

Abstract
Several lines of evidence indicate that glutamate plays a crucial role in the initiation of seizures and their propagation; abnormal glutamate release causes synchronous firing of large populations of neurons, leading to seizures. In the present study, we investigated whether enhanced glutamate uptake by increased glial glutamate transporter EAAT2, the major glutamate transporter, could prevent seizure activity and reduce epileptogenic processes. EAAT2 transgenic mice, which have a 1.5-2 fold increase in EAAT2 protein levels as compared to their non-transgenic counterparts, were tested in a pilocarpine-induced status epilepticus (SE) model. Several striking phenomena were observed in EAAT2 transgenic mice compared with their non-transgenic littermates. First, the post-SE mortality rate and chronic seizure frequency were significantly decreased. Second, neuronal degeneration in hippocampal subfields after SE were significantly reduced. Third, the SE-induced neurogenesis and mossy fiber sprouting were significantly decreased. The severity of cell loss in epileptic mice was positively correlated with that of mossy fiber sprouting and chronic seizure frequency. Our results suggest that increased EAAT2 expression can protect mice against SE-induced death, neuropathological changes, and chronic seizure development. This study suggests that enhancing EAAT2 protein expression is a potential therapeutic approach.
Keywords: Excitotoxicity, Glutamate transporter, EAAT2, Temporal lobe epilepsy, Pilocarpine, Epileptogenesis
Introduction
Epilepsy is one of the most frequent neurological disorders (Strine et al., 2005). Over 40 types of epilepsies have been identified and classified into two categories: partial epilepsies and generalized epilepsies (Engel, 2001). Partial epilepsies account for about 60% of all adult cases while temporal lobe epilepsy (TLE) is the most common form of partial epilepsy causing refractory epilepsy (Wiebe, 2000). TLE is characterized by spontaneous recurrent motor seizures (SRSs) originating from temporal lobe foci. Patients with TLE often show impairments in learning, memory, and other cognitive functions (Devinsky, 2004). TLE is usually preceded by an initial precipitating injury such as status epilepticus (SE), head trauma, childhood febrile seizures, or hypoxia (Acharya et al., 2008). There is commonly a latent period of several years between the initial injury and the emergence of chronic TLE. The initial injury is thought to cause complex molecular, biochemical, and structural changes that, over time, result in the development of SRSs. This process is referred to as epileptogenesis (McNamara et al., 2006).
Among the events that occur in response to the initial injury, neuronal death is believed to be an important propagating factor that links the initial injury with the epileptic condition. Neuronal death may be both a cause, as well as a consequence, of epileptic seizures. The intense seizure activity associated with SE can cause significant neurodegeneration in the hippocampus regions CA1, CA3, and dentate hilus (Rao et al., 2006; Sloviter, 1999; Sloviter and Dempster, 1985). This neuronal loss is most likely secondary to sustained excitation and subsequent glutamate-mediated excitotoxicity.
Glutamate plays a crucial role in the initiation and propagation of seizures. In vivo microdialysis studies of patients with epilepsy show a sustained increase in extracellular glutamate levels, which reach neurotoxic concentrations in the epileptogenic hippocampus before and during seizure onset (Cavus et al., 2005; During and Spencer, 1993). Abnormal enhanced glutamate released by astrocytes is considered a causal role in the synchronous firing of large populations of neurons during seizures (Benarroch, 2009; Binder and Steinhauser, 2006; Tian et al., 2005; Wetherington et al., 2008). Additionally, dysfunction of glutamate transport may contribute to high extracellular glutamate in the epileptogenic hippocampus. Impaired glutamate transport function has been reported in human epilepsy, but remains controversial (Bjornsen et al., 2007; Mathern et al., 1999; Proper et al., 2002; Sarac et al., 2009; Tessler et al., 1999). Therefore, reduction of glutamate-mediated excitotoxicity is a potential strategy to prevent seizure-induced neuronal death and subsequent recurrent seizures.
The glial glutamate transporter EAAT2 is expressed mainly in glial cells and is responsible for 80-90% of all glutamate transport (Rothstein et al., 1996). Our lab previously generated a transgenic mouse line, which moderately expresses human EAAT2 with a 1.5-2-fold increase in EAAT2 protein levels and the associated glutamate uptake (Guo et al., 2003). The purpose of this study is to use EAAT2 transgenic mice to investigate whether enhanced glutamate uptake by increased EAAT2 can prevent seizure-induced neuronal death, epileptogenesis, and subsequent recurrent seizures. The pilocarpine model of limbic epilepsy, which involves inducing SE with the subsequent development of SRSs, was used in this study.
Materials and methods
Genotype analysis of EAAT2 mice
EAAT2 transgenic mice were previously generated in our laboratory (Guo et al., 2003) in FVB/N mouse strain. Upregulation of EAAT2 driven by the human glial fibrillary acidic protein (hGFAP) promoter was restricted in astrocytes. Integration of the transgene was determined by PCR using genomic DNA extracted from tail biopsies at 3 weeks of age with EAAT2 transgene-specific primers (5′-ggc aac tgg gga tgt aca-3′ and 5′-acg ctg ggg agt tta ttc aag aat-3′). PCR conditions were as follows: 94°C for 3 min; 94°C for 30 s, 55°C for 30 s, 72°C for 2 min for 30 cycles followed by 10-min extension at 68°C.
Animals and pilocarpine model
Mice were housed in a 12-hr light/dark cycle with free access to food and water. All experiments were approved by the Institutional Animal Care and Use Committee of the Ohio State University and with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Pilocarpine-induced SE model was used to induce the chronic seizures (Shapiro et al., 2007). Briefly, 8- to 10-week-old male EAAT2 transgenic (EAAT2) and non-transgenic (WT) littermate mice (22–32 g) were first intraperitoneally (i.p.) injected with (-) Scopolamine methyl nitrate (1.5 mg/kg, Sigma, St. Louis, MO) to reduce peripheral cholinergic effects without interfering with the development of SE and chronic seizures, and 30 min later, were injected with pilocarpine hydrochloride (290 mg/kg, i.p., Sigma). A total of 28 independent experiments were performed; 240 EAAT2 mice and 268 WT mice in total. Control animals were injected with Scopolamine followed by saline, instead of pilocarpine.
The acute seizure score (maximal seizure stage that each mouse reached) and latency period were evaluated by observation directly after pilocarpine injections for 2 hr. The time points at which mice reached each stage of seizures were recorded and the SE latencies were calculated based on time intervals between the injection point and SE point. The scale to evaluate seizure score was based on the following features modified from the Racine scale (Racine, 1972): normal (zero), seizure consisted of immobility and occasional facial clonus (I) (“wet-dog shakes’), seizure with head nodding, unilateral forelimb clonus, frequent “wet-dog shakes” (II), seizure with bilateral forelimb clonus (III), rearing (IV), rearing and falling, salivation (V), repeated rearing and falling with tonic limbic extension for at least 60 min (SE).
Only the mice that reached SE and maintained in tonic limbic extension for at least 60 min were kept for pathological studies and observation of chronic seizure development. EAAT2 SE mice and wild-type SE littermates were carefully matched based on the body weight, seizure latency, seizure severity, and post-SE sickness. Therefore, the nature of the SE (i.e. seizure length and severity) in those mice was comparable between EAAT2 groups and wild-type littermate groups. Mice that developed SE were fed with food soaked in a high sucrose (10%) saline solution to help recovery in the first 3 days after SE.
