

Experiments with human cancer glioblastoma multiforme cell lines, primary patient samples, and preclinical mouse models are performed to show that αvβ8 integrin and RhoGDI1 are components of a signaling axis that drives brain tumor cell invasion via regulation of Rho GTPase activation.
Abstract
The malignant brain cancer glioblastoma multiforme (GBM) displays invasive growth behaviors that are regulated by extracellular cues within the neural microenvironment. The adhesion and signaling pathways that drive GBM cell invasion remain largely uncharacterized. Here we use human GBM cell lines, primary patient samples, and preclinical mouse models to demonstrate that integrin αvβ8 is a major driver of GBM cell invasion. β8 integrin is overexpressed in many human GBM cells, with higher integrin expression correlating with increased invasion and diminished patient survival. Silencing β8 integrin in human GBM cells leads to impaired tumor cell invasion due to hyperactivation of the Rho GTPases Rac1 and Cdc42. β8 integrin coimmunoprecipitates with Rho-GDP dissociation inhibitor 1 (RhoGDI1), an intracellular signaling effector that sequesters Rho GTPases in their inactive GDP-bound states. Silencing RhoGDI1 expression or uncoupling αvβ8 integrin–RhoGDI1 protein interactions blocks GBM cell invasion due to Rho GTPase hyperactivation. These data reveal for the first time that αvβ8 integrin, via interactions with RhoGDI1, regulates activation of Rho proteins to promote GBM cell invasiveness. Hence targeting the αvβ8 integrin–RhoGDI1 signaling axis might be an effective strategy for blocking GBM cell invasion.
INTRODUCTION
Grade IV astrocytoma, or glioblastoma multiforme (GBM), is a primary brain cancer displaying infiltrative growth properties that are tightly coupled to the vasculature (Gilbertson and Rich, 2007). For example, stem-like GBM cells home to perivascular niches (Calabrese et al., 2007; Hjelmeland et al., 2011), and invasive GBM cells commonly disperse through the brain microenvironment via extracellular matrix (ECM)–rich vascular basement membranes (Nakada et al., 2007). Antiangiogenesis compounds that target the vascular endothelial growth factor pathway improve progression-free survival; however, in many patients these benefits are often transient due to invasive cells forming satellite lesions that display resistance to most second-line therapies (Ellis and Reardon, 2009).
GBM cell invasion is influenced by a milieu of extrinsic cues, including growth factors and ECM proteins within the brain microenvironment (Farin et al., 2006; Hoelzinger et al., 2007). Most mammalian cells adhere to ECM proteins via integrins, a family of heterodimeric cell surface receptors consisting of α and β subunits (Hynes, 2009; Desgrosellier and Cheresh, 2010). Various integrins, their ECM protein ligands, and intracellular signaling effectors play important roles in brain development and physiology (Milner and Campbell, 2002; McCarty, 2009), as well as cancer (Hu et al., 2003; Kanamori et al., 2004; Shi et al., 2007; Lathia et al., 2010). In particular, integrin αvβ8 is highly expressed in neural progenitor cells of the embryonic and postnatal brain (McCarty et al., 2002, 2005b; Mobley et al., 2009; Mobley and McCarty, 2011). Cell type–specific ablation of αv or β8 integrin genes in neural cells causes CNS-specific angiogenesis phenotypes (McCarty et al., 2005b; Proctor et al., 2005; Hirota et al., 2011; Arnold et al., 2012) that are very similar to those that develop in GBM. αvβ8 integrin is also expressed in GBM cells (Riemenschneider et al., 2005), and in preclinical models of GBM, αvβ8 integrin can suppress tumor angiogenesis via activation of ECM-bound latent transforming growth factor βs (TGFβs; Tchaicha et al., 2010, 2011). Impaired integrin-mediated latent TGFβ activation leads to defective TGFβ receptor signaling in endothelial cells, causing developmental vascular pathologies resembling those in GBM (Hirota et al., 2011; Nguyen et al., 2011).
Here we report that αvβ8 integrin drives GBM cell invasion via associations with the intracellular signaling effector Rho-GDP dissociation inhibitor 1 (RhoGDI1). The αvβ8 integrin–RhoGDI1 protein complexes control activation of Rho GTPases in invasive GBM cells, with cells lacking these protein complexes displaying diminished invasiveness due to elevated levels of GTP-bound Rac1 and Cdc42. These data reveal that αvβ8 integrin and RhoGDI1 spatially regulate patterns of GTPase activation to promote tumor cell invasion and suggest that components of this signaling axis may be therapeutic targets for inhibiting invasive GBM cell growth.
RESULTS
During our prior analysis of αvβ8 integrin in GBM-induced angiogenesis, we found that LN229 and SNB19 human GBM cells, which display invasive growth behaviors in vivo, express robust levels of αvβ8 integrin proteins (Tchaicha et al., 2011). To further investigate functions for αvβ8 integrin in GBM growth and invasion, we targeted β8 integrin gene (ITGB8) expression by RNA interference (RNAi) using the genetically engineered pLB lentivirus, which contains a U6 promoter that drives expression of short hairpin RNAs (shRNAs) and a cytomegalovirus (CMV) promoter driving green fluorescent protein (GFP) expression (Kissler et al., 2006). LN229 or SNB19 cells were infected with lentiviruses expressing shRNAs targeting β8 integrin or nontargeting (scrambled) shRNA sequences, and 72–96 h later cells were sorted based on GFP expression. In addition, we analyzed wild-type and β8−/− murine astroglial progenitors that were transformed with oncogenes targeting pathways commonly altered in GBM (Sonoda et al., 2001; Tchaicha et al., 2010). As shown in Figure 1, A and C, in comparison to human GBM cells expressing scrambled shRNAs, a significant reduction in β8 integrin protein expression was detected in GBM cells expressing β8 shRNAs. Integrin protein was completely absent from β8−/− -transformed astrocytes owing to gene ablation (Figure 1E). Next we analyzed roles for αvβ8 integrin in tumor cell invasion using three-dimensional Matrigel assays. As shown in Figure 1, B and D, human GBM cells expressing β8 shRNAs displayed significantly reduced invasiveness. Two other lentiviral-delivered shRNAs targeting different regions of ITGB8 also resulted in diminished integrin expression and LN229 cell invasion defects (Supplemental Figure S1). The β8−/−-transformed mouse astroglial progenitors also showed invasion defects (Figure 1F). Similarly, β8−/− primary mouse astrocytes displayed defective invasion in vitro (Supplemental Figure S2). We detected reduced surface expression of αvβ8 integrin protein in GBM cells expressing β8 shRNAs and a complete loss of αvβ8 integrin protein in β8−/−-transformed astroglial progenitors (Supplemental Figure S3). Silencing β8 integrin did not result in abnormal dimerization between αv integrin and other β integrin subunits (Supplemental Figure S3), similar to our previous reports (Tchaicha et al., 2010, 2011).
