the activity of the A1T1-His6•NocI and A4T4-His6•NocI co-expressed proteins by combinations of individually expressed NocI, A1T1-His6, and A4T4-His6 was also investigated, based on the HPLC response factors for each protein. The activity of reconstituted A4T4-His6 and NocI was ~50% of the co-expressed A4T4-His6•NocI sample. For A1T1-His6, the activity of the reconstituted proteins was ~70% of the co-expressed A1T1-His6•NocI sample.
To investigate the ability of an MbtH family protein generated outside the nocardicin cluster to complement NocI, NocP was heterologously expressed in E. coli as an N-terminal His6 fusion protein from pET28b(+) and as an untagged form from pCDFDuet. Several experiments were carried out to compare the activity of His6-NocI to His6-NocP with A1T1-His6, MBP-A2T2-His6 and A4T4-His6. Interestingly, His6-NocP activated ATP/PPi exchange activity in A1T1-His6 and A4T4-His6, but not in MBP-A2T2-His6. Furthermore, analysis of the co-expression product of NocP (untagged) with MBP-A2T2-His6 by HPLC indicated that NocP does not form a strong interaction with MBP-A2T2-His6 yet NocI was pulled down by MBP-A2T2-His6. Together, these results indicate that NocP cannot fully complement NocI.
Phylogenic Analysis of N. uniformis ATCC 21806
To evaluate the phylogenic similarity of N. uniformis to A. mirum, 16S rDNA was amplified from N. uniformis for sequencing and submitted to GenBank for comparison. The 16S rDNA (1508 nt) isolated from N. uniformis was found to be identical to A. mirum 16S rDNA (GenBank accession number CP001630.1) except for one polymorphism and are thus identical based on standard classification criteria.27,28
Genome Mining for Trypsin Proteases in A. mirum
The predicted pentapeptide formed by NocA + NocB, L-pHPG–L-Arg–D-pHPG–L-Ser–L-pHPG requires trimming at the C-terminus of L-Arg during nocardicin A biosynthesis. Trypsins specifically cleave peptides at the C-terminus of L-Lys and L-Arg and have been isolated from bacteria as well as mammals.29 However, BLASTP30 analysis of the A. mirum complete genome revealed at least five probable trypsins, three of which have ≥ 38% identity ≥ 50% similarity to well characterized trypsins from Streptomyces griseus and Saccharopolyspora erythraeus.31,32
To confirm the presence of the typsin encoding genes mined from the A. mirum genome in our strain, three trypsin proteases, ACU39320, ACU39665, and ACU39678, were selected for PCR amplification and sequence comparison. All three were successfully amplified from N. unformis gDNA and confirmed by sequencing analysis to be identical to the corresponding A. mirum genes.
Trypsin Proteolysis of Pentapeptide Precursor
The predicted product of the nocardicin NRPSs, L-pHPG–L-Arg–D-pHPG–L-Ser–L-pHPG, was purchased for evaluation. HPLC analysis of a pentapeptide trypsin reaction mixture showed the disappearance of the pentapeptide peak and the corresponding appearance of two new peaks with retention times significantly shorter than the starting material. (Supplemental Information Figure S1.) LC-MS/TOF analysis of the earliest eluting peak indicated a mass of 324.17 Da, which corresponds to the (M+H) of the L-pHPG–L-Arg dipeptide. The second peak has a mass of 404.15 Da consistent with the (M+H) of the predicted D-pHPG–L-Ser–L-pHPG tripeptide. Thus, this analysis indicates complete cleavage at the C-terminus of the L-Arg residue, as expected for trypsinolysis, despite its being flanked by two nonproteinogenic amino acids, one in the D-configuration.
Discussion
NRPS A-domains constitute a sub-family of the ANL superfamily of adenylating enzymes, which also includes acyl and aryl CoA synthetases and firefly luciferase. ANL enzymes catalyze two reactions, the adenylation of a substrate carboxylate to form a high energy acyl-AMP intermediate followed by an acyl substitution reaction with the pantetheinyl side chain of a T- domain in the NRPS A-domain subfamily, reaction with CoA by members of the acyl and aryl CoA synthetases subfamily, or oxidative decarboxylation followed by the generation of light in the firefly luciferase subfamily.33 Structure studies of the PheA domain, the initial A-domain in gramicidin S synthetase, show a two-domain structure with a larger N-terminal domain (~430 residues) and a smaller C-terminal domain (~100 residues) with a connecting loop (A8 loop) between the two domains – a theme repeated throughout the ANL superfamily.12 The PheA domain, crystallized with phenylalanine and AMP in the active site, is in the adenylation conformation, in which the conserved and essential A10 lysine forms hydrogen bonds with the oxygen atom in the ribose ring and the 5′ bridging oxygen of AMP and the carboxylate oxygen of phenylalanine.12 The alternation of conformational states during the catalytic cycle of the A-domain was first supported by crystal structure studies of DltA, a D-alanyl carrier protein ligase from Bacillus cereus that catalyzes the adenylation and thioesterification reaction of D-alanine in cell wall biosynthesis, which has been captured in several conformations.34–36 This model is supported by recent crystal structure studies of PA1221, an two-domain (A-T) NRPS in both the adenylation and thioester-forming conformations.37 Experimental evidence suggests that ANLs undergo a major ~140° conformational change between the adenylation and thioester reaction conformations, an observation that is supported by the recent solution of 4-chlorobenzoate•coenzyme A ligase crystal structure captured in both the adenylate-forming and the thioester-forming conformations.38 Lacking a crystal structure of firefly luciferase trapped in the second, light producing configuration, Brachini et al. performed an experiment in which the N- and C-terminal domains were trapped in this arrangement by chemical crosslinking of two cysteine residues, one from each domain, with 1,2-bis(maleimidoethane). In the trapped configurational state, bioluminescence was not observed unless synthetic dehydroluciferyl-AMP substrate was added to bypass the inactivated adenylation half-step.39 Analysis of the recently solved structure of this cross-linked firefly luciferase finds it locked into a conformation similar to the thioester formation step in acyl CoA synthetases.40
Based on these studies, it has been proposed that A-domains may adopt at least three conformations: an open conformation in the absence of substrates, the adenylation conformation upon the binding of an amino acid substrate and ATP, and the thioester conformation, a 140° rotation from the adenylation conformation, that occurs upon the release of PPi.34 Both nocardicin synthetase initiation domains A1 and A4 and elongation domain A2 failed to catalyze an ATP/PPi exchange reaction in the presence of their preferred amino acid substrates until the addition of NocI.
