

Abstract
A new single-molecule approach for rapid and purely electronic discrimination among similar genes is presented. Combining solid-state nanopores and γ-modified synthetic peptide nucleic acid (γPNA) probes, we accurately barcode genes by counting the number of probes attached to each gene, and measuring their relative spacing. We illustrate our method by sensing individual genes from two highly similar human immunodeficiency virus (HIV) subtypes, demonstrating feasibility of a novel, single-molecule diagnostic platform for rapid pathogen classification.
Keywords: nanopore, γPNA, sequence detection, molecular diagnostics, single-molecule, genotyping
Molecular assays for infectious disease diagnosis have revolutionized the speed and accuracy with which viral diseases can be identified.1 Parallel advances in antiviral therapies require increasingly accurate and rapid pathogen classification on the genomic level.2 However, current molecular diagnostic platforms are based on processes so elaborate and time-consuming that they typically must be performed in centralized labs. The introduction of single-molecule based diagnostic sensing platforms could simplify these processes, resulting in shorter turnaround times, and potentially allowing the platform to be used in resource limited arenas.3 In this study, we report the application of a new type of single-molecule sensor, solid-state nanopores, to perform rapid molecular identification of viral genes and gene variants. This was achieved by combining highly sequence-specific γ-modified synthetic peptide nucleic acid (γPNA) probes4-7 with electrical sensing via solid-state nanopores, enabling rapid characterization of two nearly identical genes from two human immunodeficiency virus (HIV) subtypes (>92% similarity), which were originally extracted from unmarked human samples.
A solid-state nanopore is a synthetic device composed of a nanometer-scale pore fabricated in an ultra-thin membrane. Nanopores utilize an extremely simple electrical sensing principle: when biopolymers such as DNA or RNA are electrophoretically threaded across a pore, the ion current flowing through the pore is partially, and measurably, blocked.8, 9 It has recently been demonstrated that solid-state nanopores may be used to detect sub-molecular structural alterations in both DNA and RNA without the need for fluorescent/radio labels.10-12 Another practical feature of solid-state nanopores is their ability to actively funnel and subsequently capture extremely small copy numbers of DNA molecules, simply by establishing a salt gradient across the ultra-thin membrane.13 The DNA funneling and the highly sensitive electrical detection capabilities place nanopores among the most promising candidate platforms for molecular diagnostics, particularly for those involving limited sample quantities.10, 14 But despite these attributes, to date solid-state nanopores have seldom been utilized for molecular diagnostic sensing applications, primarily due to their inherent lack of DNA or RNA sequence specificity.
Recently, we reported that bis-PNA/DNA invasion sites can be detected with a nanopore, providing a strong basis for development of a nanopore-based genomic profiling method.15 bis-PNA, which can bind and intercalate into target regions of DNA, alters the local structure of the DNA at the point of binding.4 This structural change induces a local, detectable change in a solid-state nanopore’s ionic conductance when that region passes through the pore, allowing it to be readily discriminated from bare DNA regions. However, there are a number of major drawbacks for the utilization of bis-PNA for nanopore-based molecular diagnostic applications: First, bis-PNA forms structural isomers during the invasion process,16 leading to a noisy ion-current blockade signal. Second, bis-PNAs are restricted to purely homopurine target regions, severely limiting their general applicability for sequence-specific genotyping.4
In contrast, recently developed γPNA probes exhibit an extremely-high affinity for DNA, and can invade with high specificity without any sequence restrictions.5-7, 17 Moreover, γPNA forms a more streamlined duplex structure as compared to the bis-PNA triplex structure (Figure 1a). We speculated that the more compact γPNA /DNA duplex structure would allow us to utilize slightly smaller nanopores (as compared with those used for bis-PNA detection). By using slightly smaller nanopores, DNA translocation velocity is reduced,18 substantially improving the temporal resolution for multiple γPNA molecules bound to the same DNA target. To demonstrate the feasibility of this concept we first evaluated the ion current signals recorded for a ~ 3kbp DNA molecule, which includes three binding sites (15 bp long) for γPNA molecules. When the bare DNA molecule was threaded through a 3.7 nm pore crafted in a 30 nm thick silicon nitride film we obtained a single blocked-level ion current traces, as shown in Figure 1c. This blocked current level corresponds well to the expected blockade values for dsDNA considering the pore diameter used.18 Upon invasion of γPNA probes, three additional blockade episodes are observed during each translocation event, all conforming to a well-defined second blocked ion-current level (see statistical analysis of >1,500 events in Supporting Information). The results shown in Figure 1 demonstrate that in principle we can target and detect short sequences (i.e. 15 bp) in any DNA molecule of interest, forming the basis for the proposed single-molecule gene identification method.
