

Abstract
Desaktiviert und deformiert werden Enzyme, die auf magnetischen Nanopartikeln (MNPs) immobilisiert sind, beim Anlegen von magnetischen Feldern. Diese Veränderungen resultieren aus der erneuten Ausrichtung der MNPs im AC-Magnetfeld, die mit den MNP verknüpfte Polymerketten unter Belastung setzt. Für immobilisierte Enzymmoleküle auf einem MNP-Aggregat ergeben sich dadurch Deformationen und irreversible (oder lange anhaltende) Konformationsän-derungen.
Keywords: colloids, conformational analysis, enzyme catalysis, magnetic fields, magnetite nanoparticles
Weak magnetic fields (<1 T) are known to change the paths and kinetics of some radical chemical reactions at room temperature through spin conversion of short-lived radical pairs.[1] Magnetic fields were also used to tune enzyme activity (oxidation of glucose). Specifically, exposure of magnetic nanoparticles functionalized with enzymes to magnetic field allowed the movement of particles in proximity to electrode, thus activating the enzyme.[2] Moreover, radio-frequency (RF) alternating current (AC) magnetic fields can induce the heating of single-domain magnetic nanoparticles (MNPs), and this can be used in magnetic hyperthermia to kill tumors.[3]
Herein we describe a distinct effect of non-heating super-low-frequency magnetic fields as well as more conventional RF magnetic fields on the kinetics of chemical reactions catalyzed by the enzymes α-chymotrypsin (ChT) or β-galactosidase (β-GaL) immobilized on nanoscale MNP aggregates. Magnetite MNPs (7 to 12 nm diameter) were synthesized by reducing tris(acetylacetonato)iron(III), then they were coated with anionic poly(ethylene glycol)-b-polyacrylate (PEG-PAA), or poly(ethylene glycol)-b-polymethacrylate (PEG-PMA) copolymers. The polymers were bound to the magnetite surfaces by the carboxylate groups.[4] According to thermogravimetric analysis, the content of Fe3O4 in both coated MNP materials was about 35 wt%, and this was in excellent agreement with that measured by inductively coupled plasma-mass spectrometry. Upon dispersion in water (pH 6.5), they formed nanoscale, negatively charged aggregates (hydrodynamic intensity average diameters and zeta potentials of 194 nm, −70 mV and 39 nm, −49 mV for PEG-PAA/MNP and PEG-PMA/MNP, respectively, as determined by dynamic light scattering). Based on TEM, they contained clusters of 7 to 20 MNP grains. Enzymes were conjugated to these aggregates using 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide and N-hydroxysulfosuccinimide to couple amines on the protein to carboxylic acids on the copolymer (Scheme 1).
Scheme 1.
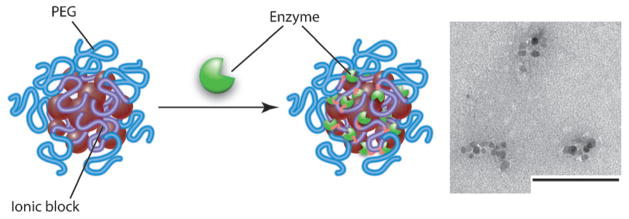
Immobilization of an enzyme on copolymer–MNP aggregates and TEM image of PEG-PMA/MNP aggregates after immobilization of ChT. Scale bar: 100 nm.