Based on the studies described above, it is postulated that these domains can be “trapped” in a state in which one or more of the substrate-binding, adenylation, or thioester-forming conformations is blocked in a way that is relieved by NocI or other compatible MbtH protein. Expanding on this hypothesis, pull-down studies reported here support a 1:1 stoichiometric relationship between NocI and an interacting A-domain. In secondary metabolism, there is a set of glycosyltransferases (GTs) whose activity requires a stoichiometric amount of a partner protein for activity. DesVII, a GT involved in macrolide biosynthesis from S. venezuelae, requires DesVIII for activity and these two proteins were shown to form a tight 1:1 complex when co-purified.41 Analogously, EryCIII a GT required for erythromycin D biosynthesis, requires partner protein EryCII for activity and a the structure of the complex formed by these two proteins, a dimer of heterodimers, has been recently solved.42 Analytical gel chromatographic analysis of the tyrosine adenylating enzyme SimH coupled to its cognate MbtH protein, SimY also indicates the formation of a heterotetrameric complex.20 The EryCIII•EryCII interface is characterized where the N-terminal helix of EryCII is sitting in a groove formed by three helices in EryCIII and stabilized by electrostatic interactions. This observation suggests two roles for the partner protein EryCII, stabilization of the GT and allosteric regulation.42 It appears that MbtH proteins also might share these two roles to varying degrees. While neither NocI nor NocP was required for the expression of modules from NocA or NocB in E. coli, the requirement for co-expression of a cognate MbtH protein with an NRPS protein domain in E. coli has been reported in several cases suggesting a stabilization role for the MbtH protein in these instances.11,43–45
The ability to reconstitute the A-domain•MbtH protein interaction in vitro with separately expressed proteins has been evaluated. Measurements of A4T4-His6 and A1T1-His6 reconstituted with NocI, indicated ATP/PPi exchange activity of 50 – 70% of the activity observed for an equivalent amount of the co-expressed proteins. These differences in activity may be due to the presence of impurities or unfolded proteins, skewing the protein quantitation. Similar reconstitution experiments on different systems have shown varying results. The co-expression of CloH and CloY resulted in a protein mixture that was ~50% less active than the reconstitution of separately expressed proteins.20 Conversely, the co-expressed proteins CmnO and CmnN resulted in a dramatically more active mixture than when the equivalent amounts of CmnO and CmnN from single expressions were combined.18
Of the three L-pHPG activating domains in NocA and NocB, only A1 is dependent on an MbtH protein for activation. Alignments of the nocardicin NRPS domains were performed to determine a conserved region that might be responsible for interaction with MbtH proteins (Supporting Information Figure S2). Based on alignment data of MbtH-dependent CloH and MbtH-independent NovH (83% identity), a single mutation, L383M was made in CloH resulting in a mutant protein with some MbtH-independent ATP/PPi exchange activity.20 However, the position corresponding to CloH L383 in all the nocardicin A-domains as well as all other MbtH protein dependent A-domains (Supporting Information Figure S3) is a positively charged arginine residue (except for lysine in NocB A4), suggesting that the analogous mutation in A-domains other than CloH would not restore MbtH protein independent A-domain activity. Identification of a residue or set of residues that may account for the binding of NocI by A1, A2, and A4 but not A3 and A5 was not fruitful. A similar analysis, alignment of MbtH-independent L-Arg activating A6 domain from the lysobactin NRPS with the similar MbtH-dependent A2 domain from NocA (Supporting Information Figure S4) also failed to suggest a MbtH protein binding site. The observation that NocP substituted for NocI for A1T1-His6 and A4T4-His6 but not for MBP-A2T2-His6 suggests variability at the A-domain•MbtH interface, complicating prediction of interaction sites.
The discovery of the MbtH protein requirement for ATP/PPi exchange activity has been crucial for furthering the understanding of nocardicin A biosynthesis. As noted earlier, the structure of the nocardicins is characterized by the nonribosomal tripeptide core D-pHPG–L-Ser–D-pHPG biosynthesized by NRPSs NocA and NocB comprising five modules, all essential for nocardicin A biosynthesis.46 The heterologous expression and ATP/PPi exchange data for the modules of NocA and NocB validates the previous assumption that the tripeptide core derives from modules 3, 4 and 5.24 The substrate specificities for MbtH-dependent A1 and A2, L-pHPG and L-Arg, respectively, have been of particular interest due to their absence in nocardicin A, and in the case of A2, the inability to predict its substrate using bioinformatics or establish it experimentally.
Both in vivo and in vitro experimental evidence collected on NocA+NocB is consistent with a linear NRPS producing pentapeptide L-pHPG–L-Arg–D-pHPG–L-Ser–L-pHPG instead of a “Type C” NRPS model in which module skipping occurs to yield a tripeptide, as suggested previously.24 The trimming of leader peptides from ribosomal protein products is well known in nature, but there are only a few examples of proteolysis in the biosynthesis of nonribosomal natural products. The trimming of a leader peptide has been observed in xenocoumarin biosynthesis by periplasmic protease XcnG47 and during didemnin biosynthesis by an unknown protease.48 The excision of an N-terminal fatty acid chain added by an NRPS in pyoverdine biosynthesis by periplasmic protease PvdQ has been demonstrated49 while a similar reaction is suspected in saframycin biosynthesis.50 Although tabtoxin is not a nonribosomal product, this β-lactam-containing pro-drug is trimmed by periplasmic peptidase, TapP to its active form.51 The backbone of caerulomycin A is biosynthesized by a PKS-NRPS hybrid in which the terminal amino acid added in the final module, L-leucine, is removed later in the biosynthesis by a metallo-dependent amidohydrolase CrmL.52
The proteases discussed in the examples above, XcnG, PvdQ, TapP and CrmL are all encoded in their respective gene clusters. The only gene candidate resembling a protease in the nocardicin A gene cluster is NocK. NocK contains a catalytic triad consistent with a serine protease 53 and an N-terminal signal sequence predicting the export of this protein via the twin arginine translocation (Tat) pathway to periplasm.54,55 However, the definitive assignment of a proteolytic role for NocK is compromised by insertional inactivation experiments showing that nocK is not essential for nocardicin A biosynthesis in N. uniformis.53 While the timing of the cleavage of the leader peptide is currently unclear as is any role the leader dipeptide might have in downstream reactions, biosynthetic logic dictates that in nocardicin A biosynthesis, trimming of the N-terminal leader dipeptide occurs prior to oxime formation (catalyzed by NocL) at the N-terminus of the remaining tripeptide. Since the P450 catalyzed oxime reaction likely occurs in the cytoplasm, it seems unlikely that during nocardicin A biosynthesis, an intermediate would be exported to the periplasm for proteolysis requiring the tripeptide intermediate to be imported back to the cytoplasm for reaction with NocL.