Figure 1.
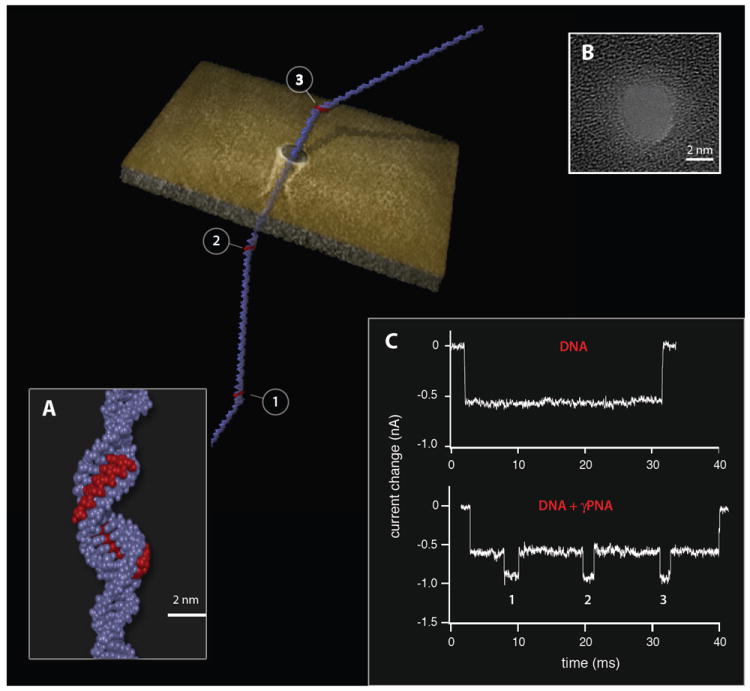
Electronic barcoding of a single DNA molecule using the nanopore/PNAs system. Illustration of a DNA molecule tagged with three equally spaced γPNA probes, translocating across a 3.7 nm solid-state nanopore sculpted in a thin silicon nitride membrane. A) Close-up approximated structure of a γPNA probe site. Given its high affinity for DNA, the single-stranded γPNA (red) binds to its complementary DNA sequence, locally displacing the second DNA strand. This process leads to a local change in the helical structure, which is detectable with the solid-state nanopore. B) High-resolution TEM image of a 3.7 nm nanopore, made in a thin silicon nitride film. C) In contrast to the translocation of an untagged or bare DNA molecule depicted by a single blockade episode for the duration of a translocation event, in the case of a γPNA tagged DNA molecule three distinct secondary blockade episodes are present, clearly marking the points in time when the γPNA-tagged region crossed through the nanopore.
