

Abstract
The limiting factor in in vivo RNA interference (RNAi) is delivery. Drug delivery methods that are effective in cell culture may not be practical in vivo for intravenous RNAi applications. Nucleic acid drugs are highly charged and do not cross cell membranes by free diffusion. Therefore, the in vivo delivery of RNAi-therapeutics must using targeting technology that enables the RNAi therapeutic to traverse biological membrane barriers in vivo. For RNAi of the brain, the nucleic acid-based drug must first cross the brain capillary endothelial wall, which forms the blood-brain barrier (BBB) in vivo, and then traverse the brain cell plasma membrane. Similar to the delivery of non-viral gene therapies, plasmid DNA encoding for short hairpin RNA (shRNA) may be delivered to brain following intravenous administration with pegylated immunoliposomes (PILs). The plasmid DNA is encapsulated in a 100 nm liposome, which is pegylated, and conjugated with receptor specific targeting monoclonal antibodies (MAb). Weekly, intravenous RNAi with PILs enables a 90% knockdown of the human epidermal growth factor receptor, which results in a 90% increase in survival time in mice with intra-cranial brain cancer. Similar to the delivery of antisense agents, short interfering RNAi (siRNA) duplexes can be delivered with the combined use of targeting MAb’s and avidin-biotin technology. The siRNA is mono-biotinylated in parallel with the production of a conjugate of the targeting MAb and streptavidin. Intravenous RNAi requires the combined use of RNAi technology and a drug targeting technology that is effective in vivo.
Keywords: blood-brain barrier, RNAi, endothelium, transferrin receptor, insulin receptor, monoclonal antibody
1. Introduction
RNA interference (RNAi) is a new strategy for the development of antisense therapeutics that knock down gene expression post-transcriptionally [1]. RNAi mechanisms may involve either the degradation of target RNA, e.g. in the case of a short interfering RNA (siRNA), or cause translation arrest of the target RNA, e.g. in the case of micro RNA (miRNA). There are two types of RNAi-based therapeutics: DNA-based RNAi and RNA-based RNAi. In DNA-based RNAi, a plasmid DNA encodes for a short hairpin RNA (shRNA). In RNA-based RNAi, a siRNA duplex is chemically synthesized without a DNA intermediate.
Drug delivery is the rate-limiting step in the translation of RNAi based therapeutics from cell culture to in vivo therapeutics in animals and humans. Many delivery methodologies that prove effective in cell culture may be difficult to implement in vivo, because biological membrane barriers exist in vivo that are absent in a Petri dish. Consequently, the examples of therapeutic effects in vivo with “intravenous RNAi” are few compared to the biological effects of RNAi demonstrated in cell culture. The goal of intravenous RNAi is to inject the RNAi therapeutic intravenously and obtain knock down of the target gene in the target organ without toxicity.
The role of drug delivery in the development of in vivo intravenous RNAi may be summarized as follows:
The problems of shRNA delivery in vivo recapitulate the problems of gene delivery in vivo.
The problems of siRNA delivery in vivo recapitulate the problems of antisense delivery in vivo.
The size of the RNAi delivery problem is illustrated by consideration of the development of antisense and gene therapeutics, both of which are >20 years in development. Presently, there are no FDA approved gene therapies, and no FDA approved drugs that work via an antisense mechanism. Yet, both gene therapy and antisense drugs have powerful effects in cell culture. Neither gene therapy nor antisense drugs have been translated into FDA approved therapeutics, because of the rate-limiting role played by in vivo drug targeting of these highly polar, large molecular weight agents. The fields of either gene therapy or antisense failed to develop effective targeting technology that enables clinically significant therapeutic effects of these agents in vivo following intravenous administration.
Many of the present-day RNAi delivery methods simply recapitulate the delivery approaches used in the past for antisense and gene therapy. If the delivery technologies were largely ineffective in vivo for antisense delivery or gene delivery, then how can these delivery systems work for in vivo RNAi? In the absence of the development of new delivery technology for nucleic acid therapeutics, it is possible that RNAi therapeutics will follow the same development path as antisense and gene therapy.
The present chapter will review the use of molecular Trojan horses (MTH) to deliver non-viral plasmid DNA therapeutics, and antisense drugs to the brain, and other organs, in vivo following intravenous administration. The chapter will review how plasmid DNA delivery technology is adapted to the problem of the delivery of shRNA encoding plasmid DNA, and how the antisense delivery technology is adapted to the problem of siRNA delivery in vivo. However, MTHs, alone, cannot easily deliver these agents to brain and other organs in vivo. The MTHs must be combined with other formulation technologies, which enable an efficient, and metabolically stable attachment of the nucleic acid therapeutic to the MTH. Therefore, this chapter will also review the use of the Trojan horse liposome (THL) technology, also called the pegylated immunoliposome (PIL) technology, which combines plasmid DNA and MTHs in a formulation that enables transport of the DNA across biological barriers in vivo following intravenous administration. In the case of antisense, and siRNA, the chapter will review avidin-biotin (AB) technology, which combines the MTH and the antisense or siRNA in a formulation that is stable in vivo.
2. Overview of blood-brain barrier molecular Trojan horses
2.1 Blood-brain barrier receptor-mediated transport (RMT)
The blood-brain barrier (BBB) is formed by the brain capillary endothelial wall [2]. Epithelial-like, high resistance tight junctions cement brain capillary endothelial cells together, eliminating endothelial pores that exist in the capillaries perfusing non-brain tissues. Therefore, there is no para-cellular pathway of solute exchange between blood and brain. There is also minimal pinocytosis in brain capillary endothelial cells. Therefore, there is no significant trans-cellular pathway for free solute exchange between blood and brain. Consequently, there are only two mechanisms by which molecules in the blood may gain access to brain interstitial fluid: (a) lipid-mediated transport of lipid soluble small molecules with a molecular weight <400 Da; and (b) catalyzed transport [3]. Catalyzed transport includes carrier-mediated transport (CMT) for small molecules and receptor-mediated transport (RMT) for large molecules. The plasma membrane of brain capillary endothelial cells express various CMTs, such as the GLUT1 glucose transporter, the LAT1 large neutral amino acid transporter, the CAT1 cationic amino acid transporter, the MCT1 monocarboxylic acid transporter, and the CNT2 adenosine transporter [4]. In addition to catalyzed transport of endogenous small molecules via the BBB CMT systems, there is also RMT of endogenous large molecules across the BBB. The brain capillary endothelial plasma membrane expresses an insulin receptor, which triggers the RMT of circulating insulin into the brain [3]. The BBB also expresses a transferrin receptor (TfR), which triggers the RMT of circulating holo-transferrin (Tf) from blood to brain. The BBB TfR also mediates the reverse transcytosis of apo-Tf in the brain to blood direction [5].
2.2 Species specific molecular Trojan horses for brain drug delivery
A molecular Trojan horse (MTH) is an endogenous peptide or peptidomimetic monoclonal antibody (MAb) that undergoes RMT across the BBB [3]. A peptidomimetic MAb binds to an exofacial epitope on the BBB receptor, which allows the MAb to undergo RMT across the BBB without interference of BBB transport of the endogenous ligand. The peptidomimetic MAb may carry across the BBB any attached drug or even plasmid DNA via the BBB RMT system.
