

Abstract
The stromal cell-derived factor-1α SDF-1α (CXCL12)/CXCR4 axis has been linked to poor prognosis in some cancers. As histone deacetylase inhibitors (HDIs) exert antitumor effects by targeting proteins affecting cell migration, we sought to evaluate the effects of the HDIs apicidin, vorinostat, entinostat (MS-275) and romidepsin on the expression and function of CXCR4 in human cancer cell lines. After treatment with romidepsin, CXCR4 mRNA expression increased 12-fold in UOK121 renal cancer cells, 16-fold in H460 non-small cell cancer cells and 4-fold in SF295 glioma cells; treatment with other HDIs yielded similar effects. CXCR4 induction was not observed in MCF7 breast cancer cells or SW620 colon cancer cells. To evaluate the corresponding functional increase, the effect of CXCR4 ligand, CXCL12, on ERK1/2, STAT3 and c-SRC activation and cell migration was examined in UOK121, SF295 and H460 cells. Alone, the HDIs increased pERK1/2, while reducing pSTAT-3 and pSRC. Following CXCL12 exposure, pERK1/2 induction was maintained, but STAT3 and SRC phosphorylation was impaired. These findings resulted in reduced basal and CXCL12-mediated cell migration. In conclusion, HDIs upregulated CXCR4 mRNA expression but impaired CXCL12-dependent signaling cascades through STAT3 and c-SRC, suggesting a potential role for HDIs in delaying or preventing metastatic processes in solid tumors.
Keywords: CXCR4, CXCL12, histone deacetylase inhibitor, romidepsin, migration
Introduction
Chemokines are 8 to 12 KDa pro-inflammatory cytokines that act through specific G-protein coupled receptors regulating cell activation, differentiation and trafficking. CXC chemokine receptor 4 (CXCR4) is expressed on hematopoietic cells, endothelial and epithelial cells. CXCR4 binds the chemokine CXCL12 (stromal cell-derived factor-1, SDF-1), which is constitutively produced by bone marrow stromal cells and epithelial cells in several other organs including lymph nodes, liver, lung, spleen, heart, skin, kidney and brain. The binding of CXCL12 to CXCR4 activates several divergent intracellular pathways regulating chemotaxis, survival, proliferation, gene transcription and intracellular calcium flux. Activation of the chemokine receptor CXCR4 through its ligand CXCL12 has been shown to induce migration and/or survival in multiple human cancer cell lines.1 A prognostic role of CXCR4 overexpression has been described in many neoplasms, including renal cancer,2,3 brain tumors,4 neuroblastoma,5 colorectal cancer,6 prostate cancer,7 melanoma,8 pancreatic tumor,9 lung cancer10 and ovarian cancer.11
The CXCR4/CXCL12 pathway may be a reasonable target in renal cancer given that overexpression of CXCR4 has been identified in RCC tumor samples.12 Pan et al. demonstrated that CXCR4 was significantly expressed on circulating cytokeratin + RCC cells from patients with metastatic RCC and that CXCR4 expression correlated with the metastatic potential of RCC.13 The loss or functional inactivation of VHL results in activation of HIF-1α and thereby enhanced CXCR4 and CXCL12 expression, presumably increasing migration and metastasis.2,3 Although a significant correlation between high levels of CXCR4 expression and tumor stage and/or differentiation grade was not detected, strong CXCR4 expression was found to correlate with poor survival.2,3,14
Histone deacetylase inhibitors represent a promising class of antineoplastic agents that affect tumor growth, differentiation and invasion.15 The hydroxamic acid derivative, vorinostat and the cyclic peptide, romidepsin, have been approved for the therapy of cutaneous T-cell lymphomas,16 although to-date, the striking activity manifested in T-cell lymphomas has not been observed in solid tumors.17,18 In vitro evidence showed that romidepsin inhibits cell growth by increasing p21WAF1 and phospho-Bcl-2 thus determining apoptosis and cell cycle arrest in RCC cell lines.19 Although significant activity in renal cancer was not confirmed after follow up, it was the one solid tumor showing major response other than T-cell lymphoma during the NCI Phase I trial of romidepsin.20,21 In related studies, another HDI, valproic acid (VPA), inhibits RCC tumor cell proliferation in vitro and in vivo.22 Thus we sought to determine the possible interaction between CXCR4 and histone deacetylase inhibition and potential significance in renal cell cancer progression and metastases.
Previous data showed that the HDIs butyrate and vorinostat reduced CXCR4 expression and migration in acute lymphoblastic leukemia (ALL),23,24 and in chronic lymphocytic leukemia (CLL) cells.24 In contrast, Gul et al. showed that VPA increased CXCR4 expression and migration toward a CXCL12 gradient in hematopoietic stem/progenitor cells (HSPCs)25; moreover, Gul reported that VPA repressed CXCR4 expression and chemotaxis in more differentiated CD34-negative AML (acute myelogenous leukemic) cells, but increased CXCR4 expression and chemotaxis in immature CD34‑positive AML cells.26 Another HDI, trichostatin A (TSA), transiently increased CXCR4 expression after 24 h treatment, but downregulated it after 48 h in melanoma cells.27
The aim of this work was to evaluate whether current epigenetic therapies might affect CXCR4 function in human renal cancer and other solid tumor cells. In this report, the effect of the HDIs romidepsin, apicidin, vorinostat and entinostat was evaluated on the CXCR4/CXCL12 axis in human renal cancer cell lines UOK108, 121, 127 and 143 as well as in H460 lung cancer cells, SF295 glioblastoma cells, MCF7 breast cancer cells and SW620 colon cancer cells. Although HDIs upregulated CXCR4 mRNA expression, downstream signaling molecules pSTAT3 and pSRC were downregulated and, correspondingly, cell migration was overall decreased.