The protein content ranged from 0.13 to 5.3 mg per mg of iron (7–270 molecules per MNP). After conjugation the aggregate sizes decreased by about 25%. The residual activity of the enzymes after conjugation was from 16–70% of the unmodified enzyme activity (the majority retained 30–40% of the initial activity). Field exposures (50 Hz, 110 kAm−1) resulted in considerable decreases of enzymatic reaction rates compared to the reference sample (Figure 1a), albeit no effect on free enzyme or on the non-conjugated enzyme-MNP mixture was observed (Supporting Information, Table S2). Repeated exposures resulted in progressive enzyme inactivation (Figure 1a; Supporting Information, Figure S1). A significant decrease in ChT reaction rate was observed after three pulse exposures of 2.5 min; this exposure regime was used in subsequent experiments unless specified differently. Notably, the number of ChT attachment points played a significant role in enzyme inactivation. ChT was covalently linked to the aggregates through either 2 or 11–15 of its 15 amines (Supporting Information, Table S1). The inactivation increased as the number of enzyme attachment points to the MNP increased from 2 to 13 amines (Figure 1b). Notably, after each field exposure, the activity of the enzyme on MNP aggregates did not restore for at least an hour. The dispersions remained stable in the field, as we did not detect any substantial change in the particle size or precipitation.
Figure 1.
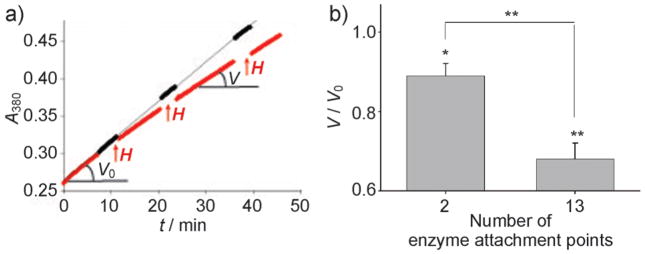
Effect of a super-low-frequency AC field (50 Hz, 110 kAm−1) on rates of enzymatic hydrolysis of N-benzoyl-L-tyrosine p-nitroanilide catalyzed by ChT immobilized on copolymer–MNP aggregates. a) Changes in enzyme kinetics after 2.5 min exposures to the field (shown as ↑↑↑H) versus no field exposure (ChT/PEG-PMA/MNP-2). b) Residual activity (V/Vo) with ChT attached to the MNP through 2 or 13 amines (ChT/PEG-PMA/MNP-1 and ChT/PEG-PMA/MNP-2) after 3 × 2.5 min field exposures compared to untreated samples (*p≤0.05, **p≤0.01, n=3 (2) or n=9 (13)). Temperature: 20 ± 0.1°C.
To demonstrate the general character of the observed effect we immobilized ChT on 85 nm diameter magnetite-core–gold-shell MNP aggregates by short L-cysteine bridges. Exposure to 3 × 2.5 min pulses of 50 Hz, 110 kAm−1 AC field resulted in 31% reduction in enzyme activity. Therefore, the effect was observed regardless of whether the enzymes were coupled to MNPs through flexible copolymer chains or attached to the rigid gold surface.
Enzyme inactivation was also observed with RF fields in the kHz range (337 kHz), which is commonly used in magnetic hyperthermia.[3a] Experiments at 337 kHz were carried out at a lower field of 21 kAm−1 to avoid solution heating. In this case, we compared effects of pulsed versus continuous RF exposure of the same overall duration. Interestingly, the extent of enzyme inactivation increased when the field was applied in pulses (Figure 2a). This was reproduced with several ChT-MNP aggregates with different polymer coatings and enzyme contents (Supporting Information, Figure S2). The same trend was observed with β-GaL immobilized on MNPs (Figure 2b). Longer field exposure resulted in greater inactivation of β-GaL. However, for the same overall duration, the shortest pulsed exposures produced the greatest inactivation.
Figure 2.
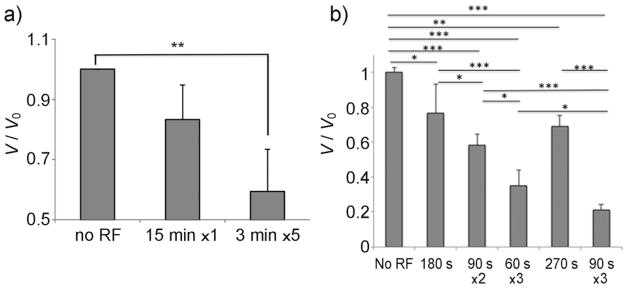
Effect of RF magnetic field (337 kHz, 21 kA m−1) on residual activity (V/Vo) of a) ChT/PEG-PAA/MNP-2 or b) β-GaL/PEG-PAA/MNP. The samples were exposed to a continuous or pulsed field. *p≤0.05, **p≤0.01, ***p≤0.005 (n=3). Temperature: 25 ±1°C.