Behavioral analysis
The chronic seizure activities were recorded by webcam (Logitech, Fremont, CA) and reviewed using Windows Live Movie Maker. The seizure activities were monitored for 8 hr/day at 28 days post-SE for 12 days. The daylight period (9 AM- 5 PM) was selected for observations because previous literature indicates a higher frequency of SRSs during the day (Arida et al., 1999; Furtado et al., 2011). The chronic seizure frequency of each mouse was represented by the average number of its spontaneous stage V seizures per 8 hr.
Tissue collection
Mice were perfused transcardially with 4% paraformaldehyde in 0.1 M PB (sodium phosphate buffer, pH 7.4) after being deeply anesthetized with tribromoethanol (Avertin; 200 μl/10 g i.p.). The mice were harvested at various time points for different staining treatments: 3 days post-SE for Fluoro-Jade C staining, 12 days post-SE for doublecortin immunostaining, and 8 weeks post-SE for cresyl violet and Timm stainings. The brains were rapidly dissected and post-fixed in 4% paraformaldehyde at 4°C for 24 hr, followed by cryoprotection with 30% sucrose for 72 hr. Twenty-micrometer thick coronal sections (− 1.8 to −2.8 mm from Bregma) were cut with a Microm cryostat at−20°C.
For Timm staining, mice were anesthetized and quickly washed with sulfide solution (0.8% Na2S in 0.1 M PB) and then perfused transcardially with 3% glutaraldehyde in 0.1 M PB followed by sulfide solution. The brains were rapidly dissected, post-fixed in 3% glutaraldehyde for 30 min, incubated in sulfide solution for 1 hr followed by cryoprotection with 30% sucrose for 72 hr.
Fluoro-Jade C staining
Fluoro-Jade C was used to detect chronic seizure-induced neurodegeneration. Coronal sections were first treated with 80% ethanol and 1% NaOH for 5 min, then 70% ethanol for 2 min followed by three washes in distilled water. Next, sections were incubated with 0.06% potassium permanganate for 10 min, distilled water for 1 min, and 0.001% FJC (Histo-Chem Inc., Jefferson, Arizona) in 0.1% acetic acid for 20 min. Sections were then washed twice in distilled water for 5 min, air dried, dipped in xylene for 2 min, and mounted with Permount (Fisher Scientific, Fair Lawn, New Jersey). Images were obtained using a Zeiss Axioskop 2 upright microscope (excitation: ~480 nm, emission: ~540 nm) with AxioVision software for viewing Fluoro-Jade C-positive cells. FJC positive-neurons in hippocampal sub-regions were counted under the microscope (200X). The means represent the average numbers of FJC-positive cell in specific hippocampal sub-regions per side of section.
Cresyl violet staining
Every fifth brain section was stained with 0.1% (w/v) cresyl violet solution. The live cells in the hilus region were analyzed by counting large neurons that were stained with cresyl violet and possessed a prominent nucleolus on each side of the hippocampus under light microscope (200×). Hilar cell numbers of each SE mouse was calculated from the mean value of live hilar cell numbers of both sides of the hippocampus. Hilar cell loss percentage in each side of the hippocampus was defined as the difference between hillar cell numbers in a control mouse and hilar cell numbers in a SE mouse divided by the hilar cell number in the control mouse multiplied by 100. For the correlation between neuronal damage and chronic seizure frequency (Fig. 4C), x and y values of each dot represent the mean hilar cell loss of one mouse from both sides of the hippocampus and the chronic seizure frequency of this mouse, respectively.
Figure 4.
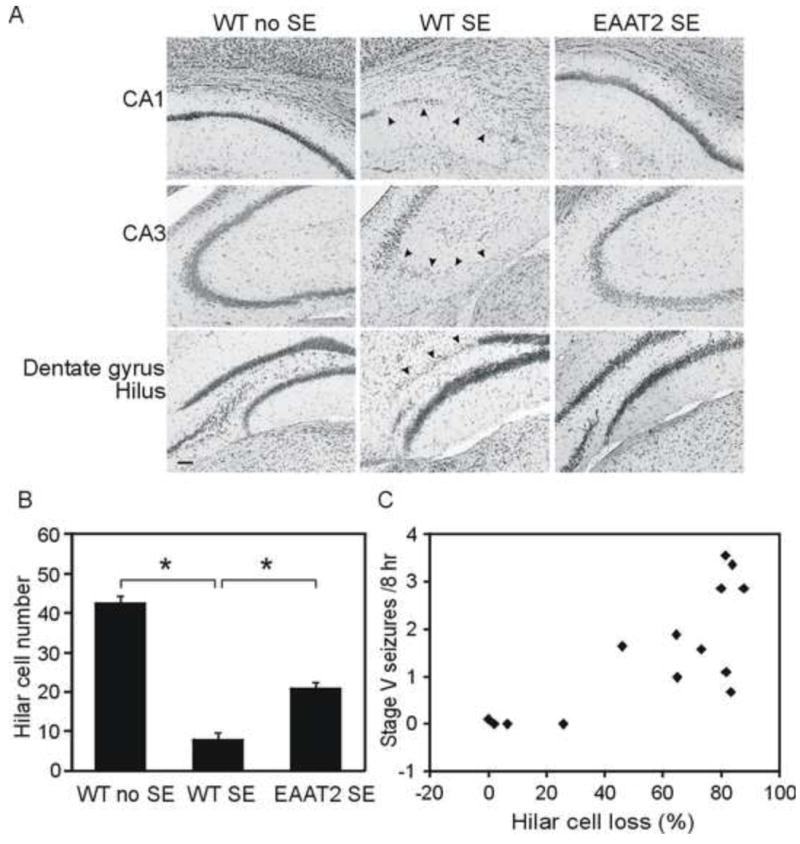
Hippocampal damage was significantly reduced in epileptic EAAT2 mice. Coronal sections from eight sets of brains at 8 weeks after SE were stained with cresyl violet for neuronal loss. Live neurons in the hilus were counted under the microscope. (A) Representative hippocampal damage were presented. Arrow heads indicate severe neuronal loss. Scale bar, 50 μm. (B) Compared to control mice, both wild-type SE and EAAT2 SE mice had significant neuronal loss in hilus (*p<0.05, Turkey test). More importantly, increased EAAT2 significantly attenuated the damages (*p<0.05, Turkey test). (C) A strong positive correlation was detected between hilar cell loss and chronic seizure frequencies. Hilar cell loss was defined as (cell number in control hilus – live cell number in SE hilus)/cell number in control hilus×100. Correlation coefficient=0.69, p<0.01, Spearman rank order correlation test.
Immunohistochemistry for doublecortin
Brain sections were blocked in 5% normal goat serum (NGS; Vector Laboratories, Burlingame, CA) of 0.2% Triton X-100 in Tris-buffered saline (TBS) for 60 min and incubated in rabbit polyclonal doublecortin (DCX) antibody overnight at 4°C. After thorough washing with TBS, the sections were incubated with goat anti-rabbit Alexa Fluoro488 secondary antibodies in TBST containing 2% NGS for 60 min followed by thorough washing with TBS. Images were obtained using a Zeiss Axioskop 2 upright microscope with AxioVision software. DCX positive-neurons in the dentate gyrus were counted under the Zeiss Axioskop 2 plus microscope with a 20X objective lens and AxioCam MRm camera. The analyses of hilar basal dendrites were performed as previously described (Shapiro et al., 2006). Hilar basal dendrites were defined as DCX-labeled processes from the basal part of the DCX-labeled cell bodies which extended into the hilus.
