

SUMMARY
Genotype-phenotype mapping is hampered by countless genomic changes between species. We introduce a computational “forward genomics” strategy that – given only an independently lost phenotype and whole genomes – matches genomic and phenotypic loss patterns to associate specific genomic regions with this phenotype. We conducted genome-wide screens for two metabolic phenotypes. First, our approach correctly matches the inactivated Gulo gene exactly with the species that lost the ability to synthesize vitamin C. Second, we attribute naturally low biliary phospholipid levels in guinea pigs and horses to the inactivated phospholipid transporter Abcb4. Human ABCB4 mutations also result in low phospholipid levels, but lead to severe liver disease, suggesting compensatory mechanisms in guinea pig and horse. Our simulation studies, counts of independent changes in existing phenotype surveys and the forthcoming availability of many new genomes, all suggest that forward genomics can be applied to many phenotypes, including those relevant for human evolution and disease.
INTRODUCTION
Despite a wealth of information obtained by comparative genomics (Green et al., 2010; McLean et al., 2011; Pollard et al., 2006; Prabhakar et al., 2006; Zhu et al., 2007), it remains extremely difficult to link any of the millions of genomic changes with the many interesting and important phenotypic differences found between even closely related species (Cheng et al., 2005; Varki and Altheide, 2005). To overcome this difficulty, we devise a method that takes advantage of parallel evolution of phenotypic traits across a larger phylogeny. Extant species that preserve an ancestral trait should also conserve the ancestral genes or – more general, genomic regions – underlying this trait due to purifying selection (Figure 1A). In contrast, in species that lose this trait, non-pleiotropic genomic regions necessary only for the lost trait should switch to evolve neutrally following the phenotypic loss. If sufficient evolutionary time has passed since trait loss, relaxed selection will erode these DNA sequences in trait-loss lineages, resulting in elevated divergence from the ancestral sequence or even complete loss. If trait loss happens in independent lineages, these regions should specifically diverge in exactly those lineages over time, regardless of whether the initial inactivating mutations leading to trait loss are identical in the independent lineages (Figure 1, and exemplified below). Thus, independent trait loss should give a unique evolutionary sequence divergence signature that violates the expected evolutionary pattern multiple times.
Figure 1. Evolutionary model and assumptions behind our forward genomics approach.
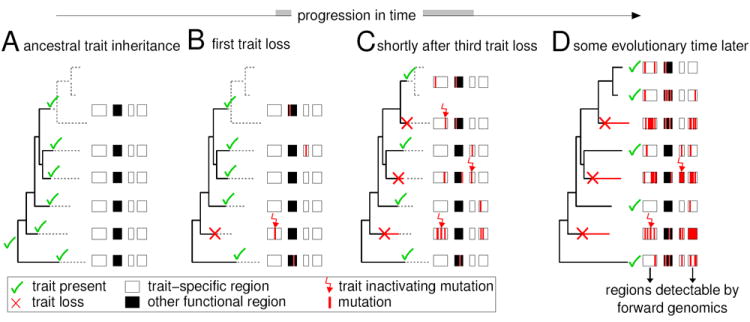
(A) An ancestral trait is passed to descendant species, along with the genomic regions required for this trait, which evolve under purifying selection.
(B) One lineage loses the ancestral trait due to an inactivating mutation in a trait-required region.
(C) Following trait loss, all trait-specific (non-pleiotropic) regions switch to evolve neutrally and begin to accumulate random mutations in the first trait-loss lineage. Meanwhile, two additional independent lineages lose this trait, due to independent mutations occurring either in the same or in other trait-required regions.
(D) All trait-specific regions continue to erode independently in the three different trait-loss lineages, while their counterparts in the trait-preserving species are conserved due to purifying selection. This characteristic evolutionary signature can be detected using forward genomics, revealing functional components of this (monogenic or polygenic) trait.
Together with approaches such as genome-wide association studies (GWAS), our strategy is conceptually similar to forward genetics, where one starts with a given phenotype and seeks the underlying mutation. We call this family of approaches “forward genomics”. With two examples and simulation studies, we show that – given a specific independently lost phenotype – a cross-species genomic screen for genes and genomic regions with this specific signature can highlight functional components of the genetic information for this trait.
