

Abstract
Background & Aims
Genetic studies indicate that distinct signaling modulators are each necessary but not individually sufficient for embryonic hepatocyte survival in vivo. Nevertheless, how signaling players are interconnected into functional circuits and how they coordinate the balance of cell survival and death in developing livers are still major unresolved issues. In the present study, we examined the modulation of the p53 pathway by HGF/Met in embryonic livers.
Methods
We combined pharmacological and genetic approaches to biochemically and functionally evaluate p53 pathway modulation in primary embryonic hepatocytes and in developing livers. RT-PCR arrays were applied to investigate the selectivity of p53 transcriptional response triggered by Met.
Results
Met recruits p53 to regulate the liver developmental program, by qualitatively modulating its transcriptional properties: turning on the Mdm2 survival gene, while keeping death and cell-cycle arrest genes Pmaip1 and p21 silent. We investigated the mechanism leading to p53 regulation by Met and found that Abl and p38MAPK are required for p53 phosphorylation on S389, Mdm2 upregulation, and hepatocyte survival. Alteration of this signaling mechanism switches p53 properties, leading to p53-dependent cell death in embryonic livers. RT-PCR array studies affirmed the ability of the Met-Abl-p53 axis to modulate the expression of distinct genes that can be regulated by p53.
Conclusions
A signaling circuit involving Abl and p38MAPK is required downstream of Met for the survival of embryonic hepatocytes, via qualitative regulation of the p53 transcriptional response, by switching its proapoptotic into survival properties.
Keywords: RTK signaling in vivo, Cell survival, Embryonic hepatocytes, HGF/Met, Abl, p53 Signaling
Introduction
Cell fate during development is regulated by environmental signals integrated through the function of several membrane-bound signaling molecules, including receptor tyrosine kinases (RTKs) [1]. RTK activation is tightly monitored during embryogenesis and in healthy adult tissues, as their aberrant function can cause degenerative pathologies or cancer. Conversely, reactivation of RTK signaling for survival and repair occurs frequently in cells undergoing degenerative processes [2]. Critical signaling circuits modulating developmental processes by RTKs, are often reemployed during regenerative processes (in a controlled manner) or in oncogenic events (through over-activation). Therefore, identification of these circuits can enlighten “druggable” signals in order to modulate RTK function in human diseases and re-establish proper tissue homeostasis.
We are interested in identifying common functional requirements of RTK signaling employed during developmental and pathological processes. The hepatocyte growth factor (HGF)/Met system is an interesting model since it regulates imperative outcomes during both development [3–10] and regenerative processes [11–14]. Our genetic studies had previously demonstrated that HGF/Met acted on selected signaling pathways to trigger qualitatively distinct biological outcomes in different tissues [5,10,15]. Intriguingly, for some developmental responses in specific cells, HGF/Met coordinates multiple signaling actors. This is illustrated by HGF/Met-mediated survival of embryonic hepatocytes, which requires a complex intracellular signaling network, as evidenced by loss-of-function genetic mutations. Using knock-in mice carrying signaling mutant versions of Met, we demonstrated that embryonic hepatocyte survival is established by distinct modulators that are each necessary, but not individually sufficient for liver development [9,15,16].
Here we have examined how Met signaling influences the p53 pathway in the developing liver and found that Met recruits p53 by qualitatively modulating its transcriptional properties. This function involves upregulation of the Mdm2 survival gene, while keeping death and cell-cycle arrest genes inactive. This regulation of p53 by Met involves a signaling circuit in which Abl activates p38MAPK, leading to p53 phosphorylation on S389. Consistent with these results, both Abl and p38αβ mutant livers show enhanced hepatocyte death, accompanied by phosphorylation of p53 on Ser18 and a switch of p53 function into pro-apoptotic properties.
materials and methods
Additional procedures are available as Supplementary material.
Protein extracts, immunoblotting, and immunoprecipitation
Cell extracts and Western blots were performed as previously described [9,16].
Mice
Experiments using animals were performed in accordance with the European Community Council Directive of 24.11.1986 on the protection of animals used for experimental purposes (86/609/EEC).
Statistical analysis
Results were expressed as mean ± s.e.m. Quantification of biological assays was analyzed by Student-t test. Statistical significance was defined as n.s., p >0.05;*p <0.05; **p <0.01; ***p <0.001.