Ultimately, a nanopore/γPNA-based genotyping method will both count the number of γPNA/DNA sites per DNA molecule and also localize their positions along a DNA molecule, effectively barcoding the target. To demonstrate that nanopores can not only count bound γPNA probes, but can also be used to accurately determine the distance between two γPNA probes along a DNA molecule, we designed six model DNA molecules, each containing two identical γPNA invasion sites to be used as ‘molecular rulers’ (see Supporting Information for the sample preparation procedure). These molecular rulers are used to calibrate the nanopore system, enabling a simple transformation from the nanopore’s time domain data (the lag time between two γPNA pulses, δt) to the DNA’s spatial coordinates (the gap in basepairs between the two γPNA sites, Δn). For these model molecules, Δn ranged from 100 to 1000 bp, while the two outer flanking regions of DNA were kept identical (~1,200 bp) for consistency, as shown in Figure 2a. Nanopore assays were conducted for each molecule to measure the average δt values for a statistical population of at least 1,000 molecules in each case. Typical examples of events for each value of Δn are shown in Figure 2b (see Supporting Information for statistical analysis). Figure 2c depicts the average value of δt (error bars represent STD of the data), plotted as a function of the actual distance Δn between γPNA sites. Figure 2c shows that δt can be accurately measured for even the shortest Δn tested (100 bp), corresponding to roughly a ~34 nm spacing. The relationship between δt and Δn shown in Figure 2c can be used empirically to estimate the spacing between two unknown γPNAs binding sites. We note that this relationship is well fitted by a power law with an exponent of 1.39 ± 0.09, in agreement with previous experiments that measured the translocation time as a function of length for untagged DNA molecules of similar length.18
Figure 2.
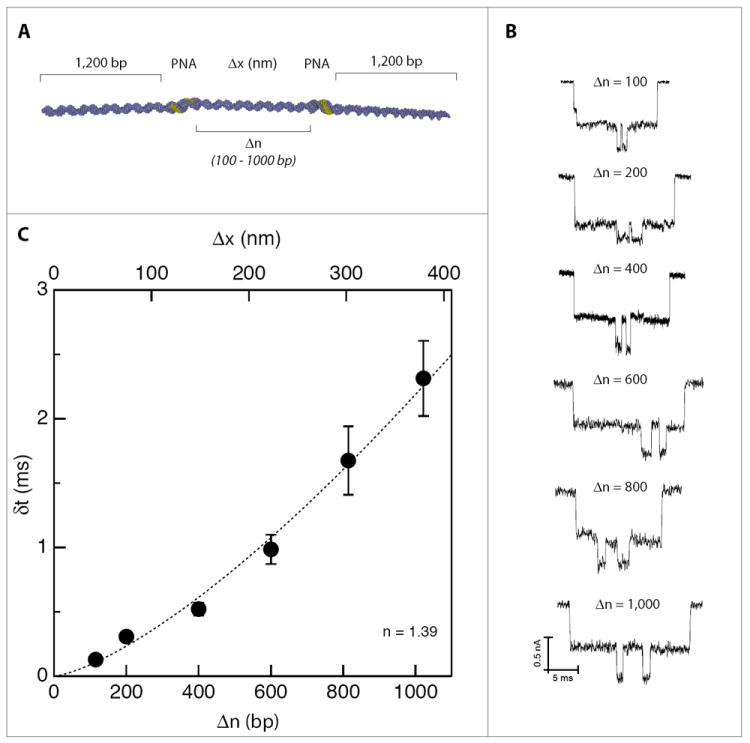
Varying the distance between two γPNA sites to create a time (δt) to distance (Δn or Δx) conversion map. A) Six nearly identical and symmetrical DNA molecules were created, consisting of identical flanking regions (1,200 bp before the first γPNA site and 1,200 bp after the second γPNA site), and incorporating varied spacing between the two γPNA sites; B) Representative single-molecule traces illustrating the time-delay differences, δt, among the various molecules; C) Statistical analysis further illuminating the relationship between δt and the γPNA site-to-site spacing (as measured in bp). The data is well approximated by a power law dependence with an exponent of 1.39 (dashed line).
Since target identification is established through a combination of both probe counting and probe spacing, it should be possible to design relatively small probe libraries that can uniquely identify a multitude of genomic targets by probing the incidence and spatial distribution of a small set of sequences. To evaluate the feasibility of this approach, we leveraged the high sequence specificity of 15-mer γPNA probes17 in a simple barcoding strategy: When a probe library set is added to the DNA sample of interest, only those probes which have sequences complementary to the designed targets will bind to the extracted DNA. In practice, this principle allows us to create maps of possible binding patterns, termed barcodes, which are specific to a given library of probes. The pattern of γPNA delay times acquired in a nanopore assay can then be directly compared against these maps to identify the presence (or absence) of DNA molecules of interest.
