

Abstract
We have previously shown that the antiprogestin and antiglucocorticoid mifepristone inhibits the growth of ovarian cancer cells. In this work, we hypothesized that cellular stress caused by mifepristone is limited to cytostasis and that cell killing is avoided as a consequence of the persistent activity of the PI3K/Akt survival pathway. To investigate the role of this pathway in mifepristone-induced growth inhibition, human ovarian cancer cells of various histological subtypes and genetic backgrounds were exposed to cytostatic doses of mifepristone in the presence or absence of the PI3K inhibitor, LY294002. The activation of Akt in ovarian cancer cells, as marked by its phosphorylation on Ser473, was not modified by cytostatic concentrations of mifepristone, but it was blocked upon treatment with LY294002. The combination mifepristone/LY294002, but not the individual drugs, killed ovarian cancer cells via apoptosis, as attested by genomic DNA fragmentation and cleavage of caspase-3, and the concomitant downregulation of antiapoptotic proteins Bcl-2 and XIAP. From a pharmacological standpoint, when assessing cell growth inhibition using a median-dose analysis algorithm, the interaction between mifepristone and LY294002 was synergistic. The lethality caused by the combination mifepristone/LY294004 in 2-dimensional cell cultures was recapitulated in organized, 3-dimensional spheroids. This study demonstrates that mifepristone and LY294002 when used individually cause cell growth arrest; yet, when combined, they cause lethality.
Keywords: mifepristone, LY294002, ovarian cancer, growth inhibition, PI3K/Akt pathway, antiprogestin, antiglucocorticoid
Introduction
Mifepristone has been used for termination of early pregnancy because of its capacity to work as an antiprogestin blocking uterine progesterone receptors (reviewed by Beal and Simmonds).1 Recently, in part due to its well-documented antiglucocorticoid properties,2 mifepristone received approval to treat endogenous Cushing’s disease;3 however, this drug possesses multipharmacological effects and has been used off-label to treat pathologies such as endometriosis, meningioma, major depression with psychotic features, and Alzheimer’s disease (reviewed by Benagiano et al).4 In oncology, mifepristone was reported to have potent antiproliferation effects in cell lines representing cancers of the breast (reviewed by Lanari et al),5 endometrium,6,7 cervix,8,9 prostate,10,11 gastrointestinal tract,12 brain,13,14 bone,14 and ovary.15
We and others demonstrated that mifepristone blocks the growth of ovarian cancer cells of different genetic backgrounds and histopathological classifications.15–17 In addition, we also showed that the growth inhibition induced by mifepristone in ovarian cancer cells is independent of the status of the p53 tumor suppressor as well as of the cellular sensitivity to standard platinum-based chemotherapy.18 From a clinical/translational perspective, we have found that cytostatic doses of mifepristone were efficient in blocking the regrowth of ovarian cancer cells that escape cisplatin or cisplatin/paclitaxel chemotherapy tailored to mimic clinically relevant conditions.19,20
When used in vitro at clinically achievable doses, the cytotoxicity of mifepristone towards ovarian cancer cells is limited to cytostasis, whereas to cause lethality, suprapharmacological concentrations of the drug are needed.21 Envisioning the translatability of mifepristone to the oncology clinic, and knowing from pharmacokinetic studies that only cytostatic concentrations of the steroid can be achieved when given orally,22–25 we questioned whether the antiovarian cancer activity of mifepristone can be potentiated by combining it with another cytostatic drug that could disengage a survival pathway operating in the presence of mifepristone and, consequently, limiting its toxicity. We focused on the phosphatidylinositol-3-kinase (PI3K)/Akt survival pathway, which is hyperactive in more than 50% of ovarian cancers.26,27 The most prevalent types of ovarian cancers are known to possess aberrations in the PI3K pathway with all three isoforms of downstream Akt highly amplified.28 Approximately 40% of low grade type I ovarian cancers composed of low grade serous, mucinous, endometrioid, and clear-cell histotypes have activating mutations of PIK3CA, whereas in high-grade type II ovarian cancers composed of high-grade serous and undifferentiated histotypes, 46% of patients have genetic amplifications consistent with increased pathway activity.28 The over-activation of the PI3K/Akt signaling pathway provides survival advantage to ovarian cancer whereas its inhibition enhances the efficiency of radiotherapies and chemotherapies.29
In this work, we hypothesized that in ovarian cancer cells, the toxicity of mifepristone is limited to cytostasis and that cell killing is avoided due to the persistent activity of the PI3K/Akt survival pathway. We postulated that simultaneous use of mifepristone and a blocker of PI3K/Akt signaling will cause a synergistic lethal interaction. By using 2-dimensional (2-D) and 3-dimensional (3-D) ovarian cancer cell culture systems, we provide preclinical proof-of-principle that a cytostatic dose of mifepristone, when combined with a cytostatic dose of the PI3K inhibitor LY294002, has a synergistic pharmacological interaction leading to ovarian cancer cell lethality.
Materials and Methods
Cell culture and reagents
The human ovarian carcinoma cell lines OV2008, A2780, A2780/CP70, and IGROV-1 were obtained in 2003 from Stephen Howell, MD (University of California, San Diego),30 and cultured as we previously described.18,19 The PC-3 prostate cancer cells were obtained from ATCC and maintained in RPMI 1640 without L-Glutamine (Mediatech, Inc., Manassas, VA), supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA), 10 mM Hepes (Mediatech), 1 mM sodium pyruvate (Mediatech), 4 mM L-Glutamine (Mediatech), 1X non-essential amino acids (Mediatech), 100 IU penicillin (Mediatech), and 100 μg/mL streptomycin (Mediatech). All cell lines were cultured at 37°C in a humidified atmosphere in the presence of 5% CO2. Treatment of cells with mifepristone (Sigma Chemical Co., St. Louis, MO) was done from a 20,000 μM stock solution in DMSO, which was maintained at −20 °C. Mifepristone was supplemented during treatments every 48 hours. LY294002 (Cayman Chemical, Ann Arbor, MI) was suspended in a stock solution of DMSO to a final concentration of 16,270 μM. LY294002 was supplemented during treatments every 12 hours directly to the media, except for cells grown in 3-D in which supplementation was every 24 hours. The maximal concentration of DMSO in medium was less than 0.1% (v/v).