FIGURE 1:
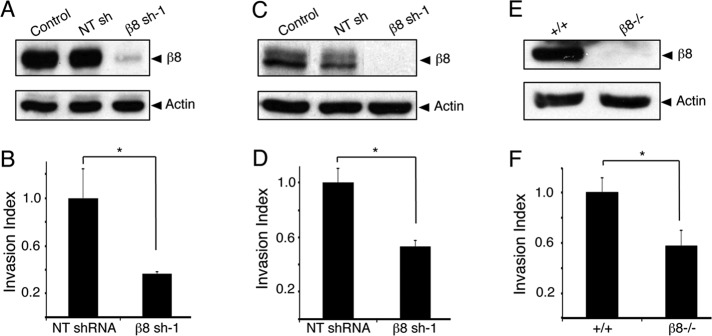
β8 integrin drives GBM cell invasion in vitro. Human LN229 cells (A), SNB19 cells (C), and transformed mouse astrocytes (E) express robust levels of β8 integrin protein. Cells genetically null for β8 integrin or expressing β8 integrin shRNAs show diminished integrin protein expression. LN229 cells (B), SNB19 cells (D), and transformed mouse astrocytes (F) expressing diminished β8 integrin proteins display significantly reduced invasiveness; *p < 0.005.
To analyze functions for β8 integrin in invasive GBM growth in vivo, we stereotactically injected LN229 cells stably expressing scrambled shRNAs or β8 shRNAs (β8 sh-1) into the striatum of NCR-nu/nu mice. Six different mice (n = 6) were injected per cell type, and all animals were killed 6 wk later to compare integrin-dependent tumor growth. As shown in Figure 2A, all LN229 tumors expressing scrambled shRNAs revealed focal, periventricular lesions that displayed infiltrative growth patterns. All intracranial tumors derived from LN229 cells expressing β8 shRNAs were larger and nearly filled the injected hemisphere. Quantitation of tumor volumes by measuring the largest cross-sectional areas in hematoxylin and eosin (H&E)–stained slides revealed 10-fold larger sizes of tumors expressing β8 shRNAs in comparison to nontargeting shRNAs (Figure 2B). H&E staining and anti-GFP immunofluorescence revealed that LN229 cells expressing nontargeting shRNAs displayed diffuse perivascular growth patterns, whereas tumor cells expressing β8 shRNAs showed minimal invasion into the surrounding brain parenchyma and displayed well-defined margins (Figure 2, C and D). Integrin-dependent differences in invasive growth were not due to differential tumor cell proliferation in vivo, since staining brain sections with anti-Ki67 antibodies revealed similar immunoreactivity in tumor cells expressing scrambled shRNAs or β8 shRNAs (Supplemental Figure 4, A and B). Furthermore, in comparison to LN229 cells expressing scrambled shRNAs, cells expressing β8 shRNAs did not display obvious differences in adherent growth in vitro (Supplemental Figure S4C). It is likely that the integrin-dependent differences in tumors volumes are due, in part, to more robust intratumoral angiogenesis and vascular permeability, as we reported previously (Tchaicha et al., 2010, 2011).
FIGURE 2:
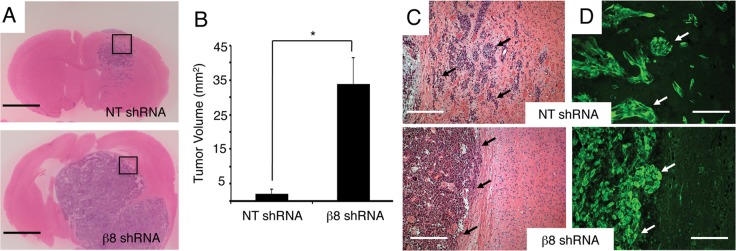
β8 integrin is essential for GBM cell invasion in vivo. (A) Coronal sections from LN229 tumors expressing scrambled shRNAs (top) or β8 shRNAs (bottom) were analyzed by H&E staining. Mice (n = 6 per cell type) were injected with GBM cells. Representative images are shown. Scale bars, 2 mm. (B) Quantitation of tumor volumes, as determined by measuring cross-sectional areas of H&E–stained sections. LN229 cells expressing β8 shRNAs generate significantly larger tumors. Sections from six different tumors per cell type were analyzed; p < 0.001. (C) High magnification H&E–stained images from boxed areas in A. Coronal sections through LN229 tumors expressing scrambled shRNAs (top) or β8 shRNAs (bottom). Scale bars, 200 μm. (D) LN229 tumors expressing scrambled shRNAs (top) or β8 shRNAs (bottom) were immunofluorescently labeled with anti-GFP antibodies (green). Note that LN229 cells expressing scrambled shRNAs display robust patterns of perivascular invasion, whereas tumor cells expressing β8 shRNAs display diminished invasiveness. Scale bars, 100 μm.
Members of the Rho GTPase protein family control cytoskeletal dynamics and play important roles in regulating cell polarity and motility (Garcia-Mata et al., 2011; Figure 3A), leading us to hypothesize that abnormal Rho GTPase activation might underlie the β8 integrin–dependent GBM cell invasion defects. Therefore we analyzed levels of GTP-bound Rho proteins after transient ITGB8 silencing (Figure 3B). In addition, levels of phosphorylated p21-activated kinase (Pak1), which undergoes autophosphorylation upon association with GTP-bound Rho proteins (Arias-Romero and Chernoff, 2008; Whale et al., 2011), were quantified. As shown in Figure 3C, elevated levels of phosphorylated Pak1 were detected in GBM cells expressing β8 small interfering RNAs (siRNAs). Lentiviral-delivered shRNAs targeting ITGB8 also showed elevated levels of Pak1 phosphorylation (Supplemental Figure S1). Increased levels of GTP-bound Rac1 and Cdc42, but not RhoA, were also evident in LN229 cells expressing diminished levels of β8 integrin (Figure 3C). Transformed mouse astrocytes genetically null for β8 integrin also displayed increased levels of phosphorylated Pak1, as well as of GTP-bound Rac1 and Cdc42 proteins (Supplemental Figure S5). Reexpression of β8 integrin in β8−/− cells resulted in reduced Rac1 and Pak1 activation and significant rescue of invasive cell behaviors (Supplemental Figure S5). Furthermore, tumors derived from LN229 cells expressing β8 shRNAs showed increased levels of phosphorylated Pak1 in situ (Figure 3, D and E).