RESULTS AND DISCUSSION
Measuring sequence divergence
To measure sequence divergence, we first reconstruct the most likely DNA sequence of the boreoeutherian ancestor (Figure 2C) separately for each mammalian conserved (coding or non-coding) region, with an estimated 98% accuracy (Blanchette et al., 2004). Then, we compute the percent of identical bases preserved by each extant species, while taking great care to distinguish assembly gaps (missing data) and low quality sequence from real mutations. For coding genes, we combine DNA sequence divergence of all coding exons. We then screen for genes or individual regions where all trait-loss species have diverged more from the ancestral sequence (lower percent of identical bases) than all trait-preserving species.
Figure 2. A forward genomics screen to match an ancestral presence/absence trait pinpoints Gulo inactivation in vitamin C non-synthesizing species.
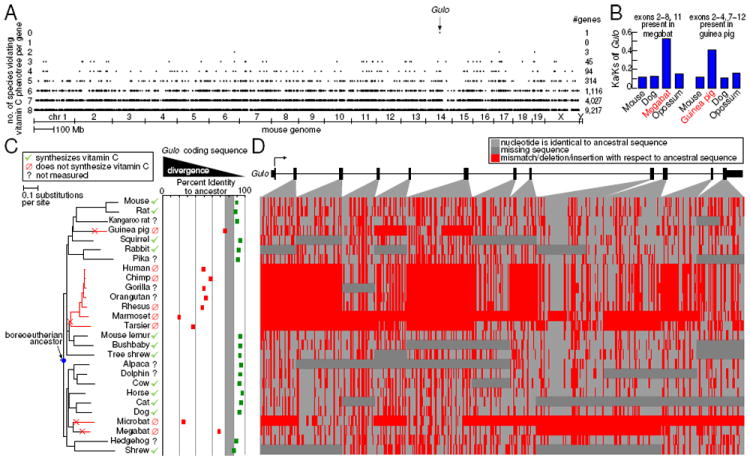
(A) For every gene (dot) in the mouse genome (x-axis) we measured how well it matches the given phenotree by counting the number of species (y-axis) whose divergence level violates the expectation of divergence or conservation based on the vitamin C phenotree shown in panel C. Gulo, with 0 violations, is the only gene that perfectly matches.
(B) Elevated ratio of non-synonymous to synonymous (Ka/Ks) substitutions show that remaining megabat and guinea pig exons evolve under relaxed pressure to preserve the Gulo protein sequence.
(C) Non-synthesizing species show elevated sequence divergence in the Gulo coding sequence, with a divergence margin (grey) that perfectly separates them from synthesizing species. Note that the microbat and megabat lineage have independently lost this trait since intermediate bat species (without a sequenced genome) were biochemically shown to synthesize vitamin C (Cui et al., 2011a).
(D) Graphical sequence alignment of the Gulo coding region. Rows match species in panel C. Large deletions (red blocks) occurred only in non-synthesizing species.
See also Figures S1 and S2.
Gulo loss perfectly matches the loss of vitamin C synthesis
The ability to synthesize the essential nutrient L-ascorbic acid (vitamin C) is an ancestral trait that arose in vertebrates. Using a biochemical assay, it has been found that – unlike most mammals – guinea pigs, certain primates including humans, and multiple bat species have lost the ability to synthesize vitamin C (Linster and Van Schaftingen, 2007). These non-synthesizing species require a dietary source of vitamin C to prevent scurvy, a disease in which defective collagen prevents the formation of strong connective tissue. Mapping the presence/absence of this phenotype to the phylogenetic tree of sequenced mammals (termed “phenotree” hereafter) shows that this trait was lost at least four times independently (Figure 2C).