Results
HDI treatment increases CXCR4 mRNA expression in human cancer cell lines
To evaluate the effect of romidepsin on CXCR4, mRNA expression was evaluated in three renal cancer cell lines UOK121, UOK143 and UOK181. Renal cancer cell lines were treated with romidepsin (10 ng/ml) for 24 h in the presence of verapamil (5 μg/ml), a P-glycoprotein (Pgp) inhibitor added to prevent Pgp-mediated efflux of romidepsin, since Pgp is readily upregulated in vitro by romidepsin.29Figure 1A shows that romidepsin treatment resulted in a 12-fold CXCR4 induction in UOK121 and a 2.5-fold induction in UOK143 cells. The expression of CXCL12, the CXCR4 ligand, was not affected (data not shown). Since UOK121 and UOK143 cells have a known hypermethylated phenotype, these cells were treated with romidepsin in the presence of decitabine, a demethylating agent; concomitant treatment with romidepsin and decitabine led to a further increase in levels of CXCR4 expression.
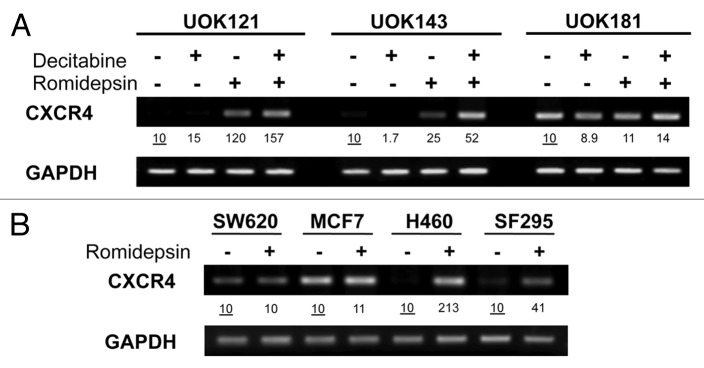
Figure 1. Romidepsin induced CXCR4 mRNA overexpression in human cancer cell lines. (A) CXCR4 expression levels were measured by semiquantitative RT-PCR in renal cancer cell lines UOK121, UOK143 and UOK181 treated with romidepsin (10 ng/ml) + verapamil (5 μg/mL) for 24 h, with or without decitabine (1 mM) daily for four days. (B) CXCR4 mRNA expression in SF295, H460, SW620 and MCF7cells treated with romidepsin (20 ng/ml for 24 h) + verapamil (5 μg/mL). GAPDH was used as the internal control. Numbers indicate fold increase of CXCR4 relative to the untreated cells. Representative results from three independent experiments are shown.
To determine whether induction of CXCR4 mRNA expression was a general phenomenon, CXCR4 expression was then evaluated in non-renal cancer cell lines H460, SF295, SW620 and MCF7 following incubation with romidepsin. Romidepsin treatment (20 ng/ml) in the presence of 5 μg/ml of verapamil for 24 h was found to induce CXCR4 expression 21.3‑fold in H460 cells and 4.1‑fold in SF295 cells, while no CXCR4 induction was detected in the MCF7 and SW620 cell lines (Fig. 1B).
Basal CXCR4 expression was compared in UOK121 and UOK143, SW620, MCF7, SF295 and H460 cells. Figure S1 shows that the basal level of CXCR4 expression was comparable in UOK121, UOK143, SF295 and H460 cells, while SW620 and MCF7 cells had a 3‑fold higher CXCR4 basal expression.
To evaluate the specificity of the romidepsin-induced CXCR4 expression, CXCR4 induction was evaluated by qPCR following treatment with romidepsin (2‑5‑10 ng/ml with verapamil) as well as other HDIs such as apicidin (1‑2‑5 μM), vorinostat (2.5‑5‑7.5 μM) and entinostat (MS-275, 2‑4‑10 μM). In UOK121 cells, CXCR4 induction was greatest, in the range of 20- to 110‑fold when treated with any of the HDIs (Fig. 2). In the non-RCC cells, CXCR4 expression was induced from 5- to 25‑fold in H460 cells, from 5- to 15‑fold in SF295 cells less than 5‑fold in MCF7 cells treated with any of the HDIs (Fig. 2).