The identification of L-Arg as the preferred substrate for A2 leads to the possibility that the pentapeptide core is trimmed during nocardicin A biosynthesis by an ordinary cytoplasmic trypsin-like protease (Scheme 1). While trypsin, a serine protease, is more commonly known as mammalian digestive enzyme, it has also been identified in a wide range of organisms including Streptomyces.29 Analysis of the nocardicin A gene cluster, 15 kbp of the upstream nucleotide sequence and ~5 kbp of the downstream nucleotide sequence failed to elucidate a gene encoding a trypsin protease. Phylogenic classification of the N. uniformis strain used in these studies based on 16S rDNA was performed to determine if N. uniformis was similar to the nocardicin A producing A. mirum strain that has been fully sequenced. Unexpectedly, the 16S rDNA of these two strains were found to be identical, supporting the previous observation that N. uniformis strain ATCC 21806 formed synnemata and zoospores highly similar to A. mirum.
Scheme 1.
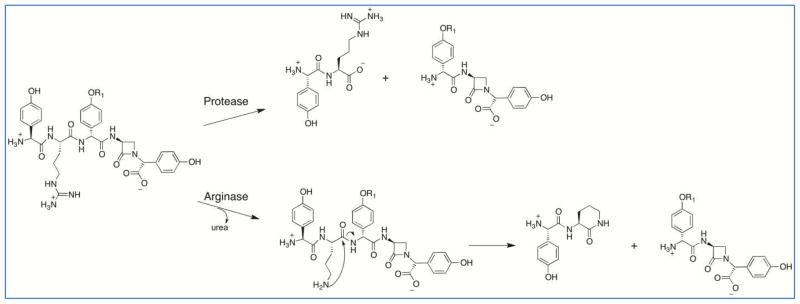
A BLAST-P search of the A. mirum genome for proteins similar to the well characterized known S. griseus and S. erythraeus trypsins identified three proteins with greater than 38% identity and greater than 50% similarity and several others of lower identity and similarity.30,32 The conserved catalytic triad and substrate specificity pocket characteristic of trypsin is observed in the protein sequence alignment of the putative A. mirum trypsin proteases.29 (Supporting Information Figure S5) These proteins are not encoded in the defined nocardicin A gene cluster, but may be available to the nocardicin A biosynthetic pathway due to their constitutive expression or cross-talk between gene clusters as observed in the erythrochelin and rhodochelin biosynthetic pathways.56–58 Cross-talk between gene clusters has been proposed to occur in A. mirum during the biosynthesis of the siderophore mirubactin.59 The presence of trypsin and the ability of bovine trypsin to cleave the predicted pentapeptide product, L-pHPG–L-Arg–D-pHPG–L-Ser–L-pHPG, at the C-terminal site of the L-Arg residue, supports this hypothesis.
Although whole cell incorporation experiments with a linear peptide would likely result in proteolysis of the peptide by nutrient uptake systems, two experiments were performed in which either peptide L-pHPG–L-Arg–D-pHPG–L-Ser–L-pHPG or its D-isomer peptide L-pHPG–LArg–D-pHPG–L-Ser–D-pHPG was added to the fermentation culture of a previously characterized N. uniformis point mutant, nocB S571A, in which the NRPS is inactivated. Unfortunately, nocardicin A production was not observed with either peptide but peaks correlated to 4-mer and 3-mer degradation products were observed, consistent with proteolytic degradation from the N-terminus.
An alternative, self-cleavage mechanism is also proposed in Scheme 1. Conversion of the second position L-Arg to L-Orn by an arginase could lead to cyclization by attack of the side chain amine of L-Orn on the amide carbonyl. This would result in self cleavage at the C-terminus of L-Arg, liberating the tripeptide core of the nocardicins as previously suggested.24
Conclusion
In conclusion, MbtH protein dependence of A-domain activity was observed in A1, A2, and A4 of the nocardicin A NRPSs, while A3 and A5 showed no such requirement. The two MbtH proteins associated with or near the cluster were found to be not completely complementary, indicating that interaction with an A-domain is more complex than simple association with the three conserved tryptophan residues characteristic of MbtH proteins. Analysis of co-expressed protein complexes indicates a 1:1 stoichiometry between A-domains A1 and A4 with NocI. This discovery enabled the determination of the substrate amino acid specificity for each A-domain using the ATP/PPi exchange assay to yield a predicted product, L-pHPG–L-Arg–D-pHPG–L-Ser–L-pHPG, for the nocardicin NRPS. The requirement for truncation of this pentapeptide and the absence of a protease in the nocardicin A gene cluster that could perform such a function led to the proposal of a self-cleavage mechanism or the action of a protease encoded outside the gene cluster. Classical morphological criteria that have suggested N. uniformis ATCC 21806 should be reclassified as A. mirum were confirmed by direct comparison of their 16S rDNA sequences and the mutual identity of three candidate trypsin-encoding genes. Cross-talk with these enzymes or other endo- or exo- proteases can be invoked to account for truncation to the tripeptide core of the nocardicins. With the identity of a putative pentapeptide precursor in hand and the availability of a sequenced genome, the way is open to address the central timing and mechanistic questions of peptide processing, β-lactam formation, and C-terminal epimerization.