We asked whether forward genomics can discover the underlying cause for loss of vitamin C synthesis, given only this phenotree and a whole genome alignment of species for which vitamin C-synthesizing ability was biochemically measured (Extended Experimental Procedures). This genomic screen finds Gulo (gulonolactone (L-) oxidase) as the only gene that perfectly matches our phenotree with substantially more divergence in all non-synthesizers (Figure 2). The detection of Gulo is extremely robust to details of our forward genomics screen. Gulo is repeatedly singled out when screening individual exons instead of genes, even when doing a genome-wide screen of 544,549 individual conserved coding and non-coding regions, when reconstructing to different ancestors, and also when measuring divergence after correcting for the different evolutionary rates of different species (Figure S1).
Gulo encodes a key enzyme responsible for vitamin C synthesis. A targeted Gulo knockout in mouse abolishes the ability to synthesize vitamin C (Maeda et al., 2000). Inactivating mutations in the Gulo gene have been identified in several non-synthesizing species (Cui et al., 2011b; Nishikimi et al., 1994; Nishikimi et al., 1992; Ohta and Nishikimi, 1999). We find Gulo inactivation in additional species (Figure S2) and show that Gulo is inactivated in all and only the sequenced non-synthesizers. Other sequenced mammals not measured for their ability to synthesize vitamin C (kangaroo rat, pika, alpaca, dolphin and hedgehog) all lack Gulo-inactivating mutations, suggesting trait retention. While Gulo is a clear candidate gene for this trait, forward genomics can detect it based on the vitamin C phenotree and genome alignments alone. Gulo also exemplifies that forward genomics does not require that the initial inactivating mutations leading to trait loss are identical in different lineages (Figure 1), as no single exon exhibits an inactivating mutation in all four non-synthesizing lineages (Figure S2A), suggesting independent gene inactivation at different genomic regions.
Abcb4 loss perfectly matches low biliary phospholipid levels
We next tested if our forward genomics approach can be applied to continuous traits that vary in magnitude between species. The composition of bile has been studied in many animals, due to its relevance in digestion, cholesterol metabolism and gall stone formation. The level of biliary phospholipids, which protect against bile acid induced cell damage (Puglielli et al., 1994), varies in mammals (Figure 3A). While most measured mammals have levels well above 1mM, including humans at 2.8mM, biliary phospholipid levels are particularly low in guinea pig (0.11mM) and horse (0.38mM) (Coleman et al., 1979; Engelking et al., 1989). We converted the continuous trait into a presence/absence trait by postulating that a subset of species with the lowest phospholipid levels may share a common associated genomic loss signature. Screening only the 11 genomes of species with measured phospholipid levels (Extended Experimental Procedures), there are 796 genes that are most diverged in guinea pig alone (our lowest value); a list too long to analyze. However, when grouping guinea pig and horse (next lowest phospholipid level), only eight genes are more diverged in these two independent lineages (Figure 3A-B). Of these eight genes, only Abcb4 has a bile-related function (Figure 3C). Abcb4 is also the strongest candidate gene when screening at the exon level (Figure S3).
Figure 3. Forward genomics implicates independent inactivation of the human disease gene ABCB4 in two species with low levels of biliary phospholipids.
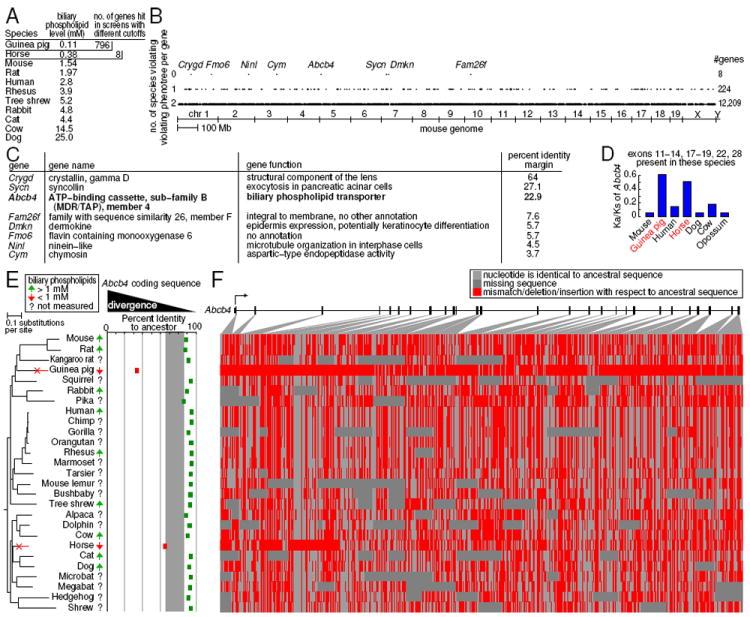
(A) The level of biliary phospholipids is a continuous trait that varies over 200 fold between mammals. 796 genes show more divergence in guinea pig than ten other measured species, but only 8 genes show elevated divergence in both guinea pig and horse, the two species with the lowest biliary phospholipid levels.