FIGURE 3:
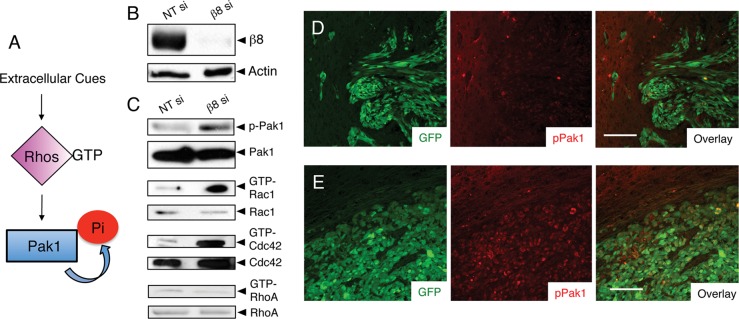
β8 integrin suppresses Rac1 and Cdc42 activation in GBM cells. (A) Schematic model for regulation of Rho GTPase and Pak1 activation by extracellular cues. (B) Lysates from LN229 cells expressing scrambled siRNAs or siRNAs targeting β8 integrin were immunoblotted for β8 integrin or actin. (C) Integrin-dependent levels of phosphorylated-Pak1 or GTP-bound Rac1, Cdc42, and RhoA were analyzed in LN229 cell lysates. Phosphorylated Pak1 levels are increased after integrin silencing. Similarly, elevated levels of GTP-bound Rac1 and Cdc42, but not RhoA, were detected in cells expressing diminished levels of β8 integrin. (D, E) Intracranial tumors generated from LN229 cells expressing scrambled shRNAs (D) or β8 shRNAs (E) were analyzed by double immunofluorescence using anti–phospho-Pak1 (red) and anti-GFP (green) antibodies. At least three different sections from four different tumors were analyzed per cell type, with representative images shown. Note the elevated levels of phosphorylated Pak1 in tumor cells expressing β8 shRNAs. Scale bars, 100 μm.
The increased levels of GTP-bound Rho proteins in cells expressing diminished β8 integrin suggested that Rho hyperactivation contributed to defects in invasion. Therefore we expressed yellow fluorescent protein (YFP)-tagged and constitutively active Rac1 variant (Q61L) in LN229 cells (Figure 4A) and then analyzed cell morphology and invasiveness in vitro. As shown in Figure 4B, in comparison to the elongated morphologies of LN229 cells expressing YFP alone, cells expressing Rac1-Q61L showed flattened morphologies. Three-dimensional Matrigel assays revealed diminished invasive capacities in cells expressing Rac1-Q61L (Figure 4C).
FIGURE 4:
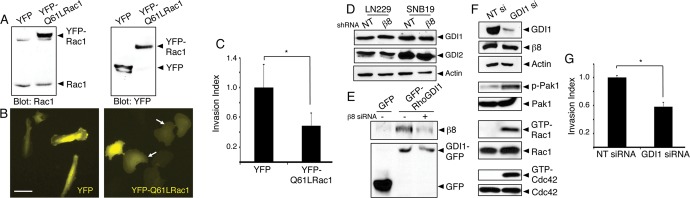
Rac1 hyperactivation or RhoGDI1 inactivation leads to impaired GBM cell invasion. (A) Lysates from LN229 cells expressing YFP or YFP-tagged Q61L-Rac1 were immunoblotted with anti-Rac1 (left) or anti-GFP (right) antibodies, revealing similar levels of expression. (B) Images of LN229 cells expressing YFP (left) or YFP-tagged Q61L-Rac1 (right). Note the flattened morphologies of cells expressing Q61L-Rac1 (arrows). Scale bar, 60 μm. (C) Quantitation of three-dimensional invasive capacities of LN229 cells expressing YFP or YFP-Q61L-Rac1; *p < 0.05. (D) SNB19 and LN229 cells expressing scrambled shRNAs or β8 shRNAs were analyzed for endogenous RhoGDI1 and RhoGDI2 protein expression. Levels of RhoGDI proteins are not altered in cells expressing β8 shRNAs. (E) LN229 cells transfected with plasmids expressing GFP or GFP-tagged RhoGDI1 were lysed and immunoprecipitated with anti-GFP monoclonal antibodies and immunoblotted with anti-β8 integrin antibodies (top) or anti-GFP polyclonal antibodies (bottom). Note that β8 integrin and RhoGDI1 proteins coimmunoprecipitate, and these associations are reduced in LN229 cells expressing β8 siRNAs. (F) LN229 cells expressing scrambled siRNAs or siRNAs targeting RhoGDI1 were analyzed for RhoGDI1 protein expression. Silencing RhoGDI1 leads to elevated levels of phosphorylated Pak1 and increased levels of GTP-bound Rac1 and Cdc42. (G) Invasive behavior of LN229 cells expressing nontargeting siRNAs or RhoGD1 siRNAs quantified in invasion assays; *p < 0.05.
In kidney glomerular cells the cytoplasmic domain of β8 integrin interacts directly with RhoGDI1, leading to release of GDP-bound Rac1 from RhoGDI1 and subsequent Rac1 activation (Lakhe-Reddy et al., 2006). Therefore we interrogated a role for RhoGDIs in integrin-mediated control of GTPase activation. Robust expression of endogenous RhoGDI1 and RhoGDI2 proteins was detected in LN229 and SNB19 GBM cells, and stably silencing β8 integrin with shRNAs did not affect RhoGDI protein levels (Figure 4D). To analyze associations between β8 integrin and RhoGDI proteins, we transfected LN229 cells with plasmids expressing GFP or GFP-tagged RhoGDI1 or RhoGDI2. Detergent-soluble cell lysates were immunoprecipitated with anti-GFP or anti–β8 integrin antibodies, revealing binding between β8 integrin and RhoGDI1 (Figure 4E and Supplemental Figure S6). Interactions between β8 integrin and RhoGDI2 were not detected by coimmunoprecipitation (unpublished data). To determine whether the β8 integrin–dependent GBM cell–invasive behaviors correlated with abnormal regulation of RhoGDI1, we used siRNAs to silence RhoGDI1 gene expression. LN229 cells expressing diminished levels of RhoGDI1 displayed elevated amounts of phosphorylated Pak1, as well as increased levels of GTP-bound Rac1 and Cdc42 (Figure 4F). We next analyzed RhoGDI1-dependent GBM cell invasion. RhoGDI1 silencing also resulted in significantly reduced LN229 and SNB19 GBM cell invasion in three-dimensional Matrigel assays (Figure 4G and Supplemental Figure S7). These results are consistent with a prior report showing that silencing RhoGDI1 resulted in hyperactivation of Rho proteins and reduced cell motility (Boulter et al., 2010).
To further explore functional links between αvβ8 integrin and RhoGDI1, we generated a β8 integrin mutant construct consisting of the extracellular and transmembrane domains but containing only 10 amino acids of the cytoplasmic tail. As a control we generated a full-length β8 integrin construct containing all 65 amino acids of the β8 integrin cytoplasmic tail (β8.FL) (Figure 5A). In transfected cells these constructs were expressed on the surface and were distinguishable by molecular weight (Supplemental Figure S8). Unlike β8.FL, β8.Trunc did not coimmunoprecipitate with RhoGDI1 in GBM cells (unpublished data). Furthermore, β8.Trunc protein was not immunoreactive with an antibody recognizing the C-terminal integrin tail but was recognized by an antibody directed against the β8 integrin extracellular domain (Figure 5B). To analyze how uncoupling β8 integrin–RhoGDI1 interactions affects Rho GTPase activation, we forcibly expressed β8.Trunc and β8.FL in β8−/−-mouse transformed astroglial cells. These β8−/− cells display high levels of GTP-bound Rac1 and phosphorylated Pak1 and are poorly invasive (Figures 1 and 2). As shown in Figure 5C, exogenous expression of β8.FL resulted in decreased Rac1 activation and Pak1 phosphorylation. In contrast, forced expression of β8.Trunc did not significantly alter levels of activated Rac1 but did partially diminish levels of phosphorylated Pak1, suggesting that β8.Trunc retains some signaling capacities via Pak1 that are RhoGDI1 independent. To determine how uncoupling β8 integrin-RhoGDI1 interactions influences invasiveness, we analyzed β8−/− cells expressing β8.FL or β8.Trunc proteins in Matrigel invasion assays. β8−/− cells expressing β8.FL, but not β8.Trunc, displayed a significant increase in invasive behaviors (Figure 5D). Similarly, invasion defects in LN229 cells expressing siRNAs targeting the endogenous ITGB8 3′ untranslated region were largely rescued after exogenous expression of β8.FL but not β8.Trunc (unpublished data). Collectively these data reveal that protein complexes between β8 integrin and its intracellular signaling effector, RhoGDI1, are essential for balancing Rho GTPase activation and GBM cell invasion.