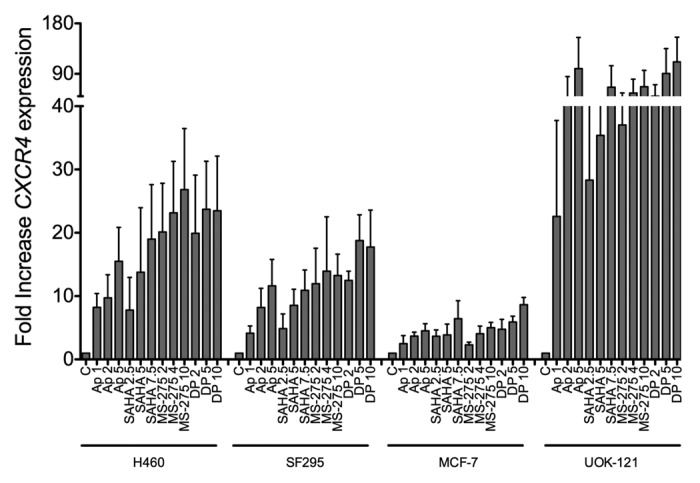
Figure 2. Other HDIs similarly induced CXCR4 mRNA in human cancer cells. CXCR4 mRNA was measured by qPCR in H460, SF295, MCF7, UOK121 cells treated with apicidin (1-2-5 μM), vorinostat (2.5-5-7.5 μM), MS-275 (2-4-10 μM) or romidepsin (1-2-5 ng/ml + verapamil 5 μg/mL) for 24 h. rRNA was used as the internal control. Results from three independent experiments are shown.
To evaluate the protein level corresponding to the mRNA induction, the effect of HDIs on CXCR4 surface expression was evaluated in UOK121, SF295 and H460 cells. Although detectable expression was observed in the Hut78 positive control cell line, basal CXCR4 expression could not be detected by flow cytometry in UOK121, SF295 or H460 cell lines and was slightly detected after treatment with romidepsin (5 ng/ml), apicidin (5 μM) and vorinostat (7.5 μM) for 24 h in UOK121 (data not shown). Functional assays were subsequently performed to detect signaling through CXCR4 in these solid tumor cell lines.
HDI treatment reduced cell migration in response to CXCL12
Since CXCR4 activation determines migration toward the specific ligand CXCL12, migration assays were conducted in HDI-pretreated cells to correlate the increased expression to function. In Figure 3 the basal and CXCL12-mediated migration of UOK121, H460 and SF295 is shown following exposure to three HDIs. Fewer UOK121 cells migrated overall, compared with H460 and SF295 cells. In general the HDIs reduced basal migration and decreased migration in the presence of CXCL12 (Fig. 3A). The one exception was romidepsin in UOK121, where basal migration was consistently increased. The percent stimulation of migration following CXCL12 is shown in the right hand panels.

Figure 3. HDIs reduced migration in human cancer cells. CXCL12-specific cell migration was assessed in romidepsin (4 ng/ml), vorinostat (7.5 μM), or apicidin (5μM) -treated UOK121 (A), SF295 (B) or H460 (C) cells. Cells were treated for 24 h with the indicated HDI and then plated on transwell in medium with 0.5% BSA in the upper well vs. CXCL12 (100 ng/ml) containing medium into the lower well. On the left, the data are given as mean ± SD of migration from three independent experiments. On the right hand panels, the percentage of migrated cells over basal migration (in the absence of CXCL12) are shown. **p < 0.05, *p < 0.01 vs control value.
HDI effects on phosphorylation of ERK1/2, STAT3, FAK and c-SRC in RCC and other cancer cell lines
Previous studies have shown that CXCR4 activation affects proliferation and migration through extracellular signal-related kinase (ERK) and AKT phosphorylation,30,31 c-SRC phosphorylation32,33 and the Janus Kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway.34,35 Moreover, CXCL12 treatment can induce the phosphorylation of focal adhesion kinase (FAK) regulating cell flexibility and migration.36
To evaluate the HDI effect on CXCR4 function, the phosphorylation status of ERK1 and 2, c-SRC, FAK and STAT3 was evaluated following HDI treatment. Figure 4 plots the densitometry values obtained from three immunoblots, normalized to a control value of 1; representative immunoblots are shown in Figure S2. Treatment with romidepsin, apicidin and vorinostat in UOK121, SF295 and H460 for 24 h in serum-free medium induced phospho-ERK1/2, while decreasing phospho-STAT3 and phospho-SRC. All but 2 points were statistically significantly different. No significant effect of HDIs was observed on FAK phoshorylation in UOK121, SF295 or H460 cells (data not shown). Taken together, these results suggest that the transducers of CXCR4 signaling, other than pERK1/2, were inhibited by the HDIs.

Figure 4. HDIs induced ERK1/2 activation but inhibited c-SRC and STAT3 activation in human cancer cells. UOK121, SF295 and H460 cells were treated with romidepsin (1-2-5 ng/ml) + verapamil (5 μg/mL), vorinostat (2.5-5-7.5 μM), or apicidin (1-2-5 μM) for 24 h and ERK1/2 phosphorylation, c-SRC phosphorylation and STAT3 phosphorylation were detected by western blot analysis. Protein levels were normalized to GAPDH and plotted as mean ± SD from three independent experiments. **p < 0.05, *p < 0.01 vs control value. Representative immunoblots are shown in Figure S2.
HDI treated-CXCL12 induced effect on phosphorylation of ERK1/2, STAT3 and c-SRC in RCC and other cancer cell lines
Next, the impact of HDI treatment on CXCL12 response was assessed. Figure 5A shows that ERK1/2 activation occurred in UOK121 cells following exposure to CXCL12. Induction in response to CXCL12 after romidepsin (5 ng/ml for 24 h) + verapamil (5 μg/ml) treatment was maintained (6.8, 2.5 and 1.4‑fold, respectively, at 2, 7 and 20 min of CXCL12 exposure). Similar results were observed following vorinostat and apicidin. In contrast, pretreatment with romidepsin, vorinostat or apicidin markedly inhibited CXCL12-induced SRC phosphorylation. STAT3 could not be detected in these cells. In Figure 5B the effect of HDIs were evaluated in SF295 cells. Romidepsin, vorinostat and apicidin treatment maintained CXCL12-dependent p- ERK1/2 activation while effects on pSTAT3 and pSRC were attenuated. Similar results were observed in H460 cells (Fig. 5C). Acetylated H3 and p21 induction confirmed the activity of all three HDIs in UOK121, SF295 and H460 cells. Together, these results suggest attenuation of CXCL12 signaling through CXCR4.