Experimental Methods
Cloning of NRPS Expression Vectors
The primers used for the PCR amplification of the coding region of each multi-domain construct are described in Table 1. Amplification from previously prepared plasmids was performed using Pfu (Agilent Technologies, Santa Clara, CA), Pfu Turbo (Agilent), or KOD (EMD Biosciences, Gibbstown, NJ) DNA polymerases. For modules 1 and 4, the PCR products were digested with Nco I and Xho I and directly ligated into the Nco I/Xho I site in pET28b(+) (EMD Biosciences). For modules 2, the PCR product was digested with Nco I and BamH I and directly ligated into the Nco I/BamH I site in pET28b(+). For cloning into pMALc2x (New England Biolabs, Ipswich, MA), the Nco I/Blp I fragment was excised from pET28b(+) blunted with a Klenow reaction and ligated into the Klenow blunted EcoR I/Hind III site of pMALc2x. The PCR product for module 3 was digested and directly ligated into the Nco I/BamH I site of pQE60 (Qiagen, Valencia, CA). The module 3-histidine tag coding sequence was digested from pQE60 with Nco I/Hind III for ligation into the Nco I/Hind III site of pET28b(+). The PCR product encoding module 5 was subcloned into pCRBlunt (Life Technologies, Grand Island, NY). The coding region was excised from pCRBlunt with a Nco I/Hind III digest and ligated into the Nco I/Hind III site of pET28b(+). All constructs were confirmed by sequencing analysis performed by the Biosynthesis and Sequencing Facility, Johns Hopkins Medical School, Baltimore, MD.
Table 1.
Oligonucleotide primers used for NRPS expression vector construction.
| Description | Label | Primer Sequence (5′ to 3′) |
|---|---|---|
| A1T1 | M1 For (Nco I) | GCGCCATGGGGGACAGCGAGGAGTGGGAGCGC |
| M1 Rev (Xho I) | GCGCTCGAGGGCCAGCTCCTCGATCCGCCTGGCCAG | |
| A2T2 | M2 For (Nco I) | GCCCATGGTTAGCACCGGCGCGCCGGTCGAACC |
| M2 Rev (BamH I) | GCGGATCCCGCGTCGTCGGCCCACGGC | |
| A3T3 E3C4 | M3 For (Nco I) | TCTTCCATGGTGCTCGGGGACGAC |
| M3 Rev (BamH I) | TATAGGATCCCGGGTTCCTTCTCCCCTCG | |
| A4T4 | M4 For (Nco I) | GCCCCATGGCCTTTCGAGTGCGCGACCTGTTCGCCG |
| M4 Rev (Xho I) | GGCCTCGAGGGTGGACGCCTCGTCGGCG | |
| C5A5T5TE | M5 For (Nco I) | GCGCTCCATGGACGAGGAGGCGCTGCTGGCG |
| M5 Rev | TATAAAGCTTCCGCTCTCCTCCCAGCGCGC |
Cloning of NocI and NocP Expression Vectors
The nocI gene was amplified by PCR from cosmid DNA using KOD DNA polymerase and the primers listed in Table 2. PCR products were subcloned into pCRBlunt or pJET1.2 (ThermoFisher Scientific, Waltham, MA), confirmed by sequencing prior to excision and ligation into pCDFDuet (EMD Biosciences) and pTYB12 (New England Biolabs), respectively, using the cut sites indicated. The nocP gene was similarly amplified by PCR from cosmid DNA using KOD DNA polymerase and the primers listed in Table 2. PCR products were directly cloned into the Nde I/Xho I sites of pET28b(+) and pCDFDuet, respectively.
Table 2.
Oligonucleotide primers used for NocI and NocP expression vector construction.
| Vector | Label | Primer Sequence (5′ to 3′) |
|---|---|---|
| pCDFDuet | NocI For (Nco I) | GCCCCATGGTGGGAGAGAACGAGGATTCCGG |
| NocI Rev (Hind III) | GCGAAGCTTTCAAGCCCGGTCCCCGGCC | |
| pTYB12 | NocI For (Bsm I) | GCCGAATGCTCTGGGAGAGAACGAGGATTCCGG |
| NocI Rev (Xho I) | GCGCTCGAGTCAAGCCCGGTCCCCGGCC | |
| pET28b(+) | NocP For (Nde I) | GCGGCATATGAACCCCTTCGACGACCACGACCGCGCT |
| NocP Rev (Xho I) | GCGCTCGAGTCAGGCGTCGGGGTCGGGCGCGCCCGCGCG | |
| pCDFDuet | NocP For (Nde I) | GCGGCATATGAACCCCTTCGACGACCACGACCGCGCT |
| NocP Rev (Xho I) | GCGCTCGAGTCAGGCGTCGGGGTCGGGCGCGCCCGCGCG |
Heterologous Expression and Purification of NRPS Modules
Seed cultures consisting of 50 mL LB medium with 25 ug/mL kanamycin for pET28 constructs or 100 ug/mL for pMALc2x constructs and 34 ug/mL chloramphenicol inoculated with E. coli BL21(DE3) Rosetta2 (EMD Biosciences) transformed with expression vector were grown overnight at 37 °C with shaking. The growth medium, 2xYT (3L) supplemented with 5 mM MgCl2, 25 ug/mL kanamycin or 100 ug/mL ampicillin, and 34 ug/mL chloramphenicol was inoculated with the seed culture at a ratio of 1:100. Cells were grown at 37 °C with shaking until the OD600 measured ~ 0.6. Growth cultures were cooled to 17.5 °C prior to induction of protein expression with the addition of IPTG (1 mM final concentration for pET28b, 0.3mM for pMALc2x). Expression was continued overnight at 17.5 °C. Cells were collected by centrifugation (5180 × g, 10 min at 4 °C) then resuspended in lysis buffer (50 mM NaH2PO4 pH = 8, 300 mM NaCl, 10 mM imidazole, 10% glycerol) and lysed by sonication. The cell debris was collected by centrifugation at 37044 × g, 4 °C for 30 min. NTA resin (Qiagen) was added to the lysate and allowed to incubate at 4 °C with turning for at least 1 h. The lysate-resin slurry was poured into an empty column. The resin was washed with lysis buffer followed by wash buffer (50 mM NaH2PO4 pH = 8, 300 mM NaCl, 20 mM imidazole, 10% glycerol). The His6 tagged protein was eluted with 4 – 10 mL elution buffer (50 mM NaH2PO4 pH = 8, 300 mM NaCl, 250 mM imidazole, 10% glycerol).