The observed effects could not be explained by elevation of the temperature for any field frequency. First, changes in the bulk temperature were either negligible (50 Hz) or <1°C (337 kHz, even in a concentrated 10 mgmL−1 MNP solution). Second, measurements in the absence of the field suggested that comparable inactivation of ChT immobilized on MNP aggregates only occurred at 45–50°C (Supporting Information, Figure S3). This is at least 25°C above the solution temperatures during the field exposures. Furthermore, due to short characteristic thermal times of t* ≈ R2MNP/4κ ≈ 10−9 s (RMNP ≈ 10 nm, κ =1.5 × 10−7 m2s−1 thermal diffusivity in aqueous solution), the heat wave would rapidly spread in the liquid to several mm and this should abolish any temperature gradients. Indeed, the upper estimate for local temperature increases in the nanoparticle surface region should not exceed 10−6 K and thus is negligible (Supporting Information). These estimates are consistent with published data.[5]
Circular dichroism (CD) showed that pulsed exposure to the AC field resulted in changes in the secondary structure of ChT covalently immobilized on MNP aggregates, whereas no changes were observed for free enzyme or a non-conjugated enzyme-MNP mixture (Table 1). There was an increase in α-helices mainly at the expense of β-sheets, thus suggesting structural condensation of the protein. A decrease in β-sheets also suggests lack of aggregation. Similar structural transitions are generally characteristic of misfolding of proteins in hydrophobic environments and were previously observed for modified ChT in an organic solvent.[6] In contrast, heat exposure (60°C) of the free enzyme and non-conjugated enzyme-MNP mixture resulted in marked decreases in α-helices with increases in β-sheets and random coils, indicating protein unfolding and aggregation. Changes in the secondary structure of the conjugated ChT after heat exposure were different than those observed for free ChT and the non-conjugated mixture (no aggregation or unfolding owing to multipoint attachment of protein to carrier occur, leading to increased thermal stability of the enzyme after immobilization). However, these changes were significantly different from those observed after RF treatment.
Table 1.
Secondary structural elements from CD spectra [%].
| Sample | α-helix | β-sheet | β-turn | Random coil |
|---|---|---|---|---|
| No treatment | ||||
| Free ChT[a] | 10.0 | 34.0 | 20.0 | 36.0 |
| Free ChT[b] | 11.3 | 33.3 | 18.9 | 34.4 |
| ChT and PEG-PMA/MNP[c] | 12.5 | 33.3 | 19.4 | 35.3 |
| ChT-PEG-PMA/MNP[d] | 11.6 | 33.4 | 19.1 | 34.9 |
| RF: 337 kHz, 21 kAm−1 2.5 minx3 | ||||
| Free ChT[b] | 11.1 | 33.5 | 19.1 | 36.3 |
| ChT and PEG-PMA/MNP[c] | 11.4 | 34.7 | 22.0 | 34.9 |
| ChT-PEG-PMA/MNP[d] | 26.8 | 21.7 | 18.5 | 33.6 |
| Heating: 60°C, 10 min | ||||
| Free ChT[b] | 3.4 | 40.4 | 18.2 | 41.4 |
| ChT and PEG-PMA/MNP[c] | 2 | 47.5 | 15.7 | 40.3 |
| ChT-PEG-PMA/MNP[d] | 12.1 | 29.8 | 13.8 | 42.6 |
Theoretical values.
Experimental values.
Non-conjugated mixture.
Covalently conjugated. ChT/PEG-PMA/MNP-3 was utilized.