A panel of species-specific MAb MTHs has been developed for brain drug delivery. For drug delivery in mice, the rat 8D3 MAb to the mouse TfR is used [6]; this MTH is not active in rats. For brain drug delivery in rats, the murine OX26 MAb to the rat TfR is used [7]; this MTH is not active in mice or other species. For brain drug delivery to Old World primates such as the Rhesus monkey, the murine 83-14 MAb to the human insulin receptor (HIR) is used [8]. The HIRMAb is not active in New World primates such as squirrel monkey. The murine HIRMAb cannot be used in humans owing to immunologic reactions to a mouse protein in humans. Therefore, genetically engineered forms of the HIRMAb, both a chimeric HIRMAb and a humanized HIRMAb, have been produced to enable future brain drug delivery in humans [9].
2.3 Brain delivery of protein therapeutics with molecular Trojan horses
The delivery of protein therapeutics to brain following intravenous administration with molecular Trojan horses has been reduced to pharmacologic practice in vivo for a number of experimental systems. The intravenous administration of vasoactive intestinal peptide (VIP) caused a 65% increase in hemispheric brain blood flow in conscious rats, providing the VIP was conjugated to a BBB MTH [10]. VIP alone had no effect on cerebral blood flow following intravenous administration because VIP does not cross the BBB. Brain derived neurotrophic factor (BDNF) caused 100% normalization of pyramidal neuron density in the CA1 sector of the hippocampus in rats subjected to transient forebrain ischemia, providing the BDNF was conjugated to a BBB molecular Trojan horse [11]. BDNF alone had no neuroprotective effect in brain ischemia following intravenous administration, because BDNF does not cross the BBB, and because the BBB is intact in the initial phases following brain ischemia when neuroprotection is still possible. The delayed intravenous administration of BDNF resulted in a 65–70% reduction in infarct volume in either permanent or reversible middle cerebral artery occlusion (MCAO), providing the BDNF was conjugated to a BBB molecular Trojan horse [12, 13]. BDNF alone had no effect on stroke volume in regional brain ischemia because BDNF does not cross the BBB and because the BBB is intact in the period following regional ischemia when neuroprotection is still possible. Fibroblast growth factor (FGF)-2 resulted in 80% reduction in stroke volume in permanent MCAO, providing the FGF-2 was conjugated to a BBB molecular Trojan horse [14]. FGF-2 alone had no effect on stroke volume because FGF-2 does not cross the BBB. An epidermal growth factor (EGF) peptide radiopharmaceutical enabled the detection of intra-cranial brain cancer following intravenous administration providing the EGF was conjugated to a BBB molecular Trojan horse [15]. This imaging allowed for the detection of the over-expression of the EGF receptor (EGFR) in brain cancer. The intravenous administration of the EGF alone resulted in no detection of intra-cranial brain cancer, because the EGF does not cross the BBB and the BBB is intact in brain cancer until the very latest stages. A model lysosomal enzyme, β-galactosidase, was delivered to brain in vivo following intravenous administration providing the β-galactosidase was conjugated to a BBB molecular Trojan horse [16]. The intravenous administration of β-galactosidase alone resulted in no increase enzyme activity in the brain because the enzyme alone does not cross the BBB.
These studies of recombinant protein delivery to brain in vivo with intravenous administration demonstrate that BBB MTHs can deliver large molecule therapeutics into brain in vivo. In future clinical applications in humans, it will possible to deliver recombinant protein therapeutics to the human brain, following the administration of fusion proteins whereby the therapeutic protein is fused to the genetically engineered molecular Trojan horse. Following the genetic engineering of fusion genes and expression in host cells, it will possible for large scale manufacturing of the fusion proteins as protein based neurotherapeutics in humans.
3. Delivery of non-viral genes and shRNA plasmid DNA to brain with Trojan horse liposomes
3.1 Formulation of Trojan horse liposomes
In order to produce a formulation whereby the MTH was associated with non-viral plasmid DNA in a way that would be stable in vivo, the Trojan horse liposome (THL) was developed [17]. As shown in Fig. 1A, a single plasmid DNA molecule is encapsulated in the interior of a 100nm liposome. The surface of the liposome was conjugated with several thousand strands of a polymer such as 2000 Da polyethylene glycol (PEG). The tips of 1–2% of the PEG strands are conjugated with a receptor (R)-specific MAb that acts as the MTH. This results in the formulation of a pegylated immunoliposome (PIL) encapsulating the plasmid DNA. The targeting MAb binds to the BBB receptor to trigger RMT from blood to the brain interstitial fluid. Subsequently, the targeting MAb binds the same receptor on brain cells to trigger receptor-mediated endocytosis into brain intracellular spaces. In the case of the insulin receptor, which normally delivers its endogenous ligand, insulin, to the nuclear compartment [18], the insulin receptor delivers the plasmid DNA to the nucleus of the brain cell, which is followed by expression of the endogenous transgene [19]. An electron micrograph of a Trojan horse liposome is shown in Fig. 1B. In this study [20], the targeting MAb was labeled with a secondary antibody that was conjugated to the 10 nm gold particles that are visible in Fig. 1B. The 10 nm gold particle is approximately the same size as an antibody molecule. This electron micrograph shows the spatial relationship of the targeting MAb with the surface of a liposome. In a typical formulation, there are 30 to 80 MAb molecules conjugated to an individual liposome [21]. Any DNA that is not fully encapsulated in the interior of the liposome is exhaustively removed by nuclease treatment. It is important to remove an exteriorized DNA from the formulation, as free DNA is pro-inflammatory in vivo [22–24].
Figure 1.
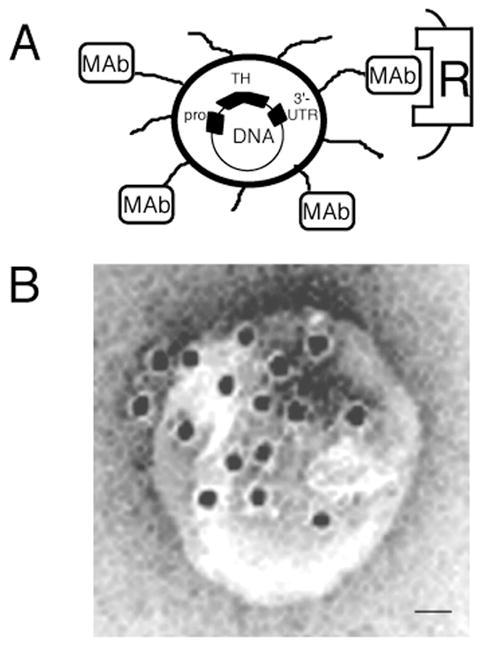
(A) Diagram of a super-coiled expression plasmid DNA encapsulated in an 85 nm pegylated immunoliposome targeted to a cell membrane receptor (R) with a receptor-specific, endocytosing monoclonal antibody (MAb). Tissue-specific expression of the plasmid is regulated by the promoter (pro), which is inserted 5′ of the gene. (B) Transmission electron microscopy of a PIL. The MAb molecule tethered to the tips of the 2000-Dalton polyethylene glycol (PEG) is bound by a conjugate of 10 nm gold and a secondary antibody. The position of the gold particles shows the relationship of the PEG extended MAb and the liposome. Magnification bar = 20 nm. From [20].