Cell number and viability
The number and viability of cultured cells were determined using microcapillary cytometry as we previously detailed.18,21 Briefly, cultures were washed with PBS, trypsinized, and suspended in the Via-Count reagent (Guava Technologies, Hayward, CA). Number of cells and their viability were then studied with the Guava ViaCount application in the Guava EasyCyte Mini cytometer (Guava Technologies). This assay allows calculation of absolute number of cells and, at the same time, discriminates viable from nonviable cells based upon a differential permeability of two DNA-binding dyes.
Live/dead viability/cytotoxicity assay
Cells were distributed equally in 8-well chamber slides and treated for the designated experimental times. The media were subsequently removed, and adherent cells were washed with PBS and incubated for 45 minutes at room temperature in the presence of 2 μM calcein AM (Molecular Probes, Eugene, OR) and 4 μM ethidium homodimer 1 (EthD-1) (Molecular Probes). Images were obtained with a confocal Olympus FV1000 microscope with FluoView® software (Olympus America, Center Valley, PA). Calcein formed from calcein AM is well retained within live cells, producing an intense uniform green fluorescence. EthD-1 undergoes enhancement of fluorescence upon binding to DNA, producing a bright red fluorescence in dead cells, but it is excluded by the intact plasma membrane of live cells. To validate the assay, cells were permeabilized with 0.1% saponin (Acros Organics, Geel, Belgium) for 5 minutes before being exposed to calcein AM and EthD-1.
Clonogenic survival assay
Clonogenicity was assessed 24 hours after exposure to the drugs using 500 cells in 6-well plates and cultured in drug-free medium for 7 days until colonies were clearly visualized. Colonies were stained and counted using an inverted microscope as we previously described.19
Determination of sub-G0/G1 DNA content
Following treatment, cells were pelleted, fixed, and stained with propidium iodide (Sigma), and cell cycle distribution was assessed by microcapillary cytometry as we previously reported in detail.19 The sub-G0/G1 regions of the cell cycle histogram were considered the hypodiploid DNA contents.
DNA fragmentation
Fragmentation of genomic DNA was studied as we previously described.31 Briefly, genomic DNA was isolated, separated in agarose gels, and images were obtained with a Typhoon Fluorescence imaging system (Amersham Biosciences Corp., Piscataway, NJ) after impregnation with SYBR Gold nuclei acid gel stain (Molecular Probes).
SDS-PAGE and western blotting
Whole cell protein extracts were obtained, separated by SDS-PAGE, transferred to PVDF membranes, and probed as previously described.19 The primary antibodies utilized were phospho-Akt (S473; 1:1,000; #4058 Cell Signaling Technology Inc., Danvers, MA), pan Akt (1:2,000; #2920 Cell Signaling Technology, Inc.), caspase 3 (#9662; 1:1000; #9662 Cell Signaling Technology, Inc.), Bcl-2 (1:1,000; sc-492 Santa Cruz Biotechnology, Santa Cruz, CA), XIAP (1:500; #2042 Cell Signaling Technology Inc.), β-actin (clone AC-15; 1:10,000; Sigma), and GAPDH (ab9485; 1:10,000; Abcam, Cambridge, MA).
Drug interaction analysis
An equal number of cells in triplicate was loaded in 6-well plates and allowed to attach for 24 hours. Treatment of cells included each agent examined alone or in a minimum of 4 combinations. At the conclusion of the experiment, the total number of cells was obtained from the various treatment groups by microcytometry.18 To characterize the pharmacological interaction between LY294002 and mifepristone, we used the CalcuSyn software (Biosoft, Cambridge, UK), which utilizes the combination index (CI) as a method for quantifying drug synergism.32,33 The CI was calculated for a two-drug combination using a variable concentration ratio and utilizing as fraction affected (Fa) level the percent growth inhibition divided by 100. To the specified drug association, CI = 0.9–1.1 denotes an additive effect, CI = 0.7–0.9 indicates slight synergism, CI = 0.3–0.7 indicates strong synergism, whereas CI > 1.1 indicates antagonism. Drug interaction between LY294002 and mifepristone was expressed as normalized isobolograms. The median effect dose of each single drug (Dm, usually depicted as IC50) and the dose reduction index (DRI) were also calculated as previously described.19
Three-dimensional cell culture
Cells were cultured using a procedure adapted from previously published protocols.34 One day prior to the plating of cells, basement membrane matrix growth factor reduced Matrigel (BD#354230, BD Biosciences, Bedford, MA) was removed from storage at −20°C and thawed on ice at 4°C. In addition, 8-well chamber glass slides were cooled at 4 °C. Forty-five μL of Matrigel were spread evenly on the surfaces of the slides that were then immediately placed for 15 minutes at 37 °C with 5% CO2 and humidity to allow the basement membrane proteins to solidify without becoming dehydrated. The cells to be plated were prepared in suspension media [RPMI 1640 without L-glutamine (Mediatech), 20% FBS (Atlanta Biologicals), and 1% penicillin/streptomycin (Mediatech)]. When the basement membrane had solidified on the slides, the cells in suspension media were centrifuged at 1000g for 3 minutes to form a pellet, the supernatant removed, and the cells resuspended in culture media. Cells were counted in a hemocytometer and suspended in the appropriate amount of medium containing 2% Matrigel and 5 ng/mL EGF (Peprotech, Rocky Hill, NJ) to yield 6000 cells/well with each well holding 400 μL of media. The cellular suspension was then plated in the 8-well chamber slide, and cells were allowed to grow for 8 to 10 days to form spheres while maintained at 37 °C with 5% CO2 and humidity. Phase contrast images of developed spheres were obtained using a Zeiss Axiovert 200 M inverted microscope with an AxioCam HRm camera (Carl Zeiss Meditec AG, Jena, Germany). For live/dead cytotoxicity assessment of spheroids, the multicellular structures were incubated, without fixation, with the fluorochromes calcein AM and EthD-1 as previously described. Fluorescence images for spheroids were obtained using an Olympus FluoView 1000 laser scanning confocal microscope (Olympus Corporation, Tokyo, Japan).
Statistical analysis
All data are reported as means ± standard error of the mean with statistical significance defined as P < 0.05. To determine statistical significance, data were entered and graphed using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA) and analyzed using one-way analysis of variance followed by Tukey’s multiple comparison test.
