

Myeloid-associated differentiation marker (MYADM) protein belongs to the MAL family and regulates raft domains in epithelial cells. Membrane rafts participate in inflammatory responses. This study shows that MYADM is expressed in endothelial cells and controls the endothelial barrier by regulating ICAM-1 expression through ezrin, radixin, and moesin proteins, connectors between plasma membrane domains and actin cytoskeleton.
Abstract
The endothelium maintains a barrier between blood and tissue that becomes more permeable during inflammation. Membrane rafts are ordered assemblies of cholesterol, glycolipids, and proteins that modulate proinflammatory cell signaling and barrier function. In epithelial cells, the MAL family members MAL, MAL2, and myeloid-associated differentiation marker (MYADM) regulate the function and dynamics of ordered membrane domains. We analyzed the expression of these three proteins in human endothelial cells and found that only MYADM is expressed. MYADM was confined in ordered domains at the plasma membrane, where it partially colocalized with filamentous actin and cell–cell junctions. Small interfering RNA (siRNA)-mediated MYADM knockdown increased permeability, ICAM-1 expression, and leukocyte adhesion, all of which are features of an inflammatory response. Barrier function decrease in MYADM-silenced cells was dependent on ICAM-1 expression. Membrane domains and the underlying actin cytoskeleton can regulate each other and are connected by ezrin, radixin, and moesin (ERM) proteins. In endothelial cells, MYADM knockdown induced ERM activation. Triple-ERM knockdown partially inhibited ICAM-1 increase induced by MYADM siRNA. Importantly, ERM knockdown also reduced ICAM-1 expression in response to the proinflammatory cytokine tumor necrosis factor-α. MYADM therefore regulates the connection between the plasma membrane and the cortical cytoskeleton and so can control the endothelial inflammatory response.
INTRODUCTION
The endothelium lines the inner side of blood vessels, forming a barrier between the blood and the surrounding tissue that is essential for vascular homeostasis. The endothelium mediates the passage of small molecules and cells from the bloodstream to the tissues, without compromising its integrity, in the presence of continuous osmotic and shear stress. The organization of the endothelial cell surface, the biological fence facing the vessel lumen, is thus essential for integrating signals from different sources that modulate selective permeability, such as mechanical forces, cytokine signaling, and cell–cell interactions (Millán and Ridley, 2005; Simionescu, 2007; Vandenbroucke et al., 2008; Reglero-Real et al., 2012).
The architecture of the endothelial surface is determined by the links between the plasma membrane, surface proteins, and underlying cytoskeleton. In the current paradigm, lipids and transmembrane proteins are connected to the underlying cytoskeleton like a picket fence that controls the stiffness of the cell surface and the ability of lipids and proteins to diffuse and signal (Ritchie et al., 2003). The 4.1 family proteins, ezrin, radixin, and moesin (ERM), are the best-known connectors between plasma membrane, transmembrane receptors, and the subcortical actin cytoskeleton (Bretscher et al., 2002; Viola and Gupta, 2007). An additional level of complexity in cell surfaces arises from the fact that the complex mixture of lipids forming the plasma membrane does not distribute homogeneously but can form microdomains with specific subsets of proteins. The model that best explains this heterogeneity, although it is still under debate, posits the existence of liquid-ordered membrane domains enriched in glycolipids and cholesterol, termed membrane rafts. Raft-mediated plasma membrane condensation would regulate signaling, protein trafficking, and compartmentalization (Lingwood and Simons, 2010; Simons and Gerl, 2010).
Some evidence suggests a mutual regulation between ERM proteins and ordered membrane rafts (Viola and Gupta, 2007). On activation, ERMs acquire an open conformation in which the N-terminal Four point one, ezrin, radixin-moesin (FERM) domain interacts directly or indirectly with the cytoplasmic tails of transmembrane receptors, and the C-terminal domain binds to actin filaments (Fehon et al., 2010). The FERM domain binds the raft lipid phosphatidylinositol 4,5-bisphosphate (PIP2) and some transmembrane proteins that also partition into ordered membranes (Wojciak-Stothard et al., 1999; Tilghman and Hoover, 2002; Johnson et al., 2008; Kwiatkowska, 2010). ERMs themselves have also been found compartmentalized into ordered membrane domains (Tomas et al., 2002; Gupta et al., 2006; Heyraud et al., 2007). Finally, active ERMs have been shown to regulate membrane raft content (Prag et al., 2007) and prevent membrane raft coalescence (Gupta et al., 2006).
Inflammatory cytokines have the ability to alter the endothelial barrier to cells and solutes by mechanisms that are not completely understood (Vandenbroucke et al., 2008). Disruption of cholesterol-rich membrane rafts has been shown to either promote or ameliorate inflammation in different cell types (Legler et al., 2003; Flemming et al., 2004; Meng et al., 2010). In addition, cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) activate ERM proteins. This activation mediates the increase in endothelial permeability or the transcriptional regulation in response to TNF-α (Kishore et al., 2005; Koss et al., 2006). However, the full role of ERM activation in the endothelial inflammatory response is still not well defined. It is of note that ERM proteins play an important role localizing receptors involved not only in leukocyte adhesion but also in the regulation of endothelial permeability in an inflammatory context, for example, intercellular adhesion molecule (ICAM)-1 or vascular cell adhesion molecule (VCAM)-1 (van Wetering et al., 2003; Clark et al., 2007; Sumagin et al., 2008).
In the current model of ordered membrane domains or rafts, protein machinery associated with these platforms is required to specify their function in the cell (Bauer and Pelkmans, 2006; Lingwood and Simons, 2010). The MAL family consists of integral proteins with several transmembrane domains involved in membrane dynamics (Sanchez-Pulido et al., 2002). Several lines of evidence support a role for these proteins in events related to organization of membrane domains, such as polarized membrane trafficking or plasma membrane reorganization in response to extracellular signaling (Puertollano et al., 1999; Anton et al., 2008; Goldstein Magal et al., 2009; Anton et al., 2011; Aranda et al., 2011). Myeloid-associated differentiation marker (MYADM) is a MAL family protein that regulates membrane order in migrating epithelial cells (Aranda et al., 2011). In this paper, we report that MYADM is a component of endothelial surface rafts. MYADM knockdown induces an inflammatory-like phenotype, altering barrier function through the increase of the adhesion receptor ICAM-1. The rise in ICAM-1 levels is mediated by MYADM-regulated activation of ERM proteins. Collectively our results strongly suggest that the interaction between the plasma membrane and the submembrane actin cytoskeleton at the endothelial surface controls the inflammatory response.