3.2 Brain delivery of reporter genes with Trojan horse liposomes
Luciferase and β-galactosidase reporter genes have been delivered to brain with Trojan horse liposomes [25]. A β-galactosidase expression plasmid was encapsulated inside Trojan horse liposomes that were targeted to mouse brain with the rat 8D3 MAb to the mouse TfR. These Trojan horse liposomes were injected intravenously into adult mice at a dose of 5 μg plasmid DNA per mouse in a volume of 0.2 ml. At 48 hours after the single intravenous injection of the Trojan horse liposome, the animals were sacrificed, the brain was removed, and coronal sections stained histochemically for β-galactosidase gene expression. As shown in Fig. 2A, there is global expression of the transgene throughout the brain following an intravenous administration of this non-viral formulation. The pattern of expression parallels the expression of the neuronal transferrin receptor, which is ubiquitous in brain. The transgene is expressed in both cortical and subcortical structures, in choroid plexus, in hippocampus, the midbrain, spinal cord, and is highly expressed in the Purkinje cell layer of the cerebellum (Fig. 2A). The β-galactosidase transgene is also globally expressed in the retinal pigmented epithelium (RPE) layer of the mouse retina (Fig. 2B). There is also gene expression in the ciliary body and iris. There is no transgene expression in the outer nuclear layer owing to the minimal expression of the TfR on the cell bodies in the photo-receptor cells [26]. However, these structures express insulin receptor and when Trojan horse liposomes are targeted to the primate eye with the HIRMAb, there is abundant expression of the transgene in the outer nuclear layer and inner segments of the photo-receptor cells of the primate retina [27].
Figure 2.

(A, B) β-Galactosidase histochemistry of brain (A) and eye (B) of the adult mouse removed 48 hours after the intravenous (IV) injection of a β-galactosidase expression plasmid encapsulated in PILs targeted with the 8D3 rat MAb to the mouse TfR. (C, D) β-Galactosidase histochemistry of brain (C) and peripheral organs (D) of the adult Rhesus monkey removed 48 hours after the IV injection of a β-galactosidase expression plasmid encapsulated in PILs targeted with the 83-14 murine MAb to the HIR. There is no gene expression in heart, skeletal muscle, or fat. (E) Tyrosine hydroxylase (TH) immunocytochemistry (left panels) or confocal microscopy (right panels) of rat brain removed 48 hours after the IV injection of a TH expression plasmid encapsulated in PILs targeted with either the OX26 mouse MAb to the rat TfR (top panels) or a mouse IgG2a isotype control antibody (bottom panels). Three weeks prior to gene administration, the rats were injected with 6-hydroxydopamine into the medial forebrain bundle on the right side, which cause a complete loss of immunoreactive TH in the striatum ipsilateral to the neurotoxin lesion. Gene therapy with PILs targeted with the TfR MAb causes a complete normalization of striatal TH (top panels). There is no restoration of striatal TH if the TH expression plasmid is encapsulated in PILs targeted with an isotype control antibody that does not target a BBB receptor (bottom panels). None of the sections in panels A, C, or D is counter-stained. Panel A from [54]; Panel B from [26]; Panels C–D from [28]; Panel E from Zhang et al [30].
Gene expression in the primate brain was observed following an intravenous administration of an expression plasmid, encoding either β-galactosidase or luciferase, which was encapsulated in Trojan horse liposomes targeted with the HIRMAb [28]. There is global expression of the transgene throughout the entire Rhesus monkey brain at 48 hours after single intravenous administration of this non-viral formulation (Fig. 2C). Confocal microscopy showed that the transgene was expressed in neurons in the primate brain [28].
There is no other delivery technology that enables global expression of a transgene in brain, such as that demonstrated in Fig. 2. The Trojan horse liposome technology enables “adult transgenics” within 24 hours after intravenous administration of non-viral formulations. The gene is delivered to virtually all cells of the brain. Real time polymerase chain reaction (PCR) was used to quantitate the number of plasmid DNA molecules delivered to each brain cell of the primate brain following the intravenous administration of 65 μg of plasmid DNA encapsulated in Trojan horse liposomes to a 6 kg Rhesus monkey [29]. At this dose of 10 μg plasmid DNA/kg body weight, approximately 3–4 plasmid DNA molecules were delivered to each brain cell of the adult primate brain. Multiple copies of the mRNA are generated from each plasmid DNA, and multiple copies of the expressed protein are generated from each expressed mRNA.
3.3 Brain delivery of tyrosine hydroxylase therapeutic gene with Trojan horse liposomes
A tyrosine hydroxylase (TH) cDNA was formulated in an expression plasmid driven by the SV40 promoter [20]. The TH expression plasmid is encapsulated in Trojan horse liposomes that were targeted to rat brain with the murine OX26 MAb to the rat TfR. Alternatively, the TH expression plasmid was encapsulated in liposomes that were targeted with the mouse IgG2a isotype control. In parallel, rats were lesioned with the intracerebral injection of the neurotoxin, 6-hydroxy-dopamine, into the medium forebrain bundle. This results in a chemical lesion of the nigral-striatal dopaminergic track and experimental Parkinson’s disease (PD). There is no detectable immunoreactive TH in the striate body ipsilateral to the lesion, as shown in Fig. 2E (bottom panel). There is no restoration of immunoreactive TH in the lesioned striatum following intravenous administration of the TH expression plasmid encapsulated in liposomes that were targeted with the mouse IgG2a isotype control (Fig. 2E, bottom panel). However, there is complete restoration of striatal TH ipsilateral to the lesion following intravenous administration of the TH expression plasmid encapsulated in Trojan horse liposomes targeted with the TfR MAb (Fig. 2E, upper panel). Confocal microscopy of striatum with antibodies to either TH, or to neuN, a neuronal cell body marker, was performed [30]. Following TH gene therapy, there is abundant immunoreactive TH in the fibers of the striatum ipsilateral to the lesion (Fig. 2E, upper panel). The efficacy of the TH gene therapy in the experimental PD rats was verified by measurements of striatal TH enzyme activity and by the rotational behavior of these rats in response to intraperitoneal apomorphine [20, 30]. TH gene therapy with the Trojan horse liposomes resulted in a complete normalization of striatal TH enzyme activity ipsilateral to the lesion and resulted in 82% reduction in abnormal rotational behavior induced by apomorphine.
3.4. Trojan horse liposomes and tissue-specific promoters
The intravenous administration of β-galactosidase transgene, under the influence of the widely read SV40 promoter, in adult Rhesus monkeys results in expression of the transgene in liver or spleen but not heart, skeletal muscle, or fat [28]. The Trojan horse liposomes administered to the Rhesus monkey were targeted with the HIRMAb. Heart, skeletal muscle, and fat have high levels of insulin receptor on the parenchymal cells in these tissues. However, these organs are perfused by capillaries with continuous endothelial barriers that do not allow for significant egress of 100 nm liposomes from the circulation into the organ interstitium. Moreover, there is no insulin receptor on the endothelium of capillaries perfusing heart, skeletal muscle, or fat, as there is in brain [3] or eye [31]. The THL cannot cross the capillary barrier in these organs, and the transgene is not expressed in heart, skeletal muscle, or fat (Figure 2D). Similarly, there is no insulin receptor on the microvessels perfusing liver or spleen. However, liver and spleen are perfused by sinusoidal microcirculatory beds, which are freely porous to 100 nm liposomes [32]. Therefore, the liposome readily escapes from the plasma compartment of liver or spleen and is available to the insulin receptor on parenchymal cells in these organs. The “ectopic” expression of the transgene in peripheral tissue such as liver or spleen would be desirable in the treatment of certain conditions such as lysosomal storage disorders. However, in the case of gene therapy of conditions such as Parkinson’s disease, it may not be desirable to have ectopic expression of the transgene in non-brain organs. The elimination of ectopic gene expression is possible with the use of tissue specific promoters. If the β-galactosidase transgene is under the influence of a brain specific promoter such has the 5′-flanking sequence (FS) of the human glial fibrillary acidic protein (GFAP) gene, then expression of the transgene in peripheral tissues is eliminated [25]. Similarly, expression of the transgene in brain, liver, or spleen of Rhesus monkeys is eliminated when the transgene is under the influence of an ocular-specific promoter such as the 5′-FS of the bovine opsin gene [27]. Therefore, the combined use of tissue-specific promoters and Trojan horse liposome delivery technology allows for localization of the expression in vivo of the therapeutic gene to the specific organ or tissue type.
