

Abstract
Endothelin-1 (ET-1) is a critical autocrine and paracrine regulator of cardiac physiology and pathology. Produced locally within the myocardium in response to diverse mechanical and neurohormonal stimuli, ET-1 acutely modulates cardiac contractility. During pathological cardiovascular conditions such as ischaemia, left ventricular hypertrophy and heart failure, myocyte expression and activity of the entire ET-1 system is enhanced, allowing the peptide to both initiate and maintain maladaptive cellular responses. Both the acute and chronic effects of ET-1 are dependent on the activation of intracellular signalling pathways, regulated by the inositol-trisphosphate and diacylglycerol produced upon activation of the ETA receptor. Subsequent stimulation of protein kinases C and D, calmodulin-dependent kinase II, calcineurin and MAPKs modifies the systolic calcium transient, myofibril function and the activity of transcription factors that coordinate cellular remodelling. The precise nature of the cellular response to ET-1 is governed by the timing, localization and context of such signals, allowing the peptide to regulate both cardiomyocyte physiology and instigate disease.
Linked Articles
This article is part of a themed section on Endothelin. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2013.168.issue-1
Keywords: endothelin, myocyte, contraction, hypertrophy, calcium, kinase
Introduction
Endothelin (ET)-1, ET-2 and ET-3 are a family of cyclic 21 amino acid peptides. The peptides are encoded within three separate yet highly conserved genes located on chromosomes 6, 1 and 20 in humans respectively (Inoue et al., 1989). ET-1 is the most well characterized member of this family. The peptide was initially isolated from the culture supernatant of porcine aortic endothelial cells and identified as a protease-sensitive vasoconstrictor. It remains the most potent vasoactive substance identified to date (Yanagisawa et al., 1988), performing an important function in the regulation of vascular smooth muscle tone. ET-1 also mediates effects on other cell types. Here, we discuss the role of ET-1 in the heart. We will focus on the actions of ET-1 on cardiac muscle and describe how the ET system contributes to cardiac regulation and autoregulation during health and disease.
ET-1 in the heart
Throughout life, ET-1 plays key roles in many aspects of cardiac physiology and pathology. It is involved in controlling aortic arch formation during development (Kurihara et al., 1995), is required for cardiomyocyte survival and prevents myocyte loss during ageing (Zhao et al., 2006). In the adult, ET-1 modulates coronary blood flow by regulation of vascular tone. Furthermore, by acting on its receptors expressed by atrial and ventricular myocytes, the peptide modulates cardiac muscle function directly (Hirata et al., 1989). The effects of ET-1 on the heart are observed over distinct and wide timescales. Acutely (within minutes), ET-1 modulates muscle contractility and induces arrhythmias (Baltogiannis et al., 2005), whereas over longer time scales (days to weeks), the peptide induces cardiomyocyte growth. The long-term effects of ET-1 are associated with maladaptive hypertrophic remodelling of the heart (Yorikane et al., 1993) and its progression to failure (Francis et al., 1990). Hence, ET-1 is considered to contribute to both the aetiology and pathology of these conditions. Indeed, the circulating plasma level of ET-1 is positively correlated with severity of cardiac disease and thus is a reliable prognostic indicator of future heart failure (Selvais et al., 2000; Zolk et al., 2002).
Paracrine/autocrine ET-1 in regulation of cardiac function
Further contributing to its significant role in modulating cardiomyocyte function, ET-1 is synthesized and secreted by the myocytes (Nunez et al., 1990; Firth and Ratcliffe, 1992; Suzuki et al., 1993), fibroblasts (Fujisaki et al., 1995) and endothelial cells (Kedzierski and Yanagisawa, 2001) of the heart. The presence of the machinery for both the production and detection of ET-1 within the myocardium generates a local autocrine and paracrine ET-1 system, providing a tightly coupled and regulated means of control (Pikkarainen et al., 2006; Higazi et al., 2009). Supporting this notion, ET-1 production/secretion is signal-responsive and contributes to both normal adaptive physiological responses and to the induction and progression of pathology. During physiology, myocyte-secreted ET-1 is reported to contribute to stretch-induced enhancement of contractility, which can occur during periods of increased workload (Pikkarainen et al., 2006). In this context, ET-1 secretion/production is either directly stimulated or induced in response to AngII, which is also secreted by the stretched myocyte (Sadoshima et al., 1993; Yamazaki et al., 1996; Pikkarainen et al., 2006; Cingolani et al., 2011). As a result, contractility is enhanced to accommodate for the change in cardiac demand. This mechanism may explain the slow force response or Anrep effect observed in stretched atrial preparations (Kockskämper et al., 2008a). Indeed, many of the cardiac effects of AngII are abrogated by ET receptor inhibition (Ito et al., 1993). Should the initiating stimulus and thus ET-1 secretion persist, hypertrophy may result (Yamazaki et al., 1996). The acute effect of ET-1 is therefore a prequel to its longer term effect on cardiac remodelling (Cingolani et al., 2011). In aged individuals, the plasma concentration of ET-1 is elevated (Maeda et al., 2003), hence the importance of ET-1-induced cardiovascular effects increases with time. Furthermore, cardiomyocyte ET-1 generation is increased following ischaemia (Serneri et al., 2000), during the cardiac hypertrophic response (Yorikane et al., 1993; Zolk et al., 2002; Mayyas et al., 2010) and as a result of heart failure (Francis et al., 1990). This increased ET-1 contributes to pathologies such as arrhythmia and the initiation and development of cardiac remodelling, the incidence of which increases with age (Lakatta and Levy, 2003b). Increased expression of ET-1 has been observed at both the mRNA level of pre-pro-ET-1 and the protein level of the mature peptide itself (Arai et al., 1995; Kobayashi et al., 1999; Zhao et al., 2006), suggesting that increased transcription contributes to enhanced ET-1 production. This is accompanied by increased activity of the processing mechanisms that produce the mature peptide. Expression and activity of endothelin converting enzyme 1a (ECE1a) is up-regulated in human failing hearts and animal models of congestive heart failure (Ergul et al., 2000a; 2000b), expediting the processing of big-ET-1 into mature ET-1. In conjunction with the increased level of ET-1 peptide, expression of the ETA receptor by ventricular myocytes is enhanced during disease (Arai et al., 1995; Pieske et al., 1999; Baltogiannis et al., 2005; Harzheim et al., 2010). Thus, under disease-associated conditions, mechanisms of ET-1 production and detection are up-regulated in a myocyte-centric manner, providing an entirely local mechanism for the pathological effects of ET-1.