RESULTS
MYADM is confined into ordered domains in the endothelial plasma membrane
Specific types of highly polarized epithelial cells express the MAL family members MAL and MAL2, which are involved in specialized, raft-dependent, apical vesicular trafficking (Puertollano et al., 1999; de Marco et al., 2002). MYADM, in contrast, is expressed ubiquitously and organizes membrane rafts at the epithelial cell surface, thereby affecting cell spreading and motility (Aranda et al., 2011). The endothelium can be considered as a simple squamous epithelium (Palade et al., 1979). Because ordered membrane domains have been shown to be central to endothelial barrier function and signaling (Muppidi et al., 2004; Heyraud et al., 2007; Chidlow and Sessa, 2010; Triantafilou et al., 2010), we wondered whether these MAL proteins are expressed and play a role in primary human endothelial cells. Detergent-resistant membrane (DRM) fractions were isolated from confluent human umbilical vein endothelial cells (HUVECs) and blotted with antibodies against MAL, MAL2, and MYADM, together with a panel of cell lines used as positive and negative controls. Endogenous MYADM was clearly detected in DRM fractions from all the analyzed cell types, including HUVECs, consistent with the ubiquitous expression pattern previously reported (Aranda et al., 2011). In contrast, MAL and MAL2 were detected only in DRM fractions from epithelial PC3 and Madin-Darby canine kidney (MDCK) cells (Figure 1A). Quantitative PCR (qPCR) assays confirmed that expression of MAL and MAL2 was negligible in HUVECs (Figure 1B). Further analysis of MYADM fractionation in HUVECs showed that this protein is almost exclusively found in DRMs (Figure 1, C and D). The 2B12 monoclonal antibody (mAb), raised against human MYADM is not suitable for immunofluorescence analysis (Aranda et al., 2011). For the study of MYADM distribution, MYADM-green fluorescent protein (GFP) was expressed in human endothelial cells and analyzed by confocal microscopy. MYADM-GFP also partitioned in DRMs, indicating that the GFP tag does not alter MYADM insertion in plasma membrane domains (Figure 1E). MYADM-GFP was localized at the plasma membrane and in cell–cell junctions in confluent endothelial cells, and this distribution overlapped with the staining for filamentous actin and junctional markers, such as vascular endothelial (VE)-cadherin (Figure 1F, insets). A partial colocalization of MYADM-GFP at cell–cell junctions was also found with platelet endothelial cell adhesion molecule (PECAM)-1, p120-catenin, occludin, and nectin-2 (Supplemental Figure S1). In contrast, MYADM-GFP showed little colocalization with caveolin-1, a raft-associated protein essential for vascular homeostasis (Chidlow and Sessa, 2010), or with CD59, a glycosylphosphatidylinositol (GPI)-anchored protein often used as a marker of membrane rafts (Millán et al., 2006), which suggests that MYADM could organize ordered membrane domains differently from caveolae or CD59 in human endothelial cells (Figure 1F).
FIGURE 1:

MYADM is a transmembrane protein associated with endothelial DRMs. (A) The indicated cell types were extracted with 1% Triton X-100 at 4ºC and centrifuged to equilibrium in sucrose density gradients. Aliquots from DRMs were immunoblotted for MYADM, MAL2, human MAL (hMAL), canine MAL (dMAL), and caveolin-1 (Cav-1). (B) RNA from the indicated cells was isolated and the expression of MYADM, MAL2 and MAL transcripts was analyzed by qPCR. Results were normalized to mRNA levels of housekeeping genes (β-actin and GADPH) and represented as percentage of PC3 mRNA levels for MYADM and MAL2 transcripts (left) or Jurkat mRNA levels for the MAL transcript (right). (C) Aliquots from soluble (SOL) and DRM fractions from HUVECs were immunoblotted for MYADM, Cav-1 (as a control of endothelial DRMs), and transferrin receptor (TfR; as a control of transmembrane proteins in the soluble fraction). (D) Quantitation of DRM segregation of the indicated protein from three independent experiments. (E) DRMs were isolated from cells expressing MYADM-GFP for 24 h and immunoblotted for MYADM with anti-MYADM mAb 2B12 and for caveolin-1. (F) MYADM-GFP distributes at the plasma membrane of HUVECs and partially colocalizes with subcortical F-actin and with VE-cadherin. Caveolin-1 and CD59 staining reveals little colocalization of MYADM-GFP with caveolae or with GPI-protein rich-rafts in endothelial cells. Scale bar: 20 μm.
MYADM knockdown induces an inflammatory-like phenotype by inducing ICAM-1 expression
To gain insight into the role of MYADM in endothelial cells, we investigated the effects of three different small interfering RNAs (siRNAs) that targeted MYADM with low (siMYADM 1), medium (siMYADM 2), and high efficiency (siMYADM 3; Figure 2, A and B). MYADM silencing caused a clear disruption of the endothelial monolayer and partially disorganized cell–cell junctions, as shown by staining of β-catenin, VE-cadherin, or PECAM-1 (Figure 2, C and D). MYADM reduction induced the appearance of intercellular gaps (Figure 2, C and D, asterisks, and E) and increased both filamentous (F)-actin (Figures 2, C and F, and S2) and permeability in Transwell assays (Figure 2G). Collectively these results indicate that MYADM knockdown caused endothelial barrier dysfunction.
FIGURE 2:

MYADM knockdown alters endothelial barrier function. (A) HUVECs were transfected in parallel with control siRNA (siControl) or with three different siRNAs targeting MYADM (siMYADM). At 72 h after transfection, MYADM levels were analyzed by immunoblotting. ERK-1/2 and caveolin-1 (Cav-1) levels are shown as loading controls. (B) Quantitation of MYADM reduction in response to siRNA transfection. The mean + SEM from three independent experiments is presented. (C and D) HUVECs were transfected with the indicated siRNAs and plated at confluence on fibronectin-coated coverslips. After 72 h, cells were fixed and stained for F-actin and β-catenin (C) or VE-cadherin or PECAM-1 (D) to detect cell–cell junctions. Asterisks in the images indicate intercellular gaps. Scale bar: 20 μm for (C) and 10 μm for (D). (E) Quantitation of intercellular gaps in cells transfected with the indicated siRNAs. (F) Cells were fixed and stained with antibody against total actin or with TRITC–phalloidin. Quantitation of the ratio F-actin/total actin (see Figure S2). (G) HUVECs were transfected with the indicated siRNAs and plated at confluence on Transwells, and permeability assays were performed 72 h after transfection. The mean + SEM from three independent experiments is shown. *, p < 0.05; **, p < 0.01.
Increased permeability and polymerization of actin are prototypical endothelial responses to several inflammatory stimuli (Pober and Sessa, 2007). We tested whether MYADM knockdown was inducing an inflammatory-like response by following the expression of different receptors previously involved in leukocyte adhesion and permeability and considered typical inflammatory markers (Albelda, 1991; Clark et al., 2007; Sumagin et al., 2008). Western blot analysis indicated that reduction of MYADM levels induced the expression of the adhesion receptor ICAM-1, but not of E-selectin or VCAM-1 (Figure 3, A and B). MYADM knockdown also increased ICAM-1 mRNA, as shown by qPCR experiments (Figure 3C). Immunofluorescence assays revealed the surface accumulation of this receptor in MYADM-depleted cells (Figure 3D). In contrast, no difference was found in the expression of different components of cell–cell junctions involved in modulation of barrier function in an inflammatory context (Fernandez-Martin et al., 2012; Reglero-Real et al., 2012), such as VE-cadherin, β-catenin, p120-catenin, and PECAM-1 (Figure 3A). The Rho GTPases RhoA, Rac1, and Cdc42 have been involved in permeability and F-actin regulation. No statistically significant changes were detected in their activity upon MYADM knockdown (Figure S3, A and B). Also, no significant alteration was detected in the phosphorylation of regulatory myosin light chain, which indicates that actomyosin-mediated contractility downstream of RhoA is not affected by MYADM depletion (Figure S3C). Although MYADM knockdown induces intercellular gaps, no increase in VE-cadherin tyrosine phosphorylation or decrease in β-catenin association to VE-cadherin was detected upon MYADM knockdown (Figure S4), which suggests the stability of adherens junctions is not regulated by MYADM expression. Collectively these experiments indicate that MYADM plays a role in barrier function and ICAM-1 transcriptional regulation.