Figure 5. Effects of HDIs on signaling patways induced by CXCL12. ERK1/2, STAT3 and c-SRC phosphorylation was detected by immunoblotting (A) UOK121, (B) SF295 and (C) H460 cells following stimulation by CXCL12 (100 ng/ml) for 2–7 and 20 min. Cells were serum starved for 24 h and then pretreated with romidepsin (5ng/ml) + verapamil (5 μg/mL), vorinostat (7.5 μM), or apicidin (5μM). Representative western blots are shown and numbers at the bottom indicate the fold variations relative to the respective starvation value. Immunoblots with anti-GAPDH antibody were used for normalization. The experiments were repeated more than three times, with similar results.
Discussion
In this manuscript, the role of HDIs in modulating the CXCR4/CXCL12 axis was evaluated. The CXCR4-CXCL12 axis is crucial in promoting cell migration and thus the metastatic process in cancer. Since HDIs widely affect gene expression, it is possible to hypothesize that HDAC inhibition could promote invasion and metastasis, as reported in hepatocellular carcinoma cells through upregulation of integrins37 and in melanoma cells through upregulation of CXCR4 and CCR7.27 Here it was shown that, although the HDIs romidepsin, vorinostat and apicidin upregulated CXCR4 mRNA expression, overall migration was reduced in renal cancer cells, NSCLC cells and glioblastoma cells. HDIs increased CXCL12-mediated ERK activation but reduced migration, STAT3 signaling and SRC phosphorylation in all cell lines studied.
The variability of the effect of the deacetylase inhibitors in the different cell lines is consistent with our experience and with published literature. In contrast to the effects of irradiation or DNA damage, the response of human cancer cell lines to deacetylase inhibitors is highly context dependent. In the study of Kanao et al., four different renal cancer cell lines—Caki-1, 769P, ACHN and 786-0—showed a different sensitivity to romidepsin in histone H3 acetylation with Caki-1 being the most sensitive and 769P the least acetylated; moreover, romidepsin induced apoptosis in Caki-1, ACHN and 786-0 while a G2 arrest was detected in 769P cells.19 Romidepsin is effective in inducing demethylation in the human lung cancer cell lines H719 and H23, human pancreatic cancer cell line PANC1 and human colon cancer cell line HT29 but not in human colon cancer cell lines HCT116 and SW480.38 In cells lines SF295 and H460, romidepsin induced CXCR4 was registered in SW620 and MCF7 cells. It is interesting to note that the two cell lines not further induced in CXCR4 mRNA expression by HDIs, MCF7 and SW620, express elevated basal CXCR4 levels. This variable gene response to romidepsin is similar to that observed for ABCG2, a drug transporter not involved in romidepsin efflux.39 In earlier studies, although global histone acetylation was detected in all cell types tested following exposure to romidepsin, the induction of ABCG2 was variable and associated with a permissive configuration of the promoter’s epigenetic code.39 We postulated that constraints in the promoter in some cell lines prevented induction of gene expression. Similarly, Baylin et al., have shown that bivalent marks present on some gene promoters represent an intermediate state between an active and inactive promoter, i.e., a promoter poised to respond to initiate transcription.40 The same gene promoter in different cell types may be differentially able to respond to HDAC inhibition. Our results with CXCR4 presented here extend that observation, again showing cell-line specific patterns of gene expression following HDAC inhibition. This fits with broad evidence regarding cell context-specific effects of HDIs.
CXCL12 binding to CXCR4 promotes activation of multiple G protein-dependent signaling pathways, resulting in diverse biological responses such as migration, adhesion, survival and/or proliferation and transcription activation. Activation of the MAP kinase cascade through Gαi can lead to the phosphorylation of ERK1/2. Phospho-ERK1/2 is an important downstream effector of proliferative, survival31 and metastatic pathways.30 Interestingly, romidepsin, vorinostat and apicidin were shown to induce ERK1/2 phosphorylation in UOK121, SF295 and H460. While the MAPK pathway is generally associated with survival, in some models activation of the pathway has resulted in cell death. Park et al. suggest that apicidin induced cell cycle arrest by activation of the ERK pathway in Ras-transformed breast epithelial cells.41 Abnormal retention of p-ERK in cytoplasm after stimulation of D1 dopamine receptors, for example, culminated in a cytotoxic response, rather than a mitogenic response in neuronal cells.42 Pettersson et al. showed that ERK activation was required for MDA-MB-231 cell death due to a retinoid-protein kinase C inhibitor combination.43
Moreover, the binding of CXCL12 to CXCR4 through G-α protein can activate c-SRC32 and Src family kinases can mediate cell proliferation via Ras/ERK/MAPK pathway and cell adhesion and migration via interaction with integrins, actins, GTPase-activating proteins and kinases as FAKs.33 CXCR4 has also been reported to activate the G-protein independent JAK/STAT3 pathway.34 Association between CXCR4 and STAT3 protein was found in hematopoietic progenitor cells and activation of JAK2 is required for SDF-1-induced migration.44 In this report we observed that HDIs such as romidepsin, vorinostat and apicidin reduced the amounts of pSTAT3 and p-SRC proteins in UOK121, SF295 and H460. These results are consistent with the key finding of inhibition of cell migration.