Results
Met selectively upregulates the p53 target gene Mdm2, but not p21 and Pmaip1, in embryonic hepatocytes
To investigate HGF/Met modulation of the p53 pathway in developing livers, we followed the expression of p53 target genes regulating survival, cell-cycle arrest, and apoptosis [17,18]. HGF treatment selectively increased mRNA levels of Mdm2 (survival), but not of p21 (cell-cycle arrest) and Pmaip1 (Noxa; apoptosis) in embryonic hepatocytes (Fig. 1A, Supplementary Fig. 1A). Upregulation of Mdm2 protein levels was also detected upon HGF stimulation and was prevented by the transcription inhibitor actinomycinD (ActD) or by the Met inhibitor SU11274 (Fig. 1B and C). The requirement of p53 for Mdm2 upregulation by Met was then assessed through pharmacological and genetic studies. Pharmacological inhibition of p53 with pifithrin-α prevented Mdm2 upregulation by HGF (Fig. 1D). Moreover, upregulation of Mdm2 mRNA and protein levels by HGF did not occur in p53−/− embryonic hepatocytes (Fig. 1E and F).
Fig. 1. Qualitative regulation of p53 target genes by HGF/Met in embryonic hepatocytes correlates with its phosphorylation on S389.

(A) Quantitative RT-PCR analysis of Mdm2, Pmaip1, and p21 transcripts with/without HGF (H) stimulation, or 5FU. (B) Mdm2, but not p21 or Pmaip1, upregulation by HGF. (C) Impaired Mdm2 protein upregulation by ActD or SU11274 (SU). (D) Dose-dependent impairment of Mdm2 upregulation by pifithrin-α. (E and F) HGF stimulation does not increase Mdm2 mRNA and protein in p53-null cells. (G) HGF treatment leads to p53 phosphorylation on S389, but not on S18. ActD + TNFα stress treatment upregulates p53, pS18-p53, and pS389-p53 levels. *p <0.05; **p <0.01; ***p <0.001.
We next addressed how p53 may be controlled to enable selective Mdm2 transcription in cells exposed to HGF. Basal levels of p53 in embryonic hepatocytes did not significantly change following HGF treatment (Fig. 1G). Post-translational modifications of p53, including multisite phosphorylation, are known to affect its transcriptional activity [17,19]. We found that HGF treatment led to p53 phosphorylation selectively on S389 (Fig. 1G), corresponding to S392 in human p53. Phosphorylation of this Ser residue, located in the carboxyl-terminal region, may lead, in specific cellular contexts, to p53 oligomerization, enhancing its DNA binding capacity and transcriptional activity [17,19,20]. In contrast, no significant changes were observed on Ser18, corresponding to Ser15 in human p53, located in the amino-terminal region, which stabilizes p53 protein predominantly in response to stress stimuli (Fig. 1G). Thus, selective Mdm2 transcriptional regulation by HGF/Met correlated with p53 phosphorylation on S389.
p38MAPK signaling is required for p53 phosphorylation on S389 and Mdm2 upregulation by Met in embryonic hepatocytes
The qualitative regulation of p53 phosphorylation and Mdm2 upregulation by Met prompted us to identify the signaling mechanisms involved. As p38MAPK regulates phosphorylation of p53 on S392 in cancer cells [21,22], we investigated whether p38MAPK signaling was involved in p53 phosphorylation on S389 and Mdm2 expression by Met in embryonic hepatocytes. HGF stimulation led to p38MAPK phosphorylation in embryonic hepatocytes (Fig. 2A, Supplementary Fig. 1B), as reported in other cell types [22,23]. Intriguingly, whereas a peak of p38MAPK phosphorylation on T180Y182 was observed shortly after HGF stimulation, enhanced phosphorylation on Y182 was detected only at later time points, when p53 phosphorylation on S389 and Mdm2 upregulation were observed (Fig. 2A, Supplementary Fig. 1B). Treatment with the p38α/β inhibitor SB202190 interfered with Met-triggered p53 phosphorylation on S389 (Fig. 2B) and Mdm2 upregulation (Fig. 2C, Supplementary Fig. 1C).
Fig. 2. Impairment of p38α; p38β interferes with p53 phosphorylation on S389 and Mdm2 upregulation by Met in embryonic hepatocytes.

(A) Time course analysis of Mdm2, pS389-p53, pY182-p38, p-T180Y182-p38, and p53 following HGF stimulation. (B and C) Inhibition of p38α; p38β signaling by SB202190 (SB) affects HGF-mediated upregulation of pS389-p53 and Mdm2. (D and E) Drastic reduction of HGF-induced (H) pS389-p53 (18 h) and Mdm2 (24 h) upregulation in p38α−/−; p38β−/− hepatocytes. (F and G) Increase in cleaved-caspase 3-positive cells parallels the rise in pS18-p53-positive cells in p38α−/−; p38β−/− livers compared to wild type. Metd/d mutants were used as controls. Quantification of cleaved-caspase 3- and pS18-p53-positive cells is shown (cleaved-caspase 3: n = 3; 2.93 ± 0.37; p <0.001; pS18-p53: n = 4; 2.84 ± 0.28; p <0.001); ***p <0.001. (This figure appears in color on the web.)
