

Abstract
Pregabalin, an anticonvulsant and anxiolytic compound that binds to α2-δ auxiliary subunit Types 1 and 2 of voltage-gated calcium channels, has been shown to reduce excitatory neurotransmission partially through modulation of glutamatergic signaling. Prepulse inhibition (PPI) of startle is an operational measure of sensorimotor gating impacted by disruption of the glutamatergic system and is reduced in schizophrenia patients. Dysregulation of the glutamatergic system has also been implicated in the pathophysiology of schizophrenia. Here we tested the hypothesis that pregabalin may ameliorate PPI in a model of deficient gating in humans and mice. In study 1, 14 healthy human subjects participated in a within subjects, cross-over study with placebo, 50 mg or 200 mg pregabalin treatment prior to undergoing a PPI task. In study 2, 24 C57BL/6 mice underwent a similar procedure with vehicle, 30 and 100 mg/kg dose treatments. In both studies, subjects were assigned to a “Low” or “High” gating group using a median split procedure based on their PPI performance during placebo/vehicle. Drug effects were then examined across these groups. In humans, pregabalin treatment significantly increased PPI performance in the “low gating” group. In mice, pregabalin treatment significantly increased PPI in the low gating group but reduced PPI in the high gating group. Across species, pregabalin treatment improves PPI in subjects with low gating. These data support further exploration of pregabalin as a potential treatment for disorders characterized by sensorimotor gating deficits and glutamatergic hypersignaling, such as schizophrenia.
Keywords: Schizophrenia, Pre-pulse inhibition, Glutamate, Pregabalin, Startle, Sensorimotor gating
1. Introduction
Recent evidence suggests that excessive glutamate transmission may be a core feature of pathology in schizophrenia, supplanting previous ‘hypoglutamatergic’ theories which were predicated on NMDA receptor dysfunction (Moghaddam and Javitt, 2012; Krystal et al., 2003). For instance, magnetic resonance spectroscopy studies of medication-naive schizophrenia patients have shown increased glutamate and glutamine levels in the prefrontal cortex (Cecil et al., 1999). Consequently, a number of novel therapies targeted toward reducing glutamate neurotransmission are being explored for treatment of schizophrenia (e.g. Chaki and Hikichi, 2011).
Pregabalin ((S)-3-(aminomethyl)-5-methylhexanoic acid) is FDA approved for use in partial seizures (French et al., 2003), neuropathic pain (Dworkin et al., 2003) and fibromyalgia (Straube et al., 2010) and has also shown efficacy in treating generalized anxiety disorder (Rickels et al., 2005) and social anxiety disorder (Pande et al., 2004). Pregabalin binds to α2-δ auxiliary subunit Types 1 and 2 of voltage-gated calcium channels (VGCC; Taylor et al., 2007), with the effect of reducing excitatory neurotransmission in “hyper-excited” neurons (Quitero et al., 2010; Kavoussi, 2006). Pregabalin has been shown to reduce levels of glutamate in the brain and spinal cord (Errante and Petroff, 2003; Fehrenbacher et al., 2003; Maneuf et al., 2001; Dooley et al., 2000). Recently, Englisch and colleagues (2010) reported on a series of 11 case studies using pregabalin as an adjunctive treatment for anxiety in schizophrenia patients. Pregabalin was effective in reducing anxiety in these patients, as well as enabling a dose decrease in antipsychotic medications. These preliminary case studies and the putative reduction of glutamatergic signaling induced by pregabalin treatment supports the further examination of its use in treatment of schizophrenia. One strategy to further examine its potential as a treatment for schizophrenia is in predictive models of antipsychotic efficacy, such as pre-pulse inhibition.
Prepulse Inhibition (PPI), or the unlearned suppression of the startle reflex to an intense acoustic stimulus when immediately preceded by a weaker acoustic pre-pulse, has been characterized as a measure of pre-attentive information processing or sensorimotor gating (Geyer et al., 1990). Specifically, PPI is thought to reflect the ability of an organism to gate out extraneous sensory information and subsequent motor response in order to allow for processing of the pre-pulse. PPI is observed across all mammals tested (Braff et al., 2001; Dulawa and Geyer, 1996; Swerdlow et al., 1986). PPI has been widely used as a model of sensorimotor gating deficits and screening tool for novel therapeutics for schizophrenia (Swerdlow et al., 2008). PPI is disrupted by infusion of glutamate into the nucleus accumbens and the ventral striatum (Klarner et al., 1998; Swerdlow et al., 1992), and infusion of the glutamate agonist NMDA into the ventral hippocampus (Wan et al., 1996), suggesting that excessive glutamate signaling in some forebrain regions can induce sensorimotor gating deficits.
Pregabalin has effects in areas of the brain implicated in the regulation of PPI, including the hippocampus, prefrontal cortex, basolateral amygdala, and striatum (Li et al., 2011; Taylor et al., 2007; Swerdlow et al., 2001). Thus, pregabalin may have the effect of regulating glutamate function in areas where excess glutamate has been shown to disrupt PPI including the prefrontal cortex, where dysregulated glutamate signaling has also been implicated in the pathophysiology of schizophrenia (Moghaddam and Javitt, 2012).
The current studies investigated the effect of pregabalin on PPI in healthy controls (experiment 1) and a sample of C57BL/6J mice (experiment 2). A median split procedure was conducted on baseline PPI in order to isolate treatment effects on subjects with low baseline gating performance. This data analytic strategy has been increasingly used in PPI research with healthy samples as a measure of gating normalization by antipsychotic medications (Holstein et al., 2011; Csomor et al., 2008; Gogos and van den Buuse, 2007; Vollenweider et al., 2006; Swerdlow et al., 2006; Bitsios et al., 2005). Such a strategy identifies a subset of healthy subjects who exhibit traits similar to those observed in patient samples, and thus facilitates translation of findings into patient populations.