FIGURE 5:
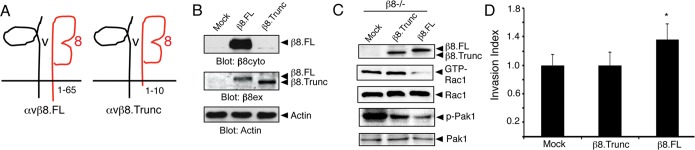
Uncoupling β8 integrin–RhoGDI1 interactions leads to enhanced Rac1 activation and diminished GBM cell invasion. (A) Diagram showing αvβ8.FL containing 65 amino acids in the β8 integrin cytoplasmic tail and αvβ8.Trunc containing only 10 amino acids in the β8 integrin tail. (B) β8.FL and β8.Trunc proteins were expressed in HEK-293T cells, and lysates were immunoblotted with antibodies recognizing the β8 integrin cytoplasmic (β8cyto) or extracellular domains (β8ex). (C) Expression of β8.FL in β8−/− transformed astroglial progenitor cells reduces Rac1 activation, whereas levels of GTP-bound Rac1 remain high in cells expressing β8.Trunc protein. (D) In contrast to β8.FL, β8.Trunc protein does not ameliorate invasion defects in β8−/−-transformed cells; *p, **p = 0.05.
We next extended our analysis of cell culture systems and mouse xenograft models to analyze β8 integrin in human clinical samples. First, mRNA expression levels in normal human brain versus GBM samples were compared using Oncomine to query publicly available databases. As shown in Figure 6A, GBM samples from two different cDNA microarray–based studies showed a significant increase in ITGB8 mRNA expression. In the first study, 27 GBM samples were compared with four normal brain samples, revealing a 2.3-fold increase in ITGB8 expression (Bredel et al., 2005). In the second study, analysis of 81 GBM samples and 27 normal brain samples detected a 2.6-fold increase in ITGB8 levels (Sun et al., 2006). Analysis of a cDNA microarray data set for GBM samples generated from The Cancer Genome Atlas (TCGA; Ovaska et al., 2010) revealed a similar twofold increase (p = 2.8 × 10−6) in ITGB8 expression in GBM samples versus normal brain tissue. Of interest, a separate TCGA-based analysis of gene expression profiles in GBM identified ITGB8 as a molecular marker for “classic” GBM subtypes (Verhaak et al., 2010).
FIGURE 6:
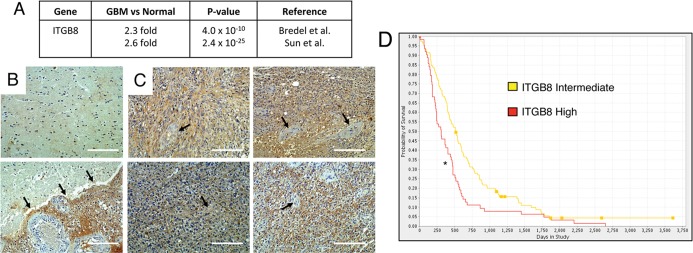
High ITGB8 expression in GBM correlates with reduced patient survival. (A) Oncomine summary of two independent transcriptome data sets comparing normal human brain samples to GBM samples. A greater-than-twofold increase in ITGB8 expression was detected in GBM samples as compared with normal brain samples. (B) Two different human brain sections containing neural tissue adjacent to the primary GBM were immunostained with anti–β8 integrin antibodies. Note the low levels of β8 integrin protein expression in the normal human brain. Bottom, a tumor margin revealing β8 integrin protein markedly upregulated in GBM cells. (C) Four different human GBM samples near the tumor core were immunolabeled with anti–β8 integrin antibodies. Note the robust levels of β8 integrin protein in tumor cells, with little if any integrin expression in intratumoral blood vessels (arrows). Scale bars, 200 μm. (D) Kaplan–Meier survival curve comparing human glioma samples with twofold or greater ITGB8 expression (n = 114) vs. samples with intermediate levels of ITGB8 expression (n = 225). Elevated ITGB8 expression correlates with diminished patient survival; *p < 0.001. Results were generated using the National Cancer Institute REMBRANDT database.
In addition, we analyzed β8 integrin protein expression in four different GBM samples by immunostaining Formalin-fixed, paraffin-embedded sections with human-specific goat anti–β8 integrin antibodies. As shown in Figure 6B, low levels of β8 integrin protein were detected in “normal” brain regions adjacent to the primary GBM mass, with higher integrin expression in GBM cells in the adjacent tumor. Within the GBM core we detected robust β8 integrin protein expression in tumor cells but not in intratumoral blood vessels (Figure 6C). The goat anti–β8 integrin antibody specificity was confirmed by immunostaining GBM samples with nonspecific control immunoglobulin Gs (Supplemental Figure S9). In addition, analysis of the National Cancer Institute Repository of Molecular Brain Neoplasia Data database (http://rembrandt.nci.nih.gov) revealed that astrocytoma samples with a twofold or greater increase in ITGB8 expression (n = 114) correlated with diminished patient survival (Figure 6D) as compared with astrocytoma samples with intermediate levels of ITGB8 expression (n = 225). Collectively these data reveal that increased β8 integrin expression predicts poor outcome, likely due to enhanced invasive GBM cell growth.
DISCUSSION
In this article we characterized β8 integrin–dependent signaling pathways that drive GBM cell invasion. Our experiments reveal the following novel findings: 1) β8 integrin is highly expressed in human GBM cells, and RNAi-mediated silencing of integrin expression significantly diminishes invasiveness in vitro and in vivo (Figures 1 and 2); 2) β8 integrin suppresses Rho GTPase activation in GBM cells, with diminished β8 integrin expression leading to elevated levels of GTP-bound Cdc42 and Rac1 (Figure 3); 3) expression of a hyperactive Rac1 variant or RNAi-mediated silencing of RhoGDI1 in GBM cells leads to diminished invasiveness (Figure 4); 4) uncoupling integrin–RhoGDI1 associations leads to Rac1 hyperactivation and diminished invasiveness (Figure 5); and 5) β8 integrin mRNA and protein are highly expressed in many human GBM samples, with elevated levels of expression correlating with reduced patient survival (Figure 6). Collectively these data reveal for the first time that β8 integrin regulates the activation status of Rho GTPases to promote GBM cell invasion within the brain microenvironment (Figure 7).