The requirement of p38MAPKs for both p53 phosphorylation and Mdm2 upregulation by Met was further investigated by genetic studies using hepatocytes from double p38α−/−;p38β−/− knock-out embryos. Notably, HGF failed to induce p53 phosphorylation on S389 and Mdm2 upregulation in p38α−/−;p38β−/− embryonic hepatocytes (Fig. 2D and E). These results demonstrated that p38MAPK signaling was required downstream of Met for p53 phosphorylation on S389 and Mdm2 upregulation in embryonic hepatocytes.
We previously showed in cancer cells that p53 phosphorylation on S392 is required for its enhanced binding to the Mdm2 promoter following HGF stimulation [21,22]. Therefore, we next assayed whether p38MAPK-dependent p53 phosphorylation on S389 enhanced its DNA binding, by performing chromatin immunoprecipitation studies in embryonic hepatocytes. HGF increased the levels of p53 bound to the Mdm2 promoter, whereas p38MAPK inhibition drastically reduced this binding (Supplementary Fig. 1D). In contrast, no differences were observed on p53 binding to the Cyclin B2 promoter (Supplementary Fig. 1D), showing the specificity of p53 DNA binding modulation.
The requirement of p38MAPK for this process was intriguing as we have recently reported that p38α−/−;p38β−/− mutant embryos show enhanced cell death in livers [24]. Consistently, we found an increased number of cleaved-caspase 3-positive cells in p38α−/−;p38β−/− embryos, similar to Metd/d signaling mutants (Fig. 2F) [5]. This was accompanied by increased levels of pS18-p53-positive cells in p38α−/−;p38β−/− embryos (Fig. 2G), indicating that altered p38MAPK signaling consequently led to a switch in p53 function towards pro-apoptosis. Together, these findings indicated that p38MAPK signaling regulated phosphorylation of p53 on S389 and embryonic hepatocyte survival by HGF/Met, thus preventing p53 stress-signaling properties.
Abl is required for p53 phosphorylation on S389 and Mdm2 upregulation by Met in embryonic hepatocytes
We next sought to investigate the pathway that connects Met to p38MAPK activation and p53-induced upregulation of Mdm2. We first explored the requirement of the MEK, Akt, mTOR, and NFκB pathways, which can all influence p53 functions in other cell types. None of them were required for Met-triggered Mdm2 upregulation (data not shown). We also excluded FAK and ENIGMA as being part of this pathway since they regulate Mdm2 ubiquitination properties and stability, respectively [25,26]. We next explored Abl involvement based mainly on two observations: Abl regulates p53 function in cell lines exposed to stress conditions [27], and contributes to cell survival downstream of RTKs in cancer cells [22,28].
We first assessed whether Abl was activated in embryonic hepatocytes following HGF stimulation and found strong Abl phosphorylation induced with a kinetics similar to that observed for increases in pY182-p38MAPK, pS389-p53, and Mdm2 levels (Figs. 2A and 3A). Inhibition of Abl by imatinib or nilotinib interfered with Met-triggered Mdm2 upregulation as well as with p38MAPK and p53 phosphorylation on S389 in embryonic hepatocytes (Fig. 3B–E, and Supplementary Fig. 1B and E). Imatinib can also inhibit PDGFR and Kit, in addition to Abl, but none of them were expressed in the cells used in our studies (Supplementary Fig. 1F and G). We then assessed Abl requirement for HGF/Met-mediated embryonic hepatocyte survival. HGF pre-incubation prevented TNFα + ActD-triggered cell death (Fig. 3F) [16], whereas Abl inhibition abolished HGF-induced survival, similar to Met inhibition (Fig. 3F). Together, these findings indicate that Abl signaling downstream of Met is required for Mdm2 expression and survival of hepatocytes in culture.
Fig. 3. Abl is required for HGF-induced Mdm2 upregulation, pS389-p53, and survival of embryonic hepatocytes.