Thus, we posit that there are non-heating factors that underlie the observed phenomena. Specifically, realignment of the MNPs along the AC magnetic field may lead to stresses in MNP-linked polymer chains. For example, a polymer attached to a MNP can experience hydrodynamic stretching forces ranging from several to dozens of pN. A chain attached to two particles in a MNP cluster can undergo tensile, contraction, twisting and tangential (grinding) forces. The amplitude of these forces (F) depends on the saturation magnetization of the single domain particle (m), radius of the magnetic domain Rm, distance between the rotation center of the magnetic particle in an AC magnetic field and the radius of the whole particle with the enzyme attached (Rr), and the field strength (H), and is expressed as F =(4/3)πμ0mH(Rm)3/Rr. For Rm = 5–10 nm, Rr = 15–20 nm, and H = 100 kAm−1, F is predicted to be up to several hundred pN. Such stresses translated to an immobilized enzyme on a MNP aggregate could induce deformation and irreversible (or long-lasting) conformational changes (or even breakage of weak chemical bonds), leading to inactivation. Similar effects are produced by force spectroscopy using optical and magnetic tweezers.[7] In this regard it is reasonable that the effect increases as the number of attachment points increases. Also, following the application of force, stress-relaxation processes can occur in the MNP aggregates, and this may at least partially explain why multiple pulses of the field result in stronger effects than continuous application.
Therefore, we have discovered non-heating effects of super-low- and medium-frequency magnetic fields on the activities and conformations of enzymes immobilized on MNPs in dispersions. The observation is unprecedented and suggests the significance of magneto-mechanochemical effects induced by realignment of MNP magnetic moments in an AC magnetic field rather than traditional heating. Such low frequency and amplitude fields are safe and are not expected to cause any damage to biological tissues.[3a] Thus, from a practical standpoint, our findings could open new directions for remote control of drug release systems, as well as modulation of biochemical functions in cells. It should be noted that several recent studies have described RF-field phenomena in MNP-based systems that may not be reasonably explained by heating. Such studies involved RF-field actuation of MNPs to kill cancer cells by disrupting epidermal growth factor receptors,[8] and release of drugs and polynucleotides from MNP-containing materials: liposomes,[9] nanospheres,[10] nanogels,[11] silica nanoparticles,[12] and dextran-coated MNPs.[13] These diverse phenomena may in fact be magneto-mechanochemical in nature. Altogether, this work may lead to magneto-mechanochemical spectroscopy of relaxation processes that can focus on the elementary acts and transition states of macromolecules immobilized on MNPs. If successful, this could profoundly influence future research directions in nanomedicine and drug delivery, and perhaps in other fields such as tissue engineering. Furthermore, this work may be of interest to other areas of life science, such as effects of super-low-frequency AC magnetic fields in biological systems with natural MNPs; for example, magnetosomes in magnetostatic bacteria.
Experimental Section
Detailed methods are described in Supporting Information.
The super-low-frequency AC field (20 Hz to 20 kHz, 10 to 220 kAm−1) was generated using a custom water-cooled coil connected to an AC power supply (see picture in the Supporting Information). A medium-frequency AC field (337 kHz, 21 kAm−1) was generated using 8 turns of a 2.5 cm-diameter water-cooled copper coil connected to a RF field generator (2.4 kW EasyHeat, Ameritherm). For ChT, the reaction rate was measured after RF field application and residual activity was calculated relative to the rate measured in the reference cuvette at the same time point. For β-GaL, the reaction rate was measured in situ and calculated relative to the reaction rate prior to RF exposure. CD spectra were recorded at 200 to 300 nm, 4 sec/measurement. Each measurement was an average of two scans. Measurements were normalized against buffer and unmodified PEG-PMA/MNP readings. Spectra were analyzed using CDNN secondary structure analysis software.