ET receptors
ET-1 elicits its effects by association with one or both of its receptor isoforms, the ETA and ETB receptors (Arai et al., 1990; Sakurai et al., 1990; Webb, 1991; Yorikane et al., 1993; Alexander et al., 2011). Both receptor isoforms are classical GPCRs comprising 7 α-helical transmembrane spanning domains, an extracellular N-terminal domain and intracellular C-terminal domain. The intracellular C-terminal domain and loops between the transmembrane segments interact with heterotrimeric G-proteins composed of Gα, β and γ subunits. Activation of these receptors stimulates multiple intracellular signalling pathways and mediates diverse cellular effects (Ishikawa et al., 1988a; 1988b; Moravec et al., 1989). The relative abundance of ET receptors varies across the chambers and regions of the normal healthy heart, with the number of ET-1 binding sites in atrial cells far exceeding those found in ventricular cells (Molenaar et al., 1993). Functionally, the ETA and ETB receptors exhibit several key differences. Although the affinity of both ET receptors for ET is subnanomolar, the ETA receptor exhibits a higher affinity for ET-1 over ET-2 or ET-3 (Hosoda et al., 1991), whilst the ETB receptor binds non-selectively to each of the three ET isoforms (Sakurai et al., 1990). The downstream signals activated following engagement of the ETA receptor and ETB receptor also differ. Both receptors can couple to multiple G-proteins but the particular G-protein repertoire that each isoform engages may vary, governing the pattern of downstream pathways activated. The ETA receptor interacts with Gαq/11 (Aramori and Nakanishi, 1992), Gαs (Eguchi et al., 1993b; Yamazaki et al., 1996) and Gα12 activating polyphosphoinositide hydrolysis and production of inositol-(1,4,5)-triphosphate (IP3), diacylglycerol (DAG) and cAMP (Griendling et al., 1989; Takuwa et al., 1990; Serneri et al., 2000). The ETB receptor also couples to Gαq/11. However, in contrast to the ETA receptor, it preferentially interacts with Gαi/o and Gα13 (Eguchi et al., 1993b; Gohla et al., 1999). Consequently, activation of the ETB receptor is associated with inhibition of cAMP accumulation as well as stimulation of polyphosphoinositide hydrolysis (Eguchi et al., 1993a). Although the heart expresses both receptor isoforms, in cardiomyocytes the ETA receptor predominates accounting for greater than 80% of ET binding sites (Allen et al., 2003).
The most well established mechanisms of ETA receptor-dependent signalling in cardiomyocytes concern those regulated downstream of Gαq/11. Inhibition of Gαq/11 prevents activation of ET-1 dependent signalling pathways in rat adult ventricular myocytes (AVM), whilst reducing the activity of either Gα12/13 or Gαi/o does not produce comparable effects (Snabaitis et al., 2005). Furthermore, ET-1-dependent signalling is enhanced by knockdown of the GTPase activating protein regulator of G-protein signalling 2 (RGS2), which negatively regulates GTP-bound Gαq/11 by enhancing its GTP hydrolysing properties (Zhang et al., 2006). The downstream effects of Gαq/11 activation are mediated through activation of PLC enzymes, which catalyse the hydrolysis of phosphatidylinositol-4,5-bisphosphate (PI-4,5-P2) to produce membrane-associated DAG and soluble IP3 (Wu et al., 1992). Cardiomyocytes express multiple PLC isoforms, with PLCβ1 and B3 showing Gαq/11-dependent activity. ET-1 stimulation of cultured neonatal rat ventricular myocytes (NRVM) leads to phosphoinositide hydrolysis within seconds (Clerk and Sugden, 1997). The rapid increase in IP3 and DAG activates multifarious downstream signalling pathways, including those regulated by calcium, small G-proteins and kinase cascades (Figure 1). In addition, ET-1 stimulates nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) (De Giusti et al., 2008) and production of reactive oxygen species (ROS) (Cheng et al., 1999). These pathways perform important roles in regulation of cardiomyocyte contractility, pacemaking and remodelling.
Figure 1.
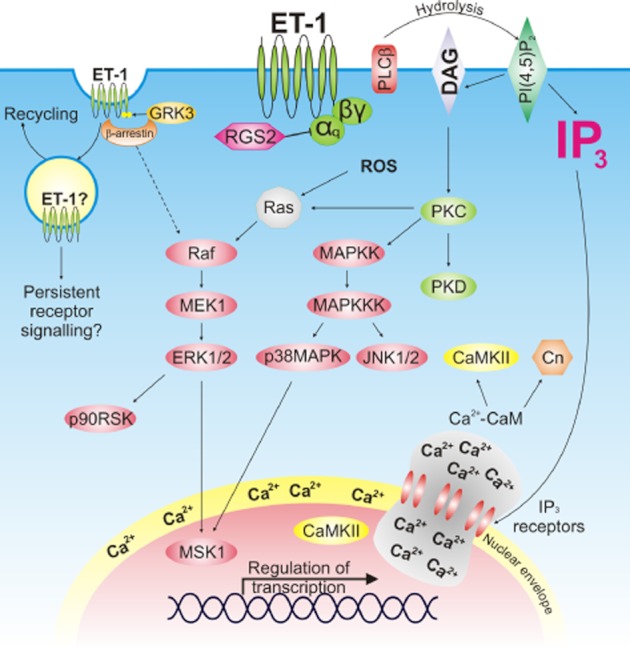
Signalling pathways activated downstream of the ETA receptor in cardiomyocytes. ET-1 binds to the Gαq-coupled ETA receptor inducing production of IP3 and DAG. These second messengers activate protein kinase cascades, ROS production and IP3-induced calcium release. Following ET-1 binding, the ET-1-ETA receptor complex is internalized in a GRK3-β-arrestin dependent manner. β-arrestin has been postulated to induce activation of MAPK cascades.
