

Abstract
Phenethyl isothiocyanate (PEITC) is a promising cancer chemopreventive agent commonly found in edible cruciferous vegetables. It has been implicated also for therapy, and is in clinical trial for lung cancer. Here, we provide evidence that the tumor suppressive effect of PEITC is related to its ability to induce expression of damaged DNA binding protein 2 (DDB2), a DNA repair protein involved also in apoptosis and premature senescence. DDB2 expression is attenuated in a wide variety of cancers including the aggressive colon cancers. We show that, in colon cancer cells, reactive oxygen species, which are induced by PEITC, augment expression of DDB2 through the p38MAPK/JNK pathway, independently of p53. PEITC-induced expression of DDB2 is critical for inhibition of tumor progression by PEITC. Tumors derived from DDB2-deficient colon cancer cells are refractory to PEITC-treatments, resulting from deficiencies in apoptosis and senescence. The DDB2-proficient tumors, on the other hand, respond effectively to PEITC. The results show that PEITC can be used to induce expression of DDB2, and that expression of DDB2 is critical for effective response of tumors to PEITC.
Keywords: DDB2, PEITC, ROS, apoptosis, colon cancer, drug resistance, senescence
Introduction
Damaged DNA-binding protein 2 (DDB2), encoded by the XPE gene, is a DNA repair protein. DDB2 also acts as a substrate receptor for Cul4a, an E3 ubiquitin ligase. XPC, p21 and DDB2 itself are some of the well-characterized targets of Cul4a-DDB1-DDB2 complex.1-3 DDB2 is important in the global genomic pathway of nucleotide excision repair (NER). There are several models that have been proposed about how DDB2 participates in NER.1,4-7 However, genetic evidence suggests that DDB2 participates in DNA repair synthesis by downregulating p21, a cyclin-dependent kinase inhibitor.4 Deletion of p21 in the DDB2−/− background restored NER activities in mouse embryonic fibroblasts (MEFs) as well as in mouse primary keratinocytes.8
DDB2 is also an important mediator of apoptosis and senescence.9,10 MEFs or human cells lacking DDB2 expression are deficient in apoptosis following DNA damage. The apoptosis-promoting function of DDB2 is related to its role in p21 regulation.2 p21 negatively regulates apoptosis, as cdk activity is required for proper functioning of caspases.11 In cells with DNA damage, DDB2 plays an important role in downregulating p21. Thereby, DDB2 ensures efficient apoptosis. The DDB2−/−p21−/− mice, on the other hand, exhibit efficient apoptosis.8 High level of p21 has been implicated in senescence.12 Despite high-level p21 accumulation, DDB2−/− MEFs were found to be deficient in replicative senescence.10 DDB2 plays a significant role in mediating senescence by inducing high-level accumulation of reactive oxygen species (ROS), as DDB2 transcriptionally inhibits expression of two important anti-oxidant enzymes MnSOD and catalase.10,13 Interestingly, ROS, in turn, also increase expression of DDB2. Thus, ROS activate DDB2 expression, which in a positive feedback loop causes persistent accumulation of ROS by inhibiting expression of the anti-oxidant enzymes MnSOD and catalase. The high level of ROS triggers the senescence response following DNA damage.
PEITC (Phenethyl Isothiocyanate) is a naturally occurring compound found in cruciferous vegetables such as watercress.14 PEITC is considered also as an anticancer compound, particularly for ovarian cancer and prostate cancer.15,16 It is also currently undergoing clinical trials for lung cancer and lympho-proliferative disorders. PEITC sensitizes cell to apoptosis, senescence and cell cycle arrest by upregulation of stress-activated protein kinase pathways and caspase pathway.17,18 Recent studies indicated that PEITC treatment resulted in cell cycle arrest and apoptosis of colon carcinoma cell HT-29 by upregulation of JNK, p38MAPK and ERK pathway.19 In vivo, in APC (Min/+) mice, chemopreventive efficacy of PEITC has been reported.20 Because PEITC increases the levels of ROS, we considered the possibility that DDB2, which is induced by ROS,10 might be an important mediator of the anticancer effects of PEITC. Here we show that, indeed PEITC increases expression of DDB2 in colon carcinoma cells, which is important for the tumor cells to undergo senescence and apoptosis following PEITC treatment. Moreover, we provide in vivo evidence that expression of DDB2 is required for PEITC-mediated tumor regression.
Results
Reactive oxygen species (ROS) stimulate expression of DDB2 by activating AP1
We showed that ROS increase the level of DDB2 in both MEFs as well as in human colon carcinoma cell HCT116. ROS increase DDB2 expression both at the RNA and protein level, suggesting that the effect is at the level of transcription. DDB2 is a p53-regulated gene in human, but not in mouse.21 However, we showed that ROS induce expression of DDB2 in mouse embryonic fibroblasts where DDB2 is not under p53 regulation, suggesting that a p53-independent mechanism of ROS mediated DDB2 upregulation. To confirm the notion, we directly examined whether ROS-mediated induction of DDB2 requires p53. HCT p53−/− cells were treated with a sub-lethal dose of hydrogen peroxide for 4 h, and total RNA was analyzed for expression of DDB2. There was a clear accumulation of DDB2 in the hydrogen peroxide-treated cells, suggesting that ROS could stimulate DDB2 expression in the absence of p53 (Fig. 1A). A major downstream effector pathway of ROS are JNK and stress-activated protein kinase.22 Therefore, we investigated whether ROS-activated MAPK pathway participates in the upregulation of DDB2. The HCT116 cells were treated with hydrogen peroxide in the presence or absence of the p38MAPK inhibitor (SB 203580) or JNK inhibitor (SP 600125). Hydrogen peroxide was shown to activate both p38MAPK and JNK.23 Moreover, SB 203580 and SP 600125 are well-characterized, commercially available inhibitors of p38MAPK and JNK, respectively. In the presence of either of these inhibitors, hydrogen peroxide treatment failed to upregulate DDB2 expression (Fig. 1B and C), suggesting a role of the MAPK pathway in the induction of DDB2.