2. Experiment 1
2.1. Methods
2.1.1. Subjects
Subjects were recruited by flyers placed around the UCSD campus and advertisements in local newspapers. 17 subjects underwent the written informed consenting process and were screened for study eligibility. Exclusionary criteria included meeting criteria for a DSM-IV Axis I disorder, current substance abuse, neurological disorders, current medication, smoking, excessive caffeine consumption (>4 cups per day), hearing threshold > 45 dB at a 500–6000 Hz range and head trauma with loss of consciousness > 5 min. Three subjects were excluded from analysis due to a lack of startle response during the procedure (mean baseline startle trials/mean no stimulus trials < 1.5). Subject characteristics are described in Table 1. All subjects gave written, informed consent and were treated in accordance with the Declaration of Helsinki. The study was approved by the University of California, San Diego Human Research Protection Program.
Table 1.
Subject characteristics by median group.
| Low PPI | High PPI | Total | |
|---|---|---|---|
| N | 7 | 7 | 14 |
| Mean age | 22.86 | 24 | 23.43 |
| Percent male | 71% | 43% | 57% |
| Ethnicity | Caucasian = 5 | Caucasian = 2 | Caucasian = 7 |
| Asian = 1 | Asian = 3 | Asian = 4 | |
| Other = 1 | Hispanic = 1 | Hispanic = 1 | |
| Other = 1 | Other = 2 | ||
| Women in follicular/luteal menstrual phase | |||
| Placebo | 1/1 | 1/3 | 2/4 |
| 50 mg | 1/1 | 2/2 | 3/3 |
| 200 mg | 0/2 | 3/1 | 3/3 |
Note. Both a t-test for Age and a Fisher’s exact test for Percent Male yielded no significant difference between PPI groups.
2.1.2. Treatment
The study consisted of a randomized double blind cross-over design with 3 testing days and a 7–10 day washout period between each test. On each testing day, subjects received either a Placebo dose, a “Low” dose (50 mg), or a “High” dose (200 mg) of pregabalin (purchased from Pfizer, Inc). This dose range covered both a sub-therapeutic dose and a dose demonstrated as therapeutic for generalized anxiety disorder (Bech, 2007). The therapeutic dose was intended to be in the low range to limit interference from sedative effects. Order of dose was randomized across subjects. Pregabalin was dissolved in a soft drink for administration, and was delivered ~150 min prior to testing. Pregabalin reaches peak plasma concentration in ~1.3 h following oral dose and has a half-life of 4.6–6.8 h in healthy subjects (Busch et al., 1998). Startle testing was part of a larger battery of tests that included psychosocial surveys and fMRI that preceded the startle study presented here (Aupperle et al., 2011).
2.1.3. Stimuli and apparatus
Startle pulses were delivered using a San Diego Instruments (SDI, San Diego, CA, USA) SR-HRLAB EMG system as previously described (Braff et al., 1992). Sound levels were measured using continuous tones and a calibrated Quest Sound Level Meter on the A scale, coupled to the headphones by an artificial ear. EMG responses were band-pass filtered (1–1000 Hz) and 60 Hz notch filtered, digitized, and recorded (1 kHz sampling frequency) using the SDI SR-HLAB EMG system coupled with a standard Dell desktop computer.
2.1.4. Experimental procedure
Subjects were seated in a comfortable lounge chair in a dimly lit testing chamber. Once seated, two electrodes (Ag/AgCl) were placed lateral to and below the left eye over the orbicularis oculi muscle. A reference electrode was also placed on the left mastoid. Subjects were fitted with standard headphones through which the startle pulses could be presented (all acoustic stimuli are presented as broadband noise; 70 dB background, 86 dB prepulses of 20 ms duration and 114 dB pulses of 40 ms duration). The session began with 5 114-dB pulses to stabilize startle responding. After this block pre-pulse trials (6 each of 3 trial types) or 114-dB pulse alone trials (10 total) were presented in a pseudorandom order. Prepulse trials consisted of 3 types, with the pre-pulse preceding the pulse at interstimulus intervals (ISI) of 30, 60 or 120 ms. The session then ended with 5 114-dB pulse trials. The intertrial interval ranged between 7 and 23 s (average 15 s) and baseline activity was recorded during each intertrial interval.
2.1.5. Data analysis
EMG responses were visually examined across each trial by a trained technician to identify and remove artifact (e.g. voluntary blinks) that were not associated with the pulse onset (e.g. a response was not counted unless it was within 100 ms of pulse onset). Data from the first and last block of 114-dB pulse-alone trials were analyzed separately from the rest of the session. This first block helps habituate startle to a stable baseline before pre-pulse trials are introduced, and comparing it to the last block at the end of the session measures habituation of the startle response across the session (e.g. Ludewig et al., 2002; Braff et al., 1992). Peak EMG response was averaged across each trial type. To assign subjects to high/low PPI groups, their average pre-pulse inhibition across all pre-pulse types was used, and subjects below and above the median (34%) were assigned to low and high PPI performance groups respectively (n = 7/group). Following median split, data were analyzed using a 2 × 3 repeated measures analysis of variance (ANOVA) with PPI group (low, high) as a between subject factor and dose (placebo, 50 mg, 200 mg) as a within subject factor. Bonferroni-corrected post-hoc tests were conducted to clarify significant main effects and interactions.
2.2. Results
2.2.1. Startle reactivity
Means and standard deviations for startle reactivity by PPI performance group and dose can be seen in Table 2. A 2 × 3 repeated-measures ANOVA showed a main effect of Dose: F(2,24) = 3.67, p <.04, partial η2 =.23. Post-hoc tests showed that independent of PPI group, startle was significantly reduced after 200 mg treatment compared to both placebo and 50 mg dose groups (ps <.05). High and low PPI groups did not differ in startle reactivity. Startle habituation was unaffected by PPI group or pregabalin (data not shown, main effect of Block: F(1,12) = 10.48, p < 0.01, no interaction with Group or Dose).
Table 2.