To illustrate this approach using genomic samples that naturally require the design and detection of multiple γPNA sequences, we chose HIV genomes as a test system. Two nearly-identical DNA fragments containing the sequences for the pol gene (3,050 bp), extracted from unmarked HIV-1/B and HIV-1/C subtypes, were prepared using standard methods.19 Four different γPNA oligomers were synthesized in parallel, designed so that only two of the PNA oligomers would be a perfect match for both subtypes (γPNA1 and γPNA2), while γPNA3 would bind to a sequence present only in subtype B and γPNA4 would bind to a sequence present only in subtype C (see Supporting Information). Binding the PNA probe libraries to each sample creates two distinct barcodes: γPNA1 and γPNA2 are spaced ~850 bp apart and bind to the same locations on both subtypes, γPNA3 binds ~850 bp after γPNA2 on subtype B only, and γPNA4 binds ~450 bp after γPNA2 on subtype C only (Figure 3). Correct γPNA binding patterns were confirmed by cutting each gene into ten fragments, each 300-400 bp, used for gel-shift binding assays to confirm binding specificity. These correct binding patterns were first demonstrated using individual types of γPNA oligos, then confirmed using the complete library set. Figure 3 shows that γPNA1 and γPNA2 bind only to fragments VIII and V, respectively, in both pol subtypes. However, γPNA3 binds to fragment II only in subtype B (and not to the corresponding fragment II from subtype C). Likewise, γPNA4 binds only to fragment IV in subtype C (and not to fragment IV from subtype B). Furthermore, the full library containing all four γPNA probes displayed identical binding patterns to those four probes bound separately. These gel-shift binding experiments confirm the sequence specificity of the γPNA probes, since the nonbinding fragments of γPNA3 and γPNA4 were nearly identical in sequence, with only a few mismatches (see Supporting Information).
Figure 3.
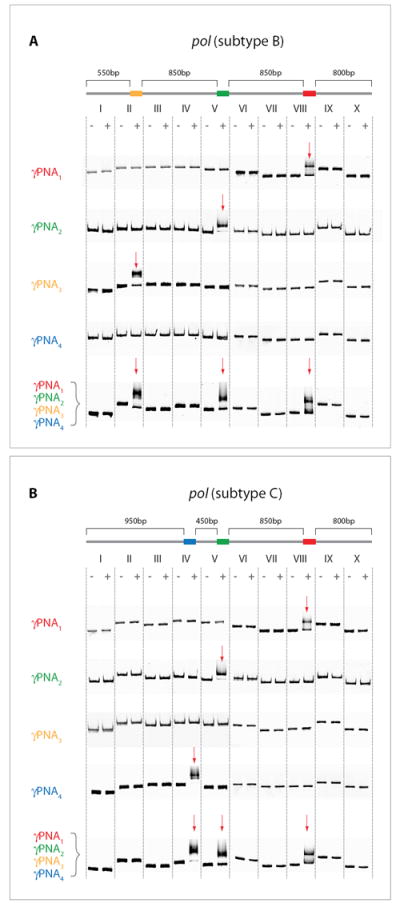
Mapping of the HIV pol gene. The 3,050 bp long pol gene - A)-subtype B, and B)-subtype C - was split into ten fragments, allowing us to determine onto which fragment a particular γPNA probe would bind using gel shift assays (see red arrow). γPNA1 and γPNA2 bound to fragments VIII and V, respectively, on both HIV subtypes. γPNA3 bound to fragment II on subtype B only, while γPNA4 bound to fragment IV on subtype C only. Mixing all four γPNA probes displayed the same binding pattern for the individual subtypes as was observed for each probe individually.