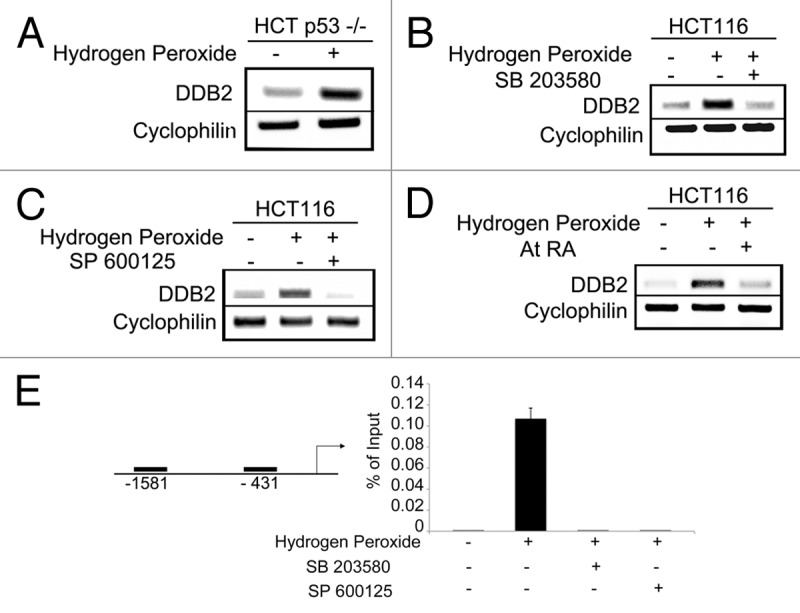
Figure 1. ROS-mediated DDB2 induction is p38MAPK/JNK-dependent. (A) HCT116 p53−/− cells were treated with 150 uM Hydrogen Peroxide for 4 h. Total RNA was analyzed by semi-quantitative PCR for the level of DDB2. Cyclophlin was used as a loading control. (B) HCT116 cells were treated with 150 uM hydrogen peroxide for 4 h with or without p38MAPK inhibitor SB 203580. Total RNA was analyzed by semi-quantitative PCR for the level of DDB2. Cyclophlin was used as a loading control. (C) HCT116 cells were treated with 150 uM hydrogen pxide for 4 h with or without JNK inhibitor SP 600125. Total RNA was analyzed by semi-quantitative PCR for the level of DDB2. Cyclophlin was used as a loading control. (D) HCT116 cells were treated with 150 uM hydrogen peroxide for 4 h with or without all-trans Retinoic Acid. Total RNA was analyzed by semi-quantitative PCR for the level of DDB2. Cyclophlin was used as a loading control. (E) HCT116 cells were treated with 150 uM hydrogen peroxide for 4 h with or without P38MAPK/JNK inhibitor. Next, cells were subjected to ChIP assay, as described in Materials and Methods. Antibody against AP-1/c-jun or IgG was used for immunoprecipitation. Filled boxes indicate the binding region of primers used to detect interaction of AP-1 with DDB2 promoter. Bar graph represents percentage of immunoprecipitation vs. input material (mean +/− standard deviation; n = 3) at the -431 site.
AP-1 is a heterodimeric transcription factor downstream of MAPK pathway.22 We examined whether AP-1 transcriptionally activates DDB2 following ROS treatment. All-trans-Retinoic Acid is an inhibitor of AP-1 transcription factor.24,25 To examine involvement of AP-1 in ROS-mediated DDB2 upregulation, HCT 116 cells were treated with hydrogen peroxide with or without Retinoic Acid. In the presence of Retinoic Acid, hydrogen peroxide failed to cause accumulation of DDB2, suggesting AP-1 is involved in the activation of DDB2 (Fig. 1D). Bioinformatics analysis revealed that there are two potential AP-1-binding sites on the DDB2 promoter, one at -431 and the other at -1581 from the start site. We performed chromatin IP on these sites following hydrogen peroxide treatment. In the absence of hydrogen peroxide treatment, there was very little binding of AP-1 on either of these sites. However, in the -431-site there was evidence of increased AP-1 binding following hydrogen peroxide treatment (Fig. 1E). We further tested whether inhibition of p38MAPK or JNK would prevent AP-1 binding to the DDB2 promoter following hydrogen peroxide treatment. Toward that, chromatin IP was performed on the -431-site with cells treated with hydrogen peroxide in the presence of p38MAPK/ JNK inhibitor. Clearly, the inhibitors reduced binding of AP-1 to the promoter of DDB2 (Fig. 1E). Together, the results suggest that ROS activates p38MAPK/ JNK that, in turn, triggers AP-1 accumulation on the promoter of DDB2 and upregulates DDB2 expression.