Startle reactivity (in arbitrary units) for both mice and humans by dose and median PPI.
| Humans
|
||||
|---|---|---|---|---|
| Low PPI (n = 7)
|
High PPI (n = 7)
|
|||
| M | SD | M | SD | |
| Placebo | 226.67 | 309.9 | 110.3 | 55.1 |
| 50 mg | 202.5 | 137.58 | 97.88 | 54.77 |
| 200 mg* | 145.63 | 129.1 | 83.98 | 65.4 |
| C57BL/6J Mice
|
||||
| Low PPI (n = 12) | High PPI (n = 12) | |||
|
|
||||
| M | SD | M | SD | |
|
| ||||
| Vehicle | 95.38 | 52.49 | 110.3 | 55.1 |
| 30 mg/kg | 95.98 | 81.6 | 97.88 | 54.77 |
| 100 mg/kg | 93.67 | 69.19 | 83.98 | 65.4 |
Note.
p < .05 vs. Placebo post-hoc test after significant main effect of dose. No main effects of Low/High group or interactions with dose were significant.
2.2.2. Prepulse inhibition
Dose effects were dependent upon PPI group [Fig. 1A; Dose × Group interaction, F(2,24) = 5.55, p <.01, partial η2 =.32]. Post-hoc tests showed that the low PPI group exhibited significant improvements in PPI after treatment with 50 or 200 mg pregabalin compared to placebo (ps <.02). There was no significant effect of Dose within the High PPI group. The effect of Dose and Group were not dependent upon the ISI, although as expected PPI performance was increased with longer ISIs across all groups (data not shown) [Main effect of ISI F(2,24) = 16.56, p < 0.001].
Fig. 1.
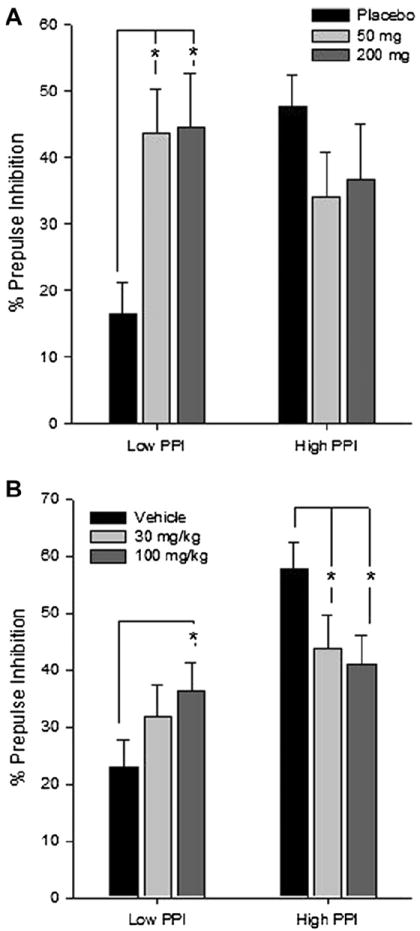
Pregabalin modulates PPI differently across high and low gating groups. (A) Healthy human subjects (n = 7/PPI group) were treated with placebo, 50 and 200 mg Pregabalin (oral) in a counterbalanced cross-over design with 1 week washout. (B) C57Bl6J mice (n = 12/PPI group) were treated with 0, 30 and 100 mg/kg (IP) in a counterbalanced cross-over design with 1 week washout. High and low PPI groups were categorized by median split of placebo treatment. Data are depicted as mean +/− SEM percentage PPI. PPI is averaged over 30−120 ms ISIs in humans and 20−120 ms ISIs in mice. *p < 0.05, post-hoc simple contrasts after Group × Pregabalin interaction. See results for details.
Both doses of pregabalin increased PPI in healthy humans with low baseline gating level. While visual inspection of Fig. 1A appears to show pregabalin decreasing PPI in the group with high baseline gating level, this difference did not reach statistical significance in the post-hoc tests. This finding is the first known demonstration of a pregabalin effect on PPI. To confirm and extend these results, we attempted to corroborate the effect in a mouse model of high and low PPI performance.
3.Experiment 2
3.1. Methods
3.1.1. Subjects
Twenty four male C57BL/6J mice obtained from Jackson Laboratories (JAX®; Bar Harbor, ME), aged 6–8 weeks on arrival. Mice were housed 4 per cage in a temperature-controlled (21–22 °C) room under a reverse 12 h light/dark cycle (lights off at 8:00 A.M.) with free access to food and water. Mice began behavioral testing one week after arrival to the vivarium. All procedures were approved by the UCSD Institutional Animal Care and Use Committee. The UCSD animal facility meets all federal and state requirements for animal care, and has been approved by the American Association for Accreditation of Laboratory Animal Care.
3.1.2. Treatment
Pregabalin (Tocris Bioscience Ellisville, Missouri) was dissolved in saline vehicle and administered 2 h before testing. On each testing day, mice received either vehicle, 30 or 100 mg/kg via interperitoneal injection. These doses were based on previous studies demonstrating anxiolytic effects in C57BL/6 mice (e.g. Lotarski et al., 2011). Order of dose was counterbalanced across mice and tests were separated by a 7 day washout period.
3.1.3. Stimuli and apparatus
Startle chambers (SR-LAB; San Diego Instruments, San Diego, CA) consisted of non-restrictive Plexiglas cylinders 5 cm in diameter resting on a Plexiglas platform in a ventilated chamber. High-frequency speakers mounted 33 cm above the cylinders produced all acoustic stimuli, which were controlled by SR-LAB software. Piezoelectric accelerometers mounted under the cylinders transduced movements of the animal, which were digitized and stored by an interface and computer assembly. Beginning at startling stimulus onset, 65 consecutive 1 ms readings were recorded to obtain the peak amplitude of the animal’s startle response. A dynamic calibration system was used to ensure comparable sensitivities across chambers. Sound levels were measured as described previously using the A weighting scale in units of decibels sound pressure level (Risbrough and Geyer, 2005). The house light remained off throughout all testing sessions.
3.1.4. Experimental procedure
The session was modified from the test used for human subjects to include a greater range of ISIs to be characterized. Broad-band noise was used for all acoustic stimuli and background noise. The background was kept constant at 65 dB. The session began with 5 pulse-alone trials at 120 dB intensity (40 ms) to stabilize startle. The pre-pulse testing block then consisted of 5 pre-pulse trial types in which the 77 dB pre-pulse (20 ms) onset preceded the 120 dB pulse (40 ms) by 20, 70, 120, 360 or 1080 ms. Prepulse trials (5 per ISI type, 25 total) and pulse alone trials (7 total) were presented in pseudorandom order. The session ended with 5 pulse alone trials. The intertrial interval was 7–23 s with average of 15 s.