Protein isolated by affinity chromatography was dialyzed in 3 × 1L dialysis buffer (50 mM Tris-HCl pH = 7.5 @ 4 °C, 50 mM NaCl, 5 mM MgCl2, 1 mM DTT, 10% glycerol), 1 h for each exchange. For further purification of modules 1, 3, 4, and 5, the protein solution was loaded onto a 5 mL HiTrapQ cartridge (GE Life Sciences, Pittsburgh, PA) pre-equilibrated with dialysis buffer (50 mM NaCl) using a ÄKTA FPLC (GE Life Sciences). Fractions were collected as the 50 – 500 mM NaCl (300mL total volume) gradient was applied to elute adsorbed proteins at a flow rate of ~1 mL/min with UV detection at 280 nm. Fractions (3 mL) were collected and analyzed by SDS-PAGE. Fractions containing the target fusion protein were collected, concentrated to > 2 mg protein/mL using a centrifugal filter (Millipore Corp., Billerica, MA), and flash frozen in liquid nitrogen for storage at −80 °C.
Heterologous Expression and Isolation of untagged NocI
Seed cultures consisting of 50 mL LB medium with 100 ug/mL ampicillin were inoculated with E. coli BL21(DE3) transformed with the pTYB12/nocI expression vector and grown overnight at 37 °C with shaking. Six flasks, each containing 0.5 L LB medium with 100 ug/mL ampicillin and inoculated with 5 mL seed culture, were grown at 37 °C, 180 rpm until OD600 ~ 0.6, then cooled to 10 °C (1 h). IPTG, final concentration 0.5 mM, was added to induce protein expression. Expression was continued at 17.5 °C overnight with shaking.
The following steps were performed at ≤ 4°C. The next day, cells were pelleted by centrifugation (5180 × g, 10 min) then resuspended in 150 mL lysis buffer (20 mM Tris-HCl pH = 8, 500 mM NaCl, 1 mM EDTA). Cells were lysed by sonication and the cell debris was removed by centrifugation. Chitin resin (New England Biolabs #E6900S), 10 mL, was conditioned with ~110 mL column buffer (50 mM Tris-HCl pH = 8, 50 mM NaCl, 10% glycerol). The clarified cell lysate was passed through the chitin bead column at a flow rate of ~1 mL/min. The column was then washed with 100 mL column buffer. When the level of the wash buffer was close to the top of the resin, 20 mL cleavage buffer (100 mM DTT in column buffer) was added to the column. The column was capped and the resin was resuspended in the cleavage buffer and incubated at 4 °C for 72 h. The column eluate was collected. An additional 10 mL cleavage buffer was applied to the column, eluted, and combined with the previous eluate. The 30 mL of column eluate was filtered using an Amicon (Millipore Corp.) Ultra 30 kDa MWCO centrifuge filter. The flow-through was collected and concentrated using an Amicon Ultra 3 kDa MWCO filter. Native NocI (untagged) was flash frozen in liquid nitrogen for storage in the −80 °C freezer.
Co-Expression of Module (His6-tagged) and NocI or NocP (untagged)
Each module expression plasmid (pET28/Mx or pMALc2x/M2) was co-transformed with pCDFDuet/nocI (untagged) into E. coli BL21(DE3) Rosetta 2 using standard electroporation protocols. Conditions for growth, expression, and isolation by NTA affinity chromatography were similar to those given for the heterologous expression of individual modules above, except that 50 ug/mL spectinomycin was added to the seed culture and growth culture medium to maintain selection for the pCDFDuet/nocI expression vector. In addition, NTA chromatography was the terminal step in the isolation, i.e. further purification by ion exchange chromatography was not done.
ATP/PPi Exchange Assay
Each 100 uL reaction consisted of 36.3 mM HEPES pH = 7.5, 0.15 mM EDTA, 7.25 mM MgCl2, 1.5 mM DTT 3.7 mM ATP, 7.3% (v/v) glycerol, 0.75 mM amino acid substrate, and 1 mM Na4P2O7 with 1 uCi 32P-labeled Na4P2O7 (NEN Perkin Elmer, Waltham, MA). The reaction was initiated by the addition of the protein (0.5–5 uM). Following incubation at room temperature for 30 min, the reaction was quenched by the addition of 400 uL 0.5M HClO4, chased with 400 uL 100 mM sodium pyrophosphate (unlabelled) and 200 uL of a 4% (w/v) suspension of activated charcoal (Norit A) was added. The charcoal was pelleted by centrifugation (14,000 rpm, 5 min), washed twice with 1 mL water and re-pelleted by centrifugation. The charcoal pellet was resuspended in 500 mL water, transferred to a 7 mL glass scintillation vial, and mixed with 5 mL of Optifluor (NEN Perkin Elmer). Each sample was measured using a Beckman model LS6500 scintillation counter (Beckman Coulter, Brea, CA).
Modified ATP/PPi Exchange Assay for Module 2
Each 200 uL reaction mixture consisted of 50 mM Tris-HCl pH = 7.5, 10 mM MgCl2, 5 mM amino acid substrate, 2.5 mM Na4P2O7 with 2 uCi 32P-labeled Na4P2O7, and 5 uM protein. Prior to the addition of ATP, the reaction mixture was incubated at 30 °C for 10 min. Following this pre-incubation, 2.5 mM ATP was added to initiate the ATP/PPi exchange reaction was incubated at 30 °C for an additional 30 min. The exchange reaction was quenched with acid and prepared for scintillation counting using the procedure outlined in the previous section.