FIGURE 3:

MYADM knockdown induces ICAM-1 expression. HUVECs were transfected with the indicated siRNAs for 72 h, and the expression of different proteins involved in endothelial barrier function was analyzed by Western blot (A). Quantitation of ICAM-1 protein (B) and mRNA by qPCR (C). The mean + SEM from three (B) and two (C) independent experiments is shown. **, p < 0.01. (D) Localization of ICAM-1 in siRNA-transfected cells by immunofluorescence analysis. (E) HUVECs expressing MYADM-GFP for 36 h were stimulated with TNF-α (10 ng/ml) for 6 h to induce detectable levels of ICAM-1. Subsequently the receptor was cross-linked with specific antibodies (X-ICAM-1). Cells were fixed, permeabilized, and stained with TRITC–phalloidin to visualize F-actin. (F) HUVECs expressing MYADM-GFP were starved, stimulated with TNF-α (10 ng/ml) for 6 h to induce expression of adhesion receptors, and then incubated with T-cells for 15 min. Cells were fixed and stained with TRITC–phalloidin to visualize F-actin (top panels) or with TRITC–phalloidin and anti–caveolin-1 antibody (bottom panels). T-cells adhering or transmigrating were morphologically distinguishable by the F-actin staining (red dotted line). Right graphs show intensity profiles across the T-cell–endothelial cell interaction area along the indicated white arrows.
MYADM did not appear to associate with ICAM-1, in either ICAM-1 immunoprecipitation (unpublished data) or cross-linking experiments (Figure 3E). In addition, MYADM did not distribute around either adhered or transmigrating leukocytes (Figure 3F), in contrast with caveolin-1, which is enriched in areas where T-cells are transmigrating transcellularly, as previously described (Carman and Springer, 2004; Millán et al., 2006; Keuschnigg et al., 2009). Altogether these data suggest that MYADM depletion induces an increase in ICAM-1 expression but that MYADM is not directly associated with ICAM-1 or involved in ICAM-1 function. To rule out the possibility of an off-target effect of the siRNA oligonucleotides, we followed a rescue-of-function strategy by transfecting siMYADM 2, which targets the 3′ untranslated region of MYADM mRNA, in wild-type HeLa cells or in HeLa cells stably expressing MYADM-GFP (Aranda et al., 2011). The MYADM-GFP transcript lacks the 3′ untranslated region and is thus resistant to siMYADM 2–mediated knockdown. siMYADM 2 transfection induced ICAM-1 expression only in parental HeLa cells, whereas, as predicted, no changes were observed in cells stably expressing MYADM-GFP (Figure 4).
FIGURE 4:
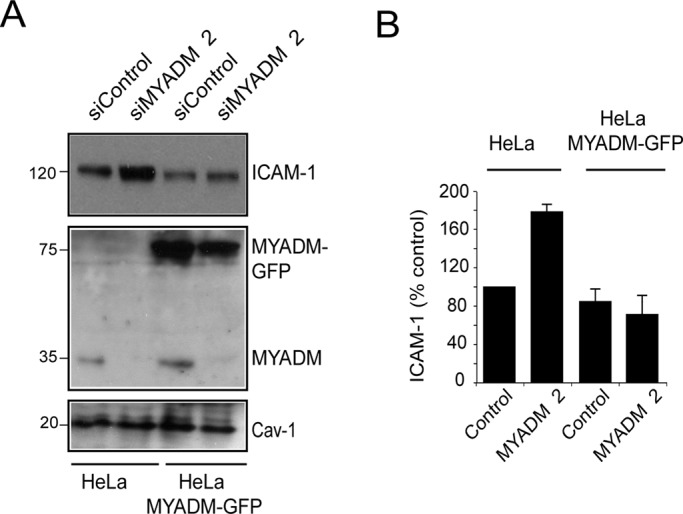
Exogenous expression of MYADM prevents ICAM-1 increase in response to MYADM siRNA transfection. (A) Parental HeLa cells or HeLa cells stably expressing MYADM-GFP were transfected with siControl or siMYADM 2 for 48 h. Cell extracts were then immunoblotted with anti–ICAM-1, anti-GFP, anti-MYADM, and anti–caveolin-1 (Cav-1) antibodies. (B) Quantitation of ICAM-1 levels in HeLa and HeLa-MYADM-GFP transfected with the indicated siRNAs. The mean + SEM is shown.
MYADM controls endothelial barrier function and ICAM-1 expression. We next addressed whether alteration of endothelial barrier function in MYADM knockdown (KD) cells is a consequence of such receptor increase. HUVECs were transfected with control siRNA, siRNA targeting either MYADM (siMYADM 2) or ICAM-1, or both simultaneously (Figure 5A), and subjected to calcium-switch experiments in an ECIS platform in order to analyze barrier dynamics (Figure 5B). Barrier formation was impaired in MYADM knockdown cells, whereas cells undergoing double silencing for MYADM and ICAM-1 restored endothelial barrier upon calcium replenishment, with dynamics comparable with control cells. In accordance, analysis of β-catenin staining showed that ICAM-1 reduction ameliorates the loss of cell–cell junction integrity in MYADM knockdown cells (Figure 5, C and D). These experiments indicate that increased ICAM-1 expression mediates endothelial barrier reduction upon MYADM knockdown.
FIGURE 5:

ICAM-1 knockdown prevents alteration of endothelial barrier function in response to MYADM reduction. HUVECs were transfected with the indicated siRNAs for 72 h. (A) Western blot analysis of ICAM-1 expression. ERK 1/2 is shown as a loading control. (B) Dynamics of barrier function in siRNA-transfected cells. Normalized transendothelial electrical resistance (TEER) of cells subjected to calcium switch in order to observe barrier formation. Right, quantitation of normalized resistance 60 min after calcium switch. The mean + SEM of three independent experiments with duplicate readings is shown. **, p > 0.02. (C) Cells were fixed and stained with β-catenin– and ICAM-1–specific antibodies. Asterisks in images indicate intercellular gaps. (D) Quantitation of intercellular gaps found in siRNA-transfected cells. Thirty images containing around 30 cells each were quantitated. The mean + SEM is shown. *, p < 0.05.
MYADM knockdown increases ERM phosphorylation
MYADM colocalizes with the cortical actin cytoskeleton and regulates ordered membrane domains at the plasma membrane (Aranda et al., 2011). We hypothesized that MYADM controls the inflammatory status by regulating proteins involved in plasma membrane and/or cortical actin organization of endothelial cells (Viola and Gupta, 2007). Activation of ERM proteins, connectors between the plasma membrane and the cytoskeleton, increases permeability in microvascular endothelial cells during the inflammatory response (Koss et al., 2006), so we tested the effect of MYADM knockdown on ERM phosphorylation at a specific threonine residue that is required for their conformational change and subsequent activation. Western blotting with an antibody recognizing phosphorylated ezrin (T567), radixin (T564), and moesin (T558) revealed that MYADM silencing increased ERM phosphorylation two- to threefold (Figure 6, A and B). Confocal analysis of MYADM-depleted cells confirmed an increase in phosphorylated ERMs at the plasma membrane (Figure 6C). Both in control and MYADM-depleted cells, pERM appeared particularly enriched in junctional membrane projections, often connecting two adjacent cells. The length of these border protrusions increased in response to MYADM knockdown (Figure 6C, arrowheads, and D). We then examined whether ERM phosphorylation can contribute to endothelial barrier dysfunction. Transfection of a constitutive active form of ERMs (moesin T558D) caused a moderate decrease of cell–cell junction integrity, whereas moesin T558A mutant, which is proposed to be in a closed, inactive conformation, did not significantly alter the distribution of the junctional marker β-catenin (Figure 6, E and F).