PEITC-induced expression of DDB2 is important for effective cellular response
PEITC is a naturally occurring compound that increases ROS accumulation in the cell.26 We confirmed the observation by measuring ROS levels in cells after treatments with PEITC (Fig. S1). To investigate whether PEITC increases DDB2 levels we treated HCT 116 cell with PEITC, and examined the DDB2 levels at different time points. Consistent with our hydrogen peroxide-mediated DDB2 response, PEITC was found to increase the level of DDB2. Moreover, that was p53-independent, as PEITC could upregulate DDB2 in HCT p53−/− cells (Fig. 2A and B). PEITC has been reported to induce apoptosis and cell cycle arrest in colon carcinoma cell, HT29.19 In HCT 116 cells we investigated whether PEITC could induce apoptosis or senescence and whether that was dependent on DDB2. HCT 116 cells expressing control-shRNA or DDB2-shRNA were treated with PEITC and TUNEL assay was performed to measure apoptosis response (Fig. 2C). Cells expressing DDB2-shRNA were significantly deficient in apoptosis response compared with the control-shRNA-expressing cells (Fig. 2D). Also, in agreement with our previous studies,2 DDB2-deficient cells were refractory to oxidative stress-induced apoptosis response (Fig. S2). Previously, we showed that DDB2-deficient cells do not undergo senescence following hydrogen peroxide treatment.10 Therefore, we investigated the senescence response following PEITC treatment, which increases oxidative stress. HCT 116 cells expressing control shRNA or DDB2 shRNA were treated with PEITC for 4 h, and after 48 h, the cells were subjected to staining for SA-β-gal, a marker for senescence. Cells expressing control-shRNA underwent senescence after PEITC treatment, but the DDB2-shRNA-expressing cells were deficient in senescence (Fig. 2E and F). To further confirm the results, clonogenicity assay was performed following treatments with PEITC. Compared with the untreated cells, PEITC-treated control-shRNA-expressing cells generated a greatly reduced number of colonies (Fig. 3A). However, the DDB2-shRNA-expressing cells generated significantly higher number of colonies following PEITC treatment (Fig. 3B). This further strengthened our observation that DDB2 is important for an effective response to PEITC.

Figure 2. PEITC induces expression of DDB2-independent of p53. (A) HCT116 cells were treated with PEITC for indicated time point. Total RNA was analyzed by semi-quantitative PCR for the level of DDB2. Cyclophlin was used as a loading control. (B) HCT116 p53−/− cells were treated with PEITC for indicated time points. Total RNA was analyzed by semi-quantitative PCR for the level of DDB2. Cyclophlin was used as a loading control. (C) HCT116 cells expressing control shRNA or DDB2 shRNA were treated with PEITC for 12 h. Next day, cells were analyzed for apoptosis by TUNEL assay. Representative images of HCT116 cells expressing control shRNA or DDB2 shRNA stained for TUNEL assay after PEITC treatment. (D) TUNEL positive cells were counted from at least 10 fields of triplicate plates. A quantification of TUNEL positive HCT116 cells after PEITC treatment is shown. (E) HCT116 cells expressing control shRNA or DDB2 shRNA were treated with PEITC for 4 h. After 48 h, cells were analyzed for SA β gal activity. Representative images of HCT116 cell expressing control shRNA or DDB2 shRNA stained for SA β gal after PEITC treatment. (F) SA β gal positive cells were counted from at least 10 fields of triplicate plates. A quantification of SA β gal positive HCT116 cells after PEITC treatment is shown.
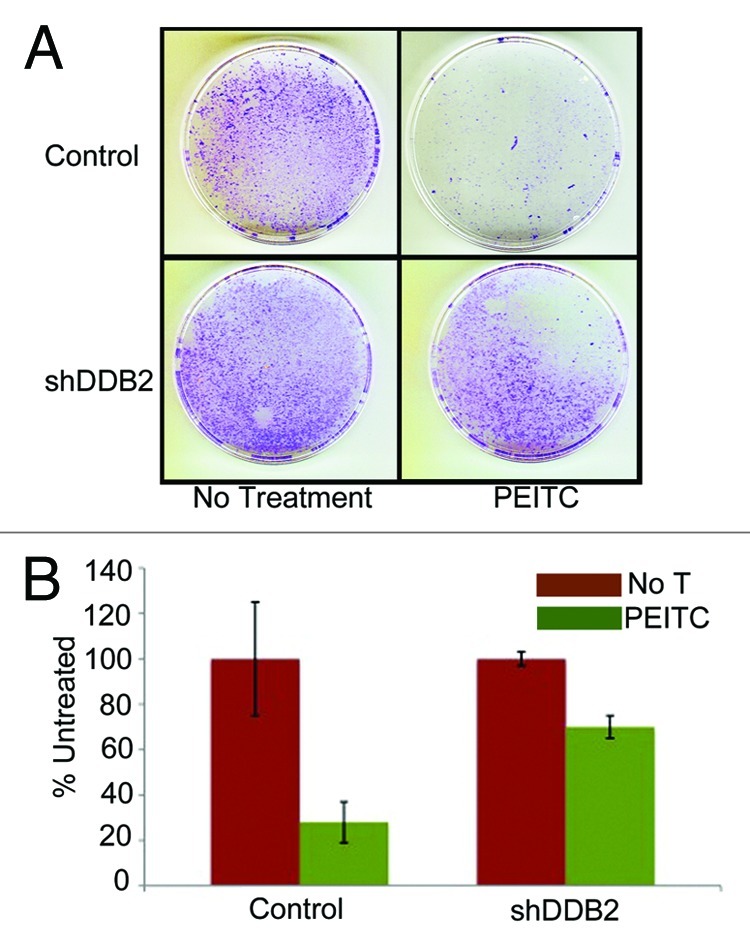
Figure 3. DDB2-deficient cells are drug resistant. (A) HCT116 cells expressing control shRNA or DDB2 shRNA were seeded (500 cells) in 6-well plates. Next day cells were treated with PEITC for 12 h followed by wash with PBS twice. Cells were grown for 12 d. Colonies with more than 50 cells were counted from triplicate plates. Representative images of colonies from HCT116 cell expressing control shRNA or DDB2 shRNA stained with crystal violet are shown. (B) A quantification of colonies formed after PEITC treatment is shown. Colonies with more than 50 cells were counted from triplicate plates.