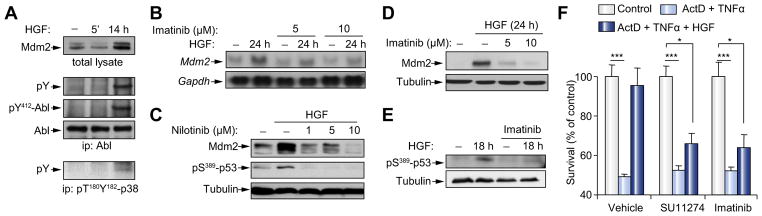
(A) Immunoprecipitation (ip) followed by Western blot analysis showing HGF-induced Abl phosphorylation on a Tyr residue (pY) in embryonic hepatocytes. Upregulation of Mdm2 and p-p38MAPK levels by HGF is shown. (B) Abl inhibition interferes with HGF-mediated upregulation of Mdm2 mRNA. (C–E) Impairment of Mdm2 upregulation and pS389-p53 by HGF in cells exposed to Abl inhibitors. (F) Pre-incubation with HGF (6 h) of embryonic hepatocytes prevents cell death triggered by TNFα + ActD treatment (18 h). HGF survival response is abolished in the presence of Met (SU11274: 1 μM) or Abl (imatinib: 10 μM) inhibitors. *p <0.05; ***p <0.001.
We next sought to clarify whether Abl regulated Mdm2 levels and hepatocyte survival in developing livers, where Mdm2 translational modulation by mTOR is required for hepatocyte survival [16]. This question was addressed in E12.5 livers dissected from imatinib-injected pregnant mice. Notably, pharmacological inhibition of Abl significantly reduced Mdm2 protein levels in E12.5 livers, caused cell death evidenced by a raise of cleaved-caspase 3-positive cells, and increased levels of pS18-p53-positive cells (Fig. 4A and B), highlighting a switch in p53 properties towards pro-apoptosis. Remarkably, Abl inhibition did not lead to cell death in p53−/− embryos (Fig. 4B, Supplementary Fig. 1H). Together, these findings demonstrated that alteration of the Met-Abl-p38MAPK axis led to cell death in a p53-dependent manner. Finally, we genetically investigated whether Abl signaling was required for hepatocyte survival in developing livers. No obvious defects were found in Abl−/− livers at developmental stages, possibly due to compensatory functions between Abl and its homologue Arg during embryogenesis. In contrast, livers of Abl−/− newborns were smaller (Fig. 4C) and often exhibited necrotic areas. Consistently, Abl−/− livers showed a higher number of cleaved-caspase 3- and pS18-p53-positive cells (Fig. 4D). Together, these findings provided pharmacological and genetic evidence that Abl signaling was required for hepatocyte survival, which depended on p53-induced Mdm2 upregulation. However, it is likely that the Abl/p38MAPK circuit is not sufficient, per se, to mediate hepatocyte survival, considering the complexity of the pathways required, as evidenced by loss-of-function genetic mutations.
Fig. 4. Abl impairment leads to Mdm2 downregulation and cell death in vivo.

(A) Mdm2, pS18-p53, and cleaved-caspase 3 levels from E12.5 livers dissected from imatinib-treated pregnant females (p = 0.03). (B) Abl inhibition increases cleaved-caspase 3- (n = 5; 4.71 ± 0.4, p <0.001), and pS18-p53-positive cells (n = 5; 3.08 ± 0.08, p <0.01). Similar levels of cleaved-caspase 3-positive cells were observed in imatinib-treated p53−/− mutants and non-injected embryos. (C) Abl−/− and wild type P4 livers. (D) Increased cleaved-caspase 3 (n = 6, 4.64 ± 1.14, p <0.01) and pS18-p53- (n = 3, 9.28 ± 1.9, p <0.01) positive cells in Abl−/− livers. (E) HGF/Met elicits qualitative transcriptional outcomes of genes known to be regulated by p53, which belong to distinct clusters. Group A: Abl-dependent HGF-regulated genes. Group B: genes inversely regulated by HGF or nilotinib/HGF. Group C: Abl-independent HGF-regulated genes. Group D: nilotinib-enhanced HGF-regulated genes. n.s., p >0.05; *p <0.05; **p <0.01. (This figure appears in color on the web.)
The Met-Abl-p53 axis regulates the expression of distinct genes known to be targeted by p53
We next assessed whether the Abl-p53 axis influenced gene expression by Met in embryonic hepatocytes. Expression levels of 84 genes, known to be p53 targets in several cellular systems, were analyzed by RT-PCR in HGF-treated or non-treated embryonic hepatocytes. We found that HGF treatment significantly modified the expression of 17 genes (greater than 50% change; p <0.05; Fig. 4E): expression of 5 genes was increased, whereas expression of 12 genes was reduced (Fig. 4E, and Supplementary Fig. 2). Consistent with the results shown in Fig. 1, the levels of Mdm2 were upregulated, those of Pmaip1 were downregulated, and those of p21 unchanged upon HGF stimulation, thus supporting the overall outcomes from the array. These results indicated that HGF/Met selectively regulated the expression of several known p53 targets.