FIGURE 7:
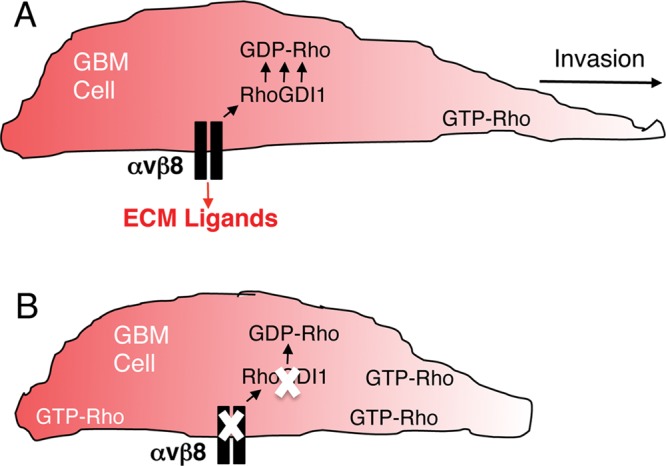
αvβ8 integrin–RhoGDI1 protein complexes regulate Rho GTPase activation and drive GBM cell invasion. (A) αvβ8 integrin is expressed in GBM cells, where it interacts with RhoGDI1 via the β8 cytoplasmic tail. αvβ8 integrin–RhoGDI1 protein complexes recruit GDP-bound Rac1 and Cdc42 to control activation of Rho proteins, thus driving GBM cell polarity and invasion. In addition, αvβ8 integrin likely binds to ECM protein ligands, including latent TGFβs, to regulate intracellular signaling pathways involved in GBM cell invasion. (B) Uncoupling αvβ8 integrin–RhoGDI1 protein complexes by mutating the integrin cytoplasmic tail or silencing β8 integrin or RhoGDI1 gene expression leads to elevated levels of GTP-bound Rho proteins, resulting in diminished GBM cell polarity and invasion.
The primary amino acid sequence of the β8 integrin cytoplasmic domain shares little homology with other β integrin subunits. For example, the NPXY peptide motif present in most integrin cytoplasmic tails, which facilitates interactions with FERM domain–containing proteins such as talins (Kim et al., 2011) and kindlins (Malinin et al., 2010), is absent in β8 integrin. These differences suggest that β8 integrin activation and/or signaling occur via mechanisms distinct from those used by other integrins. Indeed, our group and others have found that the β8 integrin cytoplasmic tail signals via intracellular proteins that are not commonly associated with other integrins (McCarty et al., 2005a; Triolo et al., 2006). In the kidney direct interactions between β8 integrin and RhoGDI1 lead to dissociation of Rac1 from RhoGDI1, enabling increased Rac1 activation and suppression of RhoA activation. Integrin-dependent balances between activated (GTP-bound) forms of Rac1 and RhoA normally inhibit mesangial cell differentiation (Lakhe-Reddy et al., 2006), with β8−/− or RhoGDI1−/− cells showing elevated levels of active RhoA, diminished levels of active Rac1, and a differentiated myofibroblast-like phenotype. In contrast, our experiments reveal that uncoupling interactions between β8 integrin and RhoGDI1 in GBM cells leads to increased levels of Rac1 and Cdc42 activation and impaired invasion, suggesting GBM cell type–specific roles for β8 integrin in regulating RhoGDI1 functions. Nonetheless, these data support a model (Figure 7) in which β8 integrin enhances the affinity of RhoGDI1 for GDP-bound Rhos, possibly resulting in increased extraction of GDP-bound Rhos from the plasma membrane or decreased delivery from the endoplasmic reticulum to the plasma membrane. The activated Rho proteins in GBM cells expressing β8 shRNAs are quite likely membrane associated, since a prior report demonstrated that silencing RhoGDI1 expression leads to enriched levels of membranous GTP-bound Rho proteins and impaired directional motility (Boulter et al., 2010).
Other proteins in addition to β8 integrin interact with RhoGDIs to maintain precise levels of GTP-bound Rho proteins (Garcia-Mata et al., 2011). For example, complexes of RhoGDI1, synectin, and syndecan 4 sequester GDP-bound RhoG during endothelial cell growth and migration (Elfenbein et al., 2009). During axonal growth the nerve growth factor receptor p75 binds directly to the RhoGDI1 complex to release GDP-bound Rho proteins (Yamashita and Tohyama, 2003). In GBM cells semaphorin 5A promotes recruitment of RhoGDI1 to plexin-B3, leading to reduced invasiveness (Li and Lee, 2010). Associations between β8 integrin and RhoGDI1 may serve to localize RhoGDI1 to membranes, thus stimulating binding of GDP-bound Cdc42 and Rac1 at the invasive front of a tumor cell to limit their activation by growth factors and β1 integrins (Del Pozo et al., 2002). Alternatively, β8 integrin–RhoGDI1 associations may occur at the trailing edge, thus facilitating retraction as the cell navigates through the ECM. In addition, integrin adhesion to ECM protein ligands likely influences intracellular signaling. The β8 integrin cytoplasmic tail is dispensable for latent TGFβ activation (Mu et al., 2002); hence it remains possible that αvβ8 integrin–mediated TGFβ activation in the ECM regulates intracellular signaling via RhoGDI1 and other effectors. Of interest, genetic interactions between αvβ8 integrin and the guanine nucleotide exchange factor β-Pix are essential for brain development (Liu et al., 2012). β-Pix facilitates the exchange of GDP to GTP in Rac1 and binds to phosphorylated Pak1 (ten Klooster et al., 2006; Arias-Romero and Chernoff, 2008). Different pools of αvβ8 integrin protein, possibly localized to different membrane domains, may interact with RhoGDI1 versus β-Pix. Alternatively, a multimeric protein complex consisting of αvβ8 integrin, RhoGDI1, β-Pix, and other signaling effectors might control the proper temporal and spatial activation of Rho proteins. Along these lines, in the rescue experiments involving β8.Trunc protein we detected a minor rescue of Pak1 signaling (Figure 5), suggesting that interactions with other effectors in addition to RhoGDI1 might control proinvasive signaling.
Our data showing that integrin regulation of Rho activation promotes GBM cell invasion might have clinical relevance. The invasive behaviors of GBM cells make them resistant to most interventions, with infiltrating cells escaping surgical resection and generating lethal secondary lesions. Furthermore, antivascular therapies such as Avastin/bevacizumab result in only modest improvements in progression-free survival (Batchelor et al., 2007; Friedman et al., 2009; Norden et al., 2009) due to compensation by vascular endothelial growth factor (VEGF)-independent angiogenic pathways, as well as by enhanced GBM cell infiltration (Lucio-Eterovic et al., 2009; Paez-Ribes et al., 2009; Gerstner et al., 2010; Keunen et al., 2011; Reardon et al., 2011). Thus, components of the β8 integrin–RhoGDI1 signaling axis might be therapeutic targets for inhibiting GBM cell invasion during cancer progression and after antivascular therapies. Finally, overexpression of ITGB8 and Rho signaling pathway genes have been reported in peripheral nerve sheath tumors, and silencing these factors diminishes invasiveness (Upadhyaya et al., 2012). In addition, αvβ8 integrin has been reported to promote invasive cell growth in nonneural cancers; for example, breast cancer cells up-regulate β8 integrin during metastatic dispersal to the lung (Landemaine et al., 2008), and silencing integrin expression leads to diminished metastatic lesions (Xu and Wu, 2012). Hence ITGB8, RhoGDI1, and related signaling components might drive invasive cell growth in cancers outside of the CNS.