PEITC-mediated tumor regression requires DDB2
We next investigated whether PEITC-mediated tumor regression in vivo requires DDB2 expression. HCT116 cells expressing control-shRNA or DDB2-shRNA were used for sub-cutaneous injection in nude mice. Once the tumor reached 10–15 mm3, the mice were subjected to PEITC or vehicle treatment up to 4 wk. Consistent with earlier reports,27 groups injected with the control-shRNA-expressing cells, treatments with PEITC caused the tumors to regress by almost 50% (Fig. 4A). However, the tumors developed from DDB2-shRNA-expressing cells were significantly less responsive to PEITC treatment (Fig. 4B), providing evidence that successful response to PEITC treatment requires expression of DDB2. We also investigated DDB2 expression in the PEITC-treated tumor from the control-shRNA cells. Tumor sections were analyzed by immunohistochemical assays for DDB2. DDB2 expression was found to be elevated in the PEITC-treated tumors compared with the vehicle-treated tumors, which is in agreement with our in vitro observation that PEITC increases expression of DDB2 (Fig. 4C).
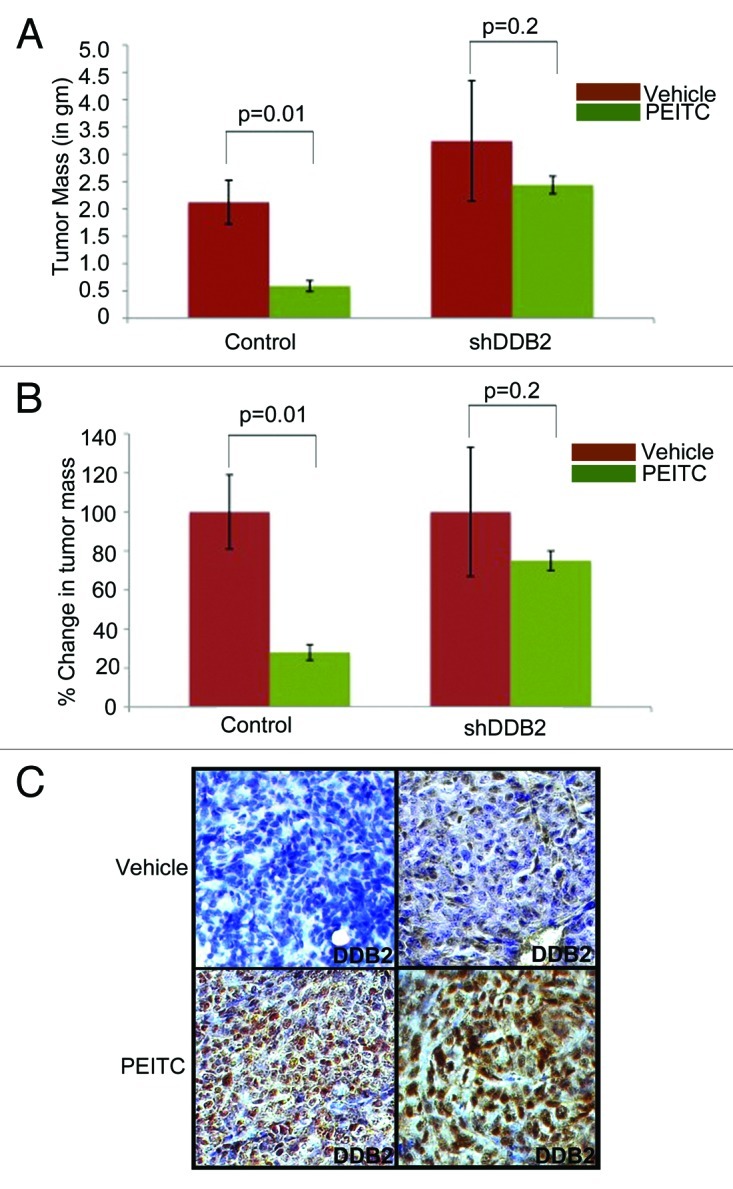
Figure 4. In vivo therapeutic activity of PEITC requires DDB2. (A and B) HCT116 cells expressing control shRNA or DDB2 shRNA were injected subcutaneously into nude mice. Mice were divided randomly in two groups for each cell line for treatment with PEITC (n = 5) or vehicle control (n = 3). Graph represents the tumor mass from control or DDB2-deficient cells line treated with PEITC or left untreated along with percent reduction in tumor mass. (C) HCT116 cells expressing control shRNA or DDB2 shRNA were injected subcutaneously into nude mice. Mice were divided randomly in two groups for each cell line for treatment with PEITC or vehicle control. Four weeks post treatment mice were sacrificed and tumor sections were fixed in 10% Formalin, processed and embedded with paraffin for sectioning. Prepared tumor section slides were then subjected to immunohistochemical analysis using DDB2 antibody. Representative images from two different tumors are shown.