(B) We plot (y-axis) the number of violations of each gene (dot) in the mouse genome (x-axis), against the biliary phospholipid level phenotree in panel E. The eight genes with 0 violations are labeled.
(C) Of the eight genes, only Abcb4 (bold) has a bile-related function.
(D) Increased Abcb4 non-synonymous to synonymous (Ka/Ks) substitution ratios for guinea pig and horse.
(E,F) Divergence from the reconstructed common ancestor (E) and a graphical sequence alignment representation (F) of the Abcb4 coding sequence reveals elevated divergence and deletions (red blocks) in trait-loss species only.
See also Figure S3 and Table S1.
Abcb4 encodes the multidrug resistance 3 P-glycoprotein, a transporter that is crucial for phospholipid secretion into bile. Interestingly, ABCB4 mutations in humans lead to biliary phospholipid levels similar to guinea pigs (0.05 - 0.13mM), resulting in gall stone formation and bile canaliculi damage that can often only be treated by liver transplantation (Davit-Spraul et al., 2010). Targeted Abcb4 knockout mice have undetectable biliary phospholipid levels (Smit et al., 1993), and are used as models for the human disease state. Our screen is the first to discover numerous Abcb4 gene-inactivating mutations, as well as relaxed selection on the defunct Abcb4 protein sequence in naturally occurring species (Figure 3, Table S1). The discovery that two well-studied mammals have naturally inactivated Abcb4 but avoid deleterious effects, suggests other genomic changes in guinea pigs and horses presumably compensate for deleterious consequences of Abcb4 loss. While horse bile is not well characterized, guinea pig bile differs from the bile of other rodents by being very dilute and highly concentrated in the less hydrophobic ursodeoxycholic acid (Hofmann et al., 2010). Further studies into nature’s own compensatory mechanisms may lead to new therapeutic targets and strategies for ameliorating the consequences of Abcb4 mutations in humans.
Simulating trait loss suggests broad forward genomics applicability
To further test if neutral evolution following independent trait loss results in an evolutionary signature that can be detected by our forward genomics approach, we simulated the independent loss of the vitamin C and biliary phospholipid traits. Using the real mammalian phylogeny, we computationally evolved a short ancestral genome containing 103,088 coding and non-coding regions. Following the assumptions in Figure 1, we simulated trait loss by evolving the 11 (27) exons of Gulo (Abcb4) neutrally in the independent trait-loss species, while evolving other regions under purifying selection. We then tested whether forward genomics can find some of the 11 (27) exons of Gulo (Abcb4) among the >100,000 other regions, based only on their specific divergence signature. We found that nearly all simulation runs detect Gulo/Abcb4 exons and that these genes most often are the only/strongest hit (Figure 4A-C). The simulated data offers support that the specific evolutionary signature of independent trait loss can be used to identify the genes for both traits by highlighting some of their exons.
Figure 4. Broad applicability of our forward genomics approach.