The nanopore method proposed here should provide a simple and efficient alternative to the laborious and time-consuming gel-based analysis described in Figure 3, only requiring a few minutes to make a single measurement for each of the two HIV genes subtypes. As expected, nanopore analysis of the two untagged subtypes produced practically indistinguishable data sets, due to the fact that the two DNA molecules are highly similar in length and sequence (see Supporting Information). In contrast, the two types of γPNA-tagged samples each produced a unique ion current signal, which could be visually distinguished: subtype B tagged molecules produced two nearly identical δt values (1.9±0.2 ms and 2.1±0.2 ms), whereas subtype C produced two different δt values (0.79±0.10 ms and 2.1±0.3 ms), corresponding to the locations of the bound PNA along the length of the DNA (see Supporting Information).
To simplify subtype classification, we defined the normalized first and second time delays as τ1 = δt1–2/(δt1–2 + δt2–3) and τ2 = δt2–3/(δt1–2 + δt2–3), respectively. This normalization facilitated the analysis of hundreds of single-molecule events, allowing for machine-based discrimination between the two subtypes. Figure 4 displays the resulting distributions of τ1 and τ2 for subtype B and subtype C (red and blue, respectively, 750 events in each case). The symmetrically-tagged subtype B variant yields two nearly identical Gaussian distributions with a single peak at 0.50±0.02 (error represents STD), reflecting nearly equal delay times between the PNA probes, as expected. In contrast, the subtype C variant yielded double peak Gaussian distributions with peaks centered at 0.27±0.02 and 0.75±0.01 for τ1, and 0.25±0.01 and 0.73±0.02 for τ2, respectively. Referring back to our calibration curve (Figure 2) we note that the expected values of the delay time associated with the 450 bp and 850 bp gaps (0.61 ms and 1.85 ms, respectively) coincide with the observed mean values of τ1 and τ2, since 0.61/(0.61+1.85) = 0.25 and 1.85/(0.61+1.85) = 0.75, reflecting consistency with the calibration experiment. The appearance of two nearly identical double-peaked distributions for subtype C may be explained simply by the asymmetry of its corresponding barcode and the fact that either end of the DNA can enter the nanopore first. We do note that the absolute values of δt in the calibration assay are slightly shorter than the values obtained in the current experiment. These small variations can be explained by slight differences in pore geometries used in these experiments. Nevertheless, the internal normalization of the delay times appears to nullify slight pore-to-pore variations, enabling a more straightforward and simpler analysis.
Figure 4.
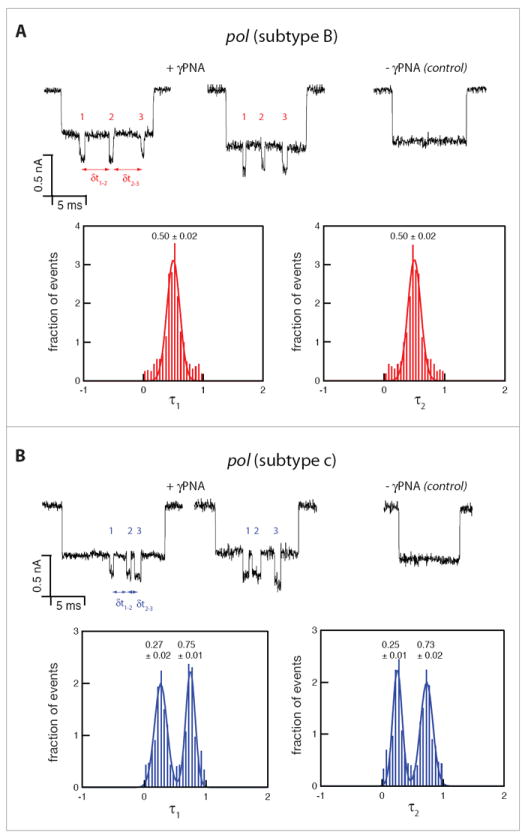
Nanopore-based analysis of γPNA-tagged HIV pol gene displaying two unique barcodes for each of the variants, while both untagged variants are indistinguishable. The distributions of the normalized delay times, τ1 and τ2 measured for the two HIV-subtypes (750 events in each case). A) Subtype B variant displays a single distribution for both τ1 and τ2, with a mean±STD value of 0.50±0.02. B) Subtype C variant, displays two distinct populations with mean±STD values of 0.27±0.02 and 0.75±0.01 for τ1, and 0.25±0.01, 0.73±0.02 for τ2.