Supplementary Material
Footnotes
The authors thank U.S. National Institutes of Health, RR021937, Russian Ministry of Science and Education, 11.G34.31.0004 (both to A.V.K.), and the U.S. National Science Foundation Materials World Network program, DMR-0805179 and the U.S. National Science Foundation contract DMR 0909065 (to J.S.R.). The authors are grateful to T. Bargar (UNMC) for carrying out TEM experiments, Dr. I. Khutsishvili (UNMC) for CD measurements, and Nazar Filonov (COBRE Nanomaterials Core Characterization) for ICP-MS measurements.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201205905.
Contributor Information
Prof. Dr. Natalia L. Klyachko, Department of Pharmaceutical Sciences and Center for Drug Delivery and Nanomedicine, University of Nebraska Medical Center 96025 Nebraska Medical Center, Omaha, NE 68198 (USA). Laboratory for Chemical Design of Bionanomaterials, Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow, 119991 (Russia).
Dr. Marina Sokolsky-Papkov, Department of Pharmaceutical Sciences and Center for Drug Delivery and Nanomedicine, University of Nebraska Medical Center 96025 Nebraska Medical Center, Omaha, NE 68198 (USA).
Nikorn Pothayee, Macromolecules and Interfaces Institute, Department of Chemistry, Virginia Polytechnic Institute and State, University Blacksburg, VA 24061 (USA).
Maria V. Efremova, Laboratory for Chemical Design of Bionanomaterials, Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow, 119991 (Russia)
Dr. Dmitry A. Gulin, Laboratory for Chemical Design of Bionanomaterials, Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow, 119991 (Russia)
Nipon Pothayee, Macromolecules and Interfaces Institute, Department of Chemistry, Virginia Polytechnic Institute and State, University Blacksburg, VA 24061 (USA).
Artem A. Kuznetsov, Laboratory for Chemical Design of Bionanomaterials, Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow, 119991 (Russia)
Dr. Alexander G. Majouga, Laboratory for Chemical Design of Bionanomaterials, Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow, 119991 (Russia)
Prof. Judy S. Riffle, Macromolecules and Interfaces Institute, Department of Chemistry, Virginia Polytechnic Institute and State, University Blacksburg, VA 24061 (USA)
Prof. Dr. Yuri I. Golovin, Laboratory for Chemical Design of Bionanomaterials, Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow, 119991 (Russia). Nanocenter of Tambov State University, Tambov State University Tambov, 392000 (Russia)
Prof. Dr. Alexander V. Kabanov, Department of Pharmaceutical Sciences and Center for Drug Delivery and Nanomedicine, University of Nebraska Medical Center 96025 Nebraska Medical Center, Omaha, NE 68198 (USA). Laboratory for Chemical Design of Bionanomaterials, Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow, 119991 (Russia)
References
- 1.a) Buchachenko AL. Chem Rev. 1995;95:2507–2528. [Google Scholar]; b) Grissom CB. Chem Phys. 1995;95:3–24. [Google Scholar]; c) Adair RK. Rep Prog Phys. 2000;63:415–456. [Google Scholar]; d) Brocklehurst B. Chem Soc Rev. 2002;31:301–311. doi: 10.1039/b107250c. [DOI] [PubMed] [Google Scholar]; e) Binhi VN. Magnitobiology: Underlying Physical Problems. Academic Press; New York: 2002. [Google Scholar]; f) Engstrom S. In: Bioengineering and Biophysical Aspects of Electromagnetic Fields. Barnes FS, Greenebaum B, editors. CRC; Boca Raton: 2007. pp. 157–168. [Google Scholar]