Results
Mifepristone blocks growth of ovarian cancer cells of different genetic backgrounds in a dose-and time-dependent manner
To study the growth inhibitory properties of mifepristone in ovarian cancer cells of different genetic makeups and sensitivities to standard chemotherapy, we selected OV2008 (p53 wt, platinum sensitive), SK-OV-3 (p53 null, platinum semiresistant), IGROV-1 (p53 wt, platinum sensitive), A2780 (p53 wt, platinum sensitive), and A2780/CP70 (p53 mut, highly resistant to platinum) cells. We exposed the cells for different times to various concentrations of mifepristone. At the end of the experiments, cell number was assessed by microcapillary cytometry. The dose-response experiments shown in Figure 1A–E illustrate that all cell lines were growth inhibited by mifepristone in a dose-related manner with a growth inhibition concentration 50% or IC50 in the 12–18 μM range. For the time-course experiment (Fig. 1F–J), cells were subjected to a one-time, fixed dose of 20 μM mifepristone and collected every day for 3 days (OV2008, IGROV-1, A2780 and A2780/CP70) or 4 days (SK-OV-3) considering their respective duplication times. In all cell lines, 24 hours or 48 hours after being exposed to mifepristone, growth was diminished and remained in decline for the time of study. With concentrations up to 20 μM mifepristone, the cultures did not grow; yet, the cells remained adherent to the plate, suggesting that the drug did not cause lethality. However, when a higher concentration of mifepristone was used (40 μM), signs of lethality were evidenced by the considerable number of floating cells. To confirm that doses of mifepristone up to 20 μM cause cytostasis whereas 40 μM causes lethality, we cultured OV2008 cells in multiwell slides in the presence of such doses of mifepristone for 48 hours and, without fixation, exposed the cells to the combination of fluorochromes calcein AM and EthD-1. Calcein AM enters the cells freely and is metabolized by live cells into a green fluorescent product, whereas EthD-1 enters only cells with a compromised plasma membrane and stains the nuclei. Figure 1K shows vehicle-treated, mostly green cells that took and metabolized calcein AM. Similar results are observed in Figure 1L where most cells exposed to 20 μM mifepristone show green fluorescence indicating that they are alive; yet their number is quite reduced when compared with vehicle-treated controls due to the cytostatic effect of mifepristone. In contrast, in Figure 1M, a large proportion of cells that were treated with 40 μM mifepristone did not metabolize calcein AM. Instead, they emit the red fluorescence of the EthD-1 that entered the dying cells, demonstrating that the higher dose of mifepristone was indeed lethal. As an intraassay control we exposed untreated healthy cells to the two fluorochromes in the presence of saponin to permeabilize the plasma membranes.16,35 Figure 1N shows that permeabilization allowed EthD-1 to enter the cells and bind the DNA (red fluorescence) while the viability of the cells is still marked by the green fluorescence mainly in the cytoplasm, denoting calcein AM metabolization. In summary, results in Figure 1 demonstrate that mifepristone is cytotoxic to ovarian cancer cells of different genetic backgrounds with cytostasis manifested at doses up to 20 μM and lethality observed at a concentration of 40 μM.
Figure 1.

Inhibition of cell growth by mifepristone in various ovarian cancer cell lines. OV2008 (A and F), SK-OV-3 (B and G), IGROV-1 (C and H), A2780 (D and I) or A2780/CP70 (E and J) cells were cultured in the presence of various concentrations of mifepristone (MF) for 48 hours (A–E) or with 20 μM MF or DMSO (VEH) for the indicated times (F–J). Results presented are the average of triplicate counts ± SEM. In (K) through (N), OV2008 ovarian cancer cells were cultured in the presence of DMSO used as vehicle (VEH) (K), 20 μM (L), or 40 μM (M) mifepristone (MF) for 48 hours. Cells were incubated with calcein AM and ethidium homodimer-1 (EthD-1) without fixation. A control to validate the assay was generated by incubating cells for 5 minutes with 0.1% saponin (SAP) prior to exposure to the two fluorochromes (N).
Notes: Images were obtained with a confocal Olympus FV1000 microscope with FluoView® software. Green fluorescence indicates live cells; red fluorescence indicates dead cells. Images presented are representative fields. Scale bar = 100 μm.
LY294002 blocks Akt phosphorylation in ovarian cancer cells
To test the hypothesis that blocking the PI3K/Akt pathway increases the potency of mifepristone in abrogating growth of ovarian cancer cells, we utilized the PI3K inhibitor LY29400236,37 and determined the dose needed to block the pathway without inducing cellular toxicity. We tailored the dose of LY294002 in SK-OV-3 cells known to have a high expression of Akt38 and used, as a positive control of Akt activation, the PC-3 prostate cancer cell line known to express elevated basal levels of activated Akt.39 Figure 2 shows that SK-OV-3 cells express phosphorylated Akt at serine 473 at similar levels as PC-3 cells and that a cytostatic, 20 μM concentration of mifepristone does not alter such phosphorylation status. LY294002 blocked phosphorylation of Akt in vehicle-as well as mifepristone-treated cells. Akt phosphorylation was only partially inhibited with 10 μM LY294002, but completely abrogated when mifepristone was also present in the culture.
Figure 2.
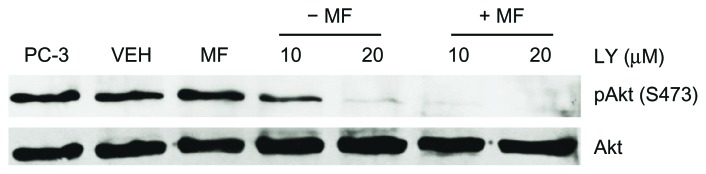
Phosphorylation of Akt in the presence of mifepristone is attenuated with the PI3K inhibitor LY294002.
Notes: SK-OV-3 cells were cultured in normal growth media (10% FBS) in the presence of DMSO (VEH), 20 μM mifepristone (MF), 10 or 20 μM LY294002 (LY), or a combination of 20 μM MF + 10 or 20 μM LY for 72 hours. LY was supplemented every 12 hours to the culture media as recommended by the provider. Whole cell extract of PC-3 prostate cancer cells was used as a positive control for Akt phosphorylation status. The experiment was repeated twice with similar outcomes. Total Akt levels did not change among the treatment groups.