In UOK121, SF295 and H460 cells, despite CXCR4 induction in romidepsin, vorinostat and apicidin-treated cells, there was a decrease in basal and CXCL12-induced migration. These findings are in accordance with previous results where HDIs affected cell motility through molecular events such as downregulation of endothelial nitric oxide synthetase,45 suppression of nuclear factor-B activity,46 inhibition of matrix metalloproteinase 2 activation through upregulation of RECK, a negative regulator of matrix metalloproteinases47 or downregulation of hepatocyte growth factor.48
Our results extend these findings by showing that critical signaling molecules in pathways involved in cell migration are downregulated. Only p-ERK is increased and, as we note above, the upregulated p-ERK has been associated with cytotoxicity in other model systems.
In conclusion, despite induction of CXCR4 and activation of pERK in three solid tumor cell lines, the net effect of HDI treatment was to reduce signal transduction. It appeared that romidepsin, vorinostat and apicidin reduced migration overall in an effect that dominated or neutralized any activation of CXCR4. These data lend support for developing therapies that employ the HDIs in solid tumors, aiming at combination strategies that exploit the unique activities of deacetylase inhibition in cancer.
Materials and Methods
Materials
Entinostat (MS-275) and romidepsin were obtained from the National Cancer Institute Anticancer Drug Screen. Vorinostat was purchased from Cayman Chemicals. Apicidin was purchased from EMD Bioscience. Verapamil was obtained from Sigma-Aldrich Co. Recombinant Feline/Human/Rhesus/Macaque CXCL12/SDF1α was purchased from R&D Systems.
Cell lines
UOK108, 121, 127 and 143 renal cell carcinoma cell lines were provided by Dr. Marston Linehan (National Institutes of Health, Bethesda, MD). SW620 colon carcinoma, SF295 human glioblastoma, H460 human NSCLC, A498 renal carcinoma and MCF7 breast cancer cell lines were obtained from the National Cancer Institute Anticancer Drug Screen. Identity was confirmed by short tandem repeat analysis (RADIL). The Hut78 T-cell lymphoma cell line was obtained from ATCC. All cells were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mmol/l L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin. Cells were grown at 37°C with 5% CO2.
Cell migration assay
Migration was assayed in 24-well Transwell chambers (Corning Inc.) using inserts with an 8-µm pore membrane. Membranes were precoated with collagen (human collagen type I/III) and fibronectin (10 µg/ml each). Test cells were placed in the upper chamber (2.0 × 105 cells/well) in DMEM containing 0.5% BSA (migration media) and 100 ng/ml CXCL12 was added to the lower chamber or to the upper and lower chamber (chemokinesis). After 16 h, cells on the upper surface of the filter were removed using a cotton wool swab. SN12C cells were used as a positive control. The cells were counted in ten different fields (original magnification × 40). For each experiment, results were expressed as the mean of three replicates ± the standard deviation (SD). The net number of migrated cells was calculated by subtracting the number of cells migrating toward CXCL12 from the number of cells migrating toward medium with 0.5% BSA.9
Immunoblot analysis
Cells were scraped into cold PBS followed by centrifugation at 1200 RPM for 10 min at 4°C. The cell pellet was then re-suspended in cold lysis buffer (40 mM Hepes pH 7.5, 120 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 0.5 mM EDTA, 1% Triton X-100) containing protease (Protease Inhibitor Cocktail, Sigma-Aldrich) and phosphatase inhibitors (50 mM NaF and PhosphoSTOP Phosphatase Inhibitor Cocktail Tablets, Roche Diagnostics). After 15 min on ice, unlysed cells and nuclei were pelleted at 14000 RPM for 20 min at 4°C. After determination of protein concentration in cell lysates with Bio-Rad’s Protein Assay Reagent (Bio-Rad), 50 µg of protein were loaded onto precast 4–12% (w/v) NuPAGE Novex Bis-Tris Mini gels (Invitrogen), subjected to electrophoresis and electrotransferred onto PVDF membranes (Millipore). Membranes were stained with 0.1% Ponceau S (Sigma) and checked for comparable loading. Blots were probed with the following primary antibodies: rabbit polyclonal anti-CXCR4 (ab2074, Abcam); rabbit polyclonal anti-phosphorylated p44/42 MAPK, rabbit polyclonal anti-phosphorylated STAT3, rabbit polyclonal anti-phosphorylated SRC (Tyr 416) (all from Cell Signaling Technology); anti p-21CIP1/WAF1 (EMD Bioscience); anti-acetyl histone H3 (Ac-K9, Millipore) and mouse monoclonal anti-GAPDH (American Research Products) and visualized with the Odyssey System (LI-COR) using a 1:4000 dilution of the IRDye 800CW goat anti-mouse secondary antibody or IRDye 680CW goat anti-rabbit secondary antibody (LI-COR).