3.5 Intravenous RNAi: brain delivery of luciferase targeted shRNA plasmid DNA
The delivery of shRNA encoding plasmid DNA to brain was initially evaluated with a luciferase brain tumor model [33]. C6 rat glioma cells were stably transfected with a luciferase expression plasmid. These cells, designated C6-790, were stereotactically implanted in the caudate-putamen nucleus of adult Fischer CD344 rats. This resulted in the growth of an intra-cranial tumor that expressed high levels of luciferase (Fig. 3A). A plasmid DNA was then genetically engineered that encoded for an shRNA that was antisense to a specific target region of the luciferase mRNA. This shRNA expression plasmid, designated clone 952, produced a shRNA directed against the luciferase mRNA and the structure and sequence of this shRNA is shown in Fig. 3B. At 5 days after implantation of the C6-790 cells, Trojan horse liposomes carrying clone 952 plasmid DNA were injected intravenously into the tumor bearing rats and luciferase enzyme activity was measured in both tumor and the contralateral brain at 7 and 10 days following implantation of the tumor cells, which is 2 and 5 days following the single intravenous injection of the shRNA encoding plasmid DNA [33]. The data in Fig. 3C (left panel) show a 90% knockdown of luciferase gene expression in the brain tumor following intravenous administration of clone 952 plasmid DNA encapsulated in Trojan horse liposomes targeted with the TfR MAb. There is no measurable luciferase enzyme activity in contralateral brain (Fig. 3C). The anti-luciferase RNAi gene therapy had no effect on the expression of a non-target gene, γ-glutamyl transpeptidase (GTP), as shown in Fig. 3D. Owing to the expression of the TfR on both the capillaries perfusing the brain tumor and on the tumor cell membrane, the TfR MAb-targeted Trojan horse liposomes deliver the anti-luciferase encoding plasmid DNA across both the BBB and the tumor cell membrane in vivo. This study was the first demonstration of intravenous RNAi in brain.
Figure 3.

(A) Coronal section of autopsy rat brain at 14 days after implantation of C6 rat glioma cells in the caudate-putamen of adult Fischer CD344 rats weighing180–200 g. The C6 cells were permanently transfected with clone 790 plasmid DNA, and produce high levels of luciferase when grown as brain tumors in vivo [33]. (B) The nucleotide sequence and secondary structure of the anti-luciferase shRNA encoded by clone 952 is shown. From [33]. (C) Luciferase (left panel) and γ-glutamyl transpeptidase (GTP, right panel) enzyme activity in C6-790 tumor and contralateral brain in animals treated either with saline or with OX26 MAb-targeted PILs carrying clone 952 plasmid DNA. Enzyme assay measurements were performed at 5 days following the single intravenous injection of saline or RNAi therapeutic. The tumor luciferase activity was no different from the saline treated animals in C6-790 tumor bearing rats treated with OX26 MAb-targeted PILs carrying clone 959 plasmid DNA, where clone 959 is the empty expression plasmid. From [33].
3.6 Intravenous RNAi: brain delivery of EGFR targeted shRNA plasmid DNA
Many brain cancers [34], similar to 70% of solid cancer in general [35], over-express the epidermal growth factor receptor (EGFR). The EGFR plays an pro-angiogenic role in cancer growth [36], and one goal of cancer therapy is to knock down the tumor EGFR. In order to examine whether it is possible to suppress expression of EGFR in intra-cranial brain cancer with RNAi-based gene therapy, an intra-cranial brain tumor model was evalulated [37]. In this model, human U87 glioma cells were stereotactically implanted in the caudate-putamen nucleus of severe combined immunodeficient (scid) mice. In parallel, an expression plasmid was engineered that encoded for an shRNA that targeted a specific sequence (nucleotides 2529–2557) of the human EGFR mRNA (Fig. 4B). The anti-EGFR shRNA was encoded by a plasmid DNA designated clone 967 (Fig. 4B). The clone 967 plasmid DNA was encapsulated in the interior of Trojan horse liposomes that were doubly targeted with two different MAb molecules (Fig. 4A). The Trojan horse liposome carried both the MAb to the HIR and the MAb to the mouse TfR (mTfR). The Trojan horse liposome was formulated with 2 targeting antibodies because (a) the targeting MAbs are species specific [3], and (b) the model involves the growth of human cancer cells in mouse brain. Following implantation of 500,000 tumor cells at day 0, the tumor grows in mouse brain following vascularization by vessels of mouse brain origin. These vessels express the mouse TfR, not the human TfR [37]. Similarly, the cancer cells express the HIR, not the murine insulin receptor. Therefore, conjugation of the HIRMAb to the THLs allowed for receptor mediated endocytosis of the Trojan horse liposome into the human brain cancer cell following transport across the mouse vascular endothelial barrier perfusing the cancer. However, the HIRMAb would not mediate the RMT of the liposome across the mouse BBB. This vascular endothelial barrier in the tumor expresses the mouse TfR, and this is targeted by conjugating the 8D3 MAb to the mouse TfR to the Trojan horse liposomes (Fig. 4A)
Figure 4.

(A) Model of pegylated immunoliposome (PIL) that is doubly targeted to both the mouse transferrin receptor (mTfR) with the 8D3 monoclonal antibody (MAb1) and to the human insulin receptor (HIR) with the 83-14 monoclonal antibody (MAb2). Encapsulated in the interior of the PIL is the plasmid DNA encoding the short hairpin RNA (shRNA) which produces the RNA interference (RNAi). The gene encoding the shRNA is driven by the U6 promoter (pro) and is followed on the 3′-end with the T5 termination sequence for the U6 RNA polymerase. (B) Nucleotide sequence of the human epidermal growth factor receptor (hEGFR) mRNA sequence between nucleotides 2529–2557 is shown on top. The sequence and secondary structure of the shRNA produced by clone 967 is shown on the bottom. The antisense strand is 5′ to the 8 nucleotide loop, and the sense strand is 3′ to the loop. The sense strand contains 4 G/U mismatches to reduce the Tm of hybridization of the stem loop structure; the sequence of the antisense strand is 100% complementary to the target mRNA sequence. (C) Intravenous RNAi gene therapy directed at the human EGFR is initiated at 5 days after implantation of 500,000 U87 cells in the caudate putamen nucleus of scid mice, and weekly intravenous gene therapy is repeated at days 12, 19, and 26 (arrows). The control group was treated with saline on the same days. There are 11 mice in each of the 2 treatment groups. The time at which 50% of the mice were dead (ED50) is 17 days and 32 days in the saline and RNAi groups, respectively. The RNAi gene therapy produces an 88% increase in survival time. From [37].