3.1.5. Data analysis
As with experiment 1, low and high baseline PPI groups were defined by median split of PPI performance across the 20–120 ms ISI trials. This grouping showed a similar median split cutoff for PPI performance as experiment 1 in human subjects (38.82%). Data were then analyzed with two separate ANOVAs. First, to best match the human PPI parameters, we examined PPI across the ascending limb of the ISI curve (20–120 ms) trials and conducted a 3 way ANOVA with performance group (low, high) as a between-groups factor and dose (0, 30, 100 mg/kg) and ISI (20–120) as a within-subject factor. Second, we conducted a 3-way repeated measures ANOVA with PPI group as a between-groups factor and dose and all ISIs on both the ascending and descending limbs of the ISI curve (20–1080 ms) as within-subject factors. Habituation effects were analyzed with a 3 way ANOVA with startle block and dose as within subject factors and PPI group as a between subject factor. Bonferroni-corrected post-hoc tests were conducted to clarify significant main effects and interactions.
3.2. Results
3.2.1. Startle reactivity
Means and standard deviations for startle reactivity by PPI group and dose can be seen in Table 2. Pregabalin had no effect on startle magnitude, nor was there an effect across PPI group. Startle habituation was also unaffected by PPI group or pregabalin (data not shown, main effect of Block: F(1,23) = 68, p < 0.0001 no interaction with Dose or Group).
3.2.2. Prepulse inhibition
Mean changes in PPI across dose by group is shown in Fig. 1B. Pregablin treatment effects on PPI were dependent upon PPI group [Dose × Group interaction F(2,44) = 6.10, p <.005, partial η2 =.22]. In the low PPI group, 100 mg/kg Pregabalin treatment significantly increased PPI compared to vehicle (p <.03). Conversely, the high PPI group exhibited reduced PPI after 30 and 100 mg/kg treatments compared to vehicle (ps <.03). The ISI (20–120 ms) did not interact with dose or group. In the second analysis that included the long ISIs in the model (380–1080 ms), pregabalin effects on PPI were dependent on ISI [Drug × Group × ISI F(8,176) = 2.33, p < 0.05]. When a post-hoc ANOVA was conducted with the long ISI trials only, there were no significant effects or interactions between ISI, dose or group (data collapsed across 360–1080 ms ISI: Low PPI group: 13 ± 6, 11 ± 8, 22 ± 8 across placebo, 30 and 100 mg respectively; High PPI group: 23 ± 4, 24 ± 8, 10 ± 9 across placebo, 30 and 100 mg respectively). These results suggest that pregabalin effects are not present at long ISI trials, likely due to the relatively variable and low PPI typically seen with very long ISI parameters (>300 ms).
4. General discussion
To our knowledge, this report represents the first investigation of pregabalin effects on sensorimotor gating in humans or mice. These experiments demonstrated that pregablin treatment increases PPI in healthy samples of both humans and mice that exhibit low baseline gating performance (lower 50% of median performance during placebo test). This median-split approach in healthy subjects has also been shown to be sensitive to atypical antipsychotics (Holstein et al., 2011; Vollenweider et al., 2006), as has an approach using healthy subjects performing within the lower quartile of normative samples (Swerdlow et al., 2006). Pregabalin effects on PPI were most robust at short ISIs in mice, which is also consistent with findings from atypical antipsychotics in healthy humans (Holstein et al., 2011; Vollenweider et al., 2006). Human subject eyeblink responses were reduced after 200 mg/kg pregabalin treatment suggesting a sedative effect at this dose, but this effect was similar across low and high gaters. Pregabalin had no significant effects on startle in mice, which may be due to differences in startle measures (eye blink vs. whole body response), plasma levels or pharmacokinetics across the human and mouse studies. These data suggest that the pregablin effects on PPI are unlikely to be an artifact of drug effects on startle reactivity overall. Startle reactivity was also not different between high and low PPI groups, consistent with other studies showing startle reactivity does not differ across subjects stratified for gating performance (Csomor et al., 2008; Vollenweider et al., 2006). Taken together, these data support further examination of pregabalin treatment in disorders linked to deficiencies in gating.
The present study also found that pregabalin treatment reduced PPI in mice and humans with high baseline gating levels. Though this decrease only reached statistical significance in mice, the same pattern is evident in humans as well, suggesting that the human subject sample may have been underpowered to detect this difference (n = 7 and 12/group for human and mouse study respectively). The pregabalin-induced decreases in PPI in high gaters are similar to the reductions in PPI observed after antipsychotic treatment in high gaters (Holstein et al., 2011; Gogos and van den Buuse, 2007). This opposing effect on PPI across low and high gaters may suggest that pregabalin is affecting a neural mechanism that modulates PPI via an inverted-U shaped curve. An analogous finding is the effects of COMT (catechol-o-methyltransferase) inhibition on PPI across individuals carrying the high or low efficiency COMT alleles (Giakoumaki et al., 2008). Subjects carrying the high efficiency alleles for the COMT gene, which presumably have lower dopamine levels in the prefrontal cortex showed low baseline PPI that was increased with treatment with the COMT inhibitor tolcapone. The opposite effect was found in individuals carrying the low efficiency COMT allele, exhibiting high baseline PPI that was reduced by tolcapone treatment. These effects were suggested to be due to the inverted U-shaped curve of cortical dopamine effects on PPI, with subjects with low dopamine tone showing increases after COMT inhibition while subjects that had higher tone were pushed into the “descending limb” of the response curve resulting in lower PPI. These effects were also mirrored in a working memory task. Thus pregabalin effects may be via a similar “inverted U” response mechanism, although the neurotransmitter and neural circuit mediating these effects is unknown.