RNA isolation and PCR analysis
Total RNA was isolated with Trizol reagent (Invitrogen). RNA (1 µg) was reverse transcribed using a commercially available cDNA synthesis kit (Bioline). CXCR4 and CXCL12 expression levels were quantified by semi-quantitative RT-PCR using primers specific for CXCR4, 5′-GGTGGTCTATGTTGGCGTCT-3′ (forward) and 5′-TGGAGTGTGACAGCTTGGAG-3′ (reverse); CXCL12, 5′-GGGCTCCTGGGTTTTGTATT-3′ (forward) and 5′-GTCCTGAGAGTCCTTTTGCG-3′ (reverse). CXCR4 and CXCL12 levels were normalized to GAPDH, 5′-ACATGTTCCAATATGATTCCA-3′ (forward) and 5′-TGGACTCCACGACGTACTCAG-3′ (forward). Each experiment was repeated 3–4 times. Representative results are shown. Induction of CXCR4 was expressed relative to the untreated control, after normalization with GAPDH. Quantitative PCR was also performed on RNA that was reverse transcribed with random primers (Invitrogen) and amplified in a LightCycler Thermocycler using probes from the Roche Universal ProbeLibrary (Roche Diagnostics) and primers for CXCR4, 5′-AGGATATAATGAAGTCACTATGGGAAA-3 (forward) and 5′-AAGGGCACAAGAGAATTAATGTAGA (reverse). Induction of CXCR4 was expressed relative to untreated control after normalization to rRNA, 5′-TTACCCTACTGATGATGTGTTGTTG-3′ (forward) and 5′-CCTGCGGTTCCTCTCGTA-3′ (reverse).
Flow cytometry analysis
To evaluate the expression of CXCR4 (CD184), adherent cancer cells at subconfluency (60−70% confluent) were detached with 2 mmol/L EDTA in PBS, washed, re-suspended in ice-cold PBS and incubated for 30 min at 4°C with anti-CD184-PE antibody (FAB 173P, clone 44717, R&D Systems) or PE-labeled mouse IgG2b as negative control. Cells to be used for staining with the antibody were first Fc-blocked by treatment with human IgG for 15 min at room temperature.28 After three washes in PBS, the cells were analyzed by FACSort flow cytometer.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Cancer Institute. This work was supported in part by a CRADA between the NCI and Celgene Corp.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/22957
References
- 1.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–31. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 2.Zagzag D, Krishnamachary B, Yee H, Okuyama H, Chiriboga L, Ali MA, et al. Stromal cell-derived factor-1alpha and CXCR4 expression in hemangioblastoma and clear cell-renal cell carcinoma: von Hippel-Lindau loss-of-function induces expression of a ligand and its receptor. Cancer Res. 2005;65:6178–88. doi: 10.1158/0008-5472.CAN-04-4406. [DOI] [PubMed] [Google Scholar]
- 3.Struckmann K, Mertz K, Steu S, Storz M, Staller P, Krek W, et al. pVHL co-ordinately regulates CXCR4/CXCL12 and MMP2/MMP9 expression in human clear-cell renal cell carcinoma. J Pathol. 2008;214:464–71. doi: 10.1002/path.2310. [DOI] [PubMed] [Google Scholar]
- 4.do Carmo A, Patricio I, Cruz MT, Carvalheiro H, Oliveira CR, Lopes MC. CXCL12/CXCR4 promotes motility and proliferation of glioma cells. Cancer Biol Ther. 2010;9:56–65. doi: 10.4161/cbt.9.1.10342. [DOI] [PubMed] [Google Scholar]
- 5.Carlisle AJ, Lyttle CA, Carlisle RY, Maris JM. CXCR4 expression heterogeneity in neuroblastoma cells due to ligand-independent regulation. Mol Cancer. 2009;8:126. doi: 10.1186/1476-4598-8-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ottaiano A, Franco R, Aiello Talamanca A, Liguori G, Tatangelo F, Delrio P, et al. Overexpression of both CXC chemokine receptor 4 and vascular endothelial growth factor proteins predicts early distant relapse in stage II-III colorectal cancer patients. Clin Cancer Res. 2006;12:2795–803. doi: 10.1158/1078-0432.CCR-05-2142. [DOI] [PubMed] [Google Scholar]
- 7.Engl T, Relja B, Marian D, Blumenberg C, Müller I, Beecken WD, et al. CXCR4 chemokine receptor mediates prostate tumor cell adhesion through alpha5 and beta3 integrins. Neoplasia. 2006;8:290–301. doi: 10.1593/neo.05694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scala S, Ottaiano A, Ascierto PA, Cavalli M, Simeone E, Giuliano P, et al. Expression of CXCR4 predicts poor prognosis in patients with malignant melanoma. Clin Cancer Res. 2005;11:1835–41. doi: 10.1158/1078-0432.CCR-04-1887. [DOI] [PubMed] [Google Scholar]
- 9.Marchesi F, Monti P, Leone BE, Zerbi A, Vecchi A, Piemonti L, et al. Increased survival, proliferation and migration in metastatic human pancreatic tumor cells expressing functional CXCR4. Cancer Res. 2004;64:8420–7. doi: 10.1158/0008-5472.CAN-04-1343. [DOI] [PubMed] [Google Scholar]