The combination of mifepristone and LY294002 is lethal to ovarian cancer cells
We studied the biological consequence of combining the effects of mifepristone while simultaneously blocking the PI3K/Akt pathway. We hypothesized that by inhibiting the PI3K/Akt survival pathway with LY294002, mifepristone would change its cytostatic action to one of lethality. The toxicities of mifepristone, when used at a dose of 20 μM, and that of LY294002, when also used at a dose of 20 μM, were limited to cytostasis without inducing lethality; however, when the drugs were combined, a significant decline in viability was observed in both OV2008 and SK-OV-3 cells (Fig. 3A and B). This lethality was further confirmed by the detection of cellular particles containing hypodiploid DNA content as represented by the sub-G0/G1 region of the cell cycle histogram in propidium iodide-stained cells, further suggesting that cell death occurred by apoptosis (Fig. 3C and D).
Figure 3.

Combination of mifepristone and LY294002 decreases viability (A and B) and increases hypo-diploid DNA content (C and D) in ovarian cancer cells. Equivalent number of cells was plated in triplicates. Following overnight incubation in media containing 0.1% FBS, cultures were exposed to DMSO (VEH), 20 μM mifepristone (MF), 20 μM LY294002 (LY), or a combination of 20 μM MF and 20 μM LY. LY was supplemented every 12 hours directly into the culture media. Cells were harvested, collected, and an aliquot was tested for viability by microcapillary cytometry using dual fluorescence (A and B).
Notes: The remaining cells were fixed with 4% paraformaldehyde, stained with propidium iodide, and analyzed for cell cycle distribution by microcytometry. Results represent the average of triplicate counts ± SEM, P < 0.05 and were analyzed using one-way ANOVA followed by the Tukey multiple comparison test.
Mifepristone and LY294002 combination therapy induces apoptotic cell death, cleavage of executor caspase-3, downregulation of survival factors, and reduced clonogenic survival
To further study the lethality induced by the combination mifepristone/LY294002, OV2008 ovarian cancer cells were treated with cytostatic concentrations of the drugs. Cells were evaluated for genomic DNA fragmentation, expression of caspase-3, and clonogenic survival. Results shown in Figure 4A illustrate the characteristic ladder of DNA denoting DNA fragmentation when the drugs were used in combination but not when they were used individually. The lethality induced by the combination mifepristone/LY294002 was associated with cleavage of executor of apoptosis caspase-3 (Fig. 4D). The ability of the combination therapy to induce cell death by an apoptotic process suggests the possible downregulation of survival factors that would otherwise prevent either drug from inducing cell death. We explored the involvement of the mitochondrial protector, Bcl-2, and the inhibitor of caspase activation, XIAP, both factors that oppose activation of apoptosis in ovarian cancer.40,41 The expression of Bcl-2 and XIAP was found to be consistent throughout treatment with vehicle, mifepristone or LY294002, yet the expression was abolished when the drugs were added together (Fig. 4C). The damage induced to the cells by the combination therapy of mifepristone/LY294002 was clearly lethal in the short term. We then questioned whether the cells that had not yet died in the experimental time period were able to recover from the damaging treatment. We treated cells for 24 hours and then harvested a total of 500 cells in triplicates, which were then used to perform a clonogenic survival assay. Pretreated cells were plated in normal growth media without further treatment for 7 days. The cells were able to recover from the cytostatic effect of either mifepristone or LY294002 when used separately, as demonstrated by the clonogenic survival being similar to that of cells never treated; however, when the two drugs were combined, we observed a substantial reduction in the number of positive colonies, indicating that the cells were not able to recover properly once the combination treatment was removed (Fig. 4B).
Figure 4.

Cell death induced by the combination mifepristone and LY294002 occurs by apoptosis and is associated with cleavage of executor caspase-3 and downregulation of survival factors Bcl-2 and XIAP. OV2008 cells were treated for 24 hours with 20 μM mifepristone (MF), the indicated concentrations of LY294002 (LY), or a combination of both drugs. (A) Cells were harvested after 24 hours of treatment, and genomic DNA was isolated and separated by electrophoresis on a 2% agarose gel, impregnated with SYBR Gold nucleic acid stain, examined with an ultraviolet transilluminator, and photographed using an Amersham Typhoon fluorescence imaging system. Western blots for caspase-3, Bcl-2, XIAP, β-actin, and GAPDH were performed with cells harvested 48 hours (C) or 24 hours (D) following treatment. In (B), cells treated with VEH, MF, LY, or MF + LY were collected after 24 hours of treatment and subjected to a clonogenic survival assay. The number of positive colonies formed after one week of plating was assessed.
Mifepristone and LY294002 have synergistic lethal interaction towards ovarian cancer cells
To study whether the presence of LY294002 potentiated the therapeutic efficacy of mifepristone, we studied cell growth in the presence of increasing concentrations of mifepristone, LY294002, or the combination of a fixed dose of LY294002 with varying doses of mifepristone. Cells receiving DMSO as vehicle were utilized for comparison. The exposure time to the drugs was based upon the replication time of each particular cell line tested, with OV2008 having the shortest replication time, thereby treated for the shortest time period of 48 hours. In contrast, SK-OV-3 ovarian cancer cells were treated for 6 days, which necessitated fresh media and drug replacement after 3 days. At the conclusion of the experiments, the cells were harvested and counted by microcapillary cytometry. Single agent IC50s ranged from 12.3 μM (OV2008 cells) to 22.1 μM (SK-OV-3 cells) for mifepristone, and from 3.19 μM (SK-OV-3 cells) to 9.8 μM (OV2008 cells) for LY294002. The presence of LY294002 decreased the concentration of mifepristone needed to achieve the IC50 (shown by a dashed line) in both cell lines (Fig. 5A and B). To determine whether the nature of the potentiation induced by LY294002 over mifepristone was additive or synergistic, we assessed the degree of interaction among the drugs using the drug combination algorithm of Chou and Talalay.32,33 The interaction between LY294002 and mifepristone was synergistic in both cell lines as suggested by CIs in the range of strong synergism (0.1–0.3) for OV2008 cells (CIs indicated for four different drug combinations in Fig. 5C) or synergism (0.3–0.7) for SK-OV-3 cells (CIs indicated for three different drug combinations in Fig. 5D). The therapeutic advantage of adding LY294002 to mifepristone was further reinforced by the calculation of the DRI, which indicates how many folds the dose of mifepristone, within the scenario of a synergistic combination, may be reduced at a given effect level compared with the dose of each drug alone and in order to achieve the IC50. In OV2008 cells, the DRI for mifepristone with 20 μM LY294002 was of 11.3 folds, whereas for LY294002 there was a 12.4 fold reduction to achieve the same effect when combined with 10 μM mifepristone. The DRI for the more resistant SK-OV-3 cell line resulted in a 2.21 fold reduction in the dose of mifepristone when 10 μM LY294002 was added and a 3.2 fold reduction in the dose of LY294002 when 20 μM mifepristone was added.