The biological efficacy of the Trojan horse liposomes encapsulated with clone 967 was initially evaluated in culture of U87 cells [37]. Upon addition of EGF to these cells, there is a rapid translocation of calcium from the endoplasmic reticulum (ER) to the cytosol. This calcium mobilization can be detected with video microscopy using a specific fluorogenic agent. As shown in Fig. 5A, there is rapid movement of calcium from the ER to the cytosol following application of EGF to the U87 glioma cells. This experiment was repeated 24 hours after exposure of the U87 cells to Trojan horse liposomes encapsulated with clone 967 and targeted with the HIRMAb. The video microscopy study shows a 95% knockdown in functional EGFR in those cells exposed to RNAi gene therapy (Fig. 5B). The knockdown of EGFR in these cells was corroborated by Western blot studies. Clone 967 was delivered to the U87 cells, and this resulted in a 75% reduction in immunoreactive EGFR in the cells (Fig. 5C). A parallel knockdown of the immunoreactive EGFR was observed with clone 882 DNA (Fig. 5C); this plasmid DNA expresses a 700 nucleotide antisense RNA against the human EGFR mRNA [38]. However, delivery of clone 952 plasmid DNA, which encodes an shRNA against luciferase, or clone 962 plasmid DNA, which is a plasmid DNA that encodes a non-functional shRNA against the EGFR [37], to the tumor cells results in no knockdown of immunoreactive EGFR (Fig. 5C).
Figure 5.

Maximum Fluo-4 fluorescence in cultured U87 human glioma cells is shown after stimulation with 200 ng/ml human epidermal growth factor (EGF). Prior to measurement of calcium induced fluorescence, the cells were pre-incubated for 24 hours with either vehicle (A) or HIRMAb-targeted PILs carrying clone 967 plasmid DNA at a dose of 0.5 μg DNA per dish (B). From [37]. (C) U87 human glioma cells were exposed to clone 967, clone 882, clone 952, or clone 962 plasmid DNA for 48 hours and harvested for EGFR Western blotting. Films were scanned and quantified with NIH Image software, arbitrary densitometric units (ADU) were computed for each treatment group. A representative scan is shown at the top of each mean ± SE (n=3–4 dishes). Clone 967 and 952 produces shRNAs against the human EGFR and the luciferase mRNAs, respectively. Clone 882 produces a 700 nt antisense RNA against the human EGFR. Clone 962 encodes a shRNA against the EGFR that produces no knockdown of EGFR expression From [37]. (D, E) Confocal microscopy of intra-cranial glioma. Sections are shown for brain tumors from RNAi treated mice (D) or saline treated mice (E). The sections are doubly labeled with the murine 528 MAb to the EGFR (green) and the rat 8D3 MAb to the mouse TfR (red). There is decreased immunoreactive EGFR in the tumor cells in the RNAi treated mice (D) relative to the saline treated mice (E). From [37].
The weekly intravenous administration of clone 967 plasmid DNA with the doubly targeted Trojan horse liposomes to the scid mice with U87 intra-cranial brain tumors resulted in a 90% knockdown in immunoreactive EGFR in the tumor in vivo, as demonstrated by confocal microscopy (Fig. 5D and 5E). The weekly intravenous administration of 5 μg per mouse of clone 967 plasmid DNA encapsulated in Trojan horse liposomes resulted in a 90% increase in survival time in the tumor brain bearing scid mice [37], as shown in Fig. 4C. Animals treated with weekly intravenous saline were 50% expired at 17 days, whereas animals treated with RNAi based gene therapy were 50% expired at 32 days (Fig. 4C). As a control experiment, an identical Trojan horse liposome formulation was produced except a luciferase expression plasmid DNA replaced the clone 967 plasmid DNA [38]; the mortality curve was virtually identical with the saline mortality curve shown in Fig. 4C. The survival study in Fig. 4C was the first demonstration of a prolongation of survival in experimental cancer with RNAi based gene therapy. This in vivo therapeutic effect of RNAi was made possible by combining RNAi technology with Trojan horse liposome targeting technology.
4. Delivery of antisense and siRNA with molecular Trojan horses and avidin-biotin technology
4.1 Avidin-biotin technology and drug delivery
The delivery of either antisense oligodeoxynucleotides (ODN), antisense peptide nucleic acids (PNA), or siRNA requires high affinity attachment of the antisense agent to the molecular Trojan horse. The bond between the MTH and the nucleic acid therapeutic must be stable in vivo in the circulation. Initially, nucleic acids were attached to targeting ligands via a polycationic bridge, such as polylysine or protamine [39]. However, the bond between the polycation and the nucleic acid is rapidly dissociated by serum proteins following in vivo administration [40]. Moreover, high doses of cationic proteins can be toxic in vivo, and lead to altered microvascular permeability [41].
Nucleic acid therapeutics may also be attached to targeting ligands with the use of avidin-biotin technology [3]. The bond between avidin or streptavidin and biotin is extremely tight, and is not disrupted by serum proteins. A conjugate of the targeting MAb and either avidin or streptavidin (SA) is formulated in one vial. In a second vial, the mono-biotinylated antisense agent or siRNA is produced. The two vials are mixed just prior to intravenous administration. Owing to the extremely high affinity of avidin or SA binding of biotin, there is immediate formation of the conjugate between antisense agent or the siRNA, and the targeting MAb. The dissociation half-time of biotin binding to avidin or SA is 89 days, and the dissociation constant is 10−15 M [42]. Therefore, the association between the antisense agent and the targeting MAb in vivo in the circulation remains intact for several hours after intravenous administration [3]. The attachment of the antisense agent to the targeting MAb through an avidin-biotin linkage has no inhibitory effect on hybridization of the antisense agent with the target RNA. This was demonstrated in previous studies by both RNAse protection assays and Northern blotting [43].
4.2 Molecular Trojan horses and antisense oligodeoxynucleotide delivery to brain
Phosphodiester (PO) ODNs are rapidly degraded by 3′ exonuclease in cell culture [44] and by endonuclease in vivo [3]. Phosphorothioate (PS) ODNs are much less sensitive to nuclease degradation as compared to PO-ODNs [45]. However, the sulfur atoms of PS-ODNs are highly reactive and bind to multiple proteins [46, 47]. PS-ODNs are strongly bound by plasma proteins including α2-macroglobulin [48], which is a protein in excess of 700,000 Da in molecular size. The attachment of a monobiotinylated PS-ODN to a BBB MTH resulted in stimulated uptake of the PS-ODN across the BBB in vivo following carotid arterial infusion in the absence of serum proteins [49]. However, when the conjugate of the MTH and the PS-ODN was administered intravenously, there was no measureable BBB transport [49]. The PS-ODN was strongly bound by plasma protein, which prevented the BBB transport of the PS-ODN attached to the MTH. A third class of antisense agents is the peptide nucleic acid (PNA). PNAs may be the preferred antisense agent. Unlike PO-ODNs, PNA are resistant to nucleases and are metabolically stable in vivo [50]. Unlike PS-ODNs, PNA lack sulfur atoms, and do not bind non-specifically to tissue or serum proteins. Therefore, mono-biotinylated PNAs were formulated and the BBB transport of these agents was examined with the molecular Trojan horse targeting technology, combined with avidin-biotin technology [51].