Terminating ET-1 signalling
In order to limit the effects of ET-1 on the heart, signalling from ET receptors must be tightly regulated. The ETA receptor forms a very high affinity interaction with ET-1 and the rate of disassociation of ligand/receptor complex is slow; hence, this process is not straightforward (Hilal-Dandan et al., 1997). Indeed, the resistance of the ET-1/ETA receptor complex to acid or exposure to inhibitor has led to the proposal that ET-1/ETA receptor binding is quasi-irreversible, possibly as a result of covalent binding of ET-1 to the receptor. Thus, it is likely that the receptor is internalized in a ligand-bound state and that internalization of this complex, rather than ET-1 dissociation, determines the duration of the ET-1 response. Indeed, an association of ET-1 with the ETA receptor within the endosome for up to 2 h has been reported (Chun et al., 1994; 1995). It is unclear whether ET-1 continues to signal via the ETA receptor when internalized in this manner but this hypothesis could explain certain of the observed effects of ET-1- stimulation. Co-localization of internalized ET-1-ETA receptor with caveolar domains may aid interaction with G-proteins, enabling ET-1-ETA receptor signalling to persist (Chun et al., 1994). Furthermore, ET-1-ETA receptor complex internalization is GRK (G-protein receptor kinase)-β-arrestin dependent (Freedman et al., 1997). GRKs 2, 3 and 5 are expressed in cardiac myocytes (Vinge et al., 2001) with GRK2 activity and expression being up-regulated in heart failure – an adaptation that limits β-adrenergic signalling (Ping et al., 1997). In AVMs, GRK3 targets the ETA receptor and inhibition of GRK3 in AVM enhances ET-1 induced signalling, suggesting that this kinase is important for attenuation of the ET-1 response (Vinge et al., 2007). Following GRK-dependent phosphorylation, recruitment of β-arrestins to activated receptors targets them for internalization. However, β-arrestin is also implicated in the activation of the MAPK cascade (Lefkowitz and Shenoy, 2005). Given these potential mechanisms of alternative and persistent ET-1-induced signalling, it appears that a simple model of ET-1-GPCR binding and agonist-induced desensitization cannot fully account for the cellular effects of ET-1.
Interestingly, internalized ETA receptor and ETB receptor are targeted to different intracellular compartments after stimulation with ET-1. The ETA receptor co-localizes with the pericentriolar recycling compartment and can be recycled to the plasmalemma so that cellular ET-1 sensitivity is maintained. In contrast, ETB receptor are targeted to lysosomes and degraded after agonist-induced internalization. The lysosomal fate of the internalized ET-1–ETB receptor complex provides a mechanism for clearance of the peptide (Attinà et al., 2005). Exemplifying this, deletion of the ETB receptor in mice impairs ET-1 clearance and selective pharmacological blockade of this isoform increases the circulating concentration of ET-1 (Gariepy et al., 2000; Kelland et al., 2010). The vasculature of the lung provides the major site of clearance of ET-1 from plasma (Dupuis et al., 1996), with approximately 80% of the peptide being removed from the bloodstream by the ETB receptors expressed by pulmonary endothelial cells. Efficient removal of the peptide via this mechanism generates a half-life for ET-1 in the circulation of less than 5 min and maintains the plasma concentration of the peptide in healthy individuals at approximately 1 pM (Kedzierski and Yanagisawa, 2001), significantly below its activity threshold. However, local ET-1 concentration at sites of ET-1 production and release is expected to be much higher, raising the level of the peptide to that which can modulate cardiac function.
ET-1 in cardiac pacemaking and contractility
Cardiac contraction during systole is initiated by an action potential generated following automatic depolarization of the pacemaking SA node. From the SA node, the action potential propagates across the atria and induces contraction. Following a pause at the atrio-ventricular node, the action potential is rapidly conducted to the apex of the heart by specialized Purkinje fibres. Subsequent spreading of the action potential throughout the ventricles induces contraction and thus expels blood into the aorta. At the molecular level, the mechanism that links membrane excitation and myocyte contraction is termed excitation-contraction coupling (ECC) (Figure 2) (Bers, 2002; Fearnley et al., 2011). This process is initiated by the depolarization-induced opening of L-type voltage-gated calcium channels (also known as dihydropyridine receptors), generating the ICa,L current. Calcium entry through these channels generates a much larger release of calcium from the sarcoplasmic reticulum (SR) via the ryanodine receptor (RyR2), in a process termed ‘calcium-induced-calcium-release’ (CICR) (Fabiato, 1983; Roderick et al., 2003). This calcium binds to the contractile apparatus and induces cellular shortening. To terminate systole and allow relaxation, calcium is extruded from the cell or re-sequestered in the SR. Synchronous release of calcium throughout the ventricular myocyte is achieved as a consequence of the highly specialized subcellular structure of the cell. The sarcolemma is extensively invaginated to form the t-tubular network; hence, depolarizing action potentials can spread into the depths of the cell and induce coordinated cellular contraction. Furthermore, the L-type calcium channel and RyR2 are co-localized within specialized structures known as the cardiac dyads, facilitating CICR.
Figure 2.