FIGURE 6:

MYADM knockdown activates ERM proteins. (A) HUVECs were transfected with the indicated siRNAs for 72 h, and ERM activation was monitored by Western blot with anti-pE(T567)R(T564)M(T558)–specific antibody (pERM) and anti-ERM protein antibody. (B) Quantitation of pERM activation was performed by measuring the ratio of anti-pERM to ERM blotting signal. The mean + SEM from three independent experiments is shown. (C and D) HUVECs were transfected as in (A) and fixed with 10% trichloroacetic acid, and pERM distribution was analyzed by immunofluorescence. Arrowheads indicate the pERM-rich protrusions (C), with their lengths quantitated in (D). (E) The HA-tagged moesin mutants, moesin T558A (TA), in an inactive conformation, or moesin T558D (TD), constitutively active, were transiently expressed in HUVECs for 48 h. Cells were fixed and stained with anti-HA and anti–β-catenin antibodies to detect transfected cells and cell–cell junctions, respectively. (F) Quantitation of β-catenin intensity of transfected cells normalized to the intensity of adjacent cells (control). Five hundred thirty regions from 53 transfected cells were analyzed in three different experiments. The mean + SEM is shown. *, p < 0.05.
ICAM-1 expression increases in response to MYADM reduction or TNF-α requires ERM expression
We then examined whether ERM proteins mediate ICAM-1 protein increase caused by MYADM knockdown. Cells were transfected with control siRNA or siRNA targeting MYADM (siMYADM 2), ERM proteins (siE+R+M), or both (Figures 7A and S5, A and B). MYADM single knockdown increased ICAM-1, whereas simultaneous silencing of MYADM and ERM proteins reduced ICAM-1 to control levels (Figure 7, A and B). In MYADM-depleted cells, single knockdown of ezrin, radixin, and moesin reduced ICAM-1 expression to a lesser extent than simultaneous triple-ERM silencing (Figure S5C). These findings indicate that the three proteins are playing additive roles. Accordingly, ERM knockdown diminished the increase in leukocyte adhesion caused by MYADM depletion (Figure 7C). Triple-ERM knockdown diminished barrier integrity in control cells, due probably to the essential role of these cross-linker proteins in connecting membrane to subcortical cytoskeleton, so no rescue of function could be performed following this strategy. Together our data indicate that MYADM reduction induces a proinflammatory-like phenotype by regulating ERM protein phosphorylation and ICAM-1 expression. Several protein kinases can potentially regulate ERM phosphorylation in this particular threonine residue. Among them, classical protein kinase C (PKC) family members, serine/threonine kinases that require a proper translocation from the cytosol to the plasma membrane and control endothelial barrier function (Hempel et al., 1997), have been shown to phosphorylate ERM proteins in different cellular contexts (Ng et al., 2001; Ivetic and Ridley, 2004; Adyshev et al., 2011). We addressed whether increased ERM phosphorylation in response to MYADM reduction is dependent on PKC by using a specific inhibitor for this family of kinases. Incubation with Gö6976 for 8 h partially reduced ERM phosphorylation increase in MYADM siRNA–transfected cells, which suggests that MYADM is involved in regulating classical PKC-mediated activation of ERM proteins at the plasma membrane (Figure 7D). Together these results are compatible with a role for MYADM organizing endothelial plasma membrane and thereby regulating the membrane targeting of proteins, such as classical PKCs, that control subcortical protein machinery. Finally, we investigated whether ERM can also regulate ICAM-1 expression in a more physiological context. TNF-α is a proinflammatory cytokine that induces ICAM-1 expression and ERM phosphorylation (Koss et al., 2006; Figure 7, E and F). Triple-ERM knockdown reduced ICAM-1 increment upon TNF-α exposure (Figure 7, G and H). In contrast, MYADM knockdown did not exacerbate ICAM-1 increase in response to this cytokine (Figure 7, I and J), and TNF-α did not significantly alter MYADM levels (Figure S6). Therefore these results indicate that ERM also regulates ICAM-1 expression in response to physiological inflammatory signaling. Collectively our data suggest that a connection between endothelial membrane and cortical actin cytoskeleton modulates the inflammatory response.
FIGURE 7:

ERM knockdown prevents ICAM-1 induction mediated by MYADM knockdown or TNF-α. (A) HUVECs were transfected with the indicated siRNAs for 72 h, and MYADM, ERM, and ICAM-1 expression was analyzed by Western blotting. (B) Quantitation of ICAM-1 levels in siRNA-transfected cells. (C) siRNA-transfected HUVECs were plated at confluence, and adhesion assays were performed 72 h after transfection. The mean + SEM from three independent experiments is shown. (D) HUVECs were transfected with the indicated siRNAs for 72 h. Eight hours before lysis, cells were incubated or not with 100 nm of the PKC inhibitor Gö6976. Subsequently MYADM, ERM, and ICAM-1 expression was analyzed by Western blotting. (E and F) HUVECs were stimulated at different times with TNF-α, lysed, and immunoblotted for the indicated antibodies. (G–J) siRNA-transfected HUVECs were stimulated at different times with TNF-α, lysed, and immunoblotted for the indicated antibodies. Graphs show the mean ± SEM from three independent experiments (H and J). *, p < 0.05; **, p < 0.01.
DISCUSSION
Membrane rafts are cholesterol-enriched ordered domains that regulate a plethora of signaling pathways. Cytokines such as TNF-α (Muppidi et al., 2004) and IL-1β (Oakley et al., 2009) or bacterial ligands of Toll-like-receptors (Soong et al., 2004; Triantafilou et al., 2010) induce proinflammatory signals through membrane rafts. Moreover, modulation of cholesterol content in these domains can induce or decrease inflammatory signaling in different cell models (Legler et al., 2003; Flemming et al., 2004; Meng et al., 2010). Collectively these observations suggest that modulation of membrane condensation may itself contribute to the endothelial inflammatory response, probably by mimicking those changes in clustering or diffusion that different stimuli can induce on protein signaling machinery associated with the plasma membrane. This could be important for chronic inflammatory diseases such as arteriosclerosis, which is triggered by deregulation of homeostasis of raft lipids, as is seen in hypercholesterolemia (Libby, 2002). However, ordered membrane microdomains require protein scaffolds to play specific roles within the cell (Bauer and Pelkmans, 2006). The MAL proteins contain one or two tetra-spanning MARVEL (MAL and related proteins for vesicle trafficking and membrane link) domains that are involved in membrane apposition (Sanchez-Pulido et al., 2002). Different experimental strategies have shown that these proteins have the ability to modulate membrane order or dynamics in a variety of cell types (Puertollano et al., 1997; Goldstein Magal et al., 2009; Aranda et al., 2011). As a consequence, these proteins control the segregation into membrane rafts of signaling proteins, such as the tyrosine kinase Lck (Anton et al., 2008) or the Rho GTPase Rac-1 (Aranda et al., 2011). In this paper, we show that, of the three MAL family proteins with a reported role in raft-mediated function, only MYADM is expressed in endothelial cells. MYADM is also the only member of the MAL family containing two MARVEL domains, rather than one. This feature seems to determine its predominant localization at the plasma membrane, rather than in vesicular compartments (Aranda et al., 2011), although MYADM trafficking has yet to be defined. On the other hand, colocalization analysis clearly shows that MYADM-containing domains are different than those of caveolae or GPI-anchored proteins in endothelial cells, in agreement with our previous studies in other cell types or with other MAL family proteins (Millán et al., 1997; Aranda et al., 2011).