We compared PCNA and smooth muscle actin expression between treated and untreated tumors from the control-shRNA or the DDB2-deficient cells. PCNA is a marker for proliferation, whereas smooth muscle actin expression indicates the extent of stromal involvement. Together, these markers indicate aggressive progression. Expression of PCNA and smooth muscle actin in tumors derived from the control-shRNA-expressing cells was inhibited by the treatments with PEITC. On the other hand, the tumors derived from DDB2-deficient cells exhibited only a modest reduction of PCNA and smooth muscle actin expression, suggesting a reduced response of the tumors to PEITC (Fig. 5). Next, we assayed for apoptosis and senescence response in the tissue sections from PEITC or vehicle-treated tumors. There was a clear accumulation of apoptotic cells as suggested by cleaved caspase 3 expression in the tumor from the control-shRNA cells (Fig. 6A and B). However, in the tumor derived from the DDB2-shRNA cells there was significantly less accumulation of apoptotic cells (Fig. 6B). The senescence response was analyzed by assaying for p16INK4a expression. Clearly, the tumors from the control-shRNA-expressing cells exhibited increased p16INK4a, a hallmark of senescence. Expression of p16INK4a was significantly lower in the tumors from the DDB2-shRNA-expressing cells (Fig. 6C). Thus, the deficiencies in apoptosis and senescence made major contributions toward the lack of tumor regression in the DDB2-depleted tumors, suggesting that DDB2 participates in tumor regression by inducing apoptosis and senescence. Taken together, the results suggest that PEITC-treated tumor regression requires DDB2 (Fig. 6D).

Figure 5. DDB2 deficiency abrogates PEITC-mediated regression in tumor aggressiveness. HCT116 cells expressing control shRNA or DDB2 shRNA were injected subcutaneously into nude mice. Mice were divided randomly in two groups for each cell line for treatment with PEITC or vehicle control. Four weeks post treatment mice were sacrificed and tumor sections were fixed in 10% Formalin, processed and embedded with paraffin for sectioning. Prepared tumor section slides were then subjected to immunohistochemical analysis using H&E, PCNA or SMA antibody. Representative images (20X magnification) are shown. (B) PCNA positive cells per 40x magnification field were counted. Six random fields were chosen for quantification. (C) SMA positive area was quantified by ImageJ. Six random fields were chosen.

Figure 6. DDB2 deficiency abrogates PEITC mediated apoptosis and senescence in vivo. (A) HCT116 cells expressing control shRNA or DDB2 shRNA were injected subcutaneously into nude mice. Mice were divided randomly in two groups for each cell line for treatment with PEITC or vehicle control. Four weeks post-treatment mice were sacrificed and tumor sections were fixed in 10% Formalin, processed and embedded with paraffin for sectioning. Prepared tumor section slides were then subjected to immunohistochemical analysis using Cleaved caspase3 or p16INK4a antibody. Representative images (at 20X magnification) are shown. (B) Cleaved caspase 3 positive cells per 40x magnification field were counted. Six random fields were chosen for quantification. (C) p16INK4a positive cells per 40x magnification field were counted. Six random fields were chosen for quantification. (D) Model depicting how PEITC mediates apoptosis and senescence response to inhibit tumor growth by augmenting DDB2 expression.
Discussion
Results shown here are significant in several ways. First, we show that ROS can activate DDB2 expression in a p53-independent manner. Along the same line, PEITC also increases DDB2 expression independently of p53. Second, PEITC induces apoptosis and senescence of colon carcinoma cells in vitro and in vivo. Moreover, the effectiveness of the PEITC-induced apoptosis and senescence depends upon DDB2.
Although initially identified as a DNA damage repair protein, other important tumor suppressor functions of DDB2 have been identified in recent times. It is involved also in apoptosis and senescence. From database search, it was obvious that DDB2 expression is downregulated in a variety of cancers.28 DDB2 is a p53-induced gene in human. Because p53 is mutated in over 50% of cancers,29,30 it is likely that the loss of p53 function would cause a deficiency in the expression of DDB2. p53 is a major player in the induction of apoptosis and senescence. Hence, loss of p53 function renders the carcinoma cells refractory to apoptosis, senescence and cell cycle arrest following chemotherapy. Therefore, there is an urgent need to find out chemotherapeutic targets which acts in a p53-independent way. DDB2 seems to fit that function. We provide evidence that ROS-mediated induction of DDB2 is not mediated by p53; rather it depends on the stress-activated protein kinase pathways. Moreover, PEITC also augments DDB2 expression independent of p53 function. Use of PEITC, exploiting the fact that ROS induces DDB2 expression, is an attractive choice.
We provided evidence that the PEITC-induced expression of DDB2 enhances apoptosis and senescence response that lead to tumor regression by PEITC. Patients with loss of p53 function exhibit compromised apoptosis and senescence response following chemotherapy.31-33 These patients can be potentially treated by PEITC to induce DDB2 expression. There is evidence for a crosstalk between DDB2 and p53. p53 stimulates DDB2 expression both at the basal level as well as following DNA damage. On the other hand, it was reported that in XP-E cells with mutated DDB2 there was deficiency in UV-induced apoptosis due to reduced basal and UV-induced p53 level.34 Moreover, apoptosis function in DDB2-deficient XP-E cells were restored with expression of p53 cDNA construct indicating that DDB2 can participate in DNA damage-induced apoptosis through p53 regulation. However, DDB2 participates in apoptosis also by downregulation of p21, a cyclin-dependent kinase inhibitor.2 p21 exhibits anti-apoptotic activity, as the activity of caspases requires cdk activity.11 In agreement with that, loss of p21 expression in DDB2-null cells restores apoptosis in both in vitro and in vivo. In our studies, we report that ROS and PEITC both can activate DDB2 expression independent of p53, and that these mechanisms would be significant in therapy of tumors harboring mutation in p53.