Figure 5.

LY294002 (LY) enhances the therapeutic efficacy of mifepristone (MF) as shown by comparative dose-response curves. In (A and B) cells were treated with various concentrations of MF in the presence or absence of 10 μM LY294002, and cell growth was calculated considering controls as 100%. In (C and D) normalized isobolograms depict the pharmacological interaction between MF and LY as calculated from the comparative dose-responses curves using CalcuSyn software. The depicted CIs in the isobolograms were calculated using the following combined concentrations: in (C), (1) 20 μM MF with 40 μM LY, (2) 5 μM MF with 20 μM LY, (3) 10 μM MF with 20 μM LY, and (4) 1.25 μM MF with 2.5 μM LY. For (D), (1) 10 μM MF with 50 μM LY, (2) 15 μM MF with 7.5 μM LY, and (3) 30 μM MF with 15 μM LY.
The lethality of the combination mifepristone and LY294002 is observed in cultures of ovarian cancer spheroids
Because there is increasing evidence that 3-D tumor cell cultures in vitro more accurately reflect the complex in vivo microenvironment than 2-D cell monolayers, we developed, from 2-D cultures, 3-D spheroids of SK-OV-3 ovarian cancer cells with the aid of extracellular matrix proteins and specific growth factors. 3-D multicellular aggregates were observed approximately 4 to 7 days after initial plating and continued to grow in diameter over time (Fig. 6A). To study the effect of the drugs on the viability of the cells composing the ovarian cancer spheroids, we performed a live/dead viability assay as done in monolayer cultures (see Fig. 1). Spheroids of SKOV-3 cells were established, and at approximately 8 to 10 days, they were treated for 72 hours with vehicle, 20 μM mifepristone, 10 μM LY294002 or the combination of 20 μM mifepristone with 10 μM LY294002. Subsequently, the spheroids were incubated without fixation with calcein AM and EthD-1. As evidenced by the green fluorescence, the spheroids consisted mostly of live cells that were not damaged by treatments with vehicle (Fig. 6B), 20 μM mifepristone (Fig. 6C), or 10 μM LY294002 (Fig. 6D). However, when the spheroids were treated with the combination of mifepristone and LY294002, dead cells indicated by red fluorescent fragmented nuclei were observed in the core as well as the periphery of the spheroids (Fig. 6E). The damage caused by the combination treatment was comparable to that observed when SK-OV-3 spheroids were treated with the DNA damaging agent cisplatin, which was used as a cell-death inducing agent (Fig. 6F).
Figure 6.

The lethality of the combination therapy of mifepristone and LY294002 is observed in cultures of ovarian cancer spheroids. In the upper panel (A), cells were plated in equivalent numbers in 8-well slides with Matrigel in media supplemented with growth factors. Media was changed every 4 days. Representative phase contrast images are shown. Scale bar = 20 μm. In the lower panels, cells were cultured for 72 hours in the presence of vehicle (VEH; B), 20 μM mifepristone (MF; C), 10 μM LY294002 (LY; D), or a combination of 20 μM MF+ 10 μM LY (E). Cells were incubated with calcein AM and EthD-1 without fixation. Live cells were defined by a bright green fluorescence and dead cells by a bright red fluorescence. (F) A positive control of spheroids containing dead cells was generated upon exposure for 1 hour to 20 μM cisplatin (CDDP). Scale bar = 50 μm.
Discussion
Ovarian cancer is the most lethal gynecologic cancer with little improvement in its survival rate over the past several decades. Despite the fact that most cases initially respond to standard cytoreductive surgery followed by platinum/taxane chemotherapy, the majority of patients relapse with chemotherapy-resistant disease for which treatment options are extremely limited. It is consequently timely to develop new treatment alternatives for this disease. A novel approach we are currently investigating in our laboratory is the use of antiprogestins as antiovarian cancer agents (reviewed by Telleria and Goyeneche).42 We have shown that mifepristone, ORG-31710, and ulipristal are efficient in hindering the growth of ovarian cancer cells of different genetic backgrounds and histological subtypes by causing G1 cell cycle arrest via inhibition of the cell cycle regulatory kinase, cyclin dependent kinase 2 (Cdk2).16,21 We have also demonstrated that mifepristone, used at clinically relevant concentrations, abrogates the repopulation of escape cells following cisplatin or cisplatin/paclitaxel combination therapy.19,20
In this work, we show that mifepristone monotherapy, when used at clinically relevant doses, blocks growth of ovarian cancer cells without inducing lethality. The doses of mifepristone used in culture have clinical relevance because pharmacokinetic studies conducted in humans demonstrate that the concentration of mifepristone reached in circulation after oral administration ranged from 2.4 to 20 μM.22–25 When combined with the PI3K inhibitor LY294002, mifepristone, used at the otherwise cytostatic concentration of 20 μM, caused ovarian cancer cell killing. The capacity of mifepristone to block Cdk2 activity while synergizing with LY294002, mechanistically supports previous studies in which androgen-independent PC-3 prostate cancer cells, which we showed can be arrested by mifepristone,14 undergo apoptosis with the combination of LY294002 and roscovitine, which operates as a cyclin dependent kinase inhibitor.43
LY294002 was the first synthetic PI3K inhibitor developed; yet, it has not been translated to the clinic due to undesirable pharmacokinetic profiles and unacceptable toxicity, as reviewed by Agarwal et al.44 Preclinical studies have, however, shown that LY294002 inhibits growth of ovarian carcinoma cells in vitro and in vivo45 and sensitizes ovarian cancer cells to carboplatin.46 Our results with LY294004 provide proof-of-concept for the blockage of the PI3K/Akt pathway as a way of creating a therapeutic window to potentiate the toxicity of mifepristone. Second generation PI3K inhibitors, which have been developed with the aim of improving their pharmacokinetic profiles, are entering clinical trials at a rapid pace (reviewed by Agarwal et al44 and Engelman),47 thus providing hope that the combination therapy of mifepristone and a PI3K inhibitor could be translated to the clinical arena in the near future.