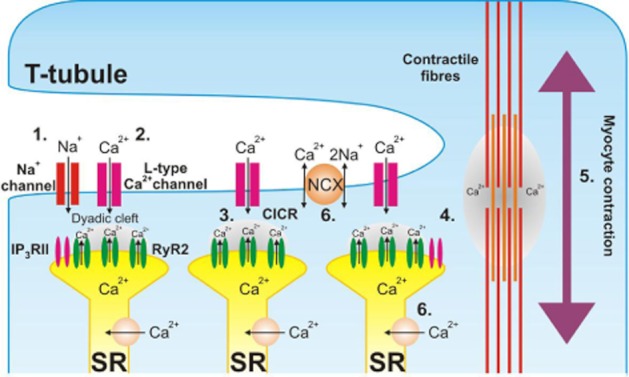
Excitation–contraction coupling in the cardiomyocyte. Opening of voltage-gated Na+-channels induces cellular depolarization (1). L-type voltage-gated calcium channels are activated in response to depolarization and mediate calcium entry (2). Increased calcium concentration within the cardiac dyad activates the closely opposed RyR2 and induces calcium-induced-calcium release (3), raising cytosolic calcium concentration. Calcium diffuses to the contractile apparatus (4), binds to the contractile filaments and stimulates contraction (5). Calcium is re-sequestered in the sarcoplasmic reticulum or extruded from the cell by NCX, returning cytosolic calcium concentration to resting levels (6). Colocalization of IP3RII within the cardiac dyad facilitates calcium-induced calcium release under stimulated conditions.
ET-1 has an established role in the regulation of cardiac contractility, in particular as a positive inotrope (Ishikawa et al., 1988b; Moravec et al., 1989; Shah et al., 1989). Other effects upon the heart include negative inotropy, arrhythmogenesis, decreased relaxation (lusitropy) and chronotropy (Ishikawa et al., 1988a; Li et al., 2005). Despite these well-described aspects of ET-1 in cardiac biology, controversy remains regarding the effects of ET on cardiac function, its mechanism(s) of action and the specific molecular targets involved. A number of factors contribute to the apparent diversity of responses reported, although variable expression levels of ET receptors account for many of the discrepancies. For example, responses are different between species and vary with age. ET-1 does not exert an inotropic effect in adult mice and dogs, which have low levels of ET receptor expression (Takanashi and Endoh, 1991; Nishimaru et al., 2007). However, in neonatal mice, where ET receptors are more abundant, inotropic responses to ET-1 are observed (Nagasaka et al., 2003). Moreover, the origin of the cardiac preparation studied is also a significant determinant of the response (Moravec et al., 1989). This is not surprising given that the distribution and abundance of ET receptor isoforms varies across the chambers of the heart (Molenaar et al., 1993; Russell and Davenport, 1996). Further diversity is provided by the experimental preparation – intact heart, chamber muscle strips or isolated cells.
Due to greater receptor abundance in the atria, the effects of ET-1 are most well established in this chamber (Ishikawa et al., 1988b; Vigne et al., 1989; Molenaar et al., 1993). The atrial response to ET-1 (e.g. in rat, rabbit and guinea pig) is characterized by an initial negative inotropic phase (first 1–2 min), followed by a slowly developing and persistent positive inotropic phase (Takanashi and Endoh, 1991; Bootman et al., 2007). Although cAMP independent, the positive inotropic effect of ET-1 in the atria can reach levels comparable to that induced by the catecholamines (Vigne et al., 1989). The cardiac ventricle also exhibits a positive inotropic response to ET-1, albeit at a substantially reduced magnitude (Takanashi and Endoh, 1991; Proven et al., 2006) and unlike the situation in the atria, a negative inotropic response is rarely observed (Proven et al., 2006). Notably, ET-1 also affects heart rate by modulating the physiology of the SA node and Purkinje fibres (Ishikawa et al., 1988a). The combined effect of ET-1-induced positive inotropy and chronotropy increases cardiac output under conditions of stress, contributing to the adaptive response of the heart. However, these actions of ET-1 also have deleterious consequences, promoting arrhythmogenic, action potential-independent calcium transients (Baltogiannis et al., 2005; Li et al., 2005; Mayyas et al., 2010).
Similarly to other cardiotonic agonists, ET-1 changes cardiac contractility by modulating components of the ECC machinery (Figure 3). These include alterations in the dynamics and amplitude of systolic calcium transients (Bootman et al., 2007; Kockskamper et al., 2008), changes in myofilament sensitivity to calcium (Yang et al., 1999; Westfall et al., 2005; Cuello et al., 2007a) or altered frequency of stimulation (Ishikawa et al., 1988a).
Figure 3.
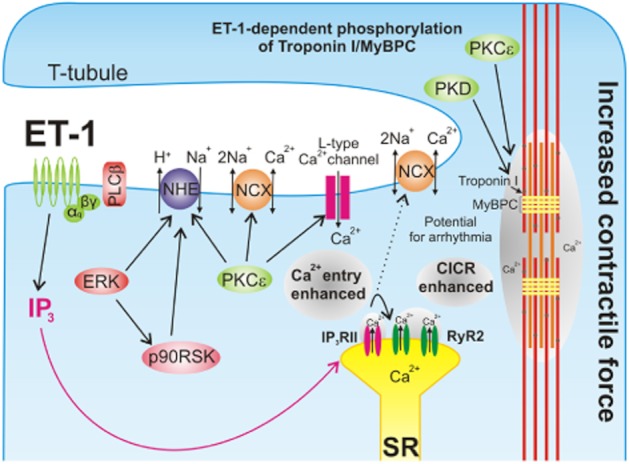
ET-1 regulates ventricular myocyte contractility. Downstream of the ETA receptor in the ventricular myocyte, IICR, PKC, PKD, p90 RSK and ERK are activated. Protein-kinase-dependent phosphorylation of NHE1 increases intracellular Na+ concentration and depolarizes the cell. Subsequent activation of NCX in reverse mode allows calcium entry to be enhanced. When combined with PKC-dependent amplification of calcium entry through the L-type calcium channel, the stimulus for CICR is increased. IP3-dependent calcium release from the SR sensitizes the closely opposed RyR2, hence facilitating CICR. The resulting elevated cytosolic calcium concentration and PKC/PKD-dependent phosphorylation of myofilament proteins allows the myocyte to produce a larger force of contraction.