We provide evidence that ROS induced DDB2 expression involves upregulated MAPK/JNK activity. Interestingly, a previous report suggested that p38MAPK mediated degradation of DDB2 following UV irradiation.35 In contrast, we find ROS induce DDB2 expression through activated p38MAPK. This apparent discrepancy possibly results from the choice of treatment and the cell fate associated with it. Cells preferably attempt to repair the DNA damage following UV irradiation. p38 MAPK is an important player in UV-induced DNA damage response. p38MAPK has been reported to facilitate nucleotide excision repair following UV irradiation through its phosphorylation of Histone H3 at serine 10 and subsequent alteration of chromatin condensation.36,37 Similarly, p38MAPK-mediated phosphorylation of DDB2 leads to its degradation that, in turn, facilitate in the recruitment of XPC in the DNA damage lesions and initiate DNA repair. In the present study, the oxidative stress treatment we employed mostly leads to apoptosis or senescence induction. In that scenario, DDB2 upregulation is important to facilitate these tumor-suppressor pathways. Thus, it appears that depending upon the context, p38MAPK either inhibits or augments DDB2 expression.
In conclusion, our data indicates potential usability of DDB2 expression as a chemotherapeutic target. As a proof of principle, we have provided evidence of PEITC-induced expression of DDB2 is critical for induction of apoptosis and senescence in tumors, as well as for PEITC-mediated regression of tumors.
Materials and Methods
Western blot analysis
Cells were harvested following wash with PBS. Cells were lysed by suspension in 2 vol of buffer containing 0.02 M HEPES (pH 7.9), 0.4 M NaCl, 0.1% NP-40, 10% (vol/vol) glycerol, 1 mM NaF, 1 mM Na-orthovanadate and a protease inhibitor cocktail. Extracts were subjected to sodium dodecyl sulfate PAGE, followed by blotting to nitrocellulose. The blots were probed with antibodies to Cdk2 (Santa Cruz), DDB2 (Cell signaling), Actin (Sigma), Tubulin (Sigma).
Cell culture
Human colon carcinoma cell line HCT 116 were cultured in DMEM medium supplemented with 10% FBS and Penicillin/Streptomycin. Stable clones of HCT 116 cells expressing control shRNA or DDB2shRNA were selected using Puromycin.
Xenograft models
Eight-week-old male nude mice were subcutaneously injected with 2 million cells (HCT116 cells expressing control shRNA or DDB2 shRNA) into both flanks. Mice were equally randomized into two groups for PEITC or vehicle treatment. When tumors reached volume of 10–15 mm3, mice were treated with PEITC (35 mg/kg of body weight) for 5 d a week for 4 wk by intraperitoneal injection as described previously.27 At the end point of the experiment, mice were euthanized by CO2 followed by cervical dislocation and tumor mass was measured.
Drug and inhibitors treatment
PEITC was used at a final concentration of 10 uM for indicated time points for DDB2 expression analysis. For TUNEL assay, cells were treated at a final concentration of 50 uM for 8 h. For SA-β-gal assay, cells were treated at a final concentration of 20 uM for 6 h.
p38MAPK inhibitor (SB 203580) and JNK inhibitor (SP600125) were purchased from calbiochem. They were used at a final concentration of 20 uM and 50 uM, respectively as described previously.38
Chromatin IP
Cells were first cross-linked by addition of 37% formaldehyde (Fisher) to a final concentration of 1% and incubated for 10 min with gentle swirling at room temperature. Cross-linking was stopped by addition of 2.5 M Glycine at a final concentration of 0.125 M Glycine for 5 min with gentle swirling. Cells were washed twice with ice cold sterile PBS and then collected by adding 1 ml of ice cold sterile PBS containing 1 mM PMSF and protease inhibitors (Roche). Cells were scraped and transferred into an Eppendorf tube and centrifuged at 2,000 rpm for 5 min. The cell pellet was then resuspended in a 2 × pellet volume of SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.1) and placed on ice for 10 min. The resulting extract was sonicated, precleared and immunoprecipitation was performed with 2 ug of Antibody (AP-1/ c-jun, Santa Cruz). Cross-links were reversed on all samples, including input, by addition of 100 μl TE containing 200 mM Nacl, 0.1 mg Proteinase K/ml and then incubated overnight. DNA was extracted from the digested samples using PCR purification kit (Qiagen). Extracted DNA was amplified by PCR alongside 0.1% of the input chromatin used to carry out the immunoprecipitation. Primers were used to carry out PCR. The PCR products were separated on agarose gels and visualized by ethidium bromide staining.
Primer I (-431 site)
Forward: CCTGTAGGGACCAGCCAAT
Reverse: GCCCGGCTAATTTCTCTCTC
Primer II (-1581 site)
Forward: CACGCCTATAATCCCAGCAT
Reverse: CAGCCTCCTCACTGCTTTTT
TUNEL assay
Cells were grown on glass coverslips and treated with PEITC. Twelve hours after the treatment cells were fixed in 1% paraformaldehyde pH 7.4. The ApopTag Red In situ Apoptosis Detection Kit (S7165) was used following manufacturer’s protocol.