ERM proteins are cross-linkers between transmembrane receptors and cortical actin filaments that regulate the structure and function of specific domains at the cell surface, namely microvilli or filopodia, and fundamental signaling processes involved in cell shape, junctional stability, motility, and cytokinesis (Fehon et al., 2010). Cytokines, such as IL-1β and TNF-α, or sphingosine 1-phosphate (S1P), all of which are involved in the regulation of the inflammatory response, can also activate ERM proteins (Koss et al., 2006; Adyshev et al., 2011; Huang et al., 2011). This activation, at least for TNF-α and S1P, mediates changes in endothelial barrier function, with different outcomes depending on the stimulus. Ordered domains at the plasma membrane and ERM proteins have thus been independently implicated in inflammation. In this study, we demonstrate that siRNA-mediated modulation of MYADM is sufficient to induce activation of ERM proteins, and this can act as a signal to alter barrier function and to provoke an inflammatory-like response in the absence of inflammatory stimuli. As the immunofluorescence analysis indicates, ERM activation occurs at the plasma membrane and at cell borders. A previous example of cross-regulation between rafts and ERM proteins at the plasma membrane has been reported in B-cells, in which ezrin can modulate the dynamics of ordered domains and their ability to contribute to B-cell receptor (BCR) signaling (Gupta et al., 2006; Viola and Gupta, 2007). Gupta et al. (2006) proposed that, in resting B-cells, active ezrin maintains small raft domains that contain signaling molecules and are linked to cortical F-actin and dispersed on the cell surface. On BCR activation, ezrin becomes transiently inactivated and uncouples actin from the plasma membrane domains. These small rafts then coalesce and promote downstream B-cell signaling. It is therefore plausible that membrane rafts, which are regulated by MYADM, in coordination with the ERM proteins, which connect rafts to the subcortical cytoskeleton, could mediate clustering, diffusion or localization of proteins involved in signaling from inflammatory agonists in endothelial or epithelial cells. In accordance, we have found that, at least for TNF-α, a complete inflammatory response requires ERM protein expression. MYADM was not modulated by TNF-α, and its reduction had no effect on TNF-α–mediated ICAM-1 induction. The identification of the stimuli that act through MYADM and ERM proteins to promote or inhibit inflammation will be important to increase our understanding of the endothelial inflammatory response.
ERMs are involved in the inflammatory control of protein expression (Kishore et al., 2005) and in connecting adhesion receptors, such as the ICAM family, to subcortical actin (Viola and Gupta, 2007; Reglero-Real et al., 2012). In this paper, we show that ERM expression is required for ICAM-1 expression in response to MYADM suppression or TNF-α. ICAM-1 is a paradigmatic adhesion receptor that regulates leukocyte adhesion. However, its role as a protein essential for controlling endothelial permeability both in vivo and in vitro is emerging (Clark et al., 2007; Sumagin et al., 2008). MYADM knockdown selectively increases ICAM-1 expression, but not many other proteins involved in barrier function, which indicates this protein selectively controls some but not all of the pathways involved in the endothelial inflammatory response. ICAM-1 increase is necessary for MYADM-mediated alteration of barrier properties, which is in agreement with previous reports that link leukocyte adhesion and permeability through dual roles played by ICAM-1 (Clark et al., 2007; Sumagin et al., 2008), VCAM-1 (van Wetering et al., 2003), or PECAM-1 (Graesser et al., 2002; Fernandez-Martin et al., 2012) receptors.
Regulation of the interaction between the plasma membrane and the underlying cytoskeleton is also essential in mechanotransduction (Chien, 2007). Cholesterol-rich membrane domains are required for signaling propagated by mechanic forces induced by shear stress (Yamamoto et al., 2003) or stretch (Zeidan et al., 2003). Laminar shear stress elicits proinflammatory signals that promote ICAM-1 up-regulation in vitro (Tzima et al., 2002). Pathological alteration of these forces in vivo, such as disturbed blood flow, are proinflammatory and proatherogenic (Chien, 2007). It is therefore possible that membrane and actin reorganization induced by MYADM knockdown regulates pathways related to cell stiffness and mechanotransduction. Finally, further understanding of the structure of membrane ordered domains, their protein machinery, and their involvement in inflammatory signaling could lead to the design of new therapies for chronic inflammatory diseases.
MATERIALS AND METHODS
Materials
Monoclonal antibody to human MYADM (mAb 2B12) was generated as previously described (Aranda et al., 2011). Rabbit anti–ICAM-1, anti–caveolin-1 (N-20), and anti-ERK1/2 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse anti–ICAM-1 antibody was from R&D Systems (Abingdon, UK). Mouse monoclonal anti–VE-cadherin antibody was from BD Biosciences (San Jose, CA). Rabbit anti–β-catenin and TRITC/FITC–phalloidin was from Sigma-Aldrich (St. Louis, MO). Anti–transferrin receptor antibodies were purchased from Zymed (San Francisco, CA). Anti-ERM, anti–phospho-ERM, and specific anti-ezrin, anti-radixin, and anti-moesin antibodies were from Cell Signaling Technology (Boston, MA). Hemagglutinin (HA)-tagged moesin-TD (T558D) and moesin-TA (T558A) were from J. Dêlon (Université René Descartes, Paris, France).
Cell culture and transfection
HUVECs were obtained and cultured as previously described (Millán et al., 2010) and were always plated at confluency on fibronectin-coated dishes for 24–72 h. HeLa, MDCK, and PC3 cells were obtained from the American Type Culture Collection (Manassas, VA) and grown in DMEM supplemented with 5% fetal bovine serum. Isolation of HeLa clones stably expressing MYADM-GFP and human memory T-cells for adhesion assays has been described elsewhere (Millán et al., 2006; Aranda et al., 2011). HUVECs were transiently transfected with 1–5 μg plasmid DNA/106 cells with calcium phosphate and used for experiments 24–72 h after transfection. For siRNA transfection, we devised a protocol for delivery of siRNA with high efficiency into primary endothelial cells that is a modification of our previous method (Millán et al., 2006). HUVECs were plated at subconfluence (105 cells on each well of a six-well dish) in EBM-2 medium (Lonza, Walkersville, MD) with no antibiotics. The following day, cells were transfected by mixing 4 μl of oligofectamine (Invitrogen, Carlsbad, CA) with siRNA to a final concentration of 100 nM. At 24 h after transfection, cells were trypsinized and plated at confluence onto different dishes for parallel assays, such as permeability, adhesion, immunofluorescence, or Western blotting. Assays were performed 72 h after transfection. Sequences of siRNA duplexes targeting MYADM were 5′-CGAGATCACTGGCTATATG-3′ (siMYADM 1), 5′-GATGTAAGCTGCAGCCGCA-3′ (siMYADM 2), and 5′GGTCTAAGACTCTCCCAAG-3′ (siMYADM 3). Ezrin was targeted with the siRNA oligonucleotide 5′-CAAGAAGGCACCUGACUUU-3′, moesin with 5′-AUAAGGAAGUGCAUAAGUC-3′, and radixin with 5′-GAACUGGCAUGAAGAACAU-3′. Sequences of siRNA controls were obtained from Dharmacon (Lafayette, CO)
DRM isolation
Different cell lines and HUVECs grown to confluence in 100-mm dishes were lysed for 20 min in 1 ml of 25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 4°C, as indicated (Millán et al., 2002). DRM fraction was isolated by centrifugation to equilibrium in a sucrose discontinuous density gradient in a swinging bucket SW40 rotor (Beckman Coulter, Brea, CA) at 39,000 rpm (188,000 × g) for 20 h (Brown and Rose, 1992; Millán et al., 2006). Fractions were harvested from the bottom sucrose layer (40%; soluble) and from the above interphase between 5 and 30% sucrose layers (DRMs). Equivalent aliquots were subjected to immunoblot analysis with the appropriate antibodies.