Timm staining
Sections were developed in the dark for 45-75 minutes in a mixture of 30 ml 50% gum Arabic, 7.5 ml hydroquinone (0.425 g), 5 ml citric acid-sodium citrate buffer (1.274 g citric acid 1H2O and 1.174 g sodium citrate 2H2O), and 7.5 ml of silver lactate solution (0.055 g silver lactate). Slides were then washed, air dried, cleared in xylene, mounted, and examined with a Zeiss Axioskop 2 upright microscope under bright field optics. The staining selectively recognized zinc-containing neurons and synaptic boutons. Mossy fiber synaptic reorganization in each side of the hippocampus was examined by studying the distribution of Timm granules in the supragranular zone and evaluated in 0~5 scales as previously described (Cavazos et al., 1991; Fig. S1). The Timm score of each SE mouse came from the mean score of both sides of the hippocampus. For the correlation between mossy fiber sprouting and chronic seizure frequency (Fig. 6E), x and y values of each dot represent the mean Timm score of one mouse from both sides of the hippocampus and the chronic seizure frequency of the mouse, respectively. For the correlation between hilar cell loss and Timm score (Fig. 6F), two dots were plotted for each mouse (left lateral and right lateral) while x and y values of each dot represent the hilar cell loss and Timm score from one side of the hippocampus in the mouse, respectively.
Figure 6.
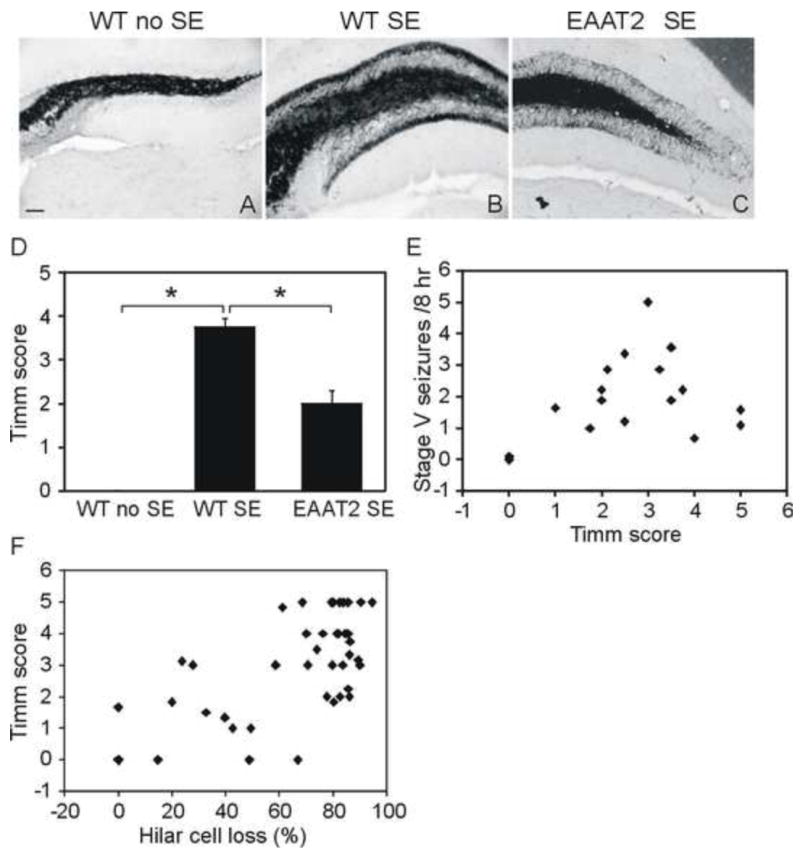
Aberrant mossy fibers sprouting decreased in the dentate gyrus of epileptic EAAT2 mice. Timm staining was performed to detect mossy fiber spouting with mice harvested at 8 weeks after SE. Sections were scored by 2 investigators independently on a scale of 0~5 (Fig. S1). (A-C) Representative images of Timm-stained dentate gyrus. scale bar, 50 μm. (D) EAAT2 mice exhibited significantly lower Timm scores compared to wild-type littermates (n=16, *p<0.05, Dunn’s method). (E) A moderate but not significant positive correlation (correlation coefficient=0.45, p=0.052, Spearman rank order correlation test) was detected between the severity of aberrant mossy fiber sprouting and chronic seizure frequency. (F) A strong positive correlation was detected between the hilar cell loss and the Timm score (correlation coefficient=0.59, p<0.001, Spearman rank order correlation test).
Statistical analysis
The continuous quantitative data in this study were expressed as the mean ± standard error. Chi-square tests were used to examine the distribution difference between wild-type and EAAT2 mice (Pitsch et al., 2007). The repeated measured seizure data were analyzed using generalized linear mixed effects models with a log link function. The correlation among Timm scores, hilar cell loss, and chronic seizure frequencies of the SE mice were examined by the Spearman rank order correlation test. For multiple groups’ comparisons, data were analyzed using one way ANOVA and the pairwise multiple comparison by Holm-Sidak method when the data passed the normality and equal variance tests. Otherwise, data were analyzed using Kruskal-Wallis test and pairwise multiple comparison by Turkey or Dunn’s method. For comparisons between two groups, a t test was performed when the normality assumptions were not violated. Otherwise, a non-parametric Mann-Whitney rank sum test was performed. Values were considered significant at p<0.05.
Results
Acute stage of pilocarpine-induced epilepsy in EAAT2 transgenic mice
The susceptibility of EAAT2 transgenic mice (EAAT2 mice) to pilocarpine-induced epilepsy was examined and compared to their non-transgenic littermates (wild-type mice). Experiments were performed in 28 independent groups, with a total of 240 EAAT2 mice and 268 wild-type mice. Only male mice were used in this study. Racine’s scale (described in the materials and methods section) was used to rate seizure severity of the pilocarpine-induced seizures. Figure 1A represents the mean seizure severity in EAAT2 mice and wild-type littermates from the 28 groups (Supplemental Table S1). During the acute stage, no significant difference in seizure severity was observed between 240 EAAT2 mice and 268 wild-type mice with respect to the maximal seizure stage (stage II and below: 17 (WT) vs. 28 (EAAT2); stage III: 9 (WT) vs. 15 (EAAT2); stage IV: 21 (WT) vs. 18 (EAAT2); stage V: 14 (WT) vs. 11 (EAAT2); and SE: 207 (WT) vs. 168 (EAAT2). p= 0.12, Chi-square test). Next, we compared the percentage of EAAT2 mice that reached SE with their wild-type littermates (last set of columns in Fig 1A; mean of SE rates from 28 independent groups: 78.7±2.8% (WT) vs. 70.0±3.4% (EAAT2)). No significant protection against the development of SE was observed in EAAT2 mice after pilocarpine injection when compared to wild-type littermates (p=0.08, Chi-square test based on the combined data). The latencies of each mouse reaching stage III and SE were not significantly affected by EAAT2 (Fig. 1B; stage III, 26±1 min (WT) vs. 28±1 min (EAAT2); SE, 33±1 min (WT) vs. 35±1 min (EAAT2); p=0.60 and 0.07, respectively; Mann-Whitney rank sum test). Furthermore, we examined the acute mortality rate as another important parameter for susceptibility, which was defined by the percentage of mice that died within 1 week after injection in each group (Fig. 1C; total 28 groups: 44.0±3.8% (WT) vs. 25.2±3.6% (EAAT2)). EAAT2 mice presented a significantly lower acute death rate (63 out of 240 EAAT2 mice) than their wild-type littermates (115 out of 268 WT mice; p<0.001, Chi-square test based on the combined data), suggesting that increased EAAT2 protein can partially prevent SE-induced acute death in mice.
Figure 1.