The data in Figure 4 demonstrates that our method can readily discriminate between these two highly similar HIV subtypes. In the case of HIV subtype B and subtype C, which display <8% sequence variance, identification is confirmed here with just four unique γPNA probes. Nanopore sensors are easily extensible to characterization of much longer DNA molecules, representing significantly more complex genomes. The method we describe here can thus be used to detect and discriminate among a wide range of highly similar genomes based on subtle variations in sequence. Improved sensitivity or broader applicability of this technique can be achieved by introducing more γPNA probes into the library, utilizing the ability to accurately resolve the spatial pattern of γPNA sites along the DNA molecule screened without significantly complicating the readout process. The effect of small pore-to-pore variations, which might slightly affect translocation velocity (and therefore δt) of tagged DNA, can be compensated for by employing the simple normalization method introduced in Figure 4.
Recent technological advances have allowed disease identification techniques to shift from antibody- to nucleic acid-based assays, enabling genotypic sensitivity, and will facilitate the continued transformation of these techniques from large laboratory-based sample testing to point-of-care diagnostics.1 Further advancements in molecular diagnostics will allow sub-classification of diseases with certain shared pathophysiological features but different responses to treatment.20 The results presented here support a new nanopore-based genomic barcoding and electronic readout method that can quickly identify genes from an unknown sample using only a small γPNA library. The sequence sensitivity of this approach is achieved through the use of specific γPNA probes, which are far more sensitive to sequence mismatches than DNA probes, and which can be incorporated into thoughtfully designed γPNA libraries to expand the range of identifiable target molecules.21 The high throughput, minute sample requirements, and spatial resolution of this approach are all achieved through the use of solid-state nanopores. By combining nanopores with γPNA probes, we were able to detect and classify genes of closely related viral subtypes, demonstrating the potential of this novel diagnostic platform.
Methods
γPNA oligomers were synthesized according to published procedures.5, 6 Upon cleavage from the resin, oligomers were purified by reverse-phase HPLC to homogeneity and verified by MALDI-TOF mass spectrometry. 3.7 ± 0.3 nm single nanopores were fabricated in 30 nm thick, low-stress LPCVD deposited SiN membranes (5×5 μm2), supported by a 380 μm thick Si chip with a 1 μm layer of SiO2, using a focused electron beam as previously described.22 Prior to use, nanopore chips were boiled for 15 min in a Piranha solution (2 parts 95% H2SO4, 1 part 35% H2O2) and assembled in a custom-designed cell under controlled atmospheric conditions as previously described.23 After assembly, nanopores were hydrated with a degassed and filtered KCl electrolyte solution (adjusted to pH 7.5 using 10 mM Tris-HCl). The nanopore cell was then placed in a custom-designed chamber, featuring thermoelectric regulation within ±0.1°C, rapid thermal equilibration (<5 min), and electromagnetic shielding. Homemade Ag/AgCl electrodes were immersed into each chamber of the cell and connected to an Axon 200B (Molecular Devices, CA, USA) headstage. All measurements were performed inside a dark Faraday cage.
All experiments used 0.2M/1M KCl solutions (cis and trans chambers, respectively) and an applied voltage of 300 mV. Asymmetric electrolyte conditions were employed to yield increased dsDNA capture rates and extended dwell times, as compared with symmetric salt concentration solutions.13 In a typical experiment, DNA solutions at 0.1 fmole/μl were added to the cis chamber, yielding a DNA capture rate of ~1,000 events/10 min. Translocation data was acquired using custom-built LabVIEW (National Instruments, TX, USA) software, which collects both continuous current data as well as individual ion current blockade pulses (“translocation events”) detected in real-time. Translocation events were identified based on two criteria: 1) A minimum dwell time of 100 μs; and 2) A minimum current blockade amplitude of 0.35 nA before returning to the original “open pore” state. The analog signal from the Axon amplifier was first low-pass filtered with a 50 kHz Butterworth filter (typical RMS noise values of 40-50 pA) before being fed into a DAQ card (PCI-6230, National Instruments, TX, USA), which sampled the data at 250 kHz/16 bit. Data was exported to Igor Pro software, where all data was analyzed. DNA and PNA/DNA blockade levels were determined by creating an all-points-histogram for each translocation event and identifying peaks for each blockade level. PNA/DNA complex sites were identified by a characteristic blockade level of > 2.5 × RMS.