ICAM-1 cross-linking and immunofluorescence
Anti–ICAM-1 antibody (1 μg/ml) was added to TNF-α–stimulated HUVECs for 45 min. Cells were rinsed and incubated with 1 μg/ml of fluorophore-coupled secondary antibody for 30 min to induce receptor clustering. Cells were rinsed and fixed with 4% paraformaldehyde for 20 min, blocked with TBS (25 mM Tris, pH 7.4, 150 mM NaCl) for 10 min, permeabilized for 5 min with phosphate-buffered saline (PBS) containing 0.2% Triton X-100 at 4°C, and incubated at 37°C with tetramethylrhodamine isothiocyanate (TRITC)-labeled phalloidin to visualize F-actin. Specimens were mounted in DAKO fluorescent mounting medium (DAKO, Ely, UK). For immunofluorescence analysis of adhered T-cells, unlabeled T-cells were added to transfected HUVECs stimulated with TNF-α for 6 h. Cells were fixed and stained with the indicated antibodies and with TRITC–phalloidin to visualize T-cells. For F-actin quantification, confocal images from cells stained for TRITC-labeled phalloidin were exported in formats compatible with ImageJ software (http://rsb.info.nih.gov/ij). ImageJ was used to obtain the mean fluorescence intensities from different images of confluent cells.
Adhesion assays
Starved memory T-cells were labeled with 2′,7′,-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester (Molecular Probes, Eugene, OR) in serum-free medium for 20 min, washed with medium containing 1% BSA, and incubated for 15 min at 37°C with confluent HUVECs previously transfected with different siRNAs. Nonadhered cells were washed off, and adhesion was determined by fluorescence measurement in a Fusion α-FS fluorimeter (Perkin Elmer-Cetus, Waltham, MA). Data were processed and statistical significance was determined using Student's t test (Microsoft Excel; Redmond, WA). For immunofluorescence analysis of adhered T-cells, unlabeled T-cells were added to transfected HUVECs stimulated with TNF-α for 6 h. Cells were fixed and stained with TRITC–phalloidin (to visualize T-cells) and with the indicated antibodies.
Permeability assays
Permeability assays were carried out as previously described (McKenzie and Ridley, 2007). siRNA-transfected HUVECs were plated at confluence on fibronectin-coated Transwells of 0.4-μm diameter. FITC-dextran (Mr 42,000; Sigma–Aldrich) was applied apically at 0.1 mg/ml and allowed to equilibrate for 60 min before a sample of the medium was removed from the lower chamber to measure fluorescence in the Fusion α-FS fluorimeter. The arithmetic mean, SE, and statistical significance corresponding to a Student's t test were calculated in Microsoft Excel. Alternatively, real-time permeability assays were performed using an electric cell-substrate impedance sensing system (ECIS 1600R; Applied Biophysics, Troy, NY) as previously described (Fernandez-Martin et al., 2012). Briefly, 2 × 105 cells/cm2, previously transfected with the indicated siRNA oligonucleotides, were plated in a well precoated with fibronectin (200 mg/ml) and containing 10 small-fold electrodes and a counter-electrode (8W10E arrays; IBIDI, Martinsried, Germany). After 48 h at confluency, transendothelial electrical resistance (TEER) was measured in each well (4000-Hz frequency). The TEER analysis included basal measurements during the first 2 to 4 h, followed by a calcium-switch assay in which cells were incubated in PBS for 20 min and the calcium levels were subsequently restored by replenishment with normal EBM-2 medium. Data were normalized to the mean resistance detected before the calcium switch.
qPCR analysis
One microgram of RNA from HUVECs, PC3, Jurkat, and HepG2 cell lines was subjected to reverse transcription with the High Capacity RNA-cDNA kit (Applied Biosystems, Bedford, MA). qPCR was performed from the resulting cDNA in a thermocycler CFX 384 (Bio-Rad, Hercules, CA) using the SsoFast EvaGreen Supermix (Bio-Rad) and the following forward and reverse primers, previously designed with Probefinder software (Roche Diagnostics, Indianapolis, IN): MAL2, 5′-GACATCCTGCGGACCTACTC-3′; 5′-AGACAAGACCCCCGAACAG-3′. MYADM, 5′-GCCATCTGCTTCATCCTAGC-3′; 5′-TAGCACGTTGGTGCACTCC-3′. MAL, 5′-AGACTTCCTGGGTCACCTTG-3′; 5′-CGTCTTGCATCGTGATGGT-3′. Parallel qPCR from β-actin and GADPH was performed to normalize data from each cell type with the following primers: β-actin, 5′-CAGGCACCAGGGCGTG-3′; 5′-GTGAGGATGCCTCTCTTGCTCT-3′. GADPH, 5′-AGCCACATCGCTCAGACAC-3′; 5′-CGCCCAATACGACCAAAT-3′.
Supplementary Material
Acknowledgments
This work was supported by grants SAF2011–22624 (to J.M.), BFU2012–32532 and CSD2009–00016 (to M.A.A.), and BFU2011-22859 (to I.C.) from the Ministerio de Ciencia e Innovación; and grant S2010/BMD-2305 from the Comunidad de Madrid (to I.C.). J.F.A. was the recipient of an EMBO short-term fellowship in A.J.R.’s laboratory. We thank Severine Gharbi for the critical reading of the manuscript. The expert technical advice of the personnel of the Optical and Confocal Microscopy and Genomic Facilities (CBMSO, Madrid) is gratefully acknowledged.
Abbreviations used:
- BCR
B-cell receptor
- DRM
detergent-resistant membrane
- ERM
ezrin, radixin, and moesin
- FERM
4.1 ezrin–radixin–moesin
- GFP
green fluorescent protein
- HA
hemagglutinin
- HUVEC
human umbilical vein endothelial cell
- ICAM-1
intercellular adhesion molecule-1
- mAb
monoclonal antibody
- MARVEL
MAL and related proteins for vesicle trafficking and membrane link
- MYADM
myeloid-associated differentiation marker
- PBS
phosphate-buffered saline
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PKC
protein kinase C
- qPCR
quantitative PCR
- S1P
sphingosine 1-phosphate
- siRNA
small interfering RNA
- TEER
transendothelial electrical resistance
- TNF-α
tumor necrosis factor-α
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-11-0914) on December 21, 2012.
*These authors contributed equally to this work.