We next asked whether transcriptional regulation of these genes required Abl signaling by analyzing their expression profile in HGF-treated cells either exposed or not to Abl inhibitor. Intriguingly, these studies showed that 5 out of the 17 genes regulated by HGF/Met, required Abl signaling (group A: Cdkn2a, Mdm2, Egr1, Jun, RelA; Fig. 4E), whereas the modulation of 6 genes was unchanged in the presence of nilotinib (group C: Bcl2, Pmaip1, Trp53bp2, Sfn, Brca1, Stat1; Fig. 4E). The remaining genes were either inversely regulated by HGF and nilotinib (group B: Gadd45a, Parc; Fig. 4E) or the effect of HGF was potentiated upon Abl inhibition (group D: Btg2, Tnf, E2f1, Ercc1; Fig. 4E). Inhibition of either Abl or p53 influenced gene expression in a similar manner (Supplementary Fig. 3A and B), further suggesting that the Abl-p53 axis may contribute to the selective gene expression regulation by HGF/Met.
By investigating the interactome network composed of the products of these p53 target genes modulated by HGF, we found that all but 3 are interconnected (Supplementary Figs. 4 and 5). By assessing their vicinity within this interactome, we noticed that proteins were very closely linked within groups A and C, as attested by the average shortest path length separating them (lA = 1.8 ± 0.74, lC = 2.33 ± 0.78, linteractome = 3.02 ± 0.8). This analysis suggested that proteins encoded by genes belonging to a single group (group A or C) might be involved in a common process. Investigating the subnetworks formed by the gene products A and C and their shared respective interactors indicated a correlation between their topological proximity and their functional homogeneity (Supplementary Fig. 4A and B). According to Biological Process Gene Ontology, the most statistically over-represented terms for subnetwork A and C are “positive regulation of gene expression” (enrichment 8,7; p val = 3.2 E−7) and “cell death” (enrichment 8,8; p val = 1.3 E−7; Supplementary Figs. 4 and 6), respectively. However, this analysis may be biased by the restricted number of p53 target genes (84) used in this study. Thus, HGF/Met signaling makes use of the Abl-p53 axis for selective gene transcriptional regulation in embryonic hepatocytes.
Discussion
Precise control of cell number by regulating their survival capability is a key step in cell fate determination. Survival deregulation can lead to developmental or degenerative diseases as well as cancer. Our data reveal a pathway that HGF/Met uses to control hepatocyte survival in developing livers. The mechanism involved highlights three key signaling aspects. First, Abl is required downstream of RTKs for cell survival during mouse embryogenesis. Second, in the absence of stress stimuli, p53 is recruited by RTKs to participate in embryonic hepatocyte survival, by qualitatively modulating p53 transcriptional properties. These findings raise the provocative idea that RTKs such as Met can instruct p53 to participate in developmental programs. Third, the genes known to be regulated by p53 that are modulated by HGF/Met belong to different clusters, which might be functionally distinct according to their interacting partners. The outcomes of our developmental studies may be relevant to liver regeneration as Met is also a key modulator of this process.
Abl functions during development: an essential player downstream of the Met RTK
Genetic studies have highlighted that the tyrosine kinases Abl and Arg play fundamental roles during mouse embryogenesis [29–31]. The uncovering of a full spectrum of their developmental functions in vertebrates has been partially limited by two main reasons. First, both Abl and Arg are widely expressed during development, with overlapping functions. Indeed, single mutants have subtle defects at birth, whereas double Abl- and Arg-deficient embryos die around E11 [29]. Second, most of the Abl mutants display mortality at perinatal stages [30–32], at the time when Arg expression becomes more restricted to neural tissues. Strategies to bypass such lethality allowed the identification of new functions of Abl during cardiac and cerebellum development [33,34]. In vitro studies have shown that Abl activation occurs downstream of PDGFR, IGFR, and ErbB family members, and contributes to proliferation, invasion, survival, cytoskeletal reorganization, cell motility, and response to oxidative stress/DNA damage, depending on cell types [35–39]. Whether Abl participates in the RTK-regulated developmental programs has not been reported, so far, although Abl is known to contribute to bone or heart development, which are modulated by RTKs [32,33]. For the first time, our findings show the direct contribution of Abl to developmental events elicited by RTKs. As Abl participates in distinct biological outcomes in cultured cells, its role during RTK-controlled embryogenesis is most likely imposed by the cellular context in which it acts.