- 10.Phillips RJ, Burdick MD, Lutz M, Belperio JA, Keane MP, Strieter RM. The stromal derived factor-1/CXCL12-CXC chemokine receptor 4 biological axis in non-small cell lung cancer metastases. Am J Respir Crit Care Med. 2003;167:1676–86. doi: 10.1164/rccm.200301-071OC. [DOI] [PubMed] [Google Scholar]
- 11.Scotton CJ, Wilson JL, Scott K, Stamp G, Wilbanks GD, Fricker S, et al. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res. 2002;62:5930–8. [PubMed] [Google Scholar]
- 12.Schrader AJ, Lechner O, Templin M, Dittmar KE, Machtens S, Mengel M, et al. CXCR4/CXCL12 expression and signalling in kidney cancer. Br J Cancer. 2002;86:1250–6. doi: 10.1038/sj.bjc.6600221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan J, Mestas J, Burdick MD, Phillips RJ, Thomas GV, Reckamp K, et al. Stromal derived factor-1 (SDF-1/CXCL12) and CXCR4 in renal cell carcinoma metastasis. Mol Cancer. 2006;5:56. doi: 10.1186/1476-4598-5-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–11. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- 15.Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009;107:600–8. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs. 2007;16:1111–20. doi: 10.1517/13543784.16.7.1111. [DOI] [PubMed] [Google Scholar]
- 17.Piekarz RL, Robey R, Sandor V, Bakke S, Wilson WH, Dahmoush L, et al. Inhibitor of histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case report. Blood. 2001;98:2865–8. doi: 10.1182/blood.V98.9.2865. [DOI] [PubMed] [Google Scholar]
- 18.Piekarz RL, Sackett DL, Bates SE. Histone deacetylase inhibitors and demethylating agents: clinical development of histone deacetylase inhibitors for cancer therapy. Cancer J. 2007;13:30–9. doi: 10.1097/PPO.0b013e31803c73cc. [DOI] [PubMed] [Google Scholar]
- 19.Kanao K, Mikami S, Mizuno R, Shinojima T, Murai M, Oya M. Decreased acetylation of histone H3 in renal cell carcinoma: a potential target of histone deacetylase inhibitors. J Urol. 2008;180:1131–6. doi: 10.1016/j.juro.2008.04.136. [DOI] [PubMed] [Google Scholar]
- 20.Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin Cancer Res. 2002;8:718–28. [PubMed] [Google Scholar]
- 21.Stadler WM, Margolin K, Ferber S, McCulloch W, Thompson JA. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clin Genitourin Cancer. 2006;5:57–60. doi: 10.3816/CGC.2006.n.018. [DOI] [PubMed] [Google Scholar]
- 22.Jones J, Juengel E, Mickuckyte A, Hudak L, Wedel S, Jonas D, et al. The histone deacetylase inhibitor valproic acid alters growth properties of renal cell carcinoma in vitro and in vivo. J Cell Mol Med. 2009;13(8B):2376–85. doi: 10.1111/j.1582-4934.2008.00436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crazzolara R, Jöhrer K, Johnstone RW, Greil R, Kofler R, Meister B, et al. Histone deacetylase inhibitors potently repress CXCR4 chemokine receptor expression and function in acute lymphoblastic leukaemia. Br J Haematol. 2002;119:965–9. doi: 10.1046/j.1365-2141.2002.03955.x. [DOI] [PubMed] [Google Scholar]
- 24.Stamatopoulos B, Meuleman N, De Bruyn C, Delforge A, Bron D, Lagneaux L. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces apoptosis, down-regulates the CXCR4 chemokine receptor and impairs migration of chronic lymphocytic leukemia cells. Haematologica. 2010;95:1136–43. doi: 10.3324/haematol.2009.013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gul H, Marquez-Curtis LA, Jahroudi N, Lo J, Turner AR, Janowska-Wieczorek A. Valproic acid increases CXCR4 expression in hematopoietic stem/progenitor cells by chromatin remodeling. Stem Cells Dev. 2009;18:831–8. doi: 10.1089/scd.2008.0235. [DOI] [PubMed] [Google Scholar]
- 26.Gul H, Marquez-Curtis LA, Jahroudi N, Larratt LM, Janowska-Wieczorek A. Valproic acid exerts differential effects on CXCR4 expression in leukemic cells. Leuk Res. 2010;34:235–42. doi: 10.1016/j.leukres.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 27.Mori T, Kim J, Yamano T, Takeuchi H, Huang S, Umetani N, et al. Epigenetic up-regulation of C-C chemokine receptor 7 and C-X-C chemokine receptor 4 expression in melanoma cells. Cancer Res. 2005;65:1800–7. doi: 10.1158/0008-5472.CAN-04-3531. [DOI] [PubMed] [Google Scholar]
- 28.Kim SW, Kim HY, Song IC, Jin SA, Lee HJ, Yun HJ, et al. Cytoplasmic trapping of CXCR4 in hepatocellular carcinoma cell lines. Cancer Res Treat. 2008;40:53–61. doi: 10.4143/crt.2008.40.2.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robey RW, Zhan Z, Piekarz RL, Kayastha GL, Fojo T, Bates SE. Increased MDR1 expression in normal and malignant peripheral blood mononuclear cells obtained from patients receiving depsipeptide (FR901228, FK228, NSC630176) Clin Cancer Res. 2006;12:1547–55. doi: 10.1158/1078-0432.CCR-05-1423. [DOI] [PubMed] [Google Scholar]