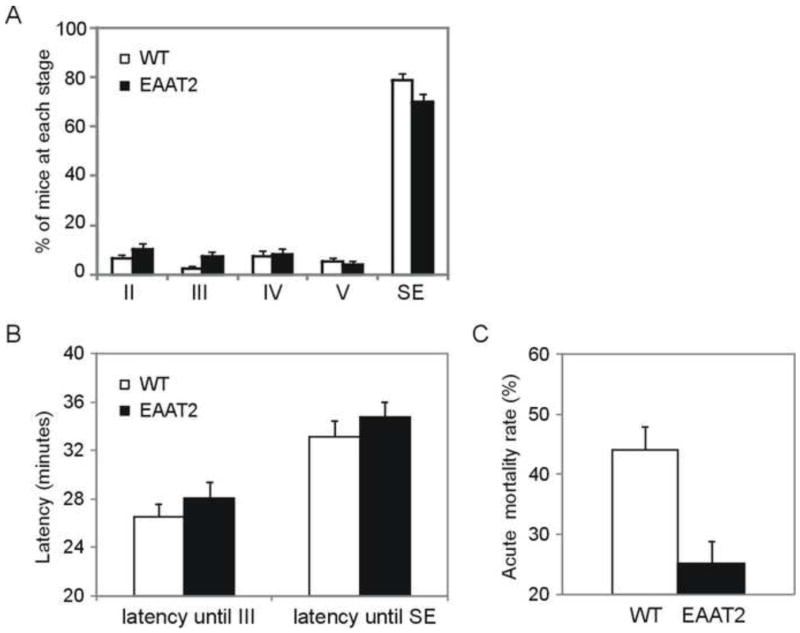
Evaluation of EAAT2 transgenic mice at the acute epileptic stage. After injections of pilocarpine (290 mg/kg, i.p.), the first seizure activity at each stage that mouse reached was noted with the corresponding time. The scale to evaluate seizure stage is described in the Methods section (Racine, 1972). A total of 240 EAAT2 mice and 268 wild-type littermates (WT) were tested in 28 independent groups. (A) The maximal seizure activity of each animal is presented. In 28 independent groups, stage II and below: 6.4±1.5% (WT) vs. 10.3±2.2% (EAAT2); stage III: 2.5±1.1% (WT) vs. 7.2±1.9% (EAAT2); stage IV: 7.2±2.4% (WT) vs. 8.2±2.3% (EAAT2); stage V: 5.2±1.6% (WT) vs. 4.2±1.3% (EAAT2); and SE: 78.7±2.8% (WT) vs. 70.0±3.4% (EAAT2). (B) Latency was defined as the time interval between the injection and the indicated stage. Neither the latency period of stage III seizure nor the latency up until SE (p=0.60 and 0.07, respectively; Mann-Whitney rank sum test) was significantly affected. (C) A reduction of acute mortality rate, defined by mean death rate within 7 days after injection, was detected in EAAT2 mice (n=28 groups, 44.0±3.8% (WT) vs. 25.2±3.6% (EAAT2)).
Chronic stage of pilocarpine-induced epilepsy in EAAT2 transgenic mice
SE-induced chronic seizures were monitored by video recording to determine the frequency and severity of seizure activities. Mice that reached the SE stage were monitored, beginning at four weeks after SE for 2 weeks (8 hr each day). The average frequency of stage V seizures that occurred per 8 hr in each mouse was quantified. As shown in Figure 2A, EAAT2 mice tended to develop fewer stage V seizures. In the 24 wild-type and 24 EAAT2 mice, 6 wild-type and 9 EAAT2 mice had no, or less than, one stage V seizure per 8 hr, 6 wild-type and 10 EAAT2 mice had 1-2 stage V seizures, 6 wild-type and 3 EAAT2 mice had 2-3 stage V seizures, 4 wild-type and 1 EAAT2 mice had more than 3 stage V seizures, 2 wild-type and 1 EAAT2 mice had stage V seizures and died during the recording period. The average seizure frequency in EAAT2 mice (1.15±0.19) was reduced to about half of that in wild-type littermates (2.01±0.26; Fig. 2B, p=0.01, t test). The repeated measurements of stage V seizure frequencies during the recording period were analyzed using generalized linear mixed effects models with a log link function. A random intercept term was included to account for the within-subject correlation among repeated measures. There is a significant difference between the WTSE and EAAT2SE groups in terms of the change of stage V seizure occurrence over time (Fig. 2C); specifically, the increased EAAT2 expression inhibited the trajectory of chronic seizure development (p=0.003, Wald test). Next, we determined the overall seizure-induced mortality rate in mice that survived 8 weeks after injection. In the 17 independent groups of experiments that included 166 non-transgenic and 143 EAAT2 transgenic mice, EAAT2 mice presented a significantly lower mortality rate when compared to wild-type littermates (Fig. 2D, 30.1±4.0% (EAAT2) vs. 52.4±4.0% (WT) from 28 groups; 47 out of 143 EAAT2 mice compared to 83 out of 166 WT littermates died in total, p=0.003, Chi-square test based on the combined data).
Figure 2.
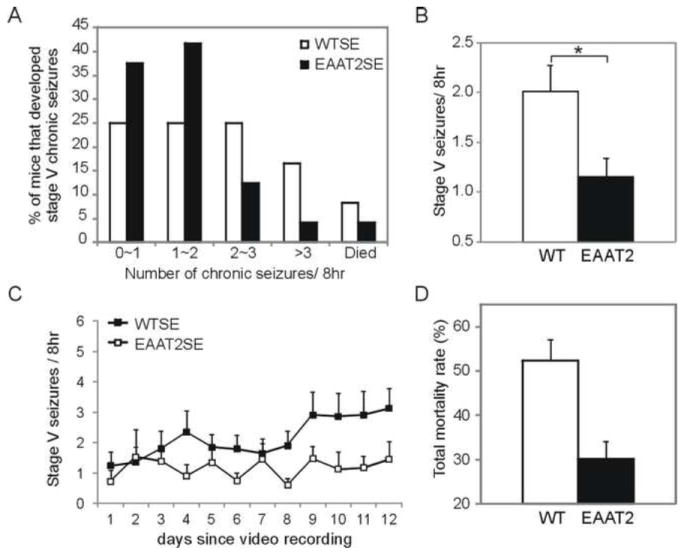
Reduced chronic seizure frequency and total mortality rate in EAAT2 mice. Recording of spontaneous seizures started at 4 weeks after SE for 2 weeks (8 hr each day). Stage V seizure frequencies (i.e., average stage V counts per 8 hr according to Racine’s scale) were determined in each mouse and compared between 22 wild-type (WT) and 20 EAAT2 mice. (A) Percentages of mice that developed fewer than 1, 1~2, 2~3, or over 3 stage V seizures per 8 hr were: 27.3% (WT) vs. 40% (EAAT2), 27.3% (WT) vs. 40% (EAAT2), 22.7% (WT) vs. 10% (EAAT2), and 13.6% (WT) vs. 5% (EAAT2), respectively. 9.1% of WT and 5% of EAAT2 mice died during the recording weeks. (B) SE-induced chronic seizures were less severe in EAAT2 mice (1.15±0.19, n=23; *p<0.05, t test) when compared with wild-type littermates (2.01±0.26, n=23). (C) Stage V seizure frequencies during each day of the recording. (D) A reduction of total mortality rate, defined by mean death rate in 17 independent groups within 8 weeks after injection, was detected in EAAT2 mice.