MATERIALS AND METHODS
GBM cell lines and human tumor samples
LN229 and SNB19 human GBM cell lines were purchased from the American Type Culture Collection (Manassas, VA). Approval for the use of human specimens was obtained from the Institutional Review Board (IRB) at the University of Texas MD Anderson Cancer Center. The IRB waived the requirement for informed consent for previously collected residual tissues from surgical procedures stripped of unique patient identifiers according to Declaration of Helsinki guidelines. Oncomine (Compendia Bioscience, Ann Arbor, MI) was used for analysis and visualization of integrin gene expression levels in normal human brain versus GBM samples. REMBRANDT (http://rembrandt.nci.nih.gov) was used to generate integrin-dependent patient survival. TCGA-based analysis of ITGB8 expression levels in GBM samples was performed at http://csbi.ltdk.helsinki.fi/anduril/tcga-gbm (Ovaska et al., 2010).
Antibodies and immunohistochemistry
The following antibodies were purchased from commercial sources: goat anti–β8 integrin (C-19; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit or mouse anti-GFP (Abcam, Cambridge, MA), rabbit anti-actin (Sigma-Aldrich, St. Louis, MO), rabbit anti-Ki67 (Dako, Carpinteria, CA), rabbit anti-Cdc42 and anti-RhoA (Cell Signaling, Beverly, MA), mouse anti-Rac1 and rabbit anti-RhoGDI1 (Millipore, Billerica, MA), rabbit anti-RhoGDI2 (Thermo Scientific, Waltham, MA), and rabbit anti-pPak1 used for immunohistochemistry (Abcam). The anti–phospho-Pak1 antibody used for immunoblotting has been reported previously (Shamah et al., 2001). The anti–αv integrin and anti-β8 rabbit polyclonal antibodies directed against the integrin cytoplasmic tail have been described elsewhere (McCarty et al., 2005a; Jung et al., 2011). Goat anti-β8 antibodies directed against the integrin extracellular domain used for immunohistochemistry (C-19) and immunoprecipitation/immunoblotting (G-17) were purchased from Santa Cruz Biotechnology. Secondary antibodies used for immunofluorescence were all conjugated to Alexa 488 or Alexa 594 (Molecular Probes, Eugene, OR). Horseradish peroxidase (HRP)–conjugated secondary antibodies were purchased from Jackson ImmunoResearch Labs (West Grove, PA).
Formalin-fixed, paraffin-embedded sections were incubated in blocking solution, and then primary antibodies were added. Sections were rinsed three times with phosphate-buffered saline (PBS), blocked with peroxidase solution, and incubated with secondary antibodies conjugated to HRP. Then VIP or DAB chromagens were added to visualize immunoreactivity. LN229 and SNB19 GBM cells were purchased from the American Type Culture Collection and cultured in DMEM/F12 (Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and antibiotics. Isolation and transformation of wild-type and β8−/− mouse astrocytes has been described previously (Tchaicha et al., 2010).
Plasmids, lentiviral vectors, and siRNAs
Plasmids expressing GFP-tagged RhoGDI1 and RhoGDI2 have been described elsewhere (Moissoglu et al., 2009). The plasmid expressing Q61L-Rac1 tagged at the N-terminus with YFP has been described previously and was purchased from Addgene (Cambridge, MA; Hoppe and Swanson, 2004). Oligonucleotide sequences and detailed methods for generation of shRNAs targeting β8 integrin have been described elsewhere (Tchaicha et al., 2011). Viruses were packaged by transfecting 293-FT cells (American Type Culture Collection) with pLB transfer vectors and plasmids encoding gag/pol and VSV-G envelope proteins. Pools of siRNAs targeting human β8 integrin or RhoGDI1 were purchased from Dharmacon (Lafayette, CO). GBM cells were transfected with siRNAs using manufacturers’ reagents. Full length (1–769) or truncated (1–714) human ITGB8 cDNAs were amplified by PCR and subcloned into EcoRI/BamHI sites of either pcDNA3.1 or the pCDH lentivirus transfer vector (pCDH-CMV-MCS-EF1-copGFP), and DNA cloning was confirmed by sequencing. Full-length human ITGB8 contains 65 amino acids of the cytoplasmic domain, whereas the truncated construct contains only the 10 juxtamembrane amino acids.
Stereotactic injections
All animal procedures were conducted under Institutional Animal Care and Use Committee–approved protocols. NCR-nu/nu male mice (Jackson Laboratory, Bar Harbor, ME) were anesthetized, and a single incision was made from the anterior pole of the skull to the posterior ridge. We targeted the striatum for cell implantation using the following stereotactic coordinates: 1.5 mm rostral, 1.5 mm anterior, and 4 mm below the pial surface. An automated micropump (Stoelting Instruments, Wood Dale, IL) was used to dispense cells in 3 μl of PBS over a 5-min period. Human intracranial tumors were generated by injecting 2 × 105 LN229 GBM cells into the striatum of mice. Animals were monitored for tumor-related neurological deficits, and moribund animals were killed with gaseous CO2. Mice were perfused with 4% paraformaldehyde/PBS, and brains were coronally sliced at 1-mm intervals; paraffin-embedded tissues were serially sectioned at 7-μm intervals.
Invasion assays
We added 5 × 104 cells in serum-free media to the upper chambers of Matrigel invasion Transwell systems with 8-μm pore sizes (BD Biosciences, San Diego, CA). Growth medium containing 10% fetal bovine serum was added to the lower wells, and invasion was quantified after 18–24 h by staining with hematoxylin and counting the number of invasive cells on the underside of the Transwell filter. For determining the invasion index, the average number of invading cells under control conditions (scrambled shRNAs or siRNAs) was set at 1 and then compared with cells expressing targeted shRNAs or siRNAs.
GTPase activation assays
GTP-bound Cdc42 and Rac1 were fractionated using glutathione S-transferase–tagged p21-binding domain of Pak1 as previously described (Tolias et al., 2007). Rhotekin conjugated to agarose (Cytoskeleton, Denver, CO) was used for fractionating GTP-bound RhoA. Cells were harvested in Mg2+ lysis buffer containing 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, 150 mM NaCl, 1% NP-40, and 10 mM MgCl2 containing 1 mM dithiothreitol, 1 mM Na3VO4, 10 mM NaF, 10 mM β-glycerol phosphate, and EDTA-free protease inhibitors (Roche, Indianapolis, IN). The lysates were incubated with PBD or Rhotekin beads for 20 min at 4°C. After precipitation, the pellets were washed three times with Mg2+ lysis buffer and then analyzed by Western blotting to determine levels of active Rac1, Cdc42, or RhoA.