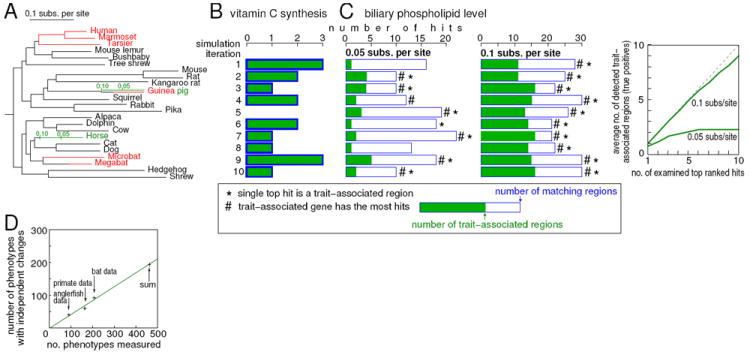
(A) We show the branches in the phylogeny that evolve neutrally for the trait-associated gene in the trait-loss simulation (red: vitamin C synthesis; green: biliary phospholipids). For biliary phospholipids, we simulated a loss that happened either 0.05 or 0.1 substitutions per site ago.
(B) Simulations suggest that the evolutionary signature of independent loss of vitamin C synthesis can highlight exons of the trait-associated gene in nine of ten iterations (iteration 5 gave no hit). We observed no false positives.
(C) Simulations of the biliary phospholipid trait show that in at least seven of ten iterations the single top-ranked hit is an exon of the trait-associated gene (shown as *) and that the trait-associated gene has often the most hits (shown as #) while false positives are scattered across the genome. The chart (right) shows that true positives (green) usually rank highly.
(D) In three very different vertebrate phenotype scoring studies, an average of 42% of phenotypes have changes in two or more independent lineages, the conditions required for forward genomics analysis.
See also Figure S4.
We further used the simulation framework to test other trait-loss scenarios. We found that performance depends on the evolutionary time since trait loss, the evolutionary rate of change of individual species, the number of independent trait losses, and the strength of purifying selection in trait-preserving species (Figure S4A-D). Importantly, despite these dependencies, forward genomics can detect some genetic trait components for a wide range of plausible trait loss scenarios, with the exception of very recent losses.
Many phenotypes are changed in independent lineages
Our forward genomics approach is constrained by the existence and accessibility of independently lost traits. Fortunately, the scientific community has been scoring traits since the days of Linnaeus (Linnaeus, 1758), with patterns of trait evolution collected into large databases such as MorphoBank (www.morphobank.org). We analyzed three vertebrate phenotype sets (selected for availability of a corresponding molecular phylogeny and for addressing distinct vertebrate clades), measuring 207, 166, and 88 different traits in ~20 species of bats, primates and anglerfish, respectively. By mapping traits to the phylogeny, we find that ~40% of scored traits show changes in independent lineages, consistent across all three sets (Figure 4D). In all cases, the sampled species make only a fraction of their clade. As expected, independently changed traits should increase as more and more species are sampled (Figure S4E). Few of the species sampled in these phenotype sets are currently sequenced but many of them are targeted for genome sequencing by the Genome10K project (Haussler et al., 2009). As these and additional genomes become available, hundreds of phenotypes can be subjected to our approach.
By requiring a longer evolutionary timescale and focusing on between-species differences, our method complements approaches to map phenotypic differences between individuals of a population such as GWAS. The biliary phospholipid example suggests potential applicability to continuous traits by testing different thresholds, to traits with changes in only two independent lineages, and to traits where phenotypic information is available only for a few of the sequenced species. Our forward genomics approach relies only on ancestral DNA sequence information loss and can therefore detect both coding and non-coding regions (Figure S4). Pleiotropic regions necessary for both the lost and unrelated traits may only experience a relaxation of selective pressure, making it harder to detect them. However, while the degree of pleiotropy in our genome is still not completely known, recent work provides evidence that many genes and quantitative trait loci (QTLs) affect only a very small number of traits (Wagner and Zhang, 2011), and regulatory non-coding regions are thought to be less-pleiotropic than the genes they regulate (Carroll, 2008). Also, future screens could improve on detecting the more subtle signatures of relaxed selection. While forward genomics may not be successful for all trait losses, the availability of thousands of vertebrate and arthropod genomes in the coming years (Haussler et al., 2009; Robinson et al., 2011) suggests applicability to many additional traits. Leveraging the unique signature of independently evolved phenotypic patterns can help to tie the study of nature’s tremendous phenotypic diversity to its underlying genomic basis. To this end, we provide a web portal at http://phenotree.stanford.edu/ to allow users to run any phenotree search against our data. The portal provides a visualization and download of the results and links to the UCSC genome browser. Finally, it is not straightforward to distinguish real mutations from genomic artifacts. Most current gene family collections focus only on collecting intact members by mapping gene structures across species. When mapping fails, no call is made to distinguish between true gene loss and an unresolved state due to low quality or missing sequence data. A systematic search for genes lost in wildtype species will likely suggest new organisms useful for studying the phenotypic consequences of human disease mutations.