Determination of Module:NocI Stoichiometry by HPLC
Samples isolated by NTA affinity chromatography were denatured by the addition of 8 M urea (Sigma-Aldrich, St. Louis, MO) to a final concentration of 6 M urea and incubated at room temperature for 30 min. Following incubation, samples were diluted with water for a final concentration of 2 M urea. Samples were analyzed by protein HPLC, employing an Agilent 1200 HPLC system equipped with a diode array detector. Filtered denatured protein solutions (100 uL) in 2 M urea were injected onto a Vydac C4 protein column 150 × 46 mm column (W.R. Grace & Co., Columbia, MD) heated to 70 °C. The flow rate was 1 mL/min. Proteins were eluted using a binary gradient in which mobile phase A consisted of 90:10 water : acetonitrile (ACN) with 0.1% trifluoroacetic acid (TFA) and mobile phase B consisted of 90:10 ACN : water with 0.1% (TFA). After 3 min at 82:18 A:B, a linear gradient to 50:50 A:B over 45 min was programmed. Proteins were detected by monitoring absorbance at 280 nm and were quantified by integration using OriginPro v.8.6 software (OriginLab, Northampton, MA). The molar absorbtivity for each protein was calculated using Vector NTI software (Life Technologies).
Conditions for Proteolysis and HPLC Analysis
Peptide L-pHPG–L-Arg–D-pHPG–L-Ser–L-pHPG was procured from Peptide 2.0, Inc. (Chantilly, VA) for testing. In a total reaction volume of 100 uL, 200 nmol of pentapeptide was reacted with 20 ug modified (TPCK-treated) trypsin (New England Biolabs) in buffer containing 50 mM Tris-HCl, pH = 8 @ 25 °C and 20 mM CaCl2. The reaction was incubated at 37 °C overnight. Trypsin was removed from the reaction by filtration through 3 kDa MWCO centrifugal filter (Millipore) and the flow-through was analyzed by an Agilent 1100 HPLC equipped with a diode array detector. Aliquots of the reaction mixture were injected onto a Luna C18(2) column (250 mm × 46 mm) (Phenomenex, Torrence, CA) using a binary gradient in which mobile phase A consisted of 0.1% trifluoroacetic acid (TFA) in water and mobile phase B consisted of 90:10 ACN : water with 0.1% (TFA). After 5 min at 94:6 A:B and a flow rate of 1 mL/min, a linear gradient to 85:15 A:B over 15 min was programmed.
LC-MS analysis was conducted using an Agilent 1200 series, 6220 LC/MS TOF analyzer equipped with a dual ESI source operated in positive mode under the following conditions: fragmentor: 135 V, skimmer: 65 V, gas temperature: 350 °C, drying gas: 12 L/min, nebulizer 40 psi and Vcap: 3500 V for mass range 100 to 1700 m/z. Separations were performed with a Zorbax C18 5 micron column (150 mm × 4.6 mm) at 30 °C using a binary gradient in which mobile phase A consisted of 0.1% formic acid in water and mobile phase B consisted of 90:10 ACN : water with 0.1% formic acid. A linear gradient elution from 6% to 85% mobile phase B over 15 min was programmed. An aliquot of the trypsin reaction mixture described above was diluted 1:100 with 0.1% formic acid in water and 5 uL was injected for LC-MS analysis.
Pentapeptide Feeding Experiment
The preparation and growth conditions for the N. uniformis nocB S571A mutant has been described previously.46 N. uniformis is maintained on ISP2 solid medium (Difco Laboratories, Detroit, MI) at 28 °C. Seed cultures were prepared in TSB medium (Difco Laboratories) and grown to saturation at 28 °C with shaking. Nocardicin fermentation medium 60 was inoculated with 2 mL of the starter seed culture and incubated at 28 °C with shaking. Following 34 h of incubation, the culture medium was supplemented with peptide L-pHPG–L-Arg–D-pHPG–L-Ser–L-pHPG or peptide L-pHPG–L-Arg–D-pHPG–L-Ser–D-pHPG (Peptide 2.0, Inc., Chantilly, VA) to a final concentration of 5 mM. The supplemented mutant N. uniformis cultures were incubated for an additional day. Upon completion, the cultures were centrifuged to separate the cell mass from the supernatant, analyzed by HPLC, and stored at −20 °C. HPLC analysis to detect nocardicin A was performed as described previously.46 A second HPLC analysis for the detection of novel metabolites was performed using the same HPLC system and Luna C18(2) column (Phenomenex, Torrance, CA). For this analysis, the binary mobile phase solutions were: A: 0.1% trifluoroacetic acid (TFA) in water and B: 90:10 acetonitrile:water with 0.1% TFA. Filtered culture supernatents (10 uL) were directly injected and eluted with a shallow gradient starting with a ratio of 94:6 (A:B) for 5 min, to a ratio of 85:15 (A:B) at 20 min and ending with a ratio of 60:40 (A:B) at 40 min. Analytes were detected at 272 nm with a diode array detector.
Phylogenic Analysis of N. uniformis ATCC 21806
The isolation of genomic DNA from Nocardia uniformis subsp. tsuyamanensis ATCC 21806 was carried out according to the standard techniques from Practical Streptomyces Genetics.61 Amplification of 16S rDNA in was performed using the universal 16S rDNA primers fD1 and rp2 62 with KOD DNA polymerase. PCR products were sub-cloned into pJET1.2 and submitted for sequencing at the Biosynthesis and Sequencing Facility, Johns Hopkins Medical School, Baltimore, MD. The resulting sequences were compared with those available in the GenBank database maintained by the National Center for Biotechnology Information (NCBI) using the BLASTN algorithm.30
PCR Amplification of Trypsin Coding Sequences from N. uniformis
Using the Primer-BLAST63 algorithm on the NCBI website, primers (Table 3) were designed to amplify regions encoding three trypsins proteases predicted to be present in N. uniformis based on the A. mirum genome. Targeted genes were amplified from gDNA isolated N. uniformis using KOD DNA polymerase. PCR products were sub-cloned into pJET1.2 and submitted for sequencing analysis.