4.3 Molecular Trojan horses and antisense peptide nucleic acid delivery to brain
The brain delivery of an antisense PNA was evaluated by attempting to image gene expression of the target mRNA in brain in vivo with sequence specific PNA radiopharmaceuticals. The formulation of the PNA radiopharmaceutical is shown in Fig. 6A. The PNA is comprised of an 18-mer that is antisense to a specific region of the caveolin-1α mRNA [43]. The amino terminus of the PNA contains a diethylenetriaminepentaacetic acid (DTPA) moiety to allow for radiolabeling with 111-indium. At the carboxyl terminus, the ε-amino group of a lysine residue is monobiotinylated, which allows for high affinity attachment to the TfR MAb via a biotin-SA linker. The carboxyl terminus of the PNA is amidated to reduce sensitivity to carboxypeptidases. The TfR MAb causes RMT of the PNA across the BBB, which is then followed by receptor-mediated endocytosis across the brain cell membrane (BCM) (Fig. 6B). Following transport across the BCM, the antisense PNA then escapes from the endosomal compartment where it can hybridize with the target mRNA (Fig. 6B).
Figure 6.

(A) Conjugation of a peptide nucleic acid (PNA) to a MAb to the TfR creates a bifunctional molecule that both accesses the TfR for transport between tissue compartments and binds to a target mRNA based on the sequence specificity of the nucleotide residues of the PNA radiopharmaceutical. The PNA has a biotin moiety at the ε-amino group of a C-terminal lysine residue to allow for capture by a conjugate of the MAb and streptavidin (SA) and has amino terminal diethylenetriaminepentaacetic acid (DTPA) moiety to allow for radiolabeling with [111In]. The sequence shown is antisense to nucleotides 10–27 of the rat caveolin-1α mRNA. From [43]. (B) Model showing the transport from blood to brain intracellular spaces of a conjugate of a peptide nucleic acid (PNA) antisense radiopharmaceutical and a monoclonal antibody (MAb) to the transferrin receptor (TfR). The transport of the PNA-MAb conjugate across either the blood brain barrier (BBB) or the brain cell membrane (BCM) is possible owing to the ability of the MAb to access the endogenous transport pathways for transferrin, which exist at both cellular barriers.
The effective delivery of antisense PNA radiopharmaceuticals to brain with molecular Trojan horses was demonstrated by quantitative autoradiography [43], as shown in Fig. 7. Adult Fischer CD344 rats were implanted with RG2 tumors. These tumors over-express the gene encoding caveolin-1α (CAV) and under-express the gene encoding for GFAP [43]. The size of the tumor is shown by the hematoxylin stains in panels B, D, F, and H of Fig. 7. The corresponding brain scans at 6 hours after intravenous administration of the anti-CAV or anti-GFAP antisense radiopharmaceutical is shown in panels A, C, E, and G of Fig. 7. The tumor bearing animals were injected with one of four different PNA antisense radiopharmaceuticals: (i) the CAV-PNA conjugated to the TfR MAb (CAV-PNA CONJ), (ii) the GFAP-PNA antisense radiopharmaceutical conjugated to the TfR MAb (GFAP-PNA CONJ), (iii) the unconjugated CAV-PNA, and (iv) the unconjugated GFAP-PNA. The radioactivity in the tumor and in the contralateral brain was quantified and these data are shown in Fig. 7I. The delivery of the CAV-PNA with the molecular Trojan horse resulted in robust imaging of caveolin-1α gene expression in the brain tumor. The brain tumor is illuminated in this study (Fig. 7G) because (a) the tumor over-expresses the gene encoding for caveolin-1α mRNA, and (b) the antisense PNA directed against the caveolin-1 α mRNA was effectively delivered to the tumor intracellular space with the molecular Trojan horse. Conversely, there was no imaging of the tumor following intravenous administration of the CAV-PNA that was not attached to the Trojan horse (Fig. 7E), because the antisense PNA does cross the BBB [51]. The tumor was not effectively imaged with the GFAP-PNA, even though the GFAP-PNA was conjugated to the BBB molecular Trojan horse (Fig. 7C), because the GFAP gene is actually under-expressed in this brain tumor [43]. The study in Fig. 7 with a targeted antisense PNA radiopharmaceutical shows it is possible to image gene expression in vivo with sequence-specific antisense radiopharmaceuticals, providing the PNA antisense agent is targeted across cell membranes in vivo with molecular Trojan horses. Moreover, the imaging paradigm demonstrates that the antisense agent specifically hybridized to the target mRNA; this hybridization causes sequestration of the CAV-PNA in the brain tumor (Fig. 7G), but not the GFAP-PNA (Fig. 7C). Imaging gene expression in brain in vivo with antisense radiopharmaceuticals would not be possible in the absence of a BBB drug targeting technology, because these molecules do not cross the BBB in vivo.
Figure 7.

(A, C, E, G) Colorized film autoradiograms of sections removed from Fisher CD344 rat brain implanted with RG2 tumors; the rats were sacrificed at six hours after intravenous injection of [111In] PNA against either the rat caveolin-1α (CAV) or rat glial fibrillary acidic protein (GFAP) mRNA. (A) RG2 tumor-bearing rat injected with unconjugated [111In]-GFAP-PNA. (C) RG2 tumor-bearing rat injected with [111In]-GFAP-PNA conjugated to the OX26 TfRMAb. (E) RG2 tumor-bearing rat injected with unconjugated [111In]-CAV-PNA. (G) RG2 tumor-bearing rat injected with [111In]-CAV-PNA conjugated to the OX26 TfRMAb. (B, D, F, H) Hematoxylin autopsy stains showing size of tumor and corresponding to panels A, C, E, and G, respectively. (I) Integrated density of the autoradiogram signal over the brain tumor (tumor) or contralateral brain (brain) for each of the four different antisense radiopharmaceutical formulations: 1,2: [111In]-CAV-PNA OX26 conjugate (CAV-PNA CONJ.); 3–4: [111In]-GFAP-PNA OX26 conjugate (GFAP-PNA CONJ.); 5–6: unconjugated [111In]-CAV-PNA (CAV-PNA); 7–8: unconjugated [111In]-GFAP-PNA (GFAP-PNA). Data are mean ± S.E. (n = four rats in each of the four groups). From [43].
4.4 Molecular Trojan horses and siRNA delivery to brain
The delivery of siRNA to brain is analogous to the delivery of antisense agents such as PNAs to brain. The studies reviewed in Figs. 6 and 7 demonstrate the delivery of PNA antisense agents to brain in vivo [43]. siRNA duplexes can be monobiotinylated [52], which allows for high efficiency attachment to molecular Trojan horses using avidin-biotin technology. In parallel with the mono-biotinylation of the siRNA, a chemical conjugate of SA and the molecular Trojan horse is produced. Alternatively, MAb-avidin fusion proteins have been genetically engineered and expressed [53]. The mono-biotinylated siRNA, and the MTH-SA or MTH-avidin conjugate, is mixed prior to intravenous administration. The MTHs may carry the siRNA molecules across the BBB and the BCM similar to that demonstrated previously for PNA antisense agents (Figure 7).