Acute hippocampal neurodegeneration in epileptic EAAT2 mice after pilocarpine application
Neuronal loss in the hippocampus has been well characterized in the pilocarpine-induced SE/TLE model. We examined neuron degeneration in eight sets of epileptic EAAT2 mice compared with the degeneration in wild-type littermates after SE (Fig. 3). Coronal hippocampal sections were stained with Fluoro-Jade C (FJC), a specific histochemical stain for dying cells. No positive signal was observed in sham control (wild-type, no SE) brains (Fig. 3, left column). Among hippocampal sub-regions, hilus interneurons were the most vulnerable as FJC-positive hilus neurons could be seen in a few hours after SE (data not shown). At 3 days after SE, significant numbers of FJC-positive neurons were observed in all hippocampal subregions of WT (Fig. 3, middle columns; CA1: 27.3±12.0 per side of section, CA3: 34.7±6.6, DGHilus: 42.9±5.5) and EAAT2 mice (Fig. 3, right columns; CA1: 10.2±4.6, CA3: 14±3.6, DGHilus: 19.8±3.4; p<0.001, Kruskal-Wallis test among three groups; p <0.05 in all subregions when compared with control group, Turkey test). FJC-positive neurons were also observed in other areas such as piriform cortex, entorhinal cortex, etc. (data not shown). Strikingly, quantification analysis showed a significant reduction of FJC-positive neurons in CA3 and dentate gyrus/ hilus sub-regions of EAAT2 mice (Fig. 3B; CA3: p <0.05, DGHilus: p <0.05, CA1: p >0.05 when compared with WTSE group, Turkey test). These findings suggest that increased EAAT2 can partially protect mice from SE-induced neurodegeneration.
Figure 3.
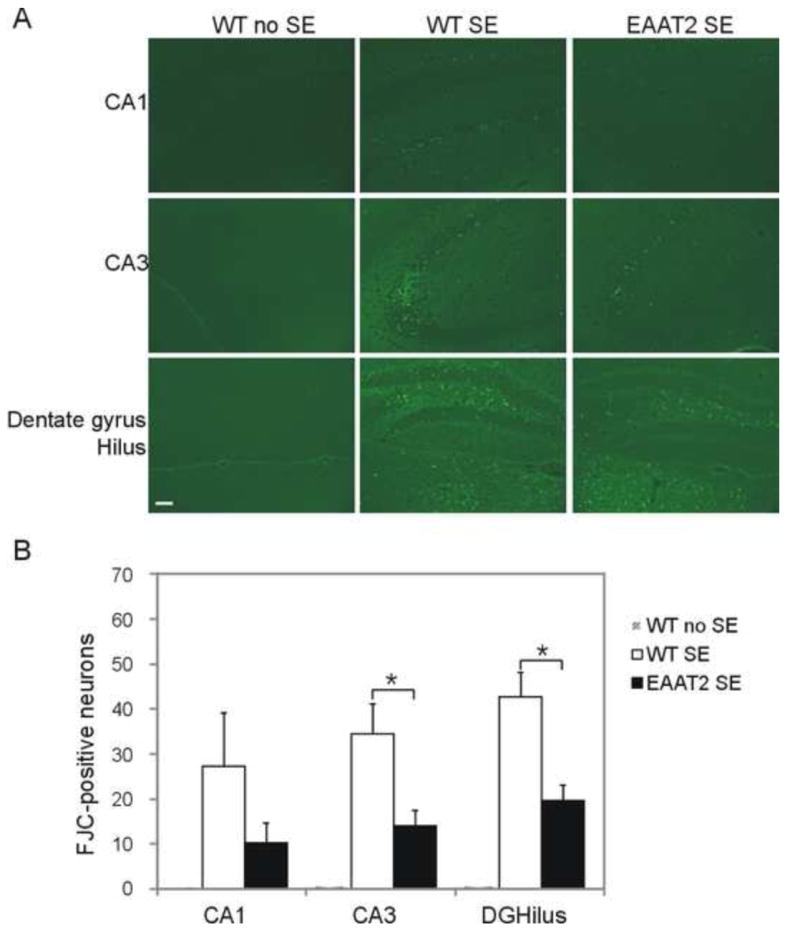
Reduced acute neurodegeneration in EAAT2 mice after SE. Coronal sections from eight sets of brains at 3 days after SE were stained with Fluoro-Jade C. Positively labeled degenerating cells were counted under the microscope in several hippocampal subfields: CA1, CA3, dentate gyrus (DG) and hilus. (A) Representative images of hippocampal neurodegeneration. Scale bar, 50 μm. (B) FJC-positive cells were significantly less abundant in EAAT2 mice. *p<0.05, Turkey test.
Hippocampal damages in epileptic EAAT2 mice at chronic stage
Next, we examined the hippocampal damage at 8 weeks after SE, when chronic seizures had developed. 14 sets of sham controls, wild-type and EAAT2 littermates were examined. Coronal sections were stained with cresyl violet for live cells. Live hilar cells were counted under light microscope (200×). No histological lesions were detected in sham controls (Fig. 4A, left column). Compared to sham controls, histological damages were observed in all hippocampal sub-regions of epileptic wild-type mice (Fig. 4A middle column), and in the hilus and CA3 regions of epileptic EAAT2 mice (Fig. 4A right column). Consistent with the above described FJC staining results at 3 days after SE (Fig. 3), hilus interneurons were the most vulnerable after SE. On average, there were about 42.4±1.9 neurons in the hilus of control mouse per section, while only 20.7±3.6 and 7.7±0.9 neurons were still alive in EAAT2 mice (p<0.001, Kruskal-Wallis test among three groups; p <0.05 when compared to sham control group, Turkey test) and wild-type littermates (p<0.05 when compared to control group, Turkey test), respectively. Importantly, more hilar neurons survived in EAAT2 mice after SE than in wild-type littermates (Fig. 4B; p<0.05 when compared to WTSE group, Turkey test). A strong positive correlation was detected between hilar cell loss and chronic seizure frequencies (Fig. 4C; correlation coefficient=0.69, p<0.01, Spearman rank order correlation test). These data indicate that increased EAAT2 protein expression partially protected the hippocampus from neuronal death to inhibit epileptogenesis.
Hippocampal neurogenesis in epileptic EAAT2 mice during SE-induced epileptogenesis
Acute neuroplastic changes were documented after pilocarpine-induced SE, which involves newly generated granule cells (Shapiro et al., 2007). Immunostaining with doublecortin, a marker of immature neurons (Francis et al., 1999), was used to determine the neurogenesis in six sets of mice at 12 days after SE (Fig. 5A-C). Two parameters were examined: 1) the number of DCX-positive neurons (Fig. 5G) and 2) the percentage of DCX-positive cells with basal dendrites (arrow heads in Fig. 5D-F). DCX-positive immature neurons appeared at the border between the subgranular zone and the granule cell layer after SE. The sham control brain had a similar distribution of DCX-positive cells as previously reported (Shapiro et al., 2007; Fig. 5A). Compared to the low abundance in control brains (19.3±3.7 cells/dentate gyrus), more newly born DCX-positive neurons were detected in both wild-type mice (59.5±7.2 cells /dentate gyrus, p<0.001, Kruskal-Wallis test among three groups; p <0.05 when compared to control group, Turkey test), and EAAT2 mice (29.6±2.5 cells /dentate gyrus, p<0.05 when compared to control group, Turkey test). A significant reduction of DCX-positive neurons was observed in EAAT2 mice (p<0.05 when compared to WTSE group, Turkey test). Moreover, at 12 days post-SE, the mean percentage of DCX-labeled cells that rapidly sprouted hilar basal dendrites was also significantly increased in both wild-type mice (56.0±3.5%, p<0.001, one way ANOVA among three groups; p<0.001 when compared to control group, Holm-Sidak method) and EAAT2 littermates (33.7±3.1%, p=0.002 when compared to control group, Holm-Sidak method) compared to the controls (19.8±2.0%). Interestingly, increased EAAT2 protein levels in EAAT2 mice were associated with a decreased rate of DCX-positive immature neurons with basal dendrites compared to wild-type littermates (p<0.001 when compared to WTSE group, Holm-Sidak method).