Semi quantitative PCR
Total RNA was extracted from the cells with Trizol. One microgram of total RNA was then subjected to DNase I treatment using RQ1 RNase free DNase I (Invitrogen). The DNase I-treated RNA was reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's protocol. PCR amplification was performed with primers for DDB2 and Cyclophilin as described previously.10
Each PCR mix contained: 5 µl 5X PCR mix, 0.5 µl dNTP, 1.5 µl MgCl2, 0.125 µl Taq Polymerase (Promega), 2 µl cDNA, 13.875 µl water and 1 µl each forward and reverse primer.
Immunohistochemistry
Tumor tissues were fixed in 10% buffer formalin and paraffin embedded. Serial sections of 5 um thicknesses were de-paraffinized in xylene, followed by re-hydration in 100%, 95% and 70% ethanol, respectively. Sections were further treated for antigen retrieval with citrate buffer pH 6.0 at 95°C for 20 min. Blocking was performed using mouse on mouse blocking reagents following manufacturer’s protocol (VectorLabs BMK-2202) or by normal horse serum. Sections were washed three times with 1 × PBS and incubated with anti-mouse/ rabbit secondary antibody (VectorLabs) and further developed with Alkaline Phosphatase Substrate (VectorLabs SK-5300) following manufacturer protocol. Nuclei were counterstained with Hematoxylin.
Immunofluorescence
Tumor tissues were fixed in 10% buffer formalin and paraffin embedded. Serial sections of 5 um thicknesses were de-paraffinized in xylene, followed by re-hydration in 100%, 95% and 70% ethanol, respectively. Sections were further treated for antigen retrieval with citrate buffer pH 6.0 at 95°C for 20 min. Blocking was performed using 5% goat serum in PBS for 1 h at room temperature. Sections were incubated overnight at 4°C using following primary antibody: p16INK4a (SC-1207). After wash with PBS, sections were incubated with anti-rabbit immunoglobulin conjugated with FITC. Coverslips were mounted in Vectashield (Vector Laboratories) containing DAPI to stain nuclei.
SA-β Gal assay
HCT 116 cells (with control shRNA or DDB2 shRNA) were plated at low density. Cells were treated with PEITC for 4 h. After 48 h, cells were washed twice with ice cold PBS and fixed in 2% formaldehyde and 0.2% glutaraldehyde solution in PBS. Cells were incubated overnight at 37°C in staining solution containing 1 mg/ml X-gal, 40 mM citric acid/sodium phosphate (pH6.0), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl and 2 mM MgCl2. They were photographed thereafter and scored by assessing 15 random fields/plate in triplicate.
Clonogenecity assay
Clonogenecity assay were performed as described previously.39 Briefly, cells were treated with PEITC. After being rinsed with PBS, fresh medium was added and they were allowed to grow for 12 d to form colonies. Colonies were stained with Crystal Violet (Sigma).
ROS measurement
ROS are measured as described previously.10
Supplementary Material
Acknowledgments
This work was supported by grants from the National Cancer Institute (CA77637 and CA 156164 to P.R. and S.B.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/22631
References
- 1.Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 2.Stoyanova T, Roy N, Kopanja D, Bagchi S, Raychaudhuri P. DDB2 decides cell fate following DNA damage. Proc Natl Acad Sci USA. 2009;106:10690–5. doi: 10.1073/pnas.0812254106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nag A, Bondar T, Shiv S, Raychaudhuri P. The xeroderma pigmentosum group E gene product DDB2 is a specific target of cullin 4A in mammalian cells. Mol Cell Biol. 2001;21:6738–47. doi: 10.1128/MCB.21.20.6738-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoyanova T, Yoon T, Kopanja D, Mokyr MB, Raychaudhuri P. The xeroderma pigmentosum group E gene product DDB2 activates nucleotide excision repair by regulating the level of p21Waf1/Cip1. Mol Cell Biol. 2008;28:177–87. doi: 10.1128/MCB.00880-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapić-Otrin V, Levine AS. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci USA. 2006;103:2588–93. doi: 10.1073/pnas.0511160103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luijsterburg MS, Lindh M, Acs K, Vrouwe MG, Pines A, van Attikum H, et al. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J Cell Biol. 2012;197:267–81. doi: 10.1083/jcb.201106074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Zhai L, Xu J, Joo HY, Jackson S, Erdjument-Bromage H, et al. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell. 2006;22:383–94. doi: 10.1016/j.molcel.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 8.Stoyanova T, Roy N, Bhattacharjee S, Kopanja D, Valli T, Bagchi S, et al. p21 cooperates with DDB2 protein in suppression of ultraviolet ray-induced skin malignancies. J Biol Chem. 2012;287:3019–28. doi: 10.1074/jbc.M111.295816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoyanova T, Roy N, Kopanja D, Raychaudhuri P, Bagchi S. DDB2 (damaged-DNA binding protein 2) in nucleotide excision repair and DNA damage response. Cell Cycle. 2009;8:4067–71. doi: 10.4161/cc.8.24.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roy N, Stoyanova T, Dominguez-Brauer C, Park HJ, Bagchi S, Raychaudhuri P. DDB2, an essential mediator of premature senescence. Mol Cell Biol. 2010;30:2681–92. doi: 10.1128/MCB.01480-09. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–49. [PubMed] [Google Scholar]