Recruiting p53 to developmental processes through its qualitative tuning by the Met RTK
In response to a wide variety of signals, p53 regulates the transcription of several genes involved in life and death cell fate decision, depending on the cell type and the extent of the stimuli [17,18]. New roles of p53, unrelated to stress, have also emerged in developmental, stemness, and reprogramming processes [40]. Thus, rather than being a simple tumor suppressor gene, p53 appears to be integrated in instructive outputs to ensure proper biological responses. The identification of p53 functions in absence of stress, such as during embryogenesis, has been limited as p53−/− mice develop normally. Such dispensable feature is consistent with the proposed “guardian” role of p53, which maintains it in an inactive state in absence of genetic damage or altered input, but makes it active when signaling networks function in an inappropriate way. We have previously shown that p53 regulates cell death in Met embryos and demonstrated that functional Mdm2 is required to restrain its stress-sensor properties [16]. The transcriptional regulation of Mdm2 via the Abl-p38MAPK pathway highlighted in this study further emphasizes the importance of fine-tuning p53 functions by Met in developmental programs such as hepatocyte survival. Intriguingly, at least in embryonic cells, dysfunction of this pathway by impairment of one of its signaling components switches p53 into an active modulator of cell stress and death. This model is supported by our findings showing: a) p53 phosphorylation on Ser18 in abl−/− or p38α−/−; p38β−/− mutants compared to controls, b) enhanced cleaved-caspase3-positive cells when Abl signaling is impaired in wild-type, but not in p53−/− mutant embryos.
Our results show that the Met-Abl-p38MAPK pathway regulates p53 phosphorylation on S389. This post-translational modification correlates with p53 binding to specific promoters (Mdm2, but not Cyclin B2) and with qualitative transcriptional read-outs of p53 target genes. RT-PCR array studies allowed us to conclude that HGF/Met modulates the expression of a restricted number of genes known to be targeted by p53 through the Abl-p53 axis. Notably, different subsets of HGF-regulated genes could be highlighted, differentially depending on Abl signaling. Based on our interactome studies, it is tempting to speculate that these groups of proteins together with their interaction partners have distinct functional characteristics. As gene activation and interactome network are redefined by the cellular context, future studies will provide mechanistic evidence of these relevant interactomes in embryonic hepatocytes.
Abl and p53 crosstalk: switching outcomes and roles according to the context
Previous studies using stressed cultured cells showed that Abl acts on p53 signaling by contributing to its pro-apoptotic function [27]. However, the oncogenic BCR-ABL leads toMdm2upregulation and survival [41], indicating that BCR-ABL has a pro-survival role in cells not under stress conditions. The role of Abl in solid tumors has remained largely unknown, mainly because mutated forms of Abl have been found only in a few cases [35]. We have recently shown that Abl interconnects oncogenic RTK signaling to p53 core pathways in cancer cells [22]. Notably, there is a significant correlation between wild type p53 phosphorylation on S392, Mdm2 upregulation, and activated Met in human hepatocellular carcinomas [22]. It is tempting to speculate that the Met-Abl-p53 circuit represents a robust signaling path for cell survival, employed during development and revisited by cancer cells. In a constant effort to identify new agents for treatment of liver pathologies, lessons from development might be instrumental to extend the effective use of available agents for molecularly targeted therapies.
Supplementary Material
Acknowledgments
Financial support
This work was funded by INCa, ARC, FRM, AFM, FdF, Fondation Bettencourt-Schueller to F.M.; by AIRC (IG10590) to D.B.; by MICINN, Fundación BBVA to A.R.N. A.F. was supported by INCa and ARC fellowships; D.B. and V.S. were also supported by Fondazione Santa Lucia, FIRC.
We are particularly grateful to R. Dono, F. Conti, F. Helmbacher, S. Soddu, G. Cesareni, and all members of our labs for helpful discussions and comments. We thank P. Dubreuil for TF1 cells, anti-Kit antibodies; S. Candeias, A. Carrier, R. Tomasini, P. N’Guessan for p53 mice; Novartis-Pharma-AG (Switzerland) for imatinib, nilotinib; V. Girod-David, L. Jullien, and staff at the IBDML for excellent help with mouse husbandry; M. Iche-Torres for help with RNA quality control; the IBDML imaging platform.
Abbreviations
- RTKs
receptor tyrosine kinases
- HGF
hepatocyte growth factor
- 5FU
5-fluoro-uracile
- RU
relative units
- ActD
actinomycinD
- AU
arbitrary units
Footnotes
Conflict of interest
The Authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jhep.2012.07.044.