ET-1 regulation of NCX activity
ET-1 effects systolic calcium predominantly via modulation of the Na+/Ca2+ exchanger (NCX) (Ballard and Schaffer, 1996; Yang et al., 1999) and L-type channel activity (Ono et al., 1994). The primary function of NCX is calcium clearance from the cytosol during diastole, accounting for up to 20% of the calcium extruded, a contribution that increases further under disease conditions (Bers, 2002; 2006). NCX uses the electrochemical gradient for Na+ between the extracellular space and the cytosol, created by the Na+K+-ATPase, to exchange 3 Na+ for every calcium ion extruded from the cell (Philipson and Nicoll, 2000). Where intracellular calcium is increased independently of an action potential, NCX-mediated Na+ entry may cause sufficient depolarization to cross the threshold for action potential generation (Gilbert et al., 1991). As well as this forward mode of activity, NCX can also operate in reverse, inducing calcium influx (Leblanc and Hume, 1990). This may be precipitated by raised intracellular Na+ concentration under conditions of membrane depolarization, for example during the initial phase of the action potential (Shattock and Bers, 1989). As a result of calcium influx at this point, calcium transients are increased in amplitude and contractility enhanced (Meyer et al., 1996; Yang et al., 1999). ET-1 regulates NCX either by engaging signalling mediators activated directly downstream of the ETA receptor such as PKC (Ballard and Schaffer, 1996; Zhang and Hancox, 2009), or by increasing activity of the sodium-hydrogen exchanger (NHE) (Cingolani et al., 2011). The NHE is the major mechanism by which intracellular pH is regulated in cardiac myocytes. It extrudes one H+ ion from the cell in exchange for 1 Na+ (Cingolani and Ennis, 2007), exploiting the Na+ gradient created by the Na+K+-ATPase. The resulting increase in intracellular Na+ activates NCX in reverse mode, increasing [Ca2+]i and producing an inotropic effect (Alvarez et al., 1999). H+ extrusion by NHE would also be expected to modulate contractility via cytosolic alkalanization, which would influence myofilament calcium sensitivity. Despite support for this hypothesis in earlier studies, it is now clear that the small changes in intracellular [H]i produced by NHE are compensated for by the bicarbonate anion transporter; providing bicarbonate is present in the extracellular medium (Alvarez et al., 1999). Therefore, this mechanism is unlikely to contribute to the ET-1 response. ET-1 increases NHE activity via kinases such as PKC, ERK1/2 and p90 RSK (Bianchini et al., 1997; Fliegel and Karmazyn, 2004; Maekawa et al., 2006; Cuello et al., 2007b; Snabaitis et al., 2008). Although ERK1/2 and therefore p90 RSK may both be activated via canonical GPCR signalling, NADPH oxidase activation and ROS formation are necessary for these kinases to phosphorylate NHE in response to ET-1 (Cingolani et al., 2011).
ET-1 regulation of L-type calcium current
L-type calcium channel activity is subject to extensive regulation by GPCRs, which elicit their effects through PKC and PKA dependent phosphorylation (Puri et al., 1997; Kamp and Hell, 2000). Phosphorylation of the pore-forming α1C and accessory α1s and β2a subunits of the channel has been detected biochemically (Puri et al., 1997). Although intensively studied, there is a lack of consensus regarding modulation of L-type calcium current (ICa,L) by ET-1, with evidence for both a positive (Lauer et al., 1992; He et al., 2000) and negative effect (Ono et al., 1994; 2001; Cheng et al., 1995). A biphasic effect of ET-1 has been reported in rabbit ventricular myocytes, consisting of a reduction followed by an enhancement of ICa,L (Watanabe and Endoh, 1999). In these cells the ET-1 stimulated augmentation of ICa,L approached a level similar to that induced by β-adrenoreceptor stimulation. However, the mechanisms by which ET-1 and sympathetic stimuli affect ICa,L are not shared. When ET-1 and sympathetic stimuli occur concurrently, ET-1 exerts an anti-adrenergic effect through a pertussis toxin-sensitive pathway (i.e. Gi/o mediated). This suppresses the enhancement of ICa,L by β-agonists and prevents their associated inotropic (Boixel et al., 2001; Munzel et al., 2005) and arrhythmic responses (Kolettis et al., 2008). In addition to the inter-species variation in the ET-1 response, the lack of a consistent effect of ET-1 has also been ascribed to the methods used for recording ICa,L (He et al., 2000). In order to mitigate this, the perforated patch technique has been used to minimize intracellular dialysis and loss of signalling mediators. Using this approach, a consistent PKC-dependent positive regulation of ICa,L by ET-1 has been reported (Kamp and Hell, 2000). In order for such a mechanism to operate efficiently, co-localization of the main players is required to facilitate interaction of the DAG generated by phosphoinositide hydrolysis with its targets (Robu et al., 2003). The importance of DAG localization is illustrated by the contrasting effects of bath-applied and intracellular-released DiC8, a DAG analogue (He et al., 2000). Bath application of DiC8 induces a PKC-independent reduction in ICa,L whereas intracellular photoactivation of caged DiC8 mimics the PKC-dependent enhancement of ICa,L observed with ET-1 application (He et al., 2000). Although PKC phosphorylates and activates L-type channels directly, the effects of PKC activation could also be transduced by calcium/calmodulin-dependent protein kinase II (Komukai et al., 2010). Furthermore, ET receptors, PLCβ and PKCε are all localized to the sarcolemma of the t-tubule in ventricular myocytes, bringing them into close proximity with L-type calcium channels (Robu et al., 2003). Interestingly, in disease situations where t-tubules are lost, ET-1 no longer elicits a positive inotropic effect (Chung et al., 2008; Smyrnias et al., 2010).