The interaction of mifepristone with LY294002 is synergistic from a pharmacological standpoint resulting in cell death as evidenced by decreased cell number, increased hypodiploid DNA content, cleavage of caspase-3, and fragmentation of genomic DNA. We found this drug interaction to be efficient in 2-D as well as 3-D ovarian cancer cell models. This is of particular relevance as it has been recently shown that 3-D spheroids of ovarian cancer cells treated with PI3K inhibitors display dead cells at the inner core and not attached to the extracellular matrix, yet show surviving cells on the outside that are adhered to extracellular matrix proteins and present with upregulation of cell survival molecules such as antiapoptotic protein Bcl-2.48 When we combined LY294002 with mifepristone, the levels of Bcl-2 were negligible coinciding with the downregulation of XIAP and the activation of caspase-3. XIAP is a survival factor for ovarian cancer cells. Its downregulation is sufficient to induce cell death,49 and its overexpression prevents apoptosis via a mechanism involving PI3K-dependent inhibition of caspases.50 Furthermore, Akt itself phosphorylates and stabilizes XIAP preventing its ubiquitin-dependent proteasome degradation and opposing cisplatin toxicity by reducing caspase-3 activation.51 Thus, the ability of mifepristone and LY294002 treatment to downregulate XIAP is of clinical relevance and could be used as adjuvant therapy either to prevent XIAP-mediated resistance to platinum or to sensitize cells to platinum-based chemotherapy.
Mifepristone and LY294002 demonstrated lethality toward ovarian cancer cells; nevertheless, of the cells that tolerated the treatment for 24 hours, we examined whether they were either able to recover from such stress or instead to undergo permanent proliferation arrest despite the provision of proper culture conditions. Clonogenic assay results revealed the number of positive colonies to be significantly reduced in the cells treated with the combination therapy, suggesting that the damage inflicted by the interaction of treatments was chronic and that the affected cells were not able to recover. This result is in sharp contrast to those obtained with either mifepristone or LY294002 when used individually, because upon drug removal, cells regained proliferation capacity, indicating that the damage inflicted by mifepristone or LY294002 was limited to cytostasis.
The synergistic lethality between mifepristone and LY294002 was observed in cells carrying wild type p53 and being platinum sensitive (OV2008), as well as in cells that are p53 null and semiresistant to platinum (SK-OV-3). These results suggest that the combination therapy would potentially be applicable to ovarian cancers of broad genetic and histopathological backgrounds. It is interesting to note the potentiation of effects between mifepristone and LY294002 in a 3-D culture of ovarian cancer spheroids: these multicellular aggregates are usually found in malignant ascites of ovarian cancer patients and can seed the mesothelium covering the peritoneum and omentum, further propagating the disease.52–54 Thus, the therapeutic efficacy of the combination mifepristone/LY294004 may have an important impact from a clinical standpoint as it opens the possibility for combining cytostatic therapies to cause lethality in multicellular aggregates surviving within the peritoneal cavity.
Our data showing cellular demise when combining mifepristone and LY294002 suggest that blocking a PI3K-driven pathway allows mifepristone to manifest higher toxicity, which would be otherwise ameliorated by overactive cell survival mechanisms. The fact that more than half of ovarian cancers have an hyperactive PI3K/Akt pathway26,27 rationalizes the existence of a window between therapeutic and toxic concentrations of specific inhibitors of this pathway, as reviewed by Mazzoletti and Broggini37 and Brugge et al.55 In summary, this work indicates that combination of the antiprogestin/antiglucocorticoid mifepristone with a PI3K/Akt inhibitor may provide a substantial clinical contribution as an alternative therapy for ovarian cancer.
Acknowledgements
We are grateful to Alicia Goyeneche, PhD for her helpful and insightful discussions.
Footnotes
Competing Interests
Author(s) disclose no potential conflicts of interest.
Author Contributions
Conceived and designed the experiments: SLW, CGL, CMT. Analyzed the data: SLW, CGL, CMT. Carried out the majority of experiments: SLW. Performed drug-interaction studies: CGL. Wrote the first draft of the manuscript: SLW. Contributed to the writing: CMT. Agree with manuscript results and conclusions: SLW, CGL, CMT. Jointly developed the structure and arguments for the paper: SLW, CMT. Made critical revisions and approved final version: SLW, CGL, CMT. Contributed reagents/materials/analysis tools: CMT. All authors reviewed and approved of the final manuscript.
Disclosures and Ethics
As a requirement of publication, author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest. Provenance: the authors were invited to submit this paper.
Funding
This study was supported by National Cancer Institute Grant K22CA121991 and R15CA164622 (to CMT) and, in part, by a South Dakota Board of Regents competitive grant award (SDBOR/USD 2011-10-06) (to CMT). The study was also supported by funds from the Department of Obstetrics and Gynecology, Sanford School of Medicine (to SLW). SLW was a recipient of a graduate student scholarship from the University of South Dakota. CGL was a recipient of a visiting scholarship from the National Council for Scientific and Technical Research (Conicet), Argentina. (CGL’s present address is Institute of Histology and Embryology of Cuyo, National Council for Scientific and Technical Research (Conicet), Mendoza, Argentina.).