References
- 1.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snider WD, Zhou FQ, Zhong J, Markus A. Signaling the pathway to regeneration. Neuron. 2002;35:13–16. doi: 10.1016/s0896-6273(02)00762-6. [DOI] [PubMed] [Google Scholar]
- 3.Knudsen BS, Vande Woude G. Showering c-MET-dependent cancers with drugs. Curr Opin Genet Dev. 2008;18:87–96. doi: 10.1016/j.gde.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 5.Maina F, Casagranda F, Audero E, Simeone A, Comoglio P, Klein R, et al. Uncoupling of Grb2 from the Met receptor in vivo reveals complex roles in muscle development. Cell. 1996;87:531–542. doi: 10.1016/s0092-8674(00)81372-0. [DOI] [PubMed] [Google Scholar]
- 6.Maina F, Hilton MC, Andres R, Wyatt S, Klein R, Davies AM. Multiple roles for hepatocyte growth factor in sympathetic neuron development. Neuron. 1998;20:835–846. doi: 10.1016/s0896-6273(00)80466-3. [DOI] [PubMed] [Google Scholar]
- 7.Maina F, Hilton MC, Ponzetto C, Davies AM, Klein R. Met receptor signaling is required for sensory nerve development and HGF promotes axonal growth and survival of sensory neurons. Genes Dev. 1997;11:3341–3350. doi: 10.1101/gad.11.24.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helmbacher F, Dessaud E, Arber S, de Lapeyriere O, Henderson CE, Klein R, et al. Met signaling is required for recruitment of motor neurons to PEA3-positive motor pools. Neuron. 2003;39:767–777. doi: 10.1016/s0896-6273(03)00493-8. [DOI] [PubMed] [Google Scholar]
- 9.Moumen A, Ieraci A, Patane S, Sole C, Comella JX, Dono R, et al. Met signals hepatocyte survival by preventing Fas-triggered FLIP degradation in a PI3k-Akt-dependent manner. Hepatology. 2007;45:1210–1217. doi: 10.1002/hep.21604. [DOI] [PubMed] [Google Scholar]
- 10.Lamballe F, Genestine M, Caruso N, Arce V, Richelme S, Helmbacher F, et al. Pool-specific regulation of motor neuron survival by neurotrophic support. J Neurosci. 2011;31:11144–11158. doi: 10.1523/JNEUROSCI.2198-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A. 2004;101:4477–4482. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borowiak M, Garratt AN, Wustefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proc Natl Acad Sci U S A. 2004;101:10608–10613. doi: 10.1073/pnas.0403412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genestine M, Caricati E, Fico A, Richelme S, Hassani H, Sunyach C, et al. Enhanced neuronal Met signalling levels in ALS mice delay disease onset. Cell Death Dis. 2011;2:e130. doi: 10.1038/cddis.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tonges L, Ostendorf T, Lamballe F, Genestine M, Dono R, Koch JC, et al. Hepatocyte growth factor protects retinal ganglion cells by increasing neuronal survival and axonal regeneration in vitro and in vivo. J Neurochem. 2011;117:892–903. doi: 10.1111/j.1471-4159.2011.07257.x. [DOI] [PubMed] [Google Scholar]
- 15.Maina F, Pante G, Helmbacher F, Andres R, Porthin A, Davies AM, et al. Coupling Met to specific pathways results in distinct developmental outcomes. Mol Cell. 2001;7:1293–1306. doi: 10.1016/s1097-2765(01)00261-1. [DOI] [PubMed] [Google Scholar]
- 16.Moumen A, Patane S, Porras A, Dono R, Maina F. Met acts on Mdm2 via mTOR to signal cell survival during development. Development. 2007;134:1443–1451. doi: 10.1242/dev.02820. [DOI] [PubMed] [Google Scholar]
- 17.Vousden KH, Lane DP. P53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 18.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto M, Furihata M, Ohtsuki Y. Posttranslational phosphorylation of mutant p53 protein in tumor development. Med Mol Morphol. 2006;39:79–87. doi: 10.1007/s00795-006-0320-0. [DOI] [PubMed] [Google Scholar]
- 20.Bruins W, Bruning O, Jonker MJ, Zwart E, van der Hoeven TV, Pennings JL, et al. The absence of S389 phosphorylation in p53 affects the basal gene expression level of many p53-dependent genes and alters the biphasic response to UV exposure in mouse embryonic fibroblasts. Mol Cell Biol. 2008;28:1974–1987. doi: 10.1128/MCB.01610-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cox ML, Meek DW. Phosphorylation of serine 392 in p53 is a common and integral event during p53 induction by diverse stimuli. Cell Signal. 2010;22:564–571. doi: 10.1016/j.cellsig.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 22.Furlan A, Stagni V, Hussain A, Richelme S, Conti F, Prodosmo A, et al. Abl interconnects oncogenic Met and p53 core pathways in cancer cells. Cell Death Differ. 2011;18:1608–1616. doi: 10.1038/cdd.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Segarra J, Balenci L, Drenth T, Maina F, Lamballe F. Combined signaling through ERK, PI3K/AKT, and RAC1/p38 is required for Met-triggered cortical neuron migration. J Biol Chem. 2006;281:4771–4778. doi: 10.1074/jbc.M508298200. [DOI] [PubMed] [Google Scholar]