ET-1 stimulated IP3-induced calcium release and cardiac contractility
As well as DAG, ET-1-dependent phosphoinositide hydrolysis in the myocytes and conduction system of the heart generates the important signalling effector IP3 (Chen et al., 2004; Remus et al., 2006; Hirose et al., 2008; Kockskämper et al., 2008b; Ju et al., 2011). Due to the relatively small increase in IP3 and low expression level of IP3 receptors (IP3Rs) in heart preparations relative to other tissues, the role, if any for IP3 in modulating intracellular [Ca2+] and contractility was for a long time unclear (Wu et al., 2006; Higazi et al., 2009). Furthermore, since expression of IP3Rs is 50–100 fold lower than that of the RyR (Moschella and Marks, 1993; Perez et al., 1997; Lipp et al., 2000), it was difficult to discern what additional function IP3Rs could perform. This contrasts with the prominent role for IP3Rs in the development and physiology of the embryonic heart. Indeed, mice in which the type 1 and type 2 IP3R isoforms (IP3RI and IP3RII) are subject to germ-line deletion are not viable and exhibit defects in cardiac development (Uchida et al., 2010). Moreover, IP3 stimulated calcium signals play a key role in pacemaker activity of the embryonic heart (Kapur and Banach, 2007). Evidence has recently accumulated which indicates that despite low expression of IP3Rs, the IP3 signalling pathway also plays a significant role in the physiology and pathophysiology of the adult heart, contributing to many of the effects of ET-1 and other Gαq coupled receptors on cardiac function (Wu et al., 2006; Kockskämper et al., 2008b; Higazi et al., 2009; Nakayama et al., 2010). As would be expected, the contribution of IP3 signalling to the action of ET-1 across the regions of the heart varies according to the level of expression and subcellular localization of the IP3R. Similarly to ETA receptors, IP3Rs are most abundant in atrial myocytes and in nodal tissue (Lipp et al., 2000; Ju et al., 2011). The IP3RII predominates in SA node, atrial and ventricular myocytes (Perez et al., 1997; Lipp et al., 2000; Ju et al., 2011), whereas in Purkinje fibres, IP3RI is most abundant (Gorza et al., 1993).
Engagement of IP3 signalling in the different cell types of the heart has varying consequences. In the SA node, ET-1 causes increases in firing rate, calcium transient amplitude, diastolic calcium levels and the frequency of elementary calcium release events (Ju et al., 2011). The recapitulation of these effects by exposure of nodal cells to cell-permeant IP3 and the decreased firing rate and loss of ET-1 responses in nodal cells from IP3R knockout mice demonstrates the importance of ET-1/IP3 signalling in this region of the heart (Ju et al., 2011). Although ET-1 also elicits a positive chronotropic effect in guinea pig SAN (Ishikawa et al., 1988a), rabbit SAN exhibits a negative chronotropic response to ET-1 (Ono et al., 2001). The effect of ET-1 in these rabbit cells was attributed to modification of calcium and potassium currents. The role of IP3 signalling in eliciting the effects of ET-1 upon the SAN in species other than mouse remains to be fully resolved. ET-1 also elicits spontaneous events in Purkinje fibres. Despite evidence for expression of functional IP3Rs that contribute to spontaneous calcium release events in these cells (Gorza et al., 1993; Wu and Tseng, 1993; Boyden et al., 2004), it is not determined whether these IP3Rs are engaged by ET-1 (Wu and Tseng, 1993). The IP3R is however stimulated in Purkinje cells by IP3 generated following activation of α-adrenergic GPCRs, resulting in wide long lasting calcium release events (Hirose et al., 2008).
In atrial myocytes, IP3 signalling contributes to the inotropic and pro-arrhythmic action of ET-1 (Li et al., 2005), and IP3-mediated calcium signals are localized to the sub-sarcolemmal and perinuclear regions (Mackenzie et al., 2004; Zima et al., 2007). Whilst perinuclear IP3-mediated calcium signals have been proposed to control transcription (see later) (Wu et al., 2006; Higazi et al., 2009), IP3-mediated calcium signals at the plasma membrane play a role in modulating atrial myocyte contractility and may underlie the enhanced automaticity of ET-1 stimulated myocytes (Mackenzie et al., 2004). A similar pattern of IP3R expression and activity is observed in cells of the SAN (Ju et al., 2011). As a result of their lack of t-tubules, ECC is restricted to the peripheral sarcolemma of atrial myocytes (Ju et al., 2011). Therefore, IP3-mediated calcium signals exert a greater than anticipated effect on the physiology of an ET-1-stimulated atrial or SAN myocyte due to their co-localization with the ECC machinery in these cells. ET-1-stimulated IP3-mediated calcium signals promote transition of the sub-sarcolemmal ring of calcium normally observed following depolarization of atrial myocytes to a global calcium elevation, enhancing ECC and recruitment of RyRs deeper in the myocyte cell volume (Mackenzie et al., 2004). As a result, contractility is increased and blood propelled with greater force into the ventricles. Since the extent of ventricular stretch determines the contractile force produced by the ventricle – the Frank Starling relationship – greater atrial contraction brings about increased cardiac output. In addition to enhancing electrically evoked calcium transients, ET-1 application also increases the incidence of elementary calcium release events in atrial myocytes (Mackenzie et al., 2001; Li et al., 2005). These events contribute to an increase in diastolic calcium levels beneath the sarcolemma, which if sufficiently elevated, activates NCX-dependent inward Na+ flux and action potential generation. As a result, a potentially arrhythmogenic ectopic event is induced (Lipp et al., 2000; Mackenzie et al., 2002).
Functional interactions between IP3Rs and RyRs are also observed in ventricular myocytes exposed to ET-1 (Proven et al., 2006; Domeier et al., 2008; Harzheim et al., 2009). However due to the lower abundance of ET and IP3 receptors in these cells (Lipp et al., 2000; Domeier et al., 2008), their interaction and influence upon myocyte physiology is not as great. Most notably, IP3 signalling does not make a prominent contribution to the inotropic action of ET-1 in the ventricular myocyte. This is not surprising given the limited effect of ET-1 upon calcium fluxes in these cells when compared with atrial myocytes (Proven et al., 2006; Domeier et al., 2008). Prolonged exposure of ventricular myocytes to ET-1 does however lead to the generation of ectopic calcium transients and IP3-dependent extra-systolic contractions (Proven et al., 2006) and IP3-dependent calcium signals also promote sodium-calcium exchange (Gilbert et al., 1991). Thus, similarly to the situation in atrial myocytes, ET-1 activated IP3 signalling has the potential to precipitate ventricular arrhythmias.