Conclusions
Antisense agents including PNAs can be delivered to brain in vivo with the use of molecular Trojan horses and avidin-biotin technology. Molecular Trojan horses may also be formulated as Trojan horse liposomes to deliver shRNA expressing plasmid DNA to brain in vivo. The genetic engineering of molecular Trojan horses has been completed, and this will enable future clinical applications in humans. Intravenous RNAi, like intravenous gene therapy, or intravenous antisense therapy, is possible, providing the RNAi technology is merged with a drug targeting technology that is effective in vivo.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat Rev Genet. 2002;(3):737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- 2.Brightman MW, Reese TS, Feder N. Assessment with the electron microscope of the permeability to peroxidase of cerebral endothelium and epithelium in mice and sharks. Capillary Permeability; Munksgaard, Copenhagen: 1970. p. 463. [Google Scholar]
- 3.Pardridge WM. Brain drug targeting; the future of brain drug development. Cambridge University Press; Cambridge, United Kingdom: 2001. pp. 1–370. [Google Scholar]
- 4.Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;(2):3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Pardridge WM. Rapid transferrin efflux from brain to blood across the blood-brain barrier. J Neurochem. 2001;(76):1597–1600. doi: 10.1046/j.1471-4159.2001.00222.x. [DOI] [PubMed] [Google Scholar]
- 6.Lee HJ, Engelhardt B, Lesley J, Bickel U, Pardridge WM. Targeting rat anti-mouse transferrin receptor monoclonal antibodies through blood-brain barrier in mouse. J Pharmacol Exp Ther. 2000;(292):1048–1052. [PubMed] [Google Scholar]
- 7.Pardridge WM, Buciak JL, Friden PM. Selective transport of an anti-transferrin receptor antibody through the blood-brain barrier in vivo. J Pharmacol Exp Ther. 1991;(259):66–70. [PubMed] [Google Scholar]
- 8.Pardridge WM, Kang YS, Buciak JL, Yang J. Human insulin receptor monoclonal antibody undergoes high affinity binding to human brain capillaries in vitro and rapid transcytosis through the blood-brain barrier in vivo in the primate. Pharm Res. 1995;(12):807–816. doi: 10.1023/a:1016244500596. [DOI] [PubMed] [Google Scholar]
- 9.Coloma MJ, et al. Transport across the primate blood-brain barrier of a genetically engineered chimeric monoclonal antibody to the human insulin receptor. Pharm Res. 2000;(17):266–274. doi: 10.1023/a:1007592720793. [DOI] [PubMed] [Google Scholar]
- 10.Wu D, Pardridge WM. Central nervous system pharmacologic effect in conscious rats after intravenous injection of a biotinylated vasoactive intestinal peptide analog coupled to a blood-brain barrier drug delivery system. J Pharmacol Exp Ther. 1996;(279):77–83. [PubMed] [Google Scholar]
- 11.Wu D, Pardridge WM. Neuroprotection with noninvasive neurotrophin delivery to the brain. Proc Natl Acad Sci U S A. 1999;(96):254–259. doi: 10.1073/pnas.96.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Pardridge WM. Conjugation of brain-derived neurotrophic factor to a blood-brain barrier drug targeting system enables neuroprotection in regional brain ischemia following intravenous injection of the neurotrophin. Brain Res. 2001;(889):49–56. doi: 10.1016/s0006-8993(00)03108-5. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Pardridge WM. Neuroprotection in transient focal brain ischemia after delayed intravenous administration of brain-derived neurotrophic factor conjugated to a blood-brain barrier drug targeting system. Stroke. 2001;(32):1378–1384. doi: 10.1161/01.str.32.6.1378. [DOI] [PubMed] [Google Scholar]
- 14.Song BW, Vinters HV, Wu D, Pardridge WM. Enhanced neuroprotective effects of basic fibroblast growth factor in regional brain ischemia after conjugation to a blood-brain barrier delivery vector. J Pharmacol Exp Ther. 2002;(301):605–610. doi: 10.1124/jpet.301.2.605. [DOI] [PubMed] [Google Scholar]
- 15.Kurihara A, Pardridge WM. Imaging brain tumors by targeting peptide radiopharmaceuticals through the blood-brain barrier. Cancer Res. 1999;(59):6159–6163. [PubMed] [Google Scholar]
- 16.Zhang Y, Pardridge WM. Delivery of beta-galactosidase to mouse brain via the blood-brain barrier transferrin receptor. J Pharmacol Exp Ther. 2005;(313):1075–1081. doi: 10.1124/jpet.104.082974. [DOI] [PubMed] [Google Scholar]
- 17.Shi N, Pardridge WM. Noninvasive gene targeting to the brain. Proc Natl Acad Sci U S A. 2000;(97):7567–7572. doi: 10.1073/pnas.130187497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah N, Zhang S, Harada S, Smith RM, Jarett L. Electron microscopic visualization of insulin translocation into the cytoplasm and nuclei of intact H35 hepatoma cells using covalently linked Nanogold-insulin. Endocrinology. 1995;(136):2825–2835. doi: 10.1210/endo.136.7.7789307. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Jeong Lee H, Boado RJ, Pardridge WM. Receptor-mediated delivery of an antisense gene to human brain cancer cells. J Gene Med. 2002;(4):183–194. doi: 10.1002/jgm.255. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Calon F, Zhu C, Boado RJ, Pardridge WM. Intravenous nonviral gene therapy causes normalization of striatal tyrosine hydroxylase and reversal of motor impairment in experimental Parkinsonism. Hum Gene Ther. 2003;(14):1–12. doi: 10.1089/10430340360464660. [DOI] [PubMed] [Google Scholar]
- 21.Pardridge WM. Gene targeting in vivo with pegylated immunoliposomes. Methods Enzymol. 2003;(373):507–528. doi: 10.1016/S0076-6879(03)73032-8. [DOI] [PubMed] [Google Scholar]
- 22.Norman J, et al. Liposome-mediated, nonviral gene transfer induces a systemic inflammatory response which can exacerbate pre-existing inflammation. Gene Ther. 2000;(7):1425–1430. doi: 10.1038/sj.gt.3301240. [DOI] [PubMed] [Google Scholar]
- 23.Loisel S, Le Gall C, Doucet L, Ferec C, Floch V. Contribution of plasmid DNA to hepatotoxicity after systemic administration of lipoplexes. Hum Gene Ther. 2001;(12):685–696. doi: 10.1089/104303401300057405. [DOI] [PubMed] [Google Scholar]
- 24.Elouahabi A, et al. Free cationic liposomes inhibit the inflammatory response to cationic lipid-DNA complex injected intravenously and enhance its transfection efficiency. Mol Ther. 2003;(7):81–88. doi: 10.1016/s1525-0016(02)00032-1. [DOI] [PubMed] [Google Scholar]
- 25.Shi N, Zhang Y, Zhu C, Boado RJ, Pardridge WM. Brain-specific expression of an exogenous gene after i.v. administration. Proc Natl Acad Sci U S A. 2001;(98):12754–12759. doi: 10.1073/pnas.221450098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu C, Zhang Y, Pardridge WM. Widespread expression of an exogenous gene in the eye after intravenous administration. Invest Ophthalmol Vis Sci. 2002;(43):3075–3080. [PubMed] [Google Scholar]