EXPERIMENTAL PROCEDURES
Alignments and input data
We extended the mouse (mm9 assembly) 30-way genome alignment provided by the University of California, Santa Cruz (UCSC) genome browser (Dreszer et al., 2012) by 13 species, using the UCSC pipeline of lastz, chaining, netting and multiz (Extended Experimental Procedures). The alignment is available through our portal. To obtain conserved regions, we downloaded PhastCons (Siepel et al., 2005) most-conserved elements from the UCSC genome browser. Elements within 30 bp of each other were merged. We retained only elements ≥70 bp after merging. Exons of “known genes” were downloaded from the UCSC genome browser. We always excluded the mitochondrial, random and haplotype chromosomes.
Ancestral sequence reconstruction and percent identity (%id) values
Given the multiple alignment of a conserved region with conservation in at least one outgroup species (elephant, rock hyrax, tenrec, armadillo, sloth, opossum, platypus, chicken, zebra finch or lizard), we used prequel (Siepel et al., 2005) (parameters --no-probs --keep-gaps) to reconstruct the ancestral sequences.
We define the percent of identical bases in the pairwise alignment between ancestor and extant species as id / (id + subs + ins + del) * 100, where id, subs, ins and del is the number of identical bases, substitutions, inserted bases and deleted bases, respectively. We ignore low quality base positions (> 1% error rate). We consider the following large-scale events: (i) Large lineage-specific insertions were counted as inserted bases. (ii) Parts of the conserved region that are un-alignable between ancestor and extant species (defined as being so diverged that no sequence alignment can be computed; un-alignable regions are annotated in the multiple alignment) were added to the number of substitutions. Specifically, we add the number of ancestral bases corresponding to this un-alignable region if the un-alignable part is at the end or we add the maximum of the number of ancestral bases and the number of extant bases in this region if the un-alignable part is flanked by aligning blocks on both sides. Un-alignable regions that map to an assembly gap in this species were ignored. If the entire conserved region is un-alignable or deleted, the %id is set to 0. (iii) If the aligning sequence is not co-linear in the extant species (different strands or different chromosomes/scaffolds), we do not compute a %id value for this species because these cases frequently arise due to incomplete genome assemblies or assembly artifacts. Percent identity ranges from 0 (complete loss) to 100 (identity to the ancestor). For gene-based screens, we computed a %id value for the entire coding region of a gene by summing the number of matches for each coding exon and dividing that number by the summed alignment lengths.
Screening for conserved regions that evolve faster in species lacking a trait
We search for regions/genes where the %id values in all trait-loss species are at least 1% lower than in all trait-preserving species. We ignored species with missing data (no %id value). We excluded regions that have missing data for many species. To get a quantitative measurement of how well a region/gene matches the phenotree, we computed the minimal number of “violating” trait-loss or trait-preserving species whose %id values have to be removed to get a 1% separation. The maximum number of violating species is the number of trait-loss species for both vitamin C and biliary phospholipids, since there are more trait-preserving than trait-loss species.
Bioinformatics analysis of the Gulo locus and re-sequencing
Standard bioinformatics analysis of the Gulo gene and its genomic locus as well as standard PCR-based re-sequencing is described in Extended Experimental Procedures.
Simulating independent trait loss
To evolve a given ancestral genome, we used Evolver (http://www.drive5.com/evolver/) with standard parameters (Extended Experimental Procedures) and the real mammalian phylogeny. To simulate independent trait loss, we randomly picked a small number of functional regions, and assumed that they encode trait-specific information (true positives). All other regions are false positives. We evolved these regions neutrally for a fixed terminal part of the branch leading to a trait-loss lineage.