Figure 5.
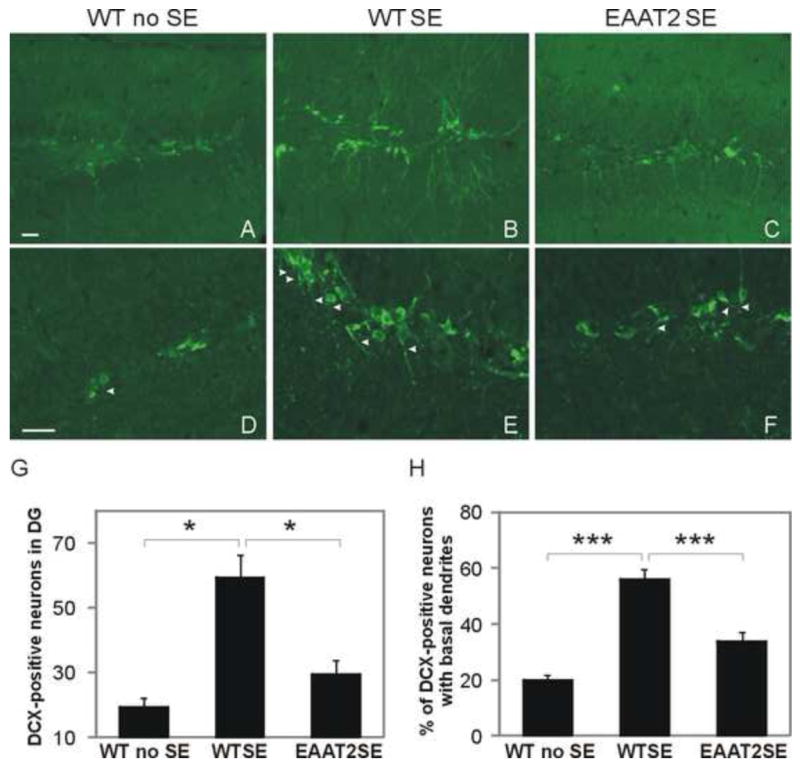
Reduced hippocamal neurogenesis in epileptic EAAT2 mice. Coronal sections from six sets of brains harvested at 12 days after SE were immunostained with a doublecortin (DCX) antibody for newly born granule cells. (A-C) Representative images of DCX-stained dentate gyrus. DCX-positive immature neurons appeared at the border between the subgranular zone and the granular layer. (D-F) Representative images of DCX-positive cells with DCX-labeled hilar basal dendrites (arrow heads). (G) There was a significant reduction of newly born DCX-positive neurons in hippocampal dentate gyrus in EAAT2 mice (*p<0.05, Turkey test). (H) The mean percentage of DCX-labeled cells with hilar basal dendrites was significantly increased in both wild-type mice (56.0±3.5%, ***p<0.001, Holm-Sidak method) and EAAT2 littermates (33.7±3.1%, **p<0.01, Holm-Sidak method) compared to control (19.8±2.0%), but the increase was significantly lower in EAAT2 mice compared to WT littermates (***p<0.001, Holm-Sidak method). Scale bar, 25 μm.
Mossy fiber reorganization in epileptic EAAT2 mice during epileptogenesis
Aberrant mossy fiber sprouting develops gradually after pilocarpine-induced SE (Mello et al., 1993). At 8 weeks post-SE, 16 sets of sham control, wild-type, and EAAT2 littermates were harvested for Timm staining. The severity of mossy fiber sprouting was evaluated by rating the distribution of supragranular Timm granules in the coronal hippocampal sections based on a 0-5 scale as previously described (Cavazos et al., 1991). Representative images for each scale are presented in Fig. S1. Aberrant mossy fiber sprouting was found in both EAAT2 (Fig. 6C; p<0.001, Kruskal-Wallis test among three groups; p <0.05 when compared to sham control group, Dunn’s method) and wild-type littermates (Fig. 6B; p<0.05 when compared to sham control group, Dunn’s method) but not in sham controls (Fig. 6A). The mean Timm score of EAAT2 mice (1.99±0.31) was reduced to less than half of the scores of their wild-type littermates (Fig. 6D; 3.75±0.21, p<0.05, Dunn’s method). At the end, Spearman rank order correlation tests were performed to check the potential correlations between the Timm scores and hilar cell loss and the stage V chronic seizure frequencies. The correlation coefficient between stage V chronic seizure frequencies and Timm scores was 0.45 (p=0.052; Fig. 6E), suggesting a moderate, but not significant, correlation between the severity of aberrant mossy fiber sprouting and chronic seizure frequency. However, a strong positive correlation was detected between the hilar cell loss and the Timm score (correlation coefficient=0.59, p<0.001; Fig. 6F).
Discussion
In the present study, we used a transgenic mouse model to investigate whether enhanced glutamate uptake function via increased EAAT2 expression has beneficial effects in a pilocarpine-induced TLE model. We demonstrated that EAAT2 transgenic mice exhibited reduced acute SE rates and mortality rates, attenuated epileptogenesis (neuronal loss, neurogenesis, mossy fiber sprouting), and reduced SRSs.
In the pilocarpine model, the initial overstimulation of cholinergic system triggers the excessive release of glutamate through calcium influx, which induces SE through synchronic firing of a set of neurons. Enhanced glutamate uptake in EAAT2 mice might impair the synchronic firing of neurons by reducing excessive extracellular glutamate levels, which subsequently protected mice against development of SE and SE-induced acute death. Previously, selective glutamate transporter inhibitors reduced the threshold for evoking epileptic activity (Demarque et al., 2004). It has also been shown that developmental changes in the expression of EAAT2 are negatively correlated with the period of increased susceptibility to seizures in humans (Lauriat et al., 2007). Our findings on decreased acute mortality rate and a mild decrease of SE rate together with these studies suggest that increased EAAT2 protein expression may prevent the generation of seizures.
Functional EAAT2 activation reduces the mortality at chronic stages and results in fewer and less severe SRSs in transgenic mice. The accumulated chronic seizures in WT mice caused greater damage as time passed and resulted in more severe chronic seizures later on. However, increased EAAT2 expression inhibited the trajectory of chronic seizure development. These results suggest a continuous anti-epileptic role of EAAT2 from the injection time of pilocarpine during the development of both acute SE phase and chronic SRS phase. Several lines of evidence have suggested a potential anti-epileptic role of EAAT2 in TLE patients and animal models. During epileptogenesis, reactive astrocytes persistently upregulate mGluR5, which could enhance glutamate release (Notenboom et al., 2006). In addition, excessive glutamate is released from astrocytes through volume-sensitive anion channels activated by cell swelling (Liu et al., 2006) and a Ca2+–dependent pathway triggered by inflammatory factors (Domercq et al., 2006). Dysfunction of glutamate transport may also contribute to high extracellular glutamate in the epileptogenic hippocampus (Cavus et al., 2005; Fernandez et al., 2009; Mathern et al., 1999; Proper et al., 2002; Tanaka et al., 1997; Zeng et al., 2010). Decreased EAAT2 levels have been reported in patients with TLE and accompanying hippocampal sclerosis (Mathern et al., 1999; Proper et al., 2002), although a controversial result was reported by Tessler et al. (Tessler et al., 1999). EAAT2, as the most abundant and primary glutamate transporter, is necessary for maintaining the extracellular glutamate concentration at low levels. Once EAAT2 expression is absence, glutamate levels are elevated and lethal chronic seizures develop in the EAAT2 knockout mice (Tanaka et al., 1997). Furthermore, upregulation of EAAT2 protein levels by ceftriaxone, a beta-lactam antibiotic reported as an EAAT2 activator (Rothstein et al., 2005), showed protective effects by decreasing seizures and reducing the mortality in a pentylenetetrazole model of epilepsy (Jelenkovic et al., 2008) and a model of Tuberous Sclerosis Complex (Zeng et al., 2010). In the pilocarpine model, the excitotoxic insult triggered by SE leads to both immediate cell damage (minutes to a few hours) and a protracted process of neurodegeneration (weeks to months) in several brain areas such as the hippocampus (Leite et al., 1990; Turski et al., 1984). Increased EAAT2 protein attenuated acute cell loss after the initial insult (SE) and protection continued during the development of chronic seizures.