REFERENCES
- Adyshev DM, Moldobaeva NK, Elangovan VR, Garcia JG, Dudek SM. Differential involvement of ezrin/radixin/moesin proteins in sphingosine 1-phosphate-induced human pulmonary endothelial cell barrier enhancement. Cell Signal. 2011;23:2086–2096. doi: 10.1016/j.cellsig.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albelda SM. Endothelial and epithelial cell adhesion molecules. Am J Respir Cell Mol Biol. 1991;4:195–203. doi: 10.1165/ajrcmb/4.3.195. [DOI] [PubMed] [Google Scholar]
- Anton O, Batista A, Millán J, Andres-Delgado L, Puertollano R, Correas I, Alonso MA. An essential role for the MAL protein in targeting Lck to the plasma membrane of human T lymphocytes. J Exp Med. 2008;205:3201–3213. doi: 10.1084/jem.20080552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton OM, Andres-Delgado L, Reglero-Real N, Batista A, Alonso MA. MAL protein controls protein sorting at the supramolecular activation cluster of human T lymphocytes. J Immunol. 2011;186:6345–6356. doi: 10.4049/jimmunol.1003771. [DOI] [PubMed] [Google Scholar]
- Aranda JF, Reglero-Real N, Kremer L, Marcos-Ramiro B, Ruiz-Saenz A, Calvo M, Enrich C, Correas I, Millán J, Alonso MA. MYADM regulates Rac1 targeting to ordered membranes required for cell spreading and migration. Mol Biol Cell. 2011;22:1252–1262. doi: 10.1091/mbc.E10-11-0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Pelkmans L. A new paradigm for membrane-organizing and -shaping scaffolds. FEBS Lett. 2006;580:5559–5564. doi: 10.1016/j.febslet.2006.08.077. [DOI] [PubMed] [Google Scholar]
- Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 2002;3:586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167:377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidlow JH, Jr., Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc Res. 2010;86:219–225. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol. 2007;292:H1209–H1224. doi: 10.1152/ajpheart.01047.2006. [DOI] [PubMed] [Google Scholar]
- Clark PR, Manes TD, Pober JS, Kluger MS. Increased ICAM-1 expression causes endothelial cell leakiness, cytoskeletal reorganization and junctional alterations. J Invest Dermatol. 2007;127:762–774. doi: 10.1038/sj.jid.5700670. [DOI] [PubMed] [Google Scholar]
- de Marco MC, Martin-Belmonte F, Kremer L, Albar JP, Correas I, Vaerman JP, Marazuela M, Byrne JA, Alonso MA. MAL2, a novel raft protein of the MAL family, is an essential component of the machinery for transcytosis in hepatoma HepG2 cells. J Cell Biol. 2002;159:37–44. doi: 10.1083/jcb.200206033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11:276–287. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Martin L, Marcos-Ramiro B, Bigarella CL, Graupera M, Cain RJ, Reglero-Real N, Jimenez A, Cernuda-Morollon E, Correas I, Cox S, Ridley AJ, Millán J. Crosstalk between reticular adherens junctions and platelet endothelial cell adhesion molecule-1 regulates endothelial barrier function. Arterioscler Thromb Vasc Biol. 2012;32:e90–e102. doi: 10.1161/ATVBAHA.112.252080. [DOI] [PubMed] [Google Scholar]
- Flemming JA, Perkins KH, Luus L, Ferguson AR, Corley RB. Disruption of membrane cholesterol stimulates MyD88-dependent NF-κB activation in immature B cells. Cell Immunol. 2004;229:68–77. doi: 10.1016/j.cellimm.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Goldstein Magal L, Yaffe Y, Shepshelovich J, Aranda JF, de Marco MD, Gaus K, Alonso MA, Hirschberg K. Clustering and lateral concentration of raft lipids by the MAL protein. Mol Biol Cell. 2009;20:3751–3762. doi: 10.1091/mbc.E09-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, Ruddle NH, Engelhardt B, Madri JA. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest. 2002;109:383–392. doi: 10.1172/JCI13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Wollscheid B, Watts JD, Scheer B, Aebersold R, DeFranco AL. Quantitative proteomic analysis of B cell lipid rafts reveals that ezrin regulates antigen receptor-mediated lipid raft dynamics. Nat Immunol. 2006;7:625–633. doi: 10.1038/ni1337. [DOI] [PubMed] [Google Scholar]
- Hempel A, Maasch C, Heintze U, Lindschau C, Dietz R, Luft FC, Haller H. High glucose concentrations increase endothelial cell permeability via activation of protein kinase Cα. Circ Res. 1997;81:363–371. doi: 10.1161/01.res.81.3.363. [DOI] [PubMed] [Google Scholar]
- Heyraud S, Jaquinod M, Durmort C, Dambroise E, Concord E, Schaal JP, Huber P, Gulino-Debrac D. Contribution of annexin 2 to the architecture of mature endothelial adherens junctions. Mol Cell Biol. 2007;28:1657–1668. doi: 10.1128/MCB.00695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, et al. Increased phosphorylation of ezrin/radixin/moesin proteins contributes to proliferation of rheumatoid fibroblast-like synoviocytes. Rheumatology (Oxford) 2011;50:1045–1053. doi: 10.1093/rheumatology/keq440. [DOI] [PubMed] [Google Scholar]
- Ivetic A, Ridley AJ. Ezrin/radixin/moesin proteins and Rho GTPase signalling in leucocytes. Immunology. 2004;112:165–176. doi: 10.1111/j.1365-2567.2004.01882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CM, Chichili GR, Rodgers W. Compartmentalization of phosphatidylinositol 4,5-bisphosphate signaling evidenced using targeted phosphatases. J Biol Chem. 2008;283:29920–29928. doi: 10.1074/jbc.M805921200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keuschnigg J, Henttinen T, Auvinen K, Karikoski M, Salmi M, Jalkanen S. The prototype endothelial marker PAL-E is a leukocyte trafficking molecule. Blood. 2009;114:478–484. doi: 10.1182/blood-2008-11-188763. [DOI] [PubMed] [Google Scholar]
- Kishore R, Qin G, Luedemann C, Bord E, Hanley A, Silver M, Gavin M, Yoon YS, Goukassian D, Losordo DW. The cytoskeletal protein ezrin regulates EC proliferation and angiogenesis via TNF-alpha-induced transcriptional repression of cyclin A. J Clin Invest. 2005;115:1785–1796. doi: 10.1172/JCI22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss M, Pfeiffer GR, II, Wang Y, Thomas ST, Yerukhimovich M, Gaarde WA, Doerschuk CM, Wang Q. Ezrin/radixin/moesin proteins are phosphorylated by TNF-α and modulate permeability increases in human pulmonary microvascular endothelial cells. J Immunol. 2006;176:1218–1227. doi: 10.4049/jimmunol.176.2.1218. [DOI] [PubMed] [Google Scholar]
- Kwiatkowska K. One lipid, multiple functions: how various pools of PI(4,5)P(2) are created in the plasma membrane. Cell Mol Life Sci. 2010;67:3927–3946. doi: 10.1007/s00018-010-0432-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legler DF, Micheau O, Doucey MA, Tschopp J, Bron C. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFα-mediated NF-κB activation. Immunity. 2003;18:655–664. doi: 10.1016/s1074-7613(03)00092-x. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- McKenzie JA, Ridley AJ. Roles of Rho/ROCK and MLCK in TNF-α-induced changes in endothelial morphology and permeability. J Cell Physiol. 2007;213:221–228. doi: 10.1002/jcp.21114. [DOI] [PubMed] [Google Scholar]