Near-full length bacterial clones of both HIV subtype variants were obtained from the AIDS Research and Reference Reagent program, NIAID, NIH. Subtypes HIV-1/B and HIV-1/C, reagent numbers p98CN009.8 and p98IS002.5, respectively, were received from Drs. Cynthia M. Rodenburg, Beatrice H. Hahn, Feng Gao, and the UNAIDS Network for HIV Isolation and Characterization.19 Clones were selectively grown on either 50 μg/ml Ampicillin or 50 μg/ml Kanamycin, and plasmids were isolated, purified, and sequenced to ensure that plasmid sequences matched those obtained from Genebank.19 Isolation of the pol gene from the two HIV subtypes was achieved through standard PCR reactions. Given the near-identical nature of the pol sequence for the two subtypes used here, identical primer sequences and PCR conditions were employed to amplify this gene. PCR amplicons were purified and sequenced to ensure sequence validity and analyzed on a 12 kbp Experion DNA chip (BioRad) (see Figure S3a in Supporting Information).
Based on our sequencing results for the two pol genes, four γPNA target regions were selected to barcode the pol gene. The first target site was placed ~800 bp from one end of the dsDNA, ensuring that the DNA blockade level would be recognizable prior to that of the first observed γPNA/DNA blockade level. The first two targets, Site 1 and Site 2, are common to both HIV-1 subtypes in both sequence and location, and are spaced ~850 bp apart. All target site sequences and their corresponding γPNA probe sequences are presented in Table 1 found in the Supporting Information.
The target sequence that is perfectly complementary to γPNA3 is located only on HIV-1/B, and is located ~850 bp upstream from Site 2. The sequence found in the same location on subtype C is shown in Table 1 as well, with the mismatched bases in red. The target sequence that is perfectly complementary to γPNA4 is located only on HIV-1/C and is located ~450 bp upstream from Site 2. Similarly, Table 1 shows the perfect complementarity of Site 4 with γPNA4, while the mismatches that prevent binding of γPNA4 to subtype B are shown in red. In all cases, ‘X’ denotes the synthetic nucleobase 9-(2-guanidinoethoxy) phenoxazine or ‘G-clamp’ as described previously.6 All other nucleobases are natural, and the γPNA oligomer was synthesized and purified as previously described.6, 7
Supplementary Material
Acknowledgments
The authors acknowledge support from Harvard’s Center for Nanoscale Systems (CNS) and financial support from National Institutes of Health award R01 HG-005871 (A.M.) and National Science Foundation awards PHY-0646637 (A.M.) and CHE-1012467 (D.H.L.), as well as the DSF Charitable Foundation (D.H.L.). The authors thank A. Squires for editing the manuscript.