EAAT2 mice also exhibited less neuroplastic changes, such as fewer DCX-positive cells, fewer hilar basal dendrites sprouting from DCX-positive cells, and reduced mossy fiber sprouting. Both acute and long-term neuroplastic changes in the dentate gyrus have been reported in the pilocarpine rodent model of TLE, such as granule cell neurogenesis (Parent et al., 1997; Shapiro et al., 2007), aberrant mossy fiber sprouting (Mello et al., 1993), and the formation of hilar basal dendrites with synapses (Spigelman et al., 1998). These neuroplastic changes are similar to features in human and primate TLE (Houser, 1990; Ribak et al., 1998; Scharfman et al., 2000). Furthermore, sprouting of Timm-stained mossy fiber axons could be targeting these basal dendrites, and mossy fiber synapses onto hilar basal dendrites form a recurrent excitatory circuitry within the epileptic dentate gyrus (Austin and Buckmaster, 2004; Mello et al., 1993; Ribak et al., 2000). By reducing the sprouting of the basal dendrites on the DCX-positive granule cells in the EAAT2 mice, these cells might become less interconnected and less functional as hubs for excitatory activity to enhance hyperexcitability in the dentate network. Therefore, reduction of basal dendrites by increased EAAT2 protein expression might impair the recurrent excitatory circuitry, and reduce the hyperexcitability, which could contribute to the protection against epileptogenesis. Our data showed that EAAT2 mice also presented less severe mossy fiber sprouting, which was strongly correlated with less hilar cell loss. The correlation analysis suggests that in the relatively more vulnerable mice, SE tended to induce both more severe neuronal damage and more neuroplastic changes that might result in more frequent chronic seizures. The frequencies of resultant chronic seizures were strongly correlated with the hilar cell loss and moderately but not significantly correlated with mossy fiber sprouting (p=0.052). These findings are consistent with previous reports by Mathern et al. and Pitkänen et al. (Mathern et al., 1995; Pitkanen et al., 2000).
This study suggests that increased EAAT2 protein expression, which enhances glutamate uptake function, is a potential therapeutic approach for treating epilepsy. Our lab and collaborators recently executed a high-throughput screening and identified several novel compounds that can specifically activate functional EAAT2 protein expression (Colton et al., 2010) at low nanomolar concentrations and in a short amount of time (Xing et al., 2011). These compounds may serve as potential therapeutic agents for protecting against excitotoxic neuronal damage. Future studies will be focused on the evaluation of these compounds in animal models of disease including the pilocarpine-induced TLE model.
Supplementary Material
Scoring scale for the severity of mossy fiber sprouting in Timm-stained sections according to the criteria previously reported by Cavazos et al (1991): 0, 1, and 2 refer to no, sparse (patchy), and more (continuous) granules distributions between the tips and crest of DG, respectively; 3 refers to prominent staining within the granule cell layer and supragranular layer; 4 refers to a prominent distribution within the granule cell layer and supragranular layer, which forms a confluent dense laminar band; and 5 refers to a confluent dense band pattern, which extends into the inner molecular layer.
Highlights.
-
➢
Mortality rates decrease in EAAT2 transgenic mice after pilocarpine injection.
-
➢
Increased EAAT2 attenuates hippocampal neuronal loss after status epilepticus (SE).
-
➢
Increased EAAT2 inhibits neurogenesis and mossy fiber sprouting after SE.
-
➢
Increased EAAT2 reduces spontaneous recurrent seizures after SE.
Acknowledgments
This work was supported by the NIH grants (R01NS064275 & U01NS074601). We thank Dr. Xin He (University of Maryland, College Park) for his work on statistical analysis and interpretation. We also thank Dr. Boyoung Lee and Dr. Karl Obrietan (Ohio State University, Columbus) for the advice on Timm staining and epilepsy model.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acharya MM, et al. Progress in neuroprotective strategies for preventing epilepsy. Prog Neurobiol. 2008;84:363–404. doi: 10.1016/j.pneurobio.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arida RM, et al. The course of untreated seizures in the pilocarpine model of epilepsy. Epilepsy Res. 1999;34:99–107. doi: 10.1016/s0920-1211(98)00092-8. [DOI] [PubMed] [Google Scholar]
- Attwell D, et al. Nonvesicular release of neurotransmitter. Neuron. 1993;11:401–7. doi: 10.1016/0896-6273(93)90145-h. [DOI] [PubMed] [Google Scholar]
- Austin JE, Buckmaster PS. Recurrent excitation of granule cells with basal dendrites and low interneuron density and inhibitory postsynaptic current frequency in the dentate gyrus of macaque monkeys. J Comp Neurol. 2004;476:205–18. doi: 10.1002/cne.20182. [DOI] [PubMed] [Google Scholar]
- Benarroch EE. Astrocyte-neuron interactions: implications for epilepsy. Neurology. 2009;73:1323–7. doi: 10.1212/WNL.0b013e3181bd432d. [DOI] [PubMed] [Google Scholar]
- Binder DK, Steinhauser C. Functional changes in astroglial cells in epilepsy. Glia. 2006;54:358–68. doi: 10.1002/glia.20394. [DOI] [PubMed] [Google Scholar]
- Bjornsen LP, et al. Changes in glial glutamate transporters in human epileptogenic hippocampus: inadequate explanation for high extracellular glutamate during seizures. Neurobiol Dis. 2007;25:319–30. doi: 10.1016/j.nbd.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, et al. Mossy fiber synaptic reorganization induced by kindling: time course of development, progression, and permanence. J Neurosci. 1991;11:2795–803. doi: 10.1523/JNEUROSCI.11-09-02795.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavus I, et al. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. 2005;57:226–35. doi: 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- Colton CK, et al. Identification of translational activators of glial glutamate transporter EAAT2 through cell-based high-throughput screening: an approach to prevent excitotoxicity. J Biomol Screen. 2010;15:653–62. doi: 10.1177/1087057110370998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarque M, et al. Glutamate transporters prevent the generation of seizures in the developing rat neocortex. J Neurosci. 2004;24:3289–94. doi: 10.1523/JNEUROSCI.5338-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O. Diagnosis and treatment of temporal lobe epilepsy. Rev Neurol Dis. 2004;1:2–9. [PubMed] [Google Scholar]
- Domercq M, et al. P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-alpha and prostaglandins. J Biol Chem. 2006;281:30684–96. doi: 10.1074/jbc.M606429200. [DOI] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341:1607–10. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- Engel J., Jr A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42:796–803. doi: 10.1046/j.1528-1157.2001.10401.x. [DOI] [PubMed] [Google Scholar]
- Fernandez JD, et al. Endovascular management of iliac rupture during endovascular aneurysm repair. J Vasc Surg. 2009;50:1293–9. doi: 10.1016/j.jvs.2009.06.020. discussion 1299-300. [DOI] [PubMed] [Google Scholar]
- Feustel PJ, et al. Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke. 2004;35:1164–8. doi: 10.1161/01.STR.0000124127.57946.a1. [DOI] [PubMed] [Google Scholar]
- Francis F, et al. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23:247–56. doi: 10.1016/s0896-6273(00)80777-1. [DOI] [PubMed] [Google Scholar]
- Furtado MA, et al. Study of spontaneous recurrent seizures and morphological alterations after status epilepticus induced by intrahippocampal injection of pilocarpine. Epilepsy Behav. 2011;20:257–66. doi: 10.1016/j.yebeh.2010.11.024. [DOI] [PubMed] [Google Scholar]