References
- 1.Beal MW, Simmonds K. Clinical uses of mifepristone: an update for women’s health practitioners. J Midwifery Womens Health. 2002;47(6):451–60. doi: 10.1016/s1526-9523(02)00331-8. [DOI] [PubMed] [Google Scholar]
- 2.Gagne D, Pons M, Philibert D. RU 38486: a potent antiglucocorticoid in vitro and in vivo. J Steroid Biochem. 1985;23(3):247–51. doi: 10.1016/0022-4731(85)90401-7. [DOI] [PubMed] [Google Scholar]
- 3.Mifepristone (Korlym) for cushing’s syndrome. Med Lett Drugs Ther. 2012;54(1392):46–7. [PubMed] [Google Scholar]
- 4.Benagiano G, Bastianelli C, Farris M. Selective progesterone receptor modulators 3: use in oncology, endocrinology and psychiatry. Expert Opin Pharmacother. 2008;9(14):2487–96. doi: 10.1517/14656566.9.14.2487. [DOI] [PubMed] [Google Scholar]
- 5.Lanari C, Wargon V, Rojas P, Molinolo AA. Antiprogestins in breast cancer treatment: are we ready? Endocr Relat Cancer. 2012;19(3):R35–50. doi: 10.1530/ERC-11-0378. [DOI] [PubMed] [Google Scholar]
- 6.Schneider CC, Gibb RK, Taylor DD, Wan T, Gercel-Taylor C. Inhibition of endometrial cancer cell lines by mifepristone (RU 486) J Soc Gynecol Investig. 1998;5(6):334–8. doi: 10.1016/s1071-5576(98)00037-9. [DOI] [PubMed] [Google Scholar]
- 7.Moe BG, Vereide AB, Orbo A, Sager G. High concentrations of progesterone and mifepristone mutually reinforce cell cycle retardation and induction of apoptosis. Anticancer Res. 2009;29(4):1053–8. [PubMed] [Google Scholar]
- 8.Jurado R, Lopez-Flores A, Alvarez A, Garcia-Lopez P. Cisplatin cytotoxicity is increased by mifepristone in cervical carcinoma: an in vitro and in vivo study. Oncol Rep. 2009;22(5):1237–45. doi: 10.3892/or_00000560. [DOI] [PubMed] [Google Scholar]
- 9.Fjelldal R, Moe BT, Orbo A, Sager G. MCF-7 cell apoptosis and cell cycle arrest: non-genomic effects of progesterone and mifepristone (RU-486) Anticancer Res. 2010;30(12):4835–40. [PubMed] [Google Scholar]
- 10.El Etreby MF, Liang Y, Johnson MH, Lewis RW. Antitumor activity of mifepristone in the human LNCaP, LNCaP-C4, and LNCaP-C4-2 prostate cancer models in nude mice. Prostate. 2000;42(2):99–106. doi: 10.1002/(sici)1097-0045(20000201)42:2<99::aid-pros3>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 11.El Etreby MF, Liang Y, Lewis RW. Induction of apoptosis by mifepristone and tamoxifen in human LNCaP prostate cancer cells in culture. Prostate. 2000;43(1):31–42. doi: 10.1002/(sici)1097-0045(20000401)43:1<31::aid-pros5>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 12.Li DQ, Wang ZB, Bai J, et al. Effects of mifepristone on invasive and metastatic potential of human gastric adenocarcinoma cell line MKN-45 in vitro and in vivo. World J Gastroenterol. 2004;10(12):1726–9. doi: 10.3748/wjg.v10.i12.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinski J, Halmos G, Shirahige Y, Wittliff JL, Schally AV. Inhibition of growth of the human malignant glioma cell line (U87MG) by the steroid hormone antagonist RU486. J Clin Endocrinol Metab. 1993;77(5):1388–92. doi: 10.1210/jcem.77.5.8077338. [DOI] [PubMed] [Google Scholar]
- 14.Tieszen CR, Goyeneche AA, Brandhagen BN, Ortbahn CT, Telleria CM. Antiprogestin mifepristone inhibits the growth of cancer cells of reproductive and non-reproductive origin regardless of progesterone receptor expression. BMC Cancer. 2011;11:207. doi: 10.1186/1471-2407-11-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rose FV, Barnea ER. Response of human ovarian carcinoma cell lines to antiprogestin mifepristone. Oncogene. 1996;12(5):999–1003. [PubMed] [Google Scholar]
- 16.Goyeneche AA, Caron RW, Telleria CM. Mifepristone inhibits ovarian cancer cell growth in vitro and in vivo. Clin Cancer Res. 2007;13(11):3370–9. doi: 10.1158/1078-0432.CCR-07-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fauvet R, Dufournet Etienne C, Poncelet C, Bringuier AF, Feldmann G, Darai E. Effects of progesterone and anti-progestin (mifepristone) treatment on proliferation and apoptosis of the human ovarian cancer cell line, OVCAR-3. Oncol Rep. 2006;15(4):743–8. [PubMed] [Google Scholar]
- 18.Freeburg EM, Goyeneche AA, Seidel EE, Telleria CM. Resistance to cisplatin does not affect sensitivity of human ovarian cancer cell lines to mifepristone cytotoxicity. Cancer Cell Int. 2009;9:4. doi: 10.1186/1475-2867-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gamarra-Luques CD, Goyeneche AA, Hapon MB, Telleria CM. Mifepristone prevents repopulation of ovarian cancer cells escaping cisplatin-paclitaxel therapy. BMC Cancer. 2012;12:200. doi: 10.1186/1471-2407-12-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeburg EM, Goyeneche AA, Telleria CM. Mifepristone abrogates repopulation of ovarian cancer cells in between courses of cisplatin treatment. Int J Oncol. 2009;34(3):743–55. doi: 10.3892/ijo_00000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goyeneche AA, Seidel EE, Telleria CM. Growth inhibition induced by antiprogestins RU-38486, ORG-31710, and CDB-2914 in ovarian cancer cells involves inhibition of cyclin dependent kinase 2. Invest New Drugs. 2012;30(3):967–80. doi: 10.1007/s10637-011-9655-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heikinheimo O, Lahteenmaki PL, Koivunen E, et al. Metabolism and serum binding of RU 486 in women after various single doses. Hum Reprod. 1987;2(5):379–85. doi: 10.1093/oxfordjournals.humrep.a136554. [DOI] [PubMed] [Google Scholar]
- 23.Heikinheimo O. Pharmacokinetics of the antiprogesterone RU 486 in women during multiple dose administration. J Steroid Biochem. 1989;32(1A):21–5. doi: 10.1016/0022-4731(89)90008-3. [DOI] [PubMed] [Google Scholar]
- 24.Shoupe D, Mishell DR, Jr, Lahteenmaki P, et al. Effects of the antiprogesterone RU 486 in normal women. I. Single-dose administration in the miduteal phase. Am J Obstet Gynecol. 1987;157(6):1415–20. doi: 10.1016/s0002-9378(87)80235-1. [DOI] [PubMed] [Google Scholar]