Table 3.
| Trypsin Primer Sets | Primer Sequence (5′ to 3′) |
|---|---|
| ACU39320 - Forward | GGACATTGCGGTGGTGGAAGACGTTC |
| ACU39320 - Reverse | GGAAGGAGATGAGCATGGCGGAAACC |
| ACU39665 - Forward | CGCGCTCAGCACGCTGTAGTAGG |
| ACU39665 - Reverse | GAAAGGAACTCCGATGGCGAAAACCCT |
| ACU39678 - Forward | CATCAGAGTCCGCATACCGGCCAAC |
| ACU39678 - Reverse | ATCGACCTGGTCTTTCAGGGCTAACCT |
Supplementary Material
Acknowledgments
This work was supported by NIH grant AI014937. The Greenberg group (JHU) is thanked for use of their FPLC and support for radiochemical experiments. Dr. W.E. Bocik performed the initial sub-cloning of nocI and we thank Prof. D. I. Barrick (Biophysics, JHU) for helpful discussions. Prof. P. F. Leadlay (Cambridge) kindly provided samples of L-δ-N-hydroxyornithine and L-δ-N-acetyl-δ-N-hydroxyornithine for the ATP/PPi exchange assays. Drs. A. C. Jacobs and I. P. Mortimer are acknowledged for their help with MALDI and ESI-MS measurements. Mr. E. Philander (Analytical Sciences Group, McCormick & Co., Inc.) kindly performed the LC-MS analyses described in this manuscript.
Footnotes
Supporting Information Available
Figure containing HPLC chromatograms and mass analysis for the trypsinolysis reaction of pentapeptide L-pHPG–L-Arg–D-pHPG–L-Ser–L-pHPG and protein sequence alignments. This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Felnagle EA, Jackson EE, Chan YA, Podevels AM, Berti AD, McMahon MD, Thomas MG. Mol Pharmaceutics. 2008;5:191–211. doi: 10.1021/mp700137g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai JR, Fischbach MA, Liu DR, Walsh CT. J Am Chem Soc. 2006;128:11002–11003. doi: 10.1021/ja063238h. [DOI] [PubMed] [Google Scholar]
- 3.Walsh C, Chen H, Keating T, Hubbard B, Losey H, Luo L, Marshall C, Miller D, Patel H. Curr Op Chem Biol. 2001;5:525–534. doi: 10.1016/s1367-5931(00)00235-0. [DOI] [PubMed] [Google Scholar]
- 4.Sattely ES, Fischbach MA, Walsh CT. Nat Prod Rep. 2008;25:757–793. doi: 10.1039/b801747f. [DOI] [PubMed] [Google Scholar]
- 5.Hahn M, Stachelhaus T. Proc Natl Acad Sci USA. 2004;101:15585–15590. doi: 10.1073/pnas.0404932101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quadri LEN, Sello J, Keating TA, Weinreb PH, Walsh CT. Chem Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]
- 7.Lautru S, Oves-Costales D, Pernodet J-L, Challis GL. Microbiology. 2007;153:1405–1412. doi: 10.1099/mic.0.2006/003145-0. [DOI] [PubMed] [Google Scholar]
- 8.Wolpert M, Gust B, Kammerer B, Heide L. Microbiology. 2007;153:1413–1423. doi: 10.1099/mic.0.2006/002998-0. [DOI] [PubMed] [Google Scholar]
- 9.Drake EJ, Cao J, Qu J, Shah MB, Straubinger RM, Gulick AM. J Biol Chem. 2007;282:20425–20434. doi: 10.1074/jbc.M611833200. [DOI] [PubMed] [Google Scholar]
- 10.Buchko GW, Kim C-Y, Terwilliger TC, Myler PJ. Tuberculosis. 2010;90:245–251. doi: 10.1016/j.tube.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heemstra JR, Walsh CT, Sattely ES. J Am Chem Soc. 2009;131:15317–15329. doi: 10.1021/ja9056008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conti E, Stachelhaus T, Marahiel MA, Brick P. The EMBO Journal. 1997;16:4174–4183. doi: 10.1093/emboj/16.14.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Challis GL, Ravel J, Townsend CA. Chem Biol. 2000;7:211–224. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 14.Stachelhaus T, Mootz HD, Marahiel MA. Chem Biol. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 15.Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH. Nucl Acids Res. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Röttig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O. Nucl Acids Res. 2011;39:W362–W367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prieto C, García-Estrada C, Lorenzana D, Martín JF. Bioinformatics. 2012;28:426–427. doi: 10.1093/bioinformatics/btr659. [DOI] [PubMed] [Google Scholar]
- 18.Felnagle EA, Barkei JJ, Park H, Podevels AM, McMahon MD, Drott DW, Thomas MG. Biochemistry. 2010;49:8815–8817. doi: 10.1021/bi1012854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang W, Heemstra JR, Walsh CT, Imker HJ. Biochemistry. 2010;49:9946–9947. doi: 10.1021/bi101539b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boll B, Taubitz T, Heide L. J Biol Chem. 2011;286:36281–36290. doi: 10.1074/jbc.M111.288092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe K, Okuda T, Yokose K, Furumai T, Maruyama HB. J Antibiot. 1983;36:321–324. doi: 10.7164/antibiotics.36.321. [DOI] [PubMed] [Google Scholar]
- 22.Hasegawa T, Lechevalier MP, Lechevalier HA. Int J Syst Bacteriol. 1978;28:304–310. [Google Scholar]
- 23.Townsend CA, Brown AM. J Am Chem Soc. 1983;105:913–918. [Google Scholar]
- 24.Gunsior M, Breazeale SD, Lind AJ, Ravel J, Janc JW, Townsend CA. Chem Biol. 2004;11:927–938. doi: 10.1016/j.chembiol.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 25.Rausch C, Hoof I, Weber T, Wohlleben W, Huson DH. BMC Evol Biol. 2007;7:78. doi: 10.1186/1471-2148-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hou J, Robbel L, Marahiel Mohamed A. Chem Biol. 2011;18:655–664. doi: 10.1016/j.chembiol.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 27.Schlaberg R, Simmon KE, Fisher MA. Emerg Infect Dis. 2012;18:422–430. doi: 10.3201/eid1803.111481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drancourt M, Bollet C, Carlioz A, Martelin R, Gayral J-P, Raoult D. J Clin Microbiol. 2000;38:3623–3630. doi: 10.1128/jcm.38.10.3623-3630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rypniewski WR, Perrakis A, Vorgias CE, Wilson KS. Protein Eng. 1994;7:57–64. doi: 10.1093/protein/7.1.57. [DOI] [PubMed] [Google Scholar]