Footnotes
Supporting Information Available: Explanation of sample preparation protocols, control experiments, and additional data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Liu Y-T. Infect Disord Drug Targets. 2008;8(3):183–188. doi: 10.2174/1871526510808030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Domiati-Saad R, Scheuermann RH. Clin Chim Acta. 2006;363(1-2):197–205. doi: 10.1016/j.cccn.2005.05.049. [DOI] [PubMed] [Google Scholar]
- 3.Kaittanis C, Santra S, Perez JM. Adv Drug Deliver Rev. 2010;62(4-5):408–423. doi: 10.1016/j.addr.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nielsen PE. Chem Biodivers. 2010;7(4):786–804. doi: 10.1002/cbdv.201000005. [DOI] [PubMed] [Google Scholar]
- 5.Sahu B, Sacui I, Rapireddy S, Zanotti KJ, Bahal R, Armitage BA, Ly DH. J Org Chem. 2011;76(14):5614–5627. doi: 10.1021/jo200482d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chenna V, Rapireddy S, Sahu B, Ausin C, Pedroso E, Ly DH. Chembiochem. 2008;9(15):2388–2391. doi: 10.1002/cbic.200800441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rapireddy S, He G, Roy S, Armitage BA, Ly DH. J Am Chem Soc. 2007;129(50):15596–15600. doi: 10.1021/ja074886j. [DOI] [PubMed] [Google Scholar]
- 8.Deamer DW, Branton D. Accounts Chem Res. 2002;35(10):817–825. doi: 10.1021/ar000138m. [DOI] [PubMed] [Google Scholar]
- 9.Bayley H, Cremer PS. Nature. 2001;413(6852):226–230. doi: 10.1038/35093038. [DOI] [PubMed] [Google Scholar]
- 10.Venkatesan BM, Bashir R. Nat Nano. 2011;6(10):615–624. doi: 10.1038/nnano.2011.129. [DOI] [PubMed] [Google Scholar]
- 11.Wanunu M, Dadosh T, Ray V, Jin J, McReynolds L, Drndic M. Nat Nano. 2010;5(11):807–814. doi: 10.1038/nnano.2010.202. [DOI] [PubMed] [Google Scholar]
- 12.Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T, Di Ventra M, Garaj S, Hibbs A, Huang XH, Jovanovich SB, Krstic PS, Lindsay S, Ling XSS, Mastrangelo CH, Meller A, Oliver JS, Pershin YV, Ramsey JM, Riehn R, Soni GV, Tabard-Cossa V, Wanunu M, Wiggin M, Schloss JA. Nat Biotechnol. 2008;26(10):1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wanunu M, Morrison W, Rabin Y, Grosberg AY, Meller A. Nat Nano. 2010;5(2):160–165. doi: 10.1038/nnano.2009.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Zheng D, Tan Q, Wang MX, Gu L-Q. Nat Nano. 2011;6(10):668–674. doi: 10.1038/nnano.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singer A, Wanunu M, Morrison W, Kuhn H, Frank-Kamenetskii M, Meller A. Nano Lett. 2010;10(2):738–742. doi: 10.1021/nl100058y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen GI, Bentin T, Larsen HJ, Nielsen PE. J Mol Biol. 2001;307(1):67–74. doi: 10.1006/jmbi.2000.4487. [DOI] [PubMed] [Google Scholar]
- 17.He GF, Rapireddy S, Bahal R, Sahu B, Ly DH. J Am Chem Soc. 2009;131(34):12088–12090. doi: 10.1021/ja900228j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wanunu M, Sutin J, McNally B, Chow A, Meller A. Biophys J. 2008;95(10):4716–4725. doi: 10.1529/biophysj.108.140475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodenburg CM, Li YY, Trask SA, Chen YL, Decker J, Robertson DL, Kalish ML, Shaw GM, Allen S, Hahn BH, Gao F, Isolati UNNH. AIDS Res Hum Retrov. 2001;17(2):161–168. doi: 10.1089/08892220150217247. [DOI] [PubMed] [Google Scholar]
- 20.Green ED, Guyer MS, Inst NHGR. Nature. 2011;470(7333):204–213. doi: 10.1038/nature09764. [DOI] [PubMed] [Google Scholar]
- 21.Zhang N, Appella DH. J Infect Dis. 2010;201:S42–S45. doi: 10.1086/650389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim MJ, Wanunu M, Bell DC, Meller A. Adv Mater. 2006;18(23):3149–3153. [Google Scholar]
- 23.Wanunu M, Meller A. Single Molecule Analysis of Nucleic Acids and DNA-protein Interactions using Nanopores. In: Selvin P, Ha T, editors. Laboratory Manual on Single Molecules. Cold Spring Harbor Press; 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.