- 27.Zhang Y, Schlachetzki F, Li JY, Boado RJ, Pardridge WM. Organ-specific gene expression in the Rhesus monkey eye following intravenous non-viral gene transfer. Mol Vis. 2003;(9):465–472. [PubMed] [Google Scholar]
- 28.Zhang Y, Schlachetzki F, Pardridge WM. Global non-viral gene transfer to the primate brain following intravenous administration. Mol Ther. 2003;(7):11–18. doi: 10.1016/s1525-0016(02)00018-7. [DOI] [PubMed] [Google Scholar]
- 29.Chu C, Zhang Y, Boado RJ, Pardridge WM. Decline in exogenous gene expression in primate brain following intravenous administration is due to plasmid degradation. Pharm Res. 2006 doi: 10.1007/s11095-006-0274-x. in press. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Schlachetzki F, Zhang YF, Boado RJ, Pardridge WM. Normalization of striatal tyrosine hydroxylase and reversal of motor impairment in experimental parkinsonism with intravenous nonviral gene therapy and a brain-specific promoter. Hum Gene Ther. 2004;(15):339–350. doi: 10.1089/104303404322959498. [DOI] [PubMed] [Google Scholar]
- 31.Naeser P. Insulin receptors in human ocular tissues. Immunohistochemical demonstration in normal and diabetic eyes. Ups J Med Sci. 1997;(102):35–40. doi: 10.3109/03009739709178930. [DOI] [PubMed] [Google Scholar]
- 32.Shi N, Boado RJ, Pardridge WM. Receptor-mediated gene targeting to tissues in vivo following intravenous administration of pegylated immunoliposomes. Pharm Res. 2001;(18):1091–1095. doi: 10.1023/a:1010910523202. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Boado RJ, Pardridge WM. In vivo knockdown of gene expression in brain cancer with intravenous RNAi in adult rats. J Gene Med. 2003;(5):1039–1045. doi: 10.1002/jgm.449. [DOI] [PubMed] [Google Scholar]
- 34.Kuan CT, Wikstrand CJ, Bigner DD. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr Relat Cancer. 2001;(8):83–96. doi: 10.1677/erc.0.0080083. [DOI] [PubMed] [Google Scholar]
- 35.Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl 4):S9–15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 36.Abe T, et al. PTEN decreases in vivo vascularization of experimental gliomas in spite of proangiogenic stimuli. Cancer Res. 2003;(63):2300–2305. [PubMed] [Google Scholar]
- 37.Zhang Y, et al. Intravenous RNA interference gene therapy targeting the human epidermal growth factor receptor prolongs survival in intracranial brain cancer. Clin Cancer Res. 2004;(10):3667–3677. doi: 10.1158/1078-0432.CCR-03-0740. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Zhu C, Pardridge WM. Antisense gene therapy of brain cancer with an artificial virus gene delivery system. Mol Ther. 2002;(6):67–72. doi: 10.1006/mthe.2002.0633. [DOI] [PubMed] [Google Scholar]
- 39.Wu CH, Wilson JM, Wu GY. Targeting genes: delivery and persistent expression of a foreign gene driven by mammalian regulatory elements in vivo. J Biol Chem. 1989;(264):16985–16987. [PubMed] [Google Scholar]
- 40.Kwoh DY, et al. Stabilization of poly-L-lysine/DNA polyplexes for in vivo gene delivery to the liver. Biochim Biophys Acta. 1999;(1444):171–190. doi: 10.1016/s0167-4781(98)00274-7. [DOI] [PubMed] [Google Scholar]
- 41.Vehaskari VM, Chang CT, Stevens JK, Robson AM. The effects of polycations on vascular permeability in the rat. A proposed role for charge sites. J Clin Invest. 1984;(73):1053–1061. doi: 10.1172/JCI111290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Green NM. Avidin. Adv Protein Chem. 1975;(29):85–133. doi: 10.1016/s0065-3233(08)60411-8. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki T, et al. Imaging endogenous gene expression in brain cancer in vivo with 111In-peptide nucleic acid antisense radiopharmaceuticals and brain drug-targeting technology. J Nucl Med. 2004;(45):1766–1775. [PubMed] [Google Scholar]
- 44.Tidd DM, Warenius HM. Partial protection of oncogene, anti-sense oligodeoxynucleotides against serum nuclease degradation using terminal methylphosphonate groups. Br J Cancer. 1989;(60):343–350. doi: 10.1038/bjc.1989.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marcus-Sekura CJ, Woerner AM, Shinozuka K, Zon G, Quinnan GV., Jr Comparative inhibition of chloramphenicol acetyltransferase gene expression by antisense oligonucleotide analogues having alkyl phosphotriester, methylphosphonate and phosphorothioate linkages. Nucleic Acids Res. 1987;(15):5749–5763. doi: 10.1093/nar/15.14.5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ho PT, Ishiguro K, Wickstrom E, Sartorelli AC. Non-sequence-specific inhibition of transferrin receptor expression in HL-60 leukemia cells by phosphorothioate oligodeoxynucleotides. Antisense Res Dev. 1991;(1):329–342. doi: 10.1089/ard.1991.1.329. [DOI] [PubMed] [Google Scholar]
- 47.Stein CA, Cheng YC. Antisense oligonucleotides as therapeutic agents--is the bullet really magical? Science. 1993;(261):1004–1012. doi: 10.1126/science.8351515. [DOI] [PubMed] [Google Scholar]
- 48.Cossum PA, et al. Disposition of the 14C-labeled phosphorothioate oligonucleotide ISIS 2105 after intravenous administration to rats. J Pharmacol Exp Ther. 1993;(267):1181–1190. [PubMed] [Google Scholar]
- 49.Wu D, Boado RJ, Pardridge WM. Pharmacokinetics and blood-brain barrier transport of [3H]-biotinylated phosphorothioate oligodeoxynucleotide conjugated to a vector-mediated drug delivery system. J Pharmacol Exp Ther. 1996;(276):206–211. [PubMed] [Google Scholar]
- 50.Garner P, Sherry B, Moilanen S, Huang Y. In vitro stability of alpha-helical peptide nucleic acids (alphaPNAs) Bioorg Med Chem Lett. 2001;(11):2315–2317. doi: 10.1016/s0960-894x(01)00422-x. [DOI] [PubMed] [Google Scholar]
- 51.Pardridge WM, Boado RJ, Kang YS. Vector-mediated delivery of a polyamide (“peptide”) nucleic acid analogue through the blood-brain barrier in vivo. Proc Natl Acad Sci U S A. 1995;(92):5592–5596. doi: 10.1073/pnas.92.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiu YL, Rana TM. RNAi in human cells: basic structural and functional features of small interfering RNA. Mol Cell. 2002;(10):549–561. doi: 10.1016/s1097-2765(02)00652-4. [DOI] [PubMed] [Google Scholar]
- 53.Penichet ML, Kang YS, Pardridge WM, Morrison SL, Shin SU. An Anti-transferrin receptor antibody-avidin fusion protein serves as a delivery vehicle for effective brain targeting in an animal model. Initial applications in antisense drug delivery to the brain. J Immunol. 1999;(163):4421–4426. [PubMed] [Google Scholar]
- 54.Zhu C, et al. Organ-specific expression of the lacZ gene controlled by the opsin promoter after intravenous gene administration in adult mice. J Gene Med. 2004;(6):906–912. doi: 10.1002/jgm.575. [DOI] [PubMed] [Google Scholar]