- 12.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 13.Minig V, Kattan Z, van Beeumen J, Brunner E, Becuwe P. Identification of DDB2 protein as a transcriptional regulator of constitutive SOD2 gene expression in human breast cancer cells. J Biol Chem. 2009;284:14165–76. doi: 10.1074/jbc.M808208200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–91. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 15.Satyan KS, Swamy N, Dizon DS, Singh R, Granai CO, Brard L. Phenethyl isothiocyanate (PEITC) inhibits growth of ovarian cancer cells by inducing apoptosis: role of caspase and MAPK activation. Gynecol Oncol. 2006;103:261–70. doi: 10.1016/j.ygyno.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Wang LG, Chiao JW. Prostate cancer chemopreventive activity of phenethyl isothiocyanate through epigenetic regulation (review) Int J Oncol. 2010;37:533–9. doi: 10.3892/ijo_00000702. [review] [DOI] [PubMed] [Google Scholar]
- 17.Xiao D, Singh SV. Phenethyl isothiocyanate-induced apoptosis in p53-deficient PC-3 human prostate cancer cell line is mediated by extracellular signal-regulated kinases. Cancer Res. 2002;62:3615–9. [PubMed] [Google Scholar]
- 18.Xiao D, Zeng Y, Choi S, Lew KL, Nelson JB, Singh SV. Caspase-dependent apoptosis induction by phenethyl isothiocyanate, a cruciferous vegetable-derived cancer chemopreventive agent, is mediated by Bak and Bax. Clin Cancer Res. 2005;11:2670–9. doi: 10.1158/1078-0432.CCR-04-1545. [DOI] [PubMed] [Google Scholar]
- 19.Hu R, Kim BR, Chen C, Hebbar V, Kong AN. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cells. Carcinogenesis. 2003;24:1361–7. doi: 10.1093/carcin/bgg092. [DOI] [PubMed] [Google Scholar]
- 20.Khor TO, Cheung WK, Prawan A, Reddy BS, Kong AN. Chemoprevention of familial adenomatous polyposis in Apc(Min/+) mice by phenethyl isothiocyanate (PEITC) Mol Carcinog. 2008;47:321–5. doi: 10.1002/mc.20390. [DOI] [PubMed] [Google Scholar]
- 21.Tan T, Chu G. p53 Binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol Cell Biol. 2002;22:3247–54. doi: 10.1128/MCB.22.10.3247-3254.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 2002;3:420–5. doi: 10.1093/embo-reports/kvf094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park HJ, Carr JR, Wang Z, Nogueira V, Hay N, Tyner AL, et al. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009;28:2908–18. doi: 10.1038/emboj.2009.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou XF, Shen XQ, Shemshedini L. Ligand-activated retinoic acid receptor inhibits AP-1 transactivation by disrupting c-Jun/c-Fos dimerization. Mol Endocrinol. 1999;13:276–85. doi: 10.1210/me.13.2.276. [DOI] [PubMed] [Google Scholar]
- 25.Benkoussa M, Brand C, Delmotte MH, Formstecher P, Lefebvre P. Retinoic acid receptors inhibit AP1 activation by regulating extracellular signal-regulated kinase and CBP recruitment to an AP1-responsive promoter. Mol Cell Biol. 2002;22:4522–34. doi: 10.1128/MCB.22.13.4522-4534.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–52. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–70. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bagchi S, Raychaudhuri P. Damaged-DNA Binding Protein-2 Drives Apoptosis Following DNA Damage. Cell Div. 2010;5:3. doi: 10.1186/1747-1028-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 30.Soussi T, Lozano G. p53 mutation heterogeneity in cancer. Biochem Biophys Res Commun. 2005;331:834–42. doi: 10.1016/j.bbrc.2005.03.190. [DOI] [PubMed] [Google Scholar]
- 31.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–87. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 32.Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–46. doi: 10.1016/S0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- 33.Vergel M, Marin JJ, Estevez P, Carnero A. Cellular senescence as a target in cancer control. J Aging Res. 2010;2011:725365. doi: 10.4061/2011/725365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itoh T, O’Shea C, Linn S. Impaired regulation of tumor suppressor p53 caused by mutations in the xeroderma pigmentosum DDB2 gene: mutual regulatory interactions between p48(DDB2) and p53. Mol Cell Biol. 2003;23:7540–53. doi: 10.1128/MCB.23.21.7540-7553.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Q, Barakat BM, Qin S, Ray A, El-Mahdy MA, Wani G, et al. The p38 mitogen-activated protein kinase augments nucleotide excision repair by mediating DDB2 degradation and chromatin relaxation. J Biol Chem. 2008;283:32553–61. doi: 10.1074/jbc.M803963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong SP, Ma WY, Dong Z. ERKs and p38 kinases mediate ultraviolet B-induced phosphorylation of histone H3 at serine 10. J Biol Chem. 2000;275:20980–4. doi: 10.1074/jbc.M909934199. [DOI] [PubMed] [Google Scholar]
- 37.Cheung P, Allis CD, Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell. 2000;103:263–71. doi: 10.1016/S0092-8674(00)00118-5. [DOI] [PubMed] [Google Scholar]
- 38.Chaveroux C, Jousse C, Cherasse Y, Maurin AC, Parry L, Carraro V, et al. Identification of a novel amino acid response pathway triggering ATF2 phosphorylation in mammals. Mol Cell Biol. 2009;29:6515–26. doi: 10.1128/MCB.00489-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–9. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.