Statistical analyses
Student's t test was performed to determine statistically significant differences between groups. The Wilcoxon rank sum test was used for analysis of Kaplan–Meier survival results. Excel (Microsoft, Redmond, WA) was used to calculate statistics.
Supplementary Material
Acknowledgments
We thank Martin Schwartz (Yale University, New Haven, CT) for providing GFP-tagged RhoGDI constructs. This research was supported by grants awarded to J.H.M. by the National Institutes of Neurological Disease and Stroke (R01NS059876 and R01NS078402) and the National Cancer Institute (P50CA127001).
Abbreviations used:
- ECM
extracellular matrix
- GBM
glioblastoma multiforme
- RhoGDI1
Rho-GDP dissociation inhibitor 1
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-07-0521) on January 2, 2013.
REFERENCES
- Arias-Romero LE, Chernoff J. A tale of two Paks. Biol Cell. 2008;100:97–108. doi: 10.1042/BC20070109. [DOI] [PubMed] [Google Scholar]
- Arnold TD, Ferrero GM, Qiu H, Phan IT, Akhurst RJ, Huang EJ, Reichardt LF. Defective retinal vascular endothelial cell development as a consequence of impaired integrin alphavbeta8-mediated activation of transforming growth factor-beta. J Neurosci. 2012;32:1197–1206. doi: 10.1523/JNEUROSCI.5648-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor TT, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, Burridge K. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol. 2010;12:477–483. doi: 10.1038/ncb2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredel M, Bredel C, Juric D, Harsh GR, Vogel H, Recht LD, Sikic BI. High-resolution genome-wide mapping of genetic alterations in human glial brain tumors. Cancer Res. 2005;65:4088–4096. doi: 10.1158/0008-5472.CAN-04-4229. [DOI] [PubMed] [Google Scholar]
- Calabrese C, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Del Pozo MA, Kiosses WB, Alderson NB, Meller N, Hahn KM, Schwartz MA. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat Cell Biol. 2002;4:232–239. doi: 10.1038/ncb759. [DOI] [PubMed] [Google Scholar]
- Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfenbein A, Rhodes JM, Meller J, Schwartz MA, Matsuda M, Simons M. Suppression of RhoG activity is mediated by a syndecan 4-synectin-RhoGDI1 complex and is reversed by PKCalpha in a Rac1 activation pathway. J Cell Biol. 2009;186:75–83. doi: 10.1083/jcb.200810179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis LM, Reardon DA. Cancer: the nuances of therapy. Nature. 2009;458:290–292. doi: 10.1038/458290a. [DOI] [PubMed] [Google Scholar]
- Farin A, Suzuki SO, Weiker M, Goldman JE, Bruce JN, Canoll P. Transplanted glioma cells migrate and proliferate on host brain vasculature: a dynamic analysis. Glia. 2006;53:799–808. doi: 10.1002/glia.20334. [DOI] [PubMed] [Google Scholar]
- Friedman HS, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata R, Boulter E, Burridge K. The “invisible hand”: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12:493–504. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner ER, Chen PJ, Wen PY, Jain RK, Batchelor TT, Sorensen G. Infiltrative patterns of glioblastoma spread detected via diffusion MRI after treatment with cediranib. Neuro Oncol. 2010;12:466–472. doi: 10.1093/neuonc/nop051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson RJ, Rich JN. Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- Hirota S, Liu Q, Lee HS, Hossain MG, Lacy-Hulbert A, McCarty JH. The astrocyte-expressed integrin alphavbeta8 governs blood vessel sprouting in the developing retina. Development. 2011;138:5157–5166. doi: 10.1242/dev.069153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmeland AB, Lathia JD, Sathornsumetee S, Rich JN. Twisted tango: brain tumor neurovascular interactions. Nat Neurosci. 2011;14:1375–1381. doi: 10.1038/nn.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst. 2007;99:1583–1593. doi: 10.1093/jnci/djm187. [DOI] [PubMed] [Google Scholar]
- Hoppe AD, Swanson JA. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol Biol Cell. 2004;15:3509–3519. doi: 10.1091/mbc.E03-11-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, et al. Angiopoietin-2 induces human glioma invasion through the activation of matrix metalloprotease-2. Proc Natl Acad Sci USA. 2003;100:8904–8909. doi: 10.1073/pnas.1533394100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y, Kissil JL, McCarty JH. beta8 integrin and band 4.1B cooperatively regulate morphogenesis of the embryonic heart. Dev Dyn. 2011;240:271–277. doi: 10.1002/dvdy.22513. [DOI] [PubMed] [Google Scholar]
- Kanamori M, Vanden Berg SR, Bergers G, Berger MS, Pieper RO. Integrin beta3 overexpression suppresses tumor growth in a human model of gliomagenesis: implications for the role of beta3 overexpression in glioblastoma multiforme. Cancer Res. 2004;64:2751–2758. doi: 10.1158/0008-5472.can-03-3354. [DOI] [PubMed] [Google Scholar]
- Keunen O, et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci USA. 2011;108:3749–3754. doi: 10.1073/pnas.1014480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol. 2011;27:321–345. doi: 10.1146/annurev-cellbio-100109-104104. [DOI] [PubMed] [Google Scholar]
- Kissler S, Stern P, Takahashi K, Hunter K, Peterson LB, Wicker LS. In vivo RNA interference demonstrates a role for Nramp1 in modifying susceptibility to type 1 diabetes. Nat Genet. 2006;38:479–483. doi: 10.1038/ng1766. [DOI] [PubMed] [Google Scholar]
- Lakhe-Reddy S, et al. Beta8 integrin binds Rho GDP dissociation inhibitor-1 and activates Rac1 to inhibit mesangial cell myofibroblast differentiation. J Biol Chem. 2006;281:19688–19699. doi: 10.1074/jbc.M601110200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landemaine T, et al. A six-gene signature predicting breast cancer lung metastasis. Canc Res. 2008;68:6092–6099. doi: 10.1158/0008-5472.CAN-08-0436. [DOI] [PubMed] [Google Scholar]
- Lathia JD, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010;6:421–432. doi: 10.1016/j.stem.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lee AY. Semaphorin 5A and plexin-B3 inhibit human glioma cell motility through RhoGDIalpha-mediated inactivation of Rac1 GTPase. J Biol Chem. 2010;285:32436–32445. doi: 10.1074/jbc.M110.120451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zeng L, Kennedy RM, Gruenig NM, Childs SJ. betaPix plays a dual role in cerebral vascular stability and angiogenesis, and interacts with integrin alphavbeta8. Dev Biol. 2012;363:95–105. doi: 10.1016/j.ydbio.2011.12.022. [DOI] [PubMed] [Google Scholar]
- Lucio-Eterovic AK, Piao Y, de Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res. 2009;15:4589–4599. doi: 10.1158/1078-0432.CCR-09-0575. [DOI] [PubMed] [Google Scholar]