- 25.Kawai S, Nieman LK, Brandon DD, Udelsman R, Loriaux DL, Chrousos GP. Pharmacokinetic properties of the antiglucocorticoid and antiprogesterone steroid RU 486 in man. J Pharmacol Exp Ther. 1987;241(2):401–6. [PubMed] [Google Scholar]
- 26.Despierre E, Lambrechts D, Neven P, Amant F, Lambrechts S, Vergote I. The molecular genetic basis of ovarian cancer and its roadmap towards a better treatment. Gynecol Oncol. 2010;117:358–65. doi: 10.1016/j.ygyno.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 27.Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–28. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bast RC, Mills GB. Dissecting “PI3Kness”: The Complexity of Personalized Therapy for Ovarian Cancer. Cancer Discov. 2012;2(1):16–8. doi: 10.1158/2159-8290.CD-11-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of Phosphatidylinositol 3′-Kinase Increases Efficacy of Paclitaxel in in Vitro and in Vivo Ovarian Cancer Models. Cancer Res. 2002;62(4):1087–92. [PubMed] [Google Scholar]
- 30.Katano K, Kondo A, Safaei R, et al. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002;62(22):6559–65. [PubMed] [Google Scholar]
- 31.Goyeneche AA, Harmon JM, Telleria CM. Cell death induced by serum deprivation in luteal cells involves the intrinsic pathway of apoptosis. Reproduction. 2006;131(1):103–11. doi: 10.1530/rep.1.00751. [DOI] [PubMed] [Google Scholar]
- 32.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–6. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 33.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 34.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5(9):675–88. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 35.Ishida H, Hirota Y, Nakazawa H. Effect of sub-skinning concentrations of saponin on intracellular Ca2+ and plasma membrane fluidity in cultured cardiac cells. Biochim Biophys Acta. 1993;1145(1):58–62. doi: 10.1016/0005-2736(93)90381-9. [DOI] [PubMed] [Google Scholar]
- 36.Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27(41):5511–26. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- 37.Mazzoletti M, Broggini M. PI3K/AKT/mTOR inhibitors in ovarian cancer. Curr Med Chem. 2010;17(36):4433–47. doi: 10.2174/092986710794182999. [DOI] [PubMed] [Google Scholar]
- 38.Chaudhuri D, Orsulic S, Ashok BT. Antiproliferative activity of sulforaphane in Akt-overexpressing ovarian cancer cells. Mol Cancer Ther. 2007;6(1):334–45. doi: 10.1158/1535-7163.MCT-06-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tiwari G, Sakaue H, Pollack JR, Roth RA. Gene expression profiling in prostate cancer cells with Akt activation reveals Fra-1 as an Akt-inducible gene. Mol Cancer Res. 2003;1(6):475–84. [PubMed] [Google Scholar]
- 40.Shaw TJ, Lacasse EC, Durkin JP, Vanderhyden BC. Downregulation of XIAP expression in ovarian cancer cells induces cell death in vitro and in vivo. Int J Cancer. 2008;122(6):1430–4. doi: 10.1002/ijc.23278. [DOI] [PubMed] [Google Scholar]
- 41.Eliopoulos AG, Kerr DJ, Herod J, et al. The control of apoptosis and drug resistance in ovarian cancer: influence of p53 and Bcl-2. Oncogene. 1995;11(7):1217–28. [PubMed] [Google Scholar]
- 42.Telleria CM, Goyeneche AA. Antiprogestins in ovarian cancer. In: Farghaly S, editor. Ovarian Cancer—Clinical and Therapeutic Perspectives. New York, NY: InTech; 2012. pp. 207–30. [Google Scholar]
- 43.Mohapatra S, Chu B, Zhao X, Djeu J, Cheng JQ, Pledger WJ. Apoptosis of metastatic prostate cancer cells by a combination of cyclin-dependent kinase and AKT inhibitors. Int J Biochem Cell Biol. 2009;41(3):595–602. doi: 10.1016/j.biocel.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 44.Agarwal R, Carey M, Hennessy B, Mills GB. PI3K pathway-directed therapeutic strategies in cancer. Curr Opin Investig Drugs. 2010;11(6):615–28. [PubMed] [Google Scholar]
- 45.Hu L, Zaloudek C, Mills GB, Gray J, Jaffe RB. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002) Clin Cancer Res. 2000;6(3):880–6. [PubMed] [Google Scholar]
- 46.Westfall SD, Skinner MK. Inhibition of phosphatidylinositol 3-kinase sensitizes ovarian cancer cells to carboplatin and allows adjunct chemotherapy treatment. Mol Cancer Ther. 2005;4(11):1764–71. doi: 10.1158/1535-7163.MCT-05-0192. [DOI] [PubMed] [Google Scholar]
- 47.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 48.Muranen T, Selfors LM, Worster DT, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21(2):227–39. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaw TJ, Lacasse EC, Durkin JP, Vandergyden BC. Downregulation of XIAP expression in ovarian cancer cells induces cell death in vitro and in vivo. Int J Cancer. 2008;122(6):1430–4. doi: 10.1002/ijc.23278. [DOI] [PubMed] [Google Scholar]
- 50.Asselin E, Mills GB, Tsang BK. XIAP regulates Akt activity and caspase-3-dependent cleavage during cisplatin-induced apoptosis in human ovarian epithelial cancer cells. Cancer Res. 2001;61(5):1862–8. [PubMed] [Google Scholar]
- 51.Dan HC, Sun M, Kaneko S, et al. Akt Phosphorylation and Stabilization of X-linked Inhibitor of Apoptosis Protein (XIAP) J Biol Chem. 2004;279(7):5405–12. doi: 10.1074/jbc.M312044200. [DOI] [PubMed] [Google Scholar]
- 52.Kenny HA, Dogan S, Zillhardt M, et al. Organotypic models of metastasis: A three-dimensional culture mimicking the human peritoneum and omentum for the study of the early steps of ovarian cancer metastasis. Cancer Treat Res. 2009;149:335–51. doi: 10.1007/978-0-387-98094-2_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iwanicki MP, Davidowitz RA, Ng MR, et al. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011;1(2):144–57. doi: 10.1158/2159-8274.CD-11-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kenny H, Nieman K, Mitra A, Lengyel E. The first line of intra-abdominal metastatic attack: breaching the mesothelial cell layer. Cancer Discov. 2011;1(2):100–2. doi: 10.1158/2159-8290.CD-11-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brugge J, Hung MC, Mills GB. A new mutational AKTivation in the PI3K pathway. Cancer Cell. 2007;12(2):104–7. doi: 10.1016/j.ccr.2007.07.014. [DOI] [PubMed] [Google Scholar]