

Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder that is characterized by progressive degeneration of motor neurons in the spinal cord, motor cortex, and brainstem. There is currently no effective treatment for ALS. Transgenic mice expressing familiar ALS‐linked mutant superoxide dismutase (SOD1) have been commonly used for preclinical trials; however, successful trails in mice mostly fail in subsequent human trials. Most preclinical trials start treatment before the onset of symptoms, while patients with ALS in clinical trials have surpassed the onset of disease. In addition, ALS is not only a multifactorial disease but also a multisystemic disease that affects several cell types 1. Therefore, therapies should be aimed at the interception of multiple mechanisms. In this study, we investigated [1] whether the therapeutic agents, which were previously tested starting before disease onset, still showed protective effects when the treatments were initiated after disease onset and [2] whether combination therapies offered better effects after disease onset.
Minocycline, which inhibits microglia activation and apoptotic cascade, was previously found to be effective at ameliorating motor impairment and increasing the lifespan of SOD1G93A mice when administered at 5 weeks of age 2. However, minocycline fails in human trials 3. We assessed whether similar beneficial effects could be seen when treatment began at 90 days of age. This time point was chosen because SOD1G93A mice 4 exhibited 10~20% decline in motor function at this age, which may be equivalent to the stage that patients with ALS first visit a doctor. The results showed no delay in impaired motor function (Figure 1A,B, Mino) and no extension in survival (Figure 1C,D, Mino). This result is consistent with a recent report 5. Ceftriaxone, which reduces excitotoxicity via activation of glial glutamate transporter EAAT2, was found to slow disease progression in SOD1G93A mice when administered at 12 weeks of age 6. Ceftriaxone is currently in clinical trials for ALS. We found that when ceftriaxone was administered starting from 90 days of age, treatment no longer slowed disease progression (Figure 1A,B, Cfx) and prolonged survival (Figure 1C,D, Cfx). We then assessed whether the combined treatments of ceftriaxone and minocycline, starting at 90 days of age, would have protective effects. The results showed no delay in impaired motor function (Figure 1A,B, Double) but a significant increase in survival by ~12 days (Figure 1C,D, Double).
Figure 1.
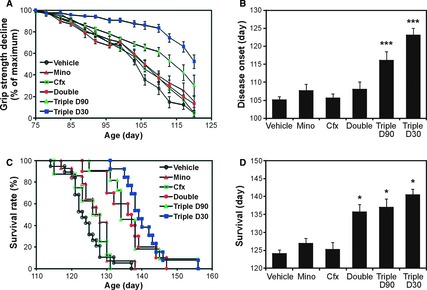
Effects of single and combination treatments on disease onset and survival of SOD1G93A mice. SOD1G93A mice (B6SJL‐Tg(SOD1‐G93A)1Gur/J with high copy number of the mutant human SOD1 gene, Jackson Laboratory) were used in this study (the cited previous trials 2, 6, 7 also used the same mouse model). Mice were divided into six groups and received various treatments: (1) minocycline alone starting from 90 days of age (Mino, 10 mg/kg per day, intraperitoneal injection, n = 14), (2) ceftriaxone alone starting from 90 days of age (Cfx, 100 mg/kg per day, intraperitoneal injection, n = 12), (3) minocycline and ceftriaxone starting from 90 days of age (Double, n = 10), (4) minocycline, ceftriaxone, and vitamin E (200 IU, administered orally) starting from 90 days of age (Triple D90, n = 11), (5) vitamin E starting from 30 days of age in combination with minocycline and ceftriaxone starting from 90 days of age (Triple D30, n = 13), and (6) vehicle control group treated with vegetable oil orally and saline injection instead (n = 25). The study followed guidelines established by the ENMC Group 10, which included use of gender‐matched mice (i.e., equal number of males and females in each experimental group), use of littermate matched mice (i.e., the littermate of a therapeutic mouse is used in the control group), use of mice with the same copy number of the SOD1 transgene, and the use of survival as the primary endpoint for analysis. Motor function was assessed by measuring grip strength using a digital grip strength meter. (A) Motor performance represented by grip strength decline. (B) Disease onset defined by 50% loss of grip strength. Declines in motor performances were significantly delayed for 18 and 11 days in Triple D30 and Triple D90 groups, respectively. ***P < 0.001. (C) Accumulative probability of survival. (D) Mean lifespan (survival time). Cocktail treatments significantly prolonged the survival of SOD1G93A mice by 12, 13, and 16 days in Double, Triple D90, and Triple D30 groups, respectively. *P < 0.05.
Vitamin E, which reduces oxidative damage, can delay disease onset when administered at 30 days of age, but has no effect in extending the lifespan of SOD1G93A mice 7, only slightly slowing disease progression in human ALS trials 8, 9. We found that when mice were treated with vitamin E starting at 30 days of age and then with ceftriaxone and minocycline starting at 90 days of age, a significant delay in motor dysfunction by ~18 days (Figure 1A,B, Triple D30) and an extension in survival by ~16 days (Figure 1C,D, Triple D30) were observed. Furthermore, we assessed the combined three treatments starting at 90 days of age. This triple treatment resulted in greater improvement of motor performance than that seen in the double treatment (ceftriaxone and minocycline) (Figure 1A,B, Triple D90). The prolonged survival for the triple treatment was similar to that seen in the double treatment (Figure 1C,D, Triple D90).