- Malinin NL, Plow EF, Byzova TV. Kindlins in FERM adhesion. Blood. 2010;115:4011–4017. doi: 10.1182/blood-2009-10-239269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty JH. Cell adhesion and signaling networks in brain neurovascular units. Curr Opin Hematol. 2009;16:209–214. doi: 10.1097/MOH.0b013e32832a07eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty JH, Cook AA, Hynes RO. An interaction between αvβ8 integrin and Band 4.1B via a highly conserved region of the Band 4.1 C-terminal domain. Proc Natl Acad Sci USA. 2005a;102:13479–13483. doi: 10.1073/pnas.0506068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty JH, Lacy-Hulbert A, Charest A, Bronson RT, Crowley D, Housman D, Savill J, Roes J, Hynes RO. Selective ablation of alphav integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development. 2005b;132:165–176. doi: 10.1242/dev.01551. [DOI] [PubMed] [Google Scholar]
- McCarty JH, et al. Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking alphav integrins. Mol Cell Biol. 2002;22:7667–7677. doi: 10.1128/MCB.22.21.7667-7677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner R, Campbell IL. The integrin family of cell adhesion molecules has multiple functions within the CNS. J Neurosci Res. 2002;69:286–291. doi: 10.1002/jnr.10321. [DOI] [PubMed] [Google Scholar]
- Mobley AK, McCarty JH. beta8 integrin is essential for neuroblast migration in the rostral migratory stream. Glia. 2011;59:1579–1587. doi: 10.1002/glia.21199. [DOI] [PubMed] [Google Scholar]
- Mobley AK, Tchaicha JH, Shin J, Hossain MG, McCarty JH. β8 integrin regulates neurogenesis and neurovascular homeostasis in the adult brain. J Cell Sci. 2009;122:1842–1851. doi: 10.1242/jcs.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moissoglu K, McRoberts KS, Meier JA, Theodorescu D, Schwartz MA. Rho GDP dissociation inhibitor 2 suppresses metastasis via unconventional regulation of RhoGTPases. Cancer Res. 2009;69:2838–2844. doi: 10.1158/0008-5472.CAN-08-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada M, Nakada S, Demuth T, Tran NL, Hoelzinger DB, Berens ME. Molecular targets of glioma invasion. Cell Mol Life Sci. 2007;64:458–478. doi: 10.1007/s00018-007-6342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HL, Lee YJ, Shin J, Lee E, Park SO, McCarty JH, Oh SP. TGF-beta signaling in endothelial cells, but not neuroepithelial cells, is essential for cerebral vascular development. Lab Invest. 2011;91:1554–1563. doi: 10.1038/labinvest.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden AD, Drappatz J, Wen PY. Antiangiogenic therapies for high-grade glioma. Nat Rev Neurol. 2009;5:610–620. doi: 10.1038/nrneurol.2009.159. [DOI] [PubMed] [Google Scholar]
- Ovaska K, et al. Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med. 2010;2:65. doi: 10.1186/gm186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor JM, Zang K, Wang D, Wang R, Reichardt LF. Vascular development of the brain requires beta8 integrin expression in the neuroepithelium. J Neurosci. 2005;25:9940–9948. doi: 10.1523/JNEUROSCI.3467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon DA, et al. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J Natl Compr Canc Netw. 2011;9:414–427. doi: 10.6004/jnccn.2011.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemenschneider MJ, Mueller W, Betensky RA, Mohapatra G, Louis DN. In situ analysis of integrin and growth factor receptor signaling pathways in human glioblastomas suggests overlapping relationships with focal adhesion kinase activation. Am J Pathol. 2005;167:1379–1387. doi: 10.1016/S0002-9440(10)61225-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamah SM, Lin MZ, Goldberg JL, Estrach S, Sahin M, Hu L, Neve RL, Corfas G, Debant A, Greenberg ME. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell. 2001;105:233–244. doi: 10.1016/s0092-8674(01)00314-2. [DOI] [PubMed] [Google Scholar]
- Shi Q, Hjelmeland AB, Keir ST, Song L, Wickman S, Jackson D, Ohmori O, Bigner DD, Friedman HS, Rich JN. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol Carcinog. 2007;46:488–496. doi: 10.1002/mc.20297. [DOI] [PubMed] [Google Scholar]
- Sonoda Y, Ozawa T, Hirose Y, Aldape KD, McMahon M, Berger MS, Pieper RO. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 2001;61:4956–4960. [PubMed] [Google Scholar]
- Sun L, et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006;9:287–300. doi: 10.1016/j.ccr.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Tchaicha JH, Mobley AK, Hossain MG, Aldape KD, McCarty JH. A mosaic mouse model of astrocytoma identifies alphavbeta8 integrin as a negative regulator of tumor angiogenesis. Oncogene. 2010;29:4460–4472. doi: 10.1038/onc.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchaicha JH, Reyes SB, Shin J, Hossain MG, Lang FF, McCarty JH. Glioblastoma angiogenesis and tumor cell invasiveness are differentially regulated by beta8 integrin. Cancer Res. 2011;71:6371–6381. doi: 10.1158/0008-5472.CAN-11-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Klooster JP, Jaffer ZM, Chernoff J, Hordijk PL. Targeting and activation of Rac1 are mediated by the exchange factor beta-Pix. J Cell Biol. 2006;172:759–769. doi: 10.1083/jcb.200509096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Kane CG, Tolias CS, Hu L, Greenberg ME. The Rac1 guanine nucleotide exchange factor Tiam1 mediates EphB receptor-dependent dendritic spine development. Proc Natl Acad Sci USA. 2007;104:7265–7270. doi: 10.1073/pnas.0702044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triolo D, Dina G, Lorenzetti I, Malaguti M, Morana P, Del Carro U, Comi G, Messing A, Quattrini A, Previtali SC. Loss of glial fibrillary acidic protein (GFAP) impairs Schwann cell proliferation and delays nerve regeneration after damage. J Cell Sci. 2006;119:3981–3993. doi: 10.1242/jcs.03168. [DOI] [PubMed] [Google Scholar]
- Upadhyaya M, Spurlock G, Thomas L, Thomas NS, Richards M, Mautner VF, Cooper DN, Guha A, Yan J. Microarray-based copy number analysis of neurofibromatosis type-1 (NF1)-associated malignant peripheral nerve sheath tumors reveals a role for Rho-GTPase pathway genes in NF1 tumorigenesis. Hum Mutat. 2012;33:763–776. doi: 10.1002/humu.22044. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whale A, Hashim FN, Fram S, Jones GE, Wells CM. Signalling to cancer cell invasion through PAK family kinases. Front Biosci. 2011;16:849–864. doi: 10.2741/3724. [DOI] [PubMed] [Google Scholar]
- Xu Z, Wu R. Alteration in metastasis potential and gene expression in human lung cancer cell lines by ITGB8 silencing. Anat Rec (Hoboken) 2012;295:1446–1454. doi: 10.1002/ar.22521. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tohyama M. The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci. 2003;6:461–467. doi: 10.1038/nn1045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.