- 30.Huang YC, Hsiao YC, Chen YJ, Wei YY, Lai TH, Tang CH. Stromal cell-derived factor-1 enhances motility and integrin up-regulation through CXCR4, ERK and NF-kappaB-dependent pathway in human lung cancer cells. Biochem Pharmacol. 2007;74:1702–12. doi: 10.1016/j.bcp.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 31.Shen X, Artinyan A, Jackson D, Thomas RM, Lowy AM, Kim J. Chemokine receptor CXCR4 enhances proliferation in pancreatic cancer cells through AKT and ERK dependent pathways. Pancreas. 2010;39:81–7. doi: 10.1097/MPA.0b013e3181bb2ab7. [DOI] [PubMed] [Google Scholar]
- 32.Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta. 2007;1768:952–63. doi: 10.1016/j.bbamem.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6:587–95. doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- 34.Ahr B, Denizot M, Robert-Hebmann V, Brelot A, Biard-Piechaczyk M. Identification of the cytoplasmic domains of CXCR4 involved in Jak2 and STAT3 phosphorylation. J Biol Chem. 2005;280:6692–700. doi: 10.1074/jbc.M408481200. [DOI] [PubMed] [Google Scholar]
- 35.Pfeiffer M, Hartmann TN, Leick M, Catusse J, Schmitt-Graeff A, Burger M. Alternative implication of CXCR4 in JAK2/STAT3 activation in small cell lung cancer. Br J Cancer. 2009;100:1949–56. doi: 10.1038/sj.bjc.6605068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandis AZ, Prasad A, Band H, Klösel R, Ganju RK. Regulation of CXCR4-mediated chemotaxis and chemoinvasion of breast cancer cells. Oncogene. 2004;23:157–67. doi: 10.1038/sj.onc.1206910. [DOI] [PubMed] [Google Scholar]
- 37.Lin KT, Yeh SH, Chen DS, Chen PJ, Jou YS. Epigenetic activation of alpha4, beta2 and beta6 integrins involved in cell migration in trichostatin A-treated Hep3B cells. J Biomed Sci. 2005;12:803–13. doi: 10.1007/s11373-005-9005-2. [DOI] [PubMed] [Google Scholar]
- 38.Wu LP, Wang X, Li L, Zhao Y, Lu S, Yu Y, et al. Histone deacetylase inhibitor depsipeptide activates silenced genes through decreasing both CpG and H3K9 methylation on the promoter. Mol Cell Biol. 2008;28:3219–35. doi: 10.1128/MCB.01516-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.To KK, Polgar O, Huff LM, Morisaki K, Bates SE. Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol Cancer Res. 2008;6:151–64. doi: 10.1158/1541-7786.MCR-07-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGarvey KM, Van Neste L, Cope L, Ohm JE, Herman JG, Van Criekinge W, et al. Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells. Cancer Res. 2008;68:5753–9. doi: 10.1158/0008-5472.CAN-08-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park H, Im JY, Kim J, Choi WS, Kim HS. Effects of apicidin, a histone deacetylase inhibitor, on the regulation of apoptosis in H-ras-transformed breast epithelial cells. Int J Mol Med. 2008;21:325–33. [PubMed] [Google Scholar]
- 42.Chen J, Rusnak M, Lombroso PJ, Sidhu A. Dopamine promotes striatal neuronal apoptotic death via ERK signaling cascades. Eur J Neurosci. 2009;29:287–306. doi: 10.1111/j.1460-9568.2008.06590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pettersson F, Couture MC, Hanna N, Miller WH. Enhanced retinoid-induced apoptosis of MDA-MB-231 breast cancer cells by PKC inhibitors involves activation of ERK. Oncogene. 2004;23:7053–66. doi: 10.1038/sj.onc.1207956. [DOI] [PubMed] [Google Scholar]
- 44.Zhang XF, Wang JF, Matczak E, Proper JA, Groopman JE. Janus kinase 2 is involved in stromal cell-derived factor-1alpha-induced tyrosine phosphorylation of focal adhesion proteins and migration of hematopoietic progenitor cells. Blood. 2001;97:3342–8. doi: 10.1182/blood.V97.11.3342. [DOI] [PubMed] [Google Scholar]
- 45.Michaelis M, Michaelis UR, Fleming I, Suhan T, Cinatl J, Blaheta RA, et al. Valproic acid inhibits angiogenesis in vitro and in vivo. Mol Pharmacol. 2004;65:520–7. doi: 10.1124/mol.65.3.520. [DOI] [PubMed] [Google Scholar]
- 46.Takada Y, Gillenwater A, Ichikawa H, Aggarwal BB. Suberoylanilide hydroxamic acid potentiates apoptosis, inhibits invasion and abolishes osteoclastogenesis by suppressing nuclear factor-kappaB activation. J Biol Chem. 2006;281:5612–22. doi: 10.1074/jbc.M507213200. [DOI] [PubMed] [Google Scholar]
- 47.Liu LT, Chang HC, Chiang LC, Hung WC. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Res. 2003;63:3069–72. [PubMed] [Google Scholar]
- 48.Matsumoto Y, Motoki T, Kubota S, Takigawa M, Tsubouchi H, Gohda E. Inhibition of tumor-stromal interaction through HGF/Met signaling by valproic acid. Biochem Biophys Res Commun. 2008;366:110–6. doi: 10.1016/j.bbrc.2007.11.089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.