We restricted the ancestral genome simulation to human chromosome 1 with its 1,603 RefSeq genes (downloaded from UCSC) as this already consumed ~50,000 CPU hours and 1.6 TB of disk space. We considered all coding exons and functional non-coding regions ≥ 70 bp that have an average accept probability of ≤ 0.7 (i.e. we ignore regions under very weak selection), giving a total of 103,088 regions. The selected trait-specific regions (≤27 in all tests) comprise only 0.026% of all regions. Evolver outputs the pairwise alignment of the evolved and ancestral genome, which we use to measure %id for all functional regions. As above, we used our approach to find regions where all trait-loss species are ≥1% more diverged than all trait-preserving species.
To simulate the loss of vitamin C synthesis, we picked a gene with 11 coding exons (like Gulo; 11 true positives). True positives evolved neutrally for the branches in the human-tarsier subtree, the final 0.07 substitutions per site of the guinea pig branch, the microbat branch and the final 0.045 substitutions per site of the megabat branch (Extended Experimental Procedures). To simulate the biliary phospholipid trait, we picked a gene with 27 coding exons (like Abcb4) and simulated two scenarios: trait loss 0.05 and 0.1 substitutions per site ago.
Supplementary Material
HIGHLIGHTS.
matching independent phenotypic losses with ancestral genomic information erosion
Gulo gene loss uniquely matches to “loss of vitamin C synthesis” in mammals
Abcb4 gene loss matches “low biliary phospholipid levels” in guinea pig & horse
broad applicability of the approach from simulation and phenotype measurements
Acknowledgments
We thank the Mammalian Genome Project for available mammalian genomes, the Broad Institute for the microbat assembly, David Ray for microbat tissue, Julie Feinstein and the Ambrose Monell Cryo Collection (AMCC) at the American Museum of Natural History for megabat tissue, Mark Batzer for advice, the UC Santa Cruz genome browser team for software and genome annotations, Hiram Clawson and Brian Raney for help with sequence quality scores and alignment annotations, Karla Neugebauer and members of the Bejerano lab for helpful discussions, and Ravi Parikh and Harendra Guturu for our web portal
This work was supported by fellowships from the German Research Foundation (Hi 1423/2-1) and Human Frontier Science Program (LT000896/2009-L) to MH; and NIH grants R01HD059862 and R01HG005058 and the NSF Center for Science of Information (CSoI) under grant agreement CCF-0939370 to GB. DMK is an investigator of the Howard Hughes Medical Institute. GB is a Packard Fellow and Microsoft Faculty Fellow.
Footnotes
Accession Numbers
The GenBank accession numbers for the Gulo re-sequencing data are JX259503-JX259509.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blanchette M, Green ED, Miller W, Haussler D. Reconstructing large regions of an ancestral mammalian genome in silico. Genome Res. 2004;14:2412–2423. doi: 10.1101/gr.2800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell. 2008;134:25–36. doi: 10.1016/j.cell.2008.06.030. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Ventura M, She X, Khaitovich P, Graves T, Osoegawa K, Church D, DeJong P, Wilson RK, Paabo S, et al. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature. 2005;437:88–93. doi: 10.1038/nature04000. [DOI] [PubMed] [Google Scholar]
- Coleman R, Iqbal S, Godfrey PP, Billington D. Membranes and bile formation. Composition of several mammalian biles and their membrane-damaging properties. Biochem J. 1979;178:201–208. doi: 10.1042/bj1780201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Pan YH, Zhang Y, Jones G, Zhang S. Progressive pseudogenization: vitamin C synthesis and its loss in bats. Mol Biol Evol. 2011a;28:1025–1031. doi: 10.1093/molbev/msq286. [DOI] [PubMed] [Google Scholar]
- Cui J, Yuan X, Wang L, Jones G, Zhang S. Recent loss of vitamin C biosynthesis ability in bats. PLoS One. 2011b;6:e27114. doi: 10.1371/journal.pone.0027114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis. 2010;30:134–146. doi: 10.1055/s-0030-1253223. [DOI] [PubMed] [Google Scholar]