As discussed earlier, both circulating and local cardiac ET-1 levels are increased with age (Maeda et al., 2003) and during cardiac pathologies such as arrhythmia, infarction and hypertrophy (Yorikane et al., 1993; Serneri et al., 2000). Although this increase in ET-1 may initially support ECC, it also has deleterious consequences and is likely to exacerbate pathology and/or cause it to persist (Tuinenburg et al., 1998; Baltogiannis et al., 2005; Mayyas et al., 2010). Furthermore, IP3Rs are increased in their abundance during cardiac pathology (Moschella and Marks, 1993; Harzheim et al., 2009; 2010). As a result, the effects of ET-1 on myocyte physiology are enhanced. Notably, the additional IP3Rs in the hypertrophied heart are not homogenously distributed throughout the cell volume but localized to the sub-sarcolemmal dyadic region, proximal to the RyRs that participate in ECC (Harzheim et al., 2009). Thus, under conditions of increased IP3, calcium emanating from these IP3Rs stimulates calcium release from their neighbouring RyRs. The effects of this activity are twofold. First, IICR may promote ECC, thereby enhancing contractility. However, second, IICR may promote potentially arrhythmogenic ectopic calcium release events (Harzheim et al., 2009). On balance, the enhancement of calcium release via RyRs by the additional IP3Rs may outweigh the potential deleterious effects of ectopic calcium signals. During hypertrophy and the progression to failure, decreased coupling efficiency between L-type channels and RyRs arises as a result of increased width of the dyadic cleft (Song et al., 2006; Xu et al., 2007; Heinzel et al., 2011). In this situation, calcium release through additional IP3Rs could provide a mechanism to overcome this defect in ECC by bringing RyRs closer to their threshold for activation. This effect is analogous to the priming of RyRs by PKA phosphorylation following β-stimulation, which sensitizes the receptors to calcium influx via LTCC (Zhou et al., 2009). In this way, increased IP3R expression may be considered an adaptive response that sustains contractile function as the heart hypertrophies.
Effects of ET-1 upon the contractile apparatus
The relationship between the enhancement in [Ca2+]i and contractility observed in cardiac preparations following exposure to ET is non-linear, indicating that the inotropic response does not solely result from enhancement of intracellular calcium levels. This non-linearity is particularly apparent in ventricular preparations where, despite a significant increase in contraction, [Ca2+]i is not substantially augmented (Wang et al., 1991; Pieske et al., 1997). This contrasts with the response to the catecholamines, which cause a robust increase in calcium transient amplitude and contractility. The mechanism by which the alteration in contraction is brought about by catecholamines is well defined and occurs primarily through PKA-dependent phosphorylation of contractile filament regulatory proteins, most notably the inhibitory subunit of troponin (TnI) and myosin binding protein C (MyBP-C) (Chen et al., 2010). Phosphorylation of TnI and MyBP-C differentially contributes to regulation of contractility. Phosphorylation of TnI reduces calcium sensitivity of the myofilaments (Pi et al., 2003), and phosphorylation of MyBP-C is responsible for enhanced cross-bridge cycling, thereby enhancing relaxation (Chen et al., 2010). GPCRs that signal via Gαq such as the ETA receptor elicit an opposing effect on myofilament action – generally increasing sensitivity to calcium and decreasing cross-bridge cycling (Pi et al., 2003; Wang et al., 2006). As such, their modulation of contractility is energetically less expensive. PKC, RSK and PKD have all been identified as ET-1-activated TnI and MyBP-C kinases (Cuello et al., 2007a; 2011; Chen et al., 2010).
Remodelling of the heart by ET-1
In addition to its diverse actions in the regulation of myocyte contractility, ET-1 has been consistently demonstrated to perform a major role in disease-associated remodelling of the heart (Russell and Molenaar, 2000; Sugden, 2003). Heart disease encompasses a spectrum of interrelated and progressive disorders, which include hypertension, coronary artery disease, arrhythmia and, most severely, heart failure (Diwan and Dorn, 2007). Pathological enlargement of the heart, termed maladaptive cardiac hypertrophy, is observed clinically in many of these conditions and can be induced as a result of increased arterial blood pressure, genetic mutations or following myocardial injury. Hypertrophic remodelling can take place within the left and/or right ventricles, depending on the nature of the initiating stimulus. It develops as a consequence of the increased size of individual ventricular myocytes, without an increase in myocyte number. When hypertrophy occurs as a consequence of persistent cardiovascular stress, an increase in heart size is a pathological and maladaptive response. As the condition progresses to heart failure, myocyte apoptosis/necrosis and myocardial fibrosis are observed, impairing cardiac contractility and reducing cardiac output. At the cellular level, hypertrophic myocytes show remodelling of gene expression profiles, altered intracellular calcium handling and reorganization of the contractile machinery. Reprogramming of cardiomyocyte gene expression profiles is responsible for many of the phenotypic changes observed in hypertrophic myocytes and is perceived to be critical for disease-associated impairment of myocyte function. Left ventricular hypertrophy produced by reprogramming under pathological conditions is one of the major risk factors for subsequent progression of patients to congestive heart failure and sudden death (Jessup and Brozena, 2003), prompting much investigation into the mechanism of action of pro-hypertrophic stimuli. Exposure of cultured cardiomyocytes to ET-1 has been shown to recapitulate many of the cellular features of hypertrophic myocytes (Ito et al., 1991). Furthermore, ET-1 stimulation of NRVM alters expression of hundreds of genes, in a manner mirroring the remodelling of gene expression observed during hypertrophy (Cullingford et al., 2008). Consequently, these observations have promoted much investigation into the role of the ET-1 system in pathological remodelling.