One candidate mechanism underlying the present study effects is regulation of glutamatergic signaling in the forebrain. PPI has been shown to be disrupted by infusion of glutamate and glutamate agonists into the nucleus accumbens, ventral striatum, and ventral hippocampus (Klarner et al., 1998; Wan et al., 1996; Swerdlow et al., 1992). Further, NMDAR antagonists MK-801, ketamine and phencyclidine disrupt PPI in rodents (Martinez et al., 2000; Mansbach and Geyer, 1991). These effects can be mimicked by direct infusion of MK-801 into the dorsal hippocampus, amygdala, or medial prefrontal cortex, but not striatum (Bakshi and Geyer, 1998). mGlu2/3R agonists reduce both glutamate release and schizophrenia-like behaviors in animal models, although the effects on NMDA antagonist-induced disruptions in PPI is inconsistent (Imre et al., 2006; Galici et al., 2005; Henry et al., 2002). Further, N-acetylaspartylglutamate peptidase inhibitors ZJ43 and 2-PMPA suppress glutamate release but failed to reverse phencyclidine-induced PPI deficits (Profaci et al., 2011). These data indicate that modulation of glutamate tone does not consistently affect PPI disruptions induced by NMDA receptor blockade. If the pregabalin-induced increases in PPI are mediated by a reduction in glutamate tone, this would suggest that pregabalin effects on PPI in low gaters would not be via normalization of a putative hypo-NMDA receptor activation state in these subjects. It is also important to note, treatment with the NMDAR antagonist ketamine in humans increases PPI, suggesting that the NMDA-modulation of PPI may not be completely conserved across species (Abel et al., 2003). In the present study, pregabalin effects were mirrored across animals and humans, suggesting that the mechanism by which it acts on PPI is conserved across species.
Pregabalin also has the effect of inhibiting the neurotransmitters norepinephrine, Substance P, and serotonin, without demonstrated effects on dopamine (Brawek et al., 2008; Fehrenbacher et al., 2003; Maneuf et al., 2001; Dooley et al., 2000). Thus, these transmitter systems must also be considered as potential pathways through which pregabalin may affect PPI. The possibility of a noradrenergic mechanism is supported by recent research which has shown that stimulation of the locus coeruleus (LC) causes disruption of PPI mediated by downstream norepinephrine (NE) release (Bakshi and Alsene, 2010). In further work, Alsene and colleagues (2011) have identified a candidate thalamocortical network (including the posterior medial prefrontal cortex (mPFC), basolateral amygdala (BLA), and mediodorsal thalamus) through which excess NE activity may disrupt PPI. Thus, pregabalin activity in the mPFC and BLA may have the effect of attenuating NE signaling and facilitating PPI in low gaters. Noradrenergic modulators such as clonidine however do not affect PPI in healthy controls, although possible differential effects were not examined across high and low gating groups (Samuels et al., 2007). Substance P has been shown to elicit excitation in the caudal pontine reticular nucleus (PnC; Kungel et al., 1994). The PnC mediates PPI as the point of convergence between excitatory glutamatergic projections and inhibitory cholinergic projections at the acoustic startle circuit (Koch and Schnitzler, 1997). Thus, excess Substance P may override cholinergic inhibition and reduce PPI. Normalization of Substance P by pregabalin may then facilitate PPI in these individuals. Finally, a substantial body of work supports modulation of PPI by the serotonin system, particularly involving the projections between the median raphe nucleus and dorsal hippocampus and 5-HT1A/1B receptor activation (Adams and van den Buuse, 2011; Geyer and Vollenweider, 2008; van den Buuse et al., 2011). The effect of on the serotonin system also represents a potential pathway through which pregabalin may modulate PPI.
Practically, the ability of pregabalin to facilitate PPI in low gaters suggests that this compound may have clinical utility as a primary or adjunctive therapy for disorders characterized by impaired sensorimotor gating. Specifically, PPI has been historically used as a screening measure for antipsychotic drugs targeting schizophrenia (Swerdlow et al., 2008; Geyer et al., 1990). The effect of pregabalin on this measure, the emerging research on the role of excess forebrain glutamate in schizophrenia (Moghaddam and Javitt, 2012), and case studies supporting the efficacy of pregabalin as an adjunctive treatment for schizophrenia (Englisch et al., 2010) suggest that this compound may have utility for treatment of this disorder. Beyond schizophrenia, pregabalin may have utility as an adjunctive treatment for other disorders characterized by impaired PPI such as obsessive-compulsive disorder (Ahmari et al., 2012). Indeed, recent research has suggested that pregabalin may be effective for OCD as an adjunct to standard SSRI/atypical antipsychotic treatment, possibly through reduction of glutamatergic neurotransmission (Oulis et al., 2011; Di Nocola et al., 2011). Further studies of pregabalin augmentation for OCD are ongoing.
The current studies contain some limitations. First, the sample size in study 1 was relatively small, however the pattern of results was confirmed in study 2 in mice. Another potential limitation of Study 1 is that menstrual cycle was not controlled for in female subjects. PPI is affected by menstrual phase in humans, with decreased inhibition observed during the luteal phase (Jovanovic et al., 2004; Swerdlow et al., 1997). However, there is no noticeable sex imbalance across median groups or phase imbalance across dose (see Table 1). Additionally, pregabalin had the same pattern of effects across a mixed sex sample of humans and male mice. Thus, it is unlikely that sex or menstrual cycle were significantly influencing the results. One may also be concerned that the median split procedure has to potential to generate a regression to the mean effect. However, we feel that this is unlikely to account for such robust effects given that PPI demonstrates very high test-retest reliability over significant lengths of time in both normal and psychiatric populations (Swerdlow et al., 2009; Talledo et al., 2009; Light et al., 2007). Thus, it is unlikely that PPI would fluctuate so dramatically over the span of 2–3 weeks nor in a dose dependent manner. Overall, this report points to a potentially novel application for gabapentenoid compounds in disorders associated with disruption in gating, including schizophrenia. Future research is needed to elucidate the mechanism and neural substrates of action underlying these effects (e.g. forebrain modulation of glutamate signaling) and to demonstrate facilitation of PPI in patient populations.
Acknowledgments
The authors thank Chelsea Wallace, Robert Voloshin, and Linda Johnson for excellent technical assistance. Funding for this study includes: NARSAD Young Investigator Award (DTA), Veterans Affairs Merit (MPP), NIH MH64122 and MH075792 (MBS), and NIMH 076497 (VBR) and MH042228 (MAG).
References
- Abel K, Allin MP, Hemsley DR, Geyer MA. Low doses of ketamine increase prepulse inhibition in healthy men. Neuropharmacology. 2003;44:729–737. doi: 10.1016/s0028-3908(03)00073-x. [DOI] [PubMed] [Google Scholar]
- Adams W, van den Buuse M. Hippocampal serotonin depletion facilitates the enhancement of prepulse inhibition by risperidone: possible role of 5-HT(2C) receptors in the dorsal hippocampus. Neuropharmacology. 2011;61:458–467. doi: 10.1016/j.neuropharm.2011.03.018. [DOI] [PubMed] [Google Scholar]
- Alsene KM, Rajbhandari AK, Ramaker MJ, Bakshi VP. Discrete forebrain neuronal networks supporting noradrenergic regulation of sensorimotor gating. Neuropsychopharmacology. 2011;36:1003–1014. doi: 10.1038/npp.2010.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmari SE, Risbrough VB, Geyer MA, Simpson HB. Impaired sensorimotor gating in unmedicated adults with obsessive-compulsive disorder. Neuropsychopharmacology. 2012 Jan 4; doi: 10.1038/npp.2011.308. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aupperle RL, Ravindran L, Tankersley D, Flagan T, Stein NR, Simmons AN, Stein MB, Paulus MP. Pregabalin influences insula and amygdala activation during anticipation of emotional images. Neuropsychopharmacology. 2011;36:1466–1477. doi: 10.1038/npp.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi VP, Geyer MA. Multiple limbic regions mediate the disruption of prepulse inhibition produced in rats by the noncompetitive NMDA antagonist dizocilpine. J Neurosci. 1998;18:8394–8401. doi: 10.1523/JNEUROSCI.18-20-08394.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi VP, Alsene KM. Locus coeruleus: a novel substrate in the regulation of sensorimotor gating. Neuropyschopharmacology. 2010;35:S292. [Google Scholar]
- Bech P. Dose-response relationship of pregabalin in patients with generalized anxiety disorder. A pooled analysis of four placebo-controlled trials. Pharmacopsychiatry. 2007;40:163–168. doi: 10.1055/s-2007-984400. [DOI] [PubMed] [Google Scholar]
- Bitsios P, Giakoumaki SG, Frangou S. The effects of dopamine agonists on prepulse inhibition in healthy men depend on baseline PPI values. Psychopharmacology. 2005;182:144–152. doi: 10.1007/s00213-005-0056-x. [DOI] [PubMed] [Google Scholar]
- Braff DL, Grillon C, Geyer MA. Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry. 1992;49:206–215. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects patient groups and pharmacological studies. Psychopharmacology. 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Brawek B, Loffler M, Dooley DJ, Weyerbrock A, Feuerstein TJ. Differential modulation of K(+)-evoked (3)H-neurotransmitter release from human neocortex by gabapentin and pregabalin. Naunyn Schmiedbergs Arch Pharmacol. 2008;376:301–307. doi: 10.1007/s00210-007-0237-8. [DOI] [PubMed] [Google Scholar]
- Busch JA, Strand JC, Posvar EL, Bockbrader HN, Radulovic LL. Pregabalin (CI-1008) single-dose pharmacokinetics and safety/tolerance in healthy subjects after oral administration of pregabalin solution or capsule doses. Epilepsia. 1998;39:58. [Google Scholar]
- Cecil KM, Lenkinski RE, Gur RE, Gur RC. Proton magnetic resonance spectroscopy in the frontal and temporal lobes of neuroleptic naïve patients with schizophrenia. Neuropsychopharmacology. 1999;20:131–140. doi: 10.1016/S0893-133X(98)00063-3. [DOI] [PubMed] [Google Scholar]
- Chaki S, Hikichi H. Targeting of metabotropic glutamate receptors for the treatment of schizophrenia. Curr Pharm Des. 2011;17:94–102. doi: 10.2174/138161211795049570. [DOI] [PubMed] [Google Scholar]
- Csomor PA, Stadler RR, Feldon J, Yee BK, Geyer MA, Vollenweider FX. Haloperidol differentially modulates prepulse inhibition and P50 suppression in healthy humans stratified for low and high gating levels. Neuropsychopharmacology. 2008;33:497–512. doi: 10.1038/sj.npp.1301421. [DOI] [PubMed] [Google Scholar]
- Di Nocola M, Tedeschi D, Martinotti G, De Vita O, Monetta M, Pozzi G, Janiri L. Pregabalin augmentation in treatment-resistant obsessive-compulsive disorder: a 16-week case series. J Clin Psychopharmacol. 2011;31:675–677. doi: 10.1097/JCP.0b013e31822c29a8. [DOI] [PubMed] [Google Scholar]
- Dooley DJ, Donovan CM, Pugsley TA. Stimulus-dependent modulation of [(3)H]norepinephrine release from rat neocortical slices by gabapentin and pregabalin. J Pharamacol Exp Ther. 2000;295:1086–1093. [PubMed] [Google Scholar]
- Dulawa SC, Geyer MA. Psychopharmacology of prepulse inhibition in mice. Chin J Physiol. 1996;39:139–146. [PubMed] [Google Scholar]
- Dworkin RH, Corbin AE, Young JP, Sharma U, LaMoreaux L, Bockbrader H, et al. Pregabalin for the treatment of postherpetic neuralgia: a randomized, placebo-controlled trail. Neurology. 2003;60:1274–1283. doi: 10.1212/01.wnl.0000055433.55136.55. [DOI] [PubMed] [Google Scholar]
- Englisch S, Eber A, Enning F, Hohmann S, Schanz H, Zink M. Augmentation with pregabalin in schizophrenia. Psychopharmacology. 2010;30:437–440. doi: 10.1097/JCP.0b013e3181e5c095. [DOI] [PubMed] [Google Scholar]
- Errante LD, Petroff OA. Acute effects of gabapentin and pregabalin on rat forebrain cellular GABA, glutamate, and glutamine concentrations. Seizure. 2003;12:300–306. doi: 10.1016/s1059-1311(02)00295-9. [DOI] [PubMed] [Google Scholar]
- Fehrenbacher JC, Taylor CP, Vasko MR. Pregabalin and gabapentin reduce release of substance P and CGRP from rat spinal tissues only after inflammation or activation of protein kinase C. Pain. 2003;105:133–141. doi: 10.1016/s0304-3959(03)00173-8. [DOI] [PubMed] [Google Scholar]
- French JA, Kugler AR, Robbins JL, Knapp LE, Garofalo EA. Dose-response trial of pregabalin adjunctive therapy in patients with partial seizures. Neurology. 2003;60:1631–1637. doi: 10.1212/01.wnl.0000068024.20285.65. [DOI] [PubMed] [Google Scholar]
- Galici R, Echemendia NG, Rodriguez AL, Conn PJ. A selective allosteric potentiator of mGluR2 receptors has similar effects to an orthosteric mGluR2/3 agonist in mouse models predictive of antipsychotic activity. J Pharmacol Exp Ther. 2005;15:1181–1187. doi: 10.1124/jpet.105.091074. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Swerdlow NR, Mansbach RS, Braff DL. Startle response models of sensorimotor gating and habituation deficits in schizophrenia. Brain Res Bull. 1990;25:485–498. doi: 10.1016/0361-9230(90)90241-q. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Vollenweider FX. Serotonin research: contributions to understanding psychoses. Trends Pharmacol Sci. 2008;29:445–453. doi: 10.1016/j.tips.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Giakoumaki SG, Roussos P, Bitsios P. Improvement of prepulse inhibition and executive function by the COMT inhibitor tolcapone depends on the COMT val158met polymorphism. Neuropsychopharmacology. 2008;33:3058–3068. doi: 10.1038/npp.2008.82. [DOI] [PubMed] [Google Scholar]
- Gogos A, van den Buuse M. The importance of baseline in identifying 8-OH-DPAT-induced effects on prepulse inhibition in rats. Br J Pharmacol. 2007;150:750–757. doi: 10.1038/sj.bjp.0707148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry SA, Lehman-Masten V, Gasparini F, Geyer MA, Markou A. The mGluR5 antagonist, MPEP, but not the mGluR2/3 agonist LY314582, augments PCP effects on prepulse inhibition and locomotor activity. Neuropharmacology. 2002;43:1199–1209. doi: 10.1016/s0028-3908(02)00332-5. [DOI] [PubMed] [Google Scholar]
- Holstein DH, Csomor PA, Geyer MA, Huber T, Brugger N, Studerus E, Vollenweider X. The effects of sertindole on sensory gating, sensorimotor gating, and cognition in healthy volunteers. Psychopharmacology. 2011 doi: 10.1177/0269881111415734. [DOI] [PubMed] [Google Scholar]
- Imre G, Salomons A, Jongsma M, Fokkema DS, Den Boer JA, Ter Horst GJ. Effects of the mGluR2/3 agonist LY379268 on ketamine-evoked behaviors and neurochemical changes in the dentate gyrus of the rat. Pharmacol Biochem Behav. 2006;84:392–399. doi: 10.1016/j.pbb.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Jovanovic T, Szilagyi S, Chakravorty S, Fiallos AM, Lewison BJ, Parwani A, et al. Menstrual cycle phase effects on prepulse inhibition of acoustic startle. Psychophysiology. 2004;41:401–406. doi: 10.1111/1469-8986.2004.00166.x. [DOI] [PubMed] [Google Scholar]
- Kavoussi R. Pregabalin: from molecule to medicine. Eur Neuropsychopharmacol. 2006;16:S128–S133. doi: 10.1016/j.euroneuro.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Klarner A, Koch M, Schnitzler HU. Induction of fos-protein in the forebrain and disruption of sensorimotor gating following N-methyl-d-aspartate infusion into the ventral hippocampus of the rat. Neuroscience. 1998;84:443–452. doi: 10.1016/s0306-4522(97)00475-2. [DOI] [PubMed] [Google Scholar]
- Koch M, Schnitzler H. The acoustic startle response in rats – circuits mediating evocation, inhibition and potentiation. Behav Brain Res. 1997;89:35–49. doi: 10.1016/s0166-4328(97)02296-1. [DOI] [PubMed] [Google Scholar]
- Krystal JH, D’Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology. 2003;169:215–233. doi: 10.1007/s00213-003-1582-z. [DOI] [PubMed] [Google Scholar]
- Kungel M, Ebert U, Hoerbert H, Ostwald J. Substance P and other putative transmitters modulate the activity of reticular pontine neurons: an electrophysiological and immunohistochemical study. Brain Res. 1994;643:29–39. doi: 10.1016/0006-8993(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Li Z, Taylor CP, Weber M, Piechan J, Prior F, Bian F, Cui M, Hoffman D, Donevan S. Pregabalin is a potent and selective ligand for α2δ-1 and α2δ-2 calcium channel subunits. Eur J Pharmacol. 2011;667:80–90. doi: 10.1016/j.ejphar.2011.05.054. [DOI] [PubMed] [Google Scholar]
- Light GA, Swerdlow NR, Cadenhead KS, Sprock J, Radant A, Braff DL. One year stability of neurophysiological and cognitive endophenotypes of schizophrenia. Proc Am Col Neuropsychopharmacology (Boca Raton, FL) 2007 [Google Scholar]
- Lotarski SM, Donevan S, El-Kattan A, Osgood S, Poe J, Taylor CP, Offord J. Anxiolytic-like activity of pregabalin in the vogel conflict test in α2δ-1 (R217A) and α2δ-2 (R279A) Mouse Mutants. J Pharmacol Exp Therapeut. 2011;338:615–621. doi: 10.1124/jpet.111.180976. [DOI] [PubMed] [Google Scholar]
- Ludewig S, Ludewig K, Geyer MA, Hell D, Vollenweider FX. Prepulse inhibition deficits in patients with panic disorder. Depress Anxiety. 2002;15:55–60. doi: 10.1002/da.10026. [DOI] [PubMed] [Google Scholar]
- Maneuf YP, Hughes J, McKnight AT. Gabapentin inhibits the substance P-facilitated K(+)-evoked release of [(3)H]glutamate from rat caudal trigeminal nucleus slices. Pain. 2001;93:191–196. doi: 10.1016/S0304-3959(01)00316-5. [DOI] [PubMed] [Google Scholar]
- Mansbach RS, Geyer MA. Parametric determinants in pre-stimulus modification of acoustic startle: interaction with ketamine. Psychopharmacology. 1991;105:162–168. doi: 10.1007/BF02244303. [DOI] [PubMed] [Google Scholar]