- 2.a) Hirsch R, Katz E, Willner I. J Am Chem Soc. 2000;122:12053–12054. [Google Scholar]; b) Willner I, Katz E. Angew Chem. 2003;115:4724– 4737. [Google Scholar]; Angew Chem Int Ed. 2003;42:4576–4588. doi: 10.1002/anie.200201602. [DOI] [PubMed] [Google Scholar]; c) Katz E, Lioubashevski O, Willner I. J Am Chem Soc. 2005;127:3979– 3988. doi: 10.1021/ja044157t. [DOI] [PubMed] [Google Scholar]
- 3.a) Gazeau F, Levy M, Wilhelm C. Nanomedicine. 2008;3:831–844. doi: 10.2217/17435889.3.6.831. [DOI] [PubMed] [Google Scholar]; b) Kikumori T, Kobayashi T, Sawaki M, Imai T. Breast Cancer Res Treat. 2009;113:435–441. doi: 10.1007/s10549-008-9948-x. [DOI] [PubMed] [Google Scholar]; c) Ito A, Honda H, Kobayashi T. Cancer Immunol Immunother. 2006;55:320– 328. doi: 10.1007/s00262-005-0049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pothayee N, Pothayee N, Jain N, Hu N, Balasubramaniam S, Johnson LM, Davis RM, Sriranganathan N, Riffle JS. Chem Mater. 2012;24:2056– 2063. [Google Scholar]
- 5.a) Rabin Y. J Int Hyperthermia. 2002;18:194–202. doi: 10.1080/02656730110116713. [DOI] [PubMed] [Google Scholar]; b) Gupta A, Kane RS, Borca-Tasciuc DA. Appl Phys. 2010;108:064901– 064907. [Google Scholar]
- 6.a) Damaschun G, Damaschun H, Gast K, Zirwer D. Mol Biol. 1999;291:715–725. doi: 10.1006/jmbi.1999.3009. [DOI] [PubMed] [Google Scholar]; b) Vinogradov AA, Kudryashova EV, Grinberg VY, Grinberg NV, Burova TV, Levashov AV. Protein Eng. 2001;14:683–689. doi: 10.1093/protein/14.9.683. [DOI] [PubMed] [Google Scholar]; c) Bessmertnaya L, Kozlov LV, Antonov VK. Eur J Biochem. 1976;63:591– 598. doi: 10.1111/j.1432-1033.1976.tb10263.x. [DOI] [PubMed] [Google Scholar]
- 7.a) Conroy R. In: Handbook Molecular Force Spectroscopy. Noy A, editor. Springer; Heidelberg: 2008. pp. 23–96. [Google Scholar]; b) Lam L, Iino R, Tabata KV, Noji H. Anal Bioanal Chem. 2008;391:2423– 2432. doi: 10.1007/s00216-008-2099-4. [DOI] [PubMed] [Google Scholar]
- 8.Creixell M, Bohorquez AC, Torres-Lugo M, Rinaldi C. ACS Nano. 2011;5:7124– 7129. doi: 10.1021/nn201822b. [DOI] [PubMed] [Google Scholar]
- 9.Amstad E, Kohlbrecher J, Muller E, Schweizer T, Textor M, Reimhult E. Nano Lett. 2011;11:1664– 1670. doi: 10.1021/nl2001499. [DOI] [PubMed] [Google Scholar]
- 10.Liu TY, Hu SH, Liu KH, Shaiu RS, Liu DM, Chen SY. Langmuir. 2008;24:13306– 13311. doi: 10.1021/la801451v. [DOI] [PubMed] [Google Scholar]
- 11.Hoare T, Timko BP, Santamaria J, Goya GF, Irusta S, Lau S, Stefanescu CF, Lin D, Langer R, Kohane DS. Nano Lett. 2011;11:1395– 1400. doi: 10.1021/nl200494t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas CR, Ferris DP, Lee JH, Choi E, Cho MH, Kim ES, Stoddart JF, Shin JS, Cheon J, Zink JI. J Am Chem Soc. 2010;132:10623– 10625. doi: 10.1021/ja1022267. [DOI] [PubMed] [Google Scholar]
- 13.Derfus AM, von Maltzahn G, Harris TJ, Duza T, Vecchio KS, Ruoslahti E, Bhatia SN. Adv Mater. 2007;19:3932–3936. [Google Scholar]
- 14.Carslaw HS, Jaeger JC. Conduction of heat in solids. Oxford Clarendon Press; Oxford: 1959. [Google Scholar]
- 15.Fields R. Biochem J. 1971;124:581– 590. doi: 10.1042/bj1240581. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.