- 24.del Barco Barrantes I, Coya JM, Maina F, Arthur JS, Nebreda AR. Genetic analysis of specific and redundant roles for p38alpha and p38beta MAPKs during mouse development. Proc Natl Acad Sci U S A. 2011;108:12764–12769. doi: 10.1073/pnas.1015013108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung CR, Lim JH, Choi Y, Kim DG, Kang KJ, Noh SM, et al. Enigma negatively regulates p53 through MDM2 and promotes tumor cell survival in mice. J Clin Invest. 2010;120:4493–4506. doi: 10.1172/JCI42674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levav-Cohen Y, Goldberg Z, Zuckerman V, Grossman T, Haupt S, Haupt Y. C-Abl as a modulator of p53. Biochem Biophys Res Commun. 2005;331:737–749. doi: 10.1016/j.bbrc.2005.03.152. [DOI] [PubMed] [Google Scholar]
- 28.Srinivasan D, Sims JT, Plattner R. Aggressive breast cancer cells are dependent on activated Abl kinases for proliferation, anchorage-independent growth and survival. Oncogene. 2008;27:1095–1105. doi: 10.1038/sj.onc.1210714. [DOI] [PubMed] [Google Scholar]
- 29.Koleske AJ, Gifford AM, Scott ML, Nee M, Bronson RT, Miczek KA, et al. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21:1259–1272. doi: 10.1016/s0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- 30.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 31.Schwartzberg PL, Stall AM, Hardin JD, Bowdish KS, Humaran T, Boast S, et al. Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell. 1991;65:1165–1175. doi: 10.1016/0092-8674(91)90012-n. [DOI] [PubMed] [Google Scholar]
- 32.Li B, Boast S, de los Santos K, Schieren I, Quiroz M, Teitelbaum SL, et al. Mice deficient in Abl are osteoporotic and have defects in osteoblast maturation. Nat Genet. 2000;24:304–308. doi: 10.1038/73542. [DOI] [PubMed] [Google Scholar]
- 33.Qiu Z, Cang Y, Goff SP. Abl family tyrosine kinases are essential for basement membrane integrity and cortical lamination in the cerebellum. J Neurosci. 2010;30:14430–14439. doi: 10.1523/JNEUROSCI.2861-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiu Z, Cang Y, Goff SP. C-Abl tyrosine kinase regulates cardiac growth and development. Proc Natl Acad Sci U S A. 2010;107:1136–1141. doi: 10.1073/pnas.0913131107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sirvent A, Benistant C, Roche S. Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biol Cell. 2008;100:617–631. doi: 10.1042/BC20080020. [DOI] [PubMed] [Google Scholar]
- 36.Woodring PJ, Hunter T, Wang JY. Regulation of F-actin-dependent processes by the Abl family of tyrosine kinases. J Cell Sci. 2003;116:2613–2626. doi: 10.1242/jcs.00622. [DOI] [PubMed] [Google Scholar]
- 37.Plattner R, Kadlec L, DeMali KA, Kazlauskas A, Pendergast AM. C-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999;13:2400–2411. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srinivasan D, Plattner R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006;66:5648–5655. doi: 10.1158/0008-5472.CAN-06-0734. [DOI] [PubMed] [Google Scholar]
- 39.Ganguly SS, Fiore LS, Sims JT, Friend JW, Srinivasan D, Thacker MA, et al. C-Abl and Arg are activated in human primary melanomas, promote melanoma cell invasion via distinct pathways, and drive metastatic progression. Oncogene. 2012;31:1804–1816. doi: 10.1038/onc.2011.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krizhanovsky V, Lowe SW. Stem cells: the promises and perils of p53. Nature. 2009;460:1085–1086. doi: 10.1038/4601085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goetz AW, van der Kuip H, Maya R, Oren M, Aulitzky WE. Requirement for Mdm2 in the survival effects of Bcr-Abl and interleukin 3 in hematopoietic cells. Cancer Res. 2001;61:7635–7641. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.