- Martinez Z, Oostwegel J, Geyer M, Swerdlow NR. Ontogeny of phencyclidine and apomorphine effects on prepulse inhibition (PPI) Pharmacol Biochem Behav. 2000;65:449–457. doi: 10.1016/s0091-3057(99)00217-8. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37:4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulis P, Mourikis I, Konstantakopoulos G. Pregabalin augementation in treatment-resistant obsessive-compulsive disorder. Int J Clin Psychopharmacol. 2011;26:221–224. doi: 10.1097/YIC.0b013e3283466657. [DOI] [PubMed] [Google Scholar]
- Pande AC, Feltner DE, Jefferson JW, Davidson JRT, Pollack M, Stein MB, Lydiard RB, Futterer R, Robinson P, et al. Efficacy of the novel anxiolytic pregabalin in social anxiety disorder. J Clin Psychopharmacol. 2004;24:141–149. doi: 10.1097/01.jcp.0000117423.05703.e7. [DOI] [PubMed] [Google Scholar]
- Profaci CP, Krolikowski KA, Olszewski RT, Neale JH. Group II mGluR agonist LY354740 and NAAG peptidase inhibitor effects on prepulse inhibition in PCP and D-amphetamine models of schizophrenia. Psychopharmacology. 2011;216:235–243. doi: 10.1007/s00213-011-2200-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quitero JE, Dooley DJ, Pomerleau F, Huettl P, Gerhardt GA. Amperometric measurement of glutamate release modulation by gabapentin and pregabalin in rat neocortical slices: role of voltage-sensitive Ca2+ α2δ-1 subunit. J Pharmacol Exp Therapeut. 2010;338:240–245. doi: 10.1124/jpet.110.178384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickels K, Pollack MH, Feltner DE, Lydiard B, Zimbroff DL, Bielski RJ, et al. Pregabalin for treatment of generalized anxiety disorder: a 4-week, multicenter, double-blind, placebo-controlled trial of pregabalin and alrpazolam. Arch Gen Psychiatry. 2005;62:1022–1030. doi: 10.1001/archpsyc.62.9.1022. [DOI] [PubMed] [Google Scholar]
- Risbrough VB, Geyer MA. Anxiogenic treatments do not increase fear-potentiated startle in mice. Biol Psychiatry. 2005;57:33–43. doi: 10.1016/j.biopsych.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Samuels ER, Hou RH, Langley RW, Szabadi E, Bradshaw CM. Modulation of the acoustic startle response by the level of arousal: comparison of clonidine and modafinil in healthy volunteers. Neuropsychopharmacology. 2007;32:2405–2421. doi: 10.1038/sj.npp.1301363. [DOI] [PubMed] [Google Scholar]
- Straube S, Derry S, Moore RA, McQuay HJ. Pregabalin in fibromyalgia: meta-analysis of efficacy and safety from company clinical trial reports. Rheumatology. 2010;49:706–715. doi: 10.1093/rheumatology/kep432. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, van Bergeijk DP, Bergsma F, Weber E, Talledo J. The effects of memantine on prepulse inhibition. Neuropsychopharmacology. 2009;34:1854–1864. doi: 10.1038/npp.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology. 2008;199:331–388. doi: 10.1007/s00213-008-1072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Talledo J, Sutherland AN, Nagy D, Shoemaker JM. Antipsychotic effects on prepulse inhibition in normal ‘low gating’ humans and rats. Neuropsychopharmacology. 2006;31:2011–2021. doi: 10.1038/sj.npp.1301043. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology. 2001;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Hartman PL, Auerbach PP. Changes in sensorimotor inhibition across the menstrual cycle: implications for neuropsychiatric disorders. Biol Psychiatry. 1997;41:452. doi: 10.1016/S0006-3223(96)00065-0. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Caine SB, Braff DL, Geyer MA. The neural substrates on sensorimotor gating of the startle reflex: a review. J Psychopharmacol. 1992;6:176–190. doi: 10.1177/026988119200600210. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA, Koob GF. Central dopamine hyperactivity in rats mimics abnormal acoustic startle response in schizophrenics. Biol Psychiatry. 1986;21:23–33. doi: 10.1016/0006-3223(86)90005-3. [DOI] [PubMed] [Google Scholar]
- Talledo JA, Owens ANS, Schortinghuis T, Swerdlow N. Amphetamine effects on startle gating in normal women and female rats. Psychopharmacology. 2009;204:165–175. doi: 10.1007/s00213-008-1446-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CP, Angelotti T, Fauman E. Pharmacology and mechanism of action of pregabalin: the calcium channel α2δ-2 subunit as a target for antiepileptic drug discovery. Epilepsy Res. 2007;73:137–150. doi: 10.1016/j.eplepsyres.2006.09.008. [DOI] [PubMed] [Google Scholar]
- van den Buuse M, Becker T, Kwek P, Martin S, Ruimschotel E, Risbrough V. Disruption of prepulse inhibition by 3,4-methylenedioxymethamphetamine (MDMA): comparison between male and female wild-type and 5-HT1A receptor knockout mice. Int J Neuropsychopharmacol. 2011;14:856–861. doi: 10.1017/S1461145711000101. [DOI] [PubMed] [Google Scholar]
- Vollenweider FX, Barro M, Csomor PA, Feldon J. Clozapine enhances prepulse inhibition in healthy humans with low but not with high prepulse inhibition levels. Biol Psychiatry. 2006;60:597–603. doi: 10.1016/j.biopsych.2006.03.058. [DOI] [PubMed] [Google Scholar]
- Wan FJ, Caine SB, Swerdlow NR. The ventral subiculum modulation of prepulse inhibition is not mediated via dopamine D2 or nucleus accumbens non-NMDA glutamate receptor activity. Eur J Pharmacol. 1996;314:9–18. doi: 10.1016/s0014-2999(96)00535-3. [DOI] [PubMed] [Google Scholar]