To confirm that the protective effects of combined treatments were attributed to the inhibition of targeted mechanisms, we performed immunofluorescent staining on lumbar spinal cord sections prepared from treated SOD1G93A mice and wild‐type littermates at 115 days of age. As shown in Figure 2A, EAAT2 protein levels were restored in treated mice (in the double and triple groups). Oxidative damage as measured RNA oxidation (15A3 staining) was also reduced 7. Astrogliosis (GFAP staining) and microglial activation (CD11b staining) in SOD1G93A mice were increased compared with those seen in wild‐type littermates. There was even greater enhanced glial activation by the cocktail treatments – the double and triple groups, which is consistent with a recent report 5. Furthermore, to confirm that the delayed disease onset and prolonged survival were associated with reduced neuronal loss, we performed cresyl violet staining on lumbar spinal cord adjacent sections. A significant reduction in motor neuron loss was observed in the triple groups (Figure 2B).
Figure 2.
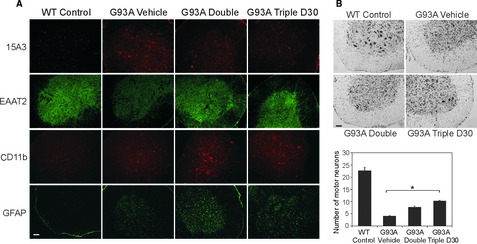
Effects of combination treatments on oxidative damage, EAAT2 expression, glial activation, and motor neuron loss in SOD1G93A mice. Immunofluorescent (A) and Cresyl violet (B) stainings were performed on lumbar spinal cord sections prepared from treated SOD1G93A mice (Vehicle, Double, and Triple D30 groups) and nontransgenic littermates (WT Control) (n = 3 each group) at 115 days of age. (A) Representative images of 15A3‐stained oxidized RNA (red, upper row), EAAT2 protein (green, second row), CD11b‐stained microglia (red, third row), and GFAP‐stained astrocytes (green, bottom row) are shown. Oxidative damage in RNA and loss of EAAT2 protein were reduced in cocktail‐treated mice. Astrogliosis and microglial activation were enhanced in cocktail‐treated mice. (B) Representative images of motor neuron loss are presented (upper panel). Triple treatment significantly protected mice against the motor neuron loss in the lumbar spinal cord (low panel, *P < 0.05). Scale bar, 50 μm.
Transition from preclinical mouse studies to human clinical trials is difficult for most diseases. As the first step, proper design of preclinical trials is very important. The results of this study suggest that inappropriate presymptomatic treatment in mice may partially provide explanations for the failure of some clinical trials in patients with ALS. In addition, combination treatments may serve as a therapeutic strategy for human trials. The agents that have been unsuccessful in human trials may prove to be more beneficial when used in combination with one another.
Acknowledgements
This work was supported by NIH (grants R01NS064275 and R21AG027797), the ALS Association, and the Neuroscience Education and Research Foundation. We thank Miss. Yuchen Lin (The Ohio State University, Columbus) for helping with manuscript preparation.
References
- 1. Boillee S, Vande Velde C, Cleveland DW. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006;52: 39–59. [DOI] [PubMed] [Google Scholar]
- 2. Zhu S, Stavrovskaya IG, Drozda M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002;417: 74–78. [DOI] [PubMed] [Google Scholar]
- 3. Gordon PH, Moore DH, Miller RG, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol 2007;6: 1045–1053. [DOI] [PubMed] [Google Scholar]
- 4. Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994;264: 1772–1775. [DOI] [PubMed] [Google Scholar]
- 5. Keller AF, Gravel M, Kriz J. Treatment with minocycline after disease onset al.ters astrocyte reactivity and increases microgliosis in SOD1 mutant mice. Exp Neurol 2011;228: 69–79. [DOI] [PubMed] [Google Scholar]
- 6. Rothstein JD, Patel S, Regan MR, et al. Beta‐lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005;433: 73–77. [DOI] [PubMed] [Google Scholar]
- 7. Chang Y, Kong Q, Shan X, et al. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS One 2008;3: e2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Desnuelle C, Dib M, Garrel C, Favier A. A double‐blind, placebo‐controlled randomized clinical trial of alpha‐tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. ALS riluzole‐tocopherol Study Group. Amyotroph Lateral Scler Other Motor Neuron Disord 2001;2: 9–18. [DOI] [PubMed] [Google Scholar]
- 9. Graf M, Ecker D, Horowski R, et al. High dose vitamin E therapy in amyotrophic lateral sclerosis as add‐on therapy to riluzole: results of a placebo‐controlled double‐blind study. J Neural Transm 2005;112: 649–660. [DOI] [PubMed] [Google Scholar]
- 10. Ludolph AC, Bendotti C, Blaugrund E, et al. Guidelines for the preclinical in vivo evaluation of pharmacological active drugs for ALS/MND: report on the 142nd ENMC international workshop. Amyotroph Lateral Scler 2007;8: 217–223. [DOI] [PubMed] [Google Scholar]