- Guo H, et al. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet. 2003;12:2519–32. doi: 10.1093/hmg/ddg267. [DOI] [PubMed] [Google Scholar]
- Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. 1990;535:195–204. doi: 10.1016/0006-8993(90)91601-c. [DOI] [PubMed] [Google Scholar]
- Jelenkovic AV, et al. Beneficial effects of ceftriaxone against pentylenetetrazole-evoked convulsions. Exp Biol Med (Maywood) 2008;233:1389–94. doi: 10.3181/0803-RM-83. [DOI] [PubMed] [Google Scholar]
- Lauriat TL, et al. Early rapid rise in EAAT2 expression follows the period of maximal seizure susceptibility in human brain. Neurosci Lett. 2007;412:89–94. doi: 10.1016/j.neulet.2006.10.037. [DOI] [PubMed] [Google Scholar]
- Leite JP, et al. Spontaneous recurrent seizures in rats: an experimental model of partial epilepsy. Neurosci Biobehav Rev. 1990;14:511–7. doi: 10.1016/s0149-7634(05)80076-4. [DOI] [PubMed] [Google Scholar]
- Liu HT, et al. Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia. 2006;54:343–57. doi: 10.1002/glia.20400. [DOI] [PubMed] [Google Scholar]
- Mathern GW, et al. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology. 1999;52:453–72. doi: 10.1212/wnl.52.3.453. [DOI] [PubMed] [Google Scholar]
- Mathern GW, et al. Quantified patterns of mossy fiber sprouting and neuron densities in hippocampal and lesional seizures. J Neurosurg. 1995;82:211–9. doi: 10.3171/jns.1995.82.2.0211. [DOI] [PubMed] [Google Scholar]
- McNamara JO, et al. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;2006:re12. doi: 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- Mello LE, et al. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–95. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Notenboom RG, et al. Up-regulation of hippocampal metabotropic glutamate receptor 5 in temporal lobe epilepsy patients. Brain. 2006;129:96–107. doi: 10.1093/brain/awh673. [DOI] [PubMed] [Google Scholar]
- Parent JM, et al. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–38. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, et al. Association between the density of mossy fiber sprouting and seizure frequency in experimental and human temporal lobe epilepsy. Epilepsia. 2000;41(Suppl 6):S24–9. doi: 10.1111/j.1528-1157.2000.tb01552.x. [DOI] [PubMed] [Google Scholar]
- Pitsch J, et al. Functional role of mGluR1 and mGluR4 in pilocarpine-induced temporal lobe epilepsy. Neurobiol Dis. 2007;26:623–33. doi: 10.1016/j.nbd.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Proper EA, et al. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain. 2002;125:32–43. doi: 10.1093/brain/awf001. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–94. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rao MS, et al. Hippocampal neurodegeneration, spontaneous seizures, and mossy fiber sprouting in the F344 rat model of temporal lobe epilepsy. J Neurosci Res. 2006;83:1088–105. doi: 10.1002/jnr.20802. [DOI] [PubMed] [Google Scholar]
- Rao VL, et al. Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J Neurosci. 2001;21:1876–83. doi: 10.1523/JNEUROSCI.21-06-01876.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribak CE, et al. Alumina gel injections into the temporal lobe of rhesus monkeys cause complex partial seizures and morphological changes found in human temporal lobe epilepsy. J Comp Neurol. 1998;401:266–90. [PubMed] [Google Scholar]
- Ribak CE, et al. Status epilepticus-induced hilar basal dendrites on rodent granule cells contribute to recurrent excitatory circuitry. J Comp Neurol. 2000;428:240–53. doi: 10.1002/1096-9861(20001211)428:2<240::aid-cne4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, et al. Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci. 2007;10:1377–86. doi: 10.1038/nn2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, et al. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–21. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–86. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–7. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Sarac S, et al. Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS. 2009;117:291–301. doi: 10.1111/j.1600-0463.2009.02443.x. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, et al. Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J Neurosci. 2000;20:6144–58. doi: 10.1523/JNEUROSCI.20-16-06144.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro LA, et al. Newly generated granule cells show rapid neuroplastic changes in the adult rat dentate gyrus during the first five days following pilocarpine-induced seizures. Eur J Neurosci. 2007;26:583–92. doi: 10.1111/j.1460-9568.2007.05662.x. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Status epilepticus-induced neuronal injury and network reorganization. Epilepsia. 1999;40(Suppl 1):S34–9. doi: 10.1111/j.1528-1157.1999.tb00876.x. discussion S40-1. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Dempster DW. “Epileptic” brain damage is replicated qualitatively in the rat hippocampus by central injection of glutamate or aspartate but not by GABA or acetylcholine. Brain Res Bull. 1985;15:39–60. doi: 10.1016/0361-9230(85)90059-0. [DOI] [PubMed] [Google Scholar]
- Spigelman I, et al. Dentate granule cells form novel basal dendrites in a rat model of temporal lobe epilepsy. Neuroscience. 1998;86:109–20. doi: 10.1016/s0306-4522(98)00028-1. [DOI] [PubMed] [Google Scholar]
- Strine TW, et al. Psychological distress, comorbidities, and health behaviors among U.S. adults with seizures: results from the 2002 National Health Interview Survey. Epilepsia. 2005;46:1133–9. doi: 10.1111/j.1528-1167.2005.01605.x. [DOI] [PubMed] [Google Scholar]
- Tanaka K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tessler S, et al. Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience. 1999;88:1083–91. doi: 10.1016/s0306-4522(98)00301-7. [DOI] [PubMed] [Google Scholar]
- Tian GF, et al. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–81. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turski WA, et al. Seizures produced by pilocarpine in mice: a behavioral, electroencephalographic and morphological analysis. Brain Res. 1984;321:237–53. doi: 10.1016/0006-8993(84)90177-x. [DOI] [PubMed] [Google Scholar]
- Wetherington J, et al. Astrocytes in the epileptic brain. Neuron. 2008;58:168–78. doi: 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiebe S. Epidemiology of temporal lobe epilepsy. Can J Neurol Sci. 2000;27(Suppl 1):S6–10. doi: 10.1017/s0317167100000561. discussion S20-1. [DOI] [PubMed] [Google Scholar]
- Xing X, et al. Structure-activity relationship study of pyridazine derivatives as glutamate transporter EAAT2 activators. Bioorg Med Chem Lett. 2011;21:5774–7. doi: 10.1016/j.bmcl.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, et al. Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of Tuberous Sclerosis Complex. Neurobiol Dis. 2010;37:764–71. doi: 10.1016/j.nbd.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–7. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scoring scale for the severity of mossy fiber sprouting in Timm-stained sections according to the criteria previously reported by Cavazos et al (1991): 0, 1, and 2 refer to no, sparse (patchy), and more (continuous) granules distributions between the tips and crest of DG, respectively; 3 refers to prominent staining within the granule cell layer and supragranular layer; 4 refers to a prominent distribution within the granule cell layer and supragranular layer, which forms a confluent dense laminar band; and 5 refers to a confluent dense band pattern, which extends into the inner molecular layer.