- 30.Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D. Nucl Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stosová Tá, Sebela M, Rehulka P, Sedo O, Havlis J, Zdráhal Z. Anal Biochem. 2008;376:94–102. doi: 10.1016/j.ab.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 32.Kiser JZ, Post M, Wang B, Miyagi M. J Proteome Res. 2009;8:1810–1817. doi: 10.1021/pr8004919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drake EJ, Nicolai DA, Gulick AM. Chem Biol. 2006;13:409–419. doi: 10.1016/j.chembiol.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 34.Yonus H, Neumann P, Zimmermann S, May JJ, Marahiel MA, Stubbs MT. J Biol Chem. 2008;283:32484–32491. doi: 10.1074/jbc.M800557200. [DOI] [PubMed] [Google Scholar]
- 35.Du L, He Y, Luo Y. Biochemistry. 2008;47:11473–11480. doi: 10.1021/bi801363b. [DOI] [PubMed] [Google Scholar]
- 36.Osman KT, Du L, He Y, Luo Y. J Mol Biol. 2009;388:345–355. doi: 10.1016/j.jmb.2009.03.040. [DOI] [PubMed] [Google Scholar]
- 37.Mitchell CA, Shi C, Aldrich CC, Gulick AM. Biochemistry. 2012;51:3252–3263. doi: 10.1021/bi300112e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reger AS, Wu R, Dunaway-Mariano D, Gulick AM. Biochemistry. 2008;47:8016–8025. doi: 10.1021/bi800696y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Branchini BR, Rosenberg JC, Fontaine DM, Southworth TL, Behney CE, Uzasci L. J Am Chem Soc. 2011;133:11088–11091. doi: 10.1021/ja2041496. [DOI] [PubMed] [Google Scholar]
- 40.Sundlov JA, Fontaine DM, Southworth TL, Branchini BR, Gulick AM. Biochemistry. 2012;51:6493–6495. doi: 10.1021/bi300934s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borisova SA, Liu H-w. Biochemistry. 2010;49:8071–8084. doi: 10.1021/bi1007657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moncrieffe MC, Fernandez M-J, Spiteller D, Matsumura H, Gay NJ, Luisi BF, Leadlay PF. J Mol Biol. 2012;415:92–101. doi: 10.1016/j.jmb.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imker HJ, Krahn D, Clerc J, Kaiser M, Walsh CT. Chem Biol. 2010;17:1077–1083. doi: 10.1016/j.chembiol.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zolova OE, Garneau-Tsodikova S. MedChemComm. 2012;3:950–955. [Google Scholar]
- 45.McMahon MD, Rush JS, Thomas MG. J Bacteriol. 2012;194:2809–2818. doi: 10.1128/JB.00088-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davidsen JM, Townsend CA. Chem Biol. 2012;19:297–306. doi: 10.1016/j.chembiol.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reimer D, Pos KM, Thines M, Grun P, Bode HB. Nat Chem Biol. 2011;7:888–890. doi: 10.1038/nchembio.688. [DOI] [PubMed] [Google Scholar]
- 48.Xu Y, Kersten RD, Nam SJ, Lu L, Al-Suwailem AM, Zheng HJ, Fenical W, Dorrestein PC, Moore BS, Qian PY. J Am Chem Soc. 2012;134:8625–8632. doi: 10.1021/ja301735a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drake EJ, Gulick AM. ACS Chem Biol. 2011;6:1277–1286. doi: 10.1021/cb2002973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koketsu K, Watanabe K, Suda H, Oguri H, Oikawa H. Nat Chem Biol. 2010;6:408–410. doi: 10.1038/nchembio.365. [DOI] [PubMed] [Google Scholar]
- 51.Levi C, Durbin RD. Physiol Mol Plant Pathol. 1986;28:345–352. [Google Scholar]
- 52.Zhu YG, Fu P, Lin QH, Zhang GT, Zhang HB, Li SM, Ju JH, Zhu WM, Zhang CS. Org Lett. 2012;14:2666–2669. doi: 10.1021/ol300589r. [DOI] [PubMed] [Google Scholar]
- 53.Kelly WL, Townsend CA. J Bacteriol. 2005;187:739–46. doi: 10.1128/JB.187.2.739-746.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petersen TN, Brunak S, von Heijne G, Nielsen H. Nat Meth. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 55.Bendtsen J, Nielsen H, Widdick D, Palmer T, Brunak S. BMC Bioinf. 2005;6:167. doi: 10.1186/1471-2105-6-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lazos O, Tosin M, Slusarczyk AL, Boakes S, Cortés J, Sidebottom PJ, Leadlay PF. Chem Biol. 2010;17:160–173. doi: 10.1016/j.chembiol.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 57.Robbel L, Helmetag V, Knappe TA, Marahiel MA. Biochemistry. 2011;50:6073–6080. doi: 10.1021/bi200699x. [DOI] [PubMed] [Google Scholar]
- 58.Bosello M, Robbel L, Linne U, Xie X, Marahiel MA. J Am Chem Soc. 2011;133:4587–4595. doi: 10.1021/ja1109453. [DOI] [PubMed] [Google Scholar]
- 59.Giessen TW, Franke KB, Knappe TA, Kraas FI, Bosello M, Xie X, Linne U, Marahiel MA. J Nat Prod. 2012;75:905–914. doi: 10.1021/np300046k. [DOI] [PubMed] [Google Scholar]
- 60.Reeve AM, Breazeale SD, Townsend CA. J Biol Chem. 1998;273:30695–30703. doi: 10.1074/jbc.273.46.30695. [DOI] [PubMed] [Google Scholar]
- 61.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. The John Innes Foundation; Norwich UK: 2000. [Google Scholar]
- 62.Weisburg WG, Barns SM, Pelletier DA, Lane DJ. J Bacteriol. 1991;173:697–703. doi: 10.1128/jb.173.2.697-703.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden T. BMC Bioinf. 2012;13:134–145. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.