- Dreszer TR, Karolchik D, Zweig AS, Hinrichs AS, Raney BJ, Kuhn RM, Meyer LR, Wong M, Sloan CA, Rosenbloom KR, et al. The UCSC Genome Browser database: extensions and updates 2011. Nucleic acids research. 2012;40:D918–923. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelking LR, Anwer MS, Hofmann AF. Basal and bile salt-stimulated bile flow and biliary lipid excretion in ponies. Am J Vet Res. 1989;50:578–582. [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussler D, O’Brien S, Ryder O, Barker F, Clamp M, Crawford A, Hanner R, Hanotte O, Johnson W, McGuire J, et al. Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J Hered. 2009;100:659–674. doi: 10.1093/jhered/esp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann AF, Hagey LR, Krasowski MD. Bile salts of vertebrates: structural variation and possible evolutionary significance. J Lipid Res. 2010;51:226–246. doi: 10.1194/jlr.R000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnaeus C. Systema naturae. Sumptibus Guiliemi Engelmann; 1758. [Google Scholar]
- Linster CL, Van Schaftingen E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- Maeda N, Hagihara H, Nakata Y, Hiller S, Wilder J, Reddick R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc Natl Acad Sci U S A. 2000;97:841–846. doi: 10.1073/pnas.97.2.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, Indjeian VB, Lim X, Menke DB, Schaar BT, et al. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature. 2011;471:216–219. doi: 10.1038/nature09774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikimi M, Fukuyama R, Minoshima S, Shimizu N, Yagi K. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-gamma-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. J Biol Chem. 1994;269:13685–13688. [PubMed] [Google Scholar]
- Nishikimi M, Kawai T, Yagi K. Guinea pigs possess a highly mutated gene for L-gulono-gamma-lactone oxidase, the key enzyme for L-ascorbic acid biosynthesis missing in this species. J Biol Chem. 1992;267:21967–21972. [PubMed] [Google Scholar]
- Ohta Y, Nishikimi M. Random nucleotide substitutions in primate nonfunctional gene for L-gulono-gamma-lactone oxidase, the missing enzyme in L-ascorbic acid biosynthesis. Biochim Biophys Acta. 1999;1472:408–411. doi: 10.1016/s0304-4165(99)00123-3. [DOI] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- Prabhakar S, Noonan JP, Paabo S, Rubin EM. Accelerated evolution of conserved noncoding sequences in humans. Science. 2006;314:786. doi: 10.1126/science.1130738. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Amigo L, Arrese M, Nunez L, Rigotti A, Garrido J, Gonzalez S, Mingrone G, Greco AV, Accatino L, et al. Protective role of biliary cholesterol and phospholipid lamellae against bile acid-induced cell damage. Gastroenterology. 1994;107:244–254. doi: 10.1016/0016-5085(94)90083-3. [DOI] [PubMed] [Google Scholar]
- Robinson GE, Hackett KJ, Purcell-Miramontes M, Brown SJ, Evans JD, Goldsmith MR, Lawson D, Okamuro J, Robertson HM, Schneider DJ. Creating a buzz about insect genomes. Science. 2011;331:1386. doi: 10.1126/science.331.6023.1386. [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, Mol CA, Ottenhoff R, van der Lugt NM, van Roon MA, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- Varki A, Altheide TK. Comparing the human and chimpanzee genomes: searching for needles in a haystack. Genome Res. 2005;15:1746–1758. doi: 10.1101/gr.3737405. [DOI] [PubMed] [Google Scholar]
- Wagner GP, Zhang J. The pleiotropic structure of the genotype-phenotype map: the evolvability of complex organisms. Nature reviews Genetics. 2011;12:204–213. doi: 10.1038/nrg2949. [DOI] [PubMed] [Google Scholar]
- Zhu J, Sanborn JZ, Diekhans M, Lowe CB, Pringle TH, Haussler D. Comparative genomics search for losses of long-established genes on the human lineage. PLoS Comput Biol. 2007;3:e247. doi: 10.1371/journal.pcbi.0030247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.