- Meng G, Liu Y, Lou C, Yang H. Emodin suppresses lipopolysaccharide-induced pro-inflammatory responses and NF-κB activation by disrupting lipid rafts in CD14-negative endothelial cells. British J Pharmacol. 2010;161:1628–1644. doi: 10.1111/j.1476-5381.2010.00993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millán J, Cain RJ, Reglero-Real N, Bigarella C, Marcos-Ramiro B, Fernandez-Martin L, Correas I, Ridley AJ. Adherens junctions connect stress fibres between adjacent endothelial cells. BMC Biol. 2010;8:11. doi: 10.1186/1741-7007-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millán J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol. 2006;8:113–123. doi: 10.1038/ncb1356. [DOI] [PubMed] [Google Scholar]
- Millán J, Montoya MC, Sancho D, Sanchez-Madrid F, Alonso MA. Lipid rafts mediate biosynthetic transport to the T lymphocyte uropod subdomain and are necessary for uropod integrity and function. Blood. 2002;99:978–984. doi: 10.1182/blood.v99.3.978. [DOI] [PubMed] [Google Scholar]
- Millán J, Puertollano R, Fan L, Alonso MA. Caveolin and MAL, two protein components of internal detergent-insoluble membranes, are in distinct lipid microenvironments in MDCK cells. Biochem Biophys Res Commun. 1997;233:707–712. doi: 10.1006/bbrc.1997.6530. [DOI] [PubMed] [Google Scholar]
- Millán J, Ridley AJ. Rho GTPases and leucocyte-induced endothelial remodelling. Biochem J. 2005;385:329–337. doi: 10.1042/BJ20041584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muppidi JR, Tschopp J, Siegel RM. Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity. 2004;21:461–465. doi: 10.1016/j.immuni.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Ng T, et al. Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility. EMBO J. 2001;20:2723–2741. doi: 10.1093/emboj/20.11.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley FD, Smith RL, Engelhardt JF. Lipid rafts and caveolin-1 coordinate interleukin-1β (IL-1β)-dependent activation of NFκB by controlling endocytosis of Nox2 and IL-1β receptor 1 from the plasma membrane. J Biol Chem. 2009;284:33255–33264. doi: 10.1074/jbc.M109.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palade GE, Simionescu M, Simionescu N. Structural aspects of the permeability of the microvascular endothelium. Acta Physiol Scand. 1979;463:11–32. [PubMed] [Google Scholar]
- Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- Prag S, et al. Activated ezrin promotes cell migration through recruitment of the GEF Dbl to lipid rafts and preferential downstream activation of Cdc42. Mol Biol Cell. 2007;18:2935–2948. doi: 10.1091/mbc.E06-11-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R, Li S, Lisanti MP, Alonso MA. Recombinant expression of the MAL proteolipid, a component of glycolipid-enriched membrane microdomains, induces the formation of vesicular structures in insect cells. J Biol Chem. 1997;272:18311–18315. doi: 10.1074/jbc.272.29.18311. [DOI] [PubMed] [Google Scholar]
- Puertollano R, Martin-Belmonte F, Millán J, de Marco MC, Albar JP, Kremer L, Alonso MA. The MAL proteolipid is necessary for normal apical transport and accurate sorting of the influenza virus hemagglutinin in Madin-Darby canine kidney cells. J Cell Biol. 1999;145:141–151. doi: 10.1083/jcb.145.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reglero-Real N, Marcos-Ramiro B, Millán J. Endothelial membrane reorganization during leukocyte extravasation. Cell Mol Life Sci. 2012;69:3079–3099. doi: 10.1007/s00018-012-0987-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie K, Iino R, Fujiwara T, Murase K, Kusumi A. The fence and picket structure of the plasma membrane of live cells as revealed by single molecule techniques (review) Mol Membr Biol. 2003;20:13–18. doi: 10.1080/0968768021000055698. [DOI] [PubMed] [Google Scholar]
- Sanchez-Pulido L, Martin-Belmonte F, Valencia A, Alonso MA. MARVEL: a conserved domain involved in membrane apposition events. Trends Biochem Sci. 2002;27:599–601. doi: 10.1016/s0968-0004(02)02229-6. [DOI] [PubMed] [Google Scholar]
- Simionescu M. Implications of early structural-functional changes in the endothelium for vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:266–274. doi: 10.1161/01.ATV.0000253884.13901.e4. [DOI] [PubMed] [Google Scholar]
- Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol. 2010;11:688–699. doi: 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- Soong G, Reddy B, Sokol S, Adamo R, Prince A. TLR2 is mobilized into an apical lipid raft receptor complex to signal infection in airway epithelial cells. J Clin Invest. 2004;113:1482–1489. doi: 10.1172/JCI20773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumagin R, Lomakina E, Sarelius IH. Leukocyte-endothelial cell interactions are linked to vascular permeability via ICAM-1-mediated signaling. Am J Physiol Heart Circulat Physiol. 2008;295:H969–H977. doi: 10.1152/ajpheart.00400.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilghman RW, Hoover RL. E-selectin and ICAM-1 are incorporated into detergent-insoluble membrane domains following clustering in endothelial cells. FEBS Lett. 2002;525:83–87. doi: 10.1016/s0014-5793(02)03070-3. [DOI] [PubMed] [Google Scholar]
- Tomas EM, Chau TA, Madrenas J. Clustering of a lipid-raft associated pool of ERM proteins at the immunological synapse upon T cell receptor or CD28 ligation. Immunol Lett. 2002;83:143–147. doi: 10.1016/s0165-2478(02)00075-5. [DOI] [PubMed] [Google Scholar]
- Triantafilou M, Lepper PM, Olden R, Dias Ide S, Triantafilou K. Location, location, location: is membrane partitioning everything when it comes to innate immune activation. Mediators Inflamm. 2010;2011:186093. doi: 10.1155/2011/186093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzima E, Del Pozo MA, Kiosses WB, Mohamed SA, Li S, Chien S, Schwartz MA. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 2002;21:6791–6800. doi: 10.1093/emboj/cdf688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann NY Acad Sci. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- van Wetering S, van den Berk N, van Buul JD, Mul FP, Lommerse I, Mous R, ten Klooster JP, Zwaginga JJ, Hordijk PL. VCAM-1-mediated Rac signaling controls endothelial cell-cell contacts and leukocyte transmigration. Am J Physiol Cell Physiol. 2003;285:C343–C352. doi: 10.1152/ajpcell.00048.2003. [DOI] [PubMed] [Google Scholar]
- Viola A, Gupta N. Tether and trap: regulation of membrane-raft dynamics by actin-binding proteins. Nat Rev Immunol. 2007;7:889–896. doi: 10.1038/nri2193. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Williams L, Ridley AJ. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J Cell Biol. 1999;145:1293–1307. doi: 10.1083/jcb.145.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Sokabe T, Ohura N, Nakatsuka H, Kamiya A, Ando J. Endogenously released ATP mediates shear stress-induced Ca2+ influx into pulmonary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2003;285:H793–H803. doi: 10.1152/ajpheart.01155.2002. [DOI] [PubMed] [Google Scholar]
- Zeidan A, Broman J, Hellstrand P, Sward K. Cholesterol dependence of vascular ERK1/2 activation and growth in response to stretch: role of endothelin-1. Arterioscler Thromb Vasc Biol. 2003;23:1528–1534. doi: 10.1161/01.ATV.0000090129.75275.C2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.