As indicated earlier, cardiomyocyte ET-1 biosynthesis and secretion and ET receptor expression are increased in heart failure patients and animal models of cardiovascular conditions (Hasegawa et al., 1996; Motte et al., 2003; McMullen and Jennings, 2007). This remodelling of the ET system thereby provides a local mechanism for the pathological effects of ET-1. The importance of a local role for ET-1 in the induction of myocardial remodelling has been demonstrated by the effects observed upon infection of an organism with the intracellular protozoan Trypanosoma cruzi. Parasite infection leads to the development of Chagas disease (Coura and Viñas, 2010), which in its chronic form induces a cardiomyopathic phenotype, consisting of cardiomyocyte hypertrophy, myocardial fibrosis and, eventually, myocyte death. Together, these changes promote the development of heart failure (Coura and Borges-Pereira, 2010). Similar to other forms of hypertrophic remodelling, plasma ET-1 concentration is high in mice and humans infected with T. cruzi and mRNA levels of both ET-1 itself and the enzymes involved in its processing are increased under these conditions (Petkova et al., 2000; Salomone et al., 2001). When the ET-1 gene is specifically deleted in cardiomyocytes, the maladaptive effects of T. cruzi infection are reduced. Such a protective effect is not observed upon deletion of ET-1 in endothelial cells, suggesting a myocyte-specific ET-1-dependent origin for the cardiomyopathic effect of the disease (Tanowitz et al., 2005).
Manipulation of the ET-1 system experimentally in murine models of hypertrophy and heart failure has provided valuable further insight into the role of the peptide in left ventricular remodelling. Cardio-specific conditional overexpression of ET-1 in mice induces a pathological hypertrophic phenotype, which can be attenuated by combined blockade of ETA and ETB receptors (Yang et al., 2004), whilst cardio-specific deletion of the ET-1 gene inhibits the hypertrophic response to treatment with the T3 thyroid hormone (Shohet et al., 2004). Infusion of the ETA receptor antagonist, BQ123, inhibits the response to a pro-hypertrophic stimulus in adult rats (Ito et al., 1994) and when administered to rats experiencing chronic heart failure, BQ123 impedes left ventricular remodelling and improves survival (Sakai et al., 1996). These observations support the conclusion that ET-1 and the ETA receptor have an important role in pathological remodelling of the heart.
Based on these findings, the signalling pathways activated downstream of ET-1 and ETA receptor activation have been extensively studied within the context of the hypertrophic response. Overexpression of PLCβ1b, a primary proximal target of the ETA receptor, is sufficient to induce a hypertrophic response in NRVM (Filtz et al., 2009). Moreover, by inducing calcium signals and activation of PKC, respectively, the IP3 and DAG generated following ET-1 stimulated PLC-mediated phosphoinositide hydrolysis have pro-hypertrophic effects.
Role of IP3-dependent calcium release in hypertrophy
The low baseline expression of the IP3RII in ventricular myocytes (Perez et al., 1997) has impeded investigation into its role in hypertrophic remodelling. Moreover, it has been unclear how IICR arising from the relatively few cardiomyocyte IP3RII could be discriminated from the cyclical global release and reuptake of calcium that mediates cellular contractility. However, it has been recognized that IP3RII expression increases during cardiovascular disease, with increased receptor expression being observed in human cardiomyopathic hearts and animal models of hypertrophic remodelling (Go et al., 1995; Harzheim et al., 2009; 2010; Nakayama et al., 2010). As well as being expressed in the dyadic region, these receptors are also found in the peri-nuclear region. IP3-dependent nuclear calcium signals occur in response to ET-1 and are required to elicit a hypertrophic response (Wu et al., 2006; Higazi et al., 2009). Furthermore, a mouse-model with cardio-specific overexpression of the IP3RII demonstrated enhanced IP3-induced calcium release (IICR) and mild baseline hypertrophy (Nakayama et al., 2010). Together, these data suggest that IICR has a pro-hypertrophic effect on the myocardium and that nuclear IICR plays a key role in the hypertrophic response to ET-1. The observation that ET-1 elicits release of calcium specifically in the nuclear domain is significant as it would allow pro-hypertrophic IP3-dependent calcium signals to be detected independently from the bulk calcium transients that elicit myocyte contractility. Furthermore, calcium elevation within the nucleus co-localizes the calcium signal with calcium-sensitive kinases and regulators of transcription, spatially coupling the stimulus with its effectors (Goonasekera and Molkentin, 2012). Such mechanisms are likely to be crucial for the remodelling of gene expression profiles observed in hypertrophy. Calcineurin-nuclear factor of activated T cells (Cn-NFAT) (Higazi et al., 2009; Nakayama et al., 2010; Rinne and Blatter, 2010) and CaMKII (Wu et al., 2006), are examples of IICR-regulated proteins that can be localized within the nucleus. Significantly, both have important roles in the hypertrophic response.
Calcineurin (Cn) is a calcium-dependent heterotrimeric serine–threonine phosphatase (Clipstone and Crabtree, 1992; Fruman et al., 1992). In response to increases in calcium concentration such as those elicited by ET-1-induced IP3 generation, IP3, the Cn autoinhibitory domain is displaced from the active site, stimulating Cn phosphatase activity. The major cellular target of activated Cn is a family of 4 Rel-homology domain containing transcription factors, NFATc1-4 (Jain et al., 1993). Subcellular localization of NFATc1-4 is controlled by the phosphorylation status of the protein. Phosphorylated NFAT is predominantly localized in the cytosol. When Cn is active, NFAT is dephosphorylated, exposing two nuclear localization sequences (NLS) and inducing translocation to the nucleus, where it can activate transcription (Shaw et al., 1995). Phosphorylation of NFAT by protein kinases such as GSK3β (Neal and Clipstone, 2001), DYRK1a (Kuhn et al., 2009), p38-MAPK (Gomez del Arco et al., 2000) and JNK (Chow et al., 1997) leads to nuclear export of the protein (Hogan et al., 2003). In response to ET-1 stimulation, multiple NFAT isoforms have been shown to translocate to the nucleus of cultured cardiomyocytes (Kakita et al., 2001; Kawamura et al., 2004; Higazi et al., 2009), with identity of the ET-1-regulated isoforms species and cell-type specific (Rinne et al., 2010). Interestingly, nuclear import of Cn has also been observed in the cardiomyocyte, promoting sustained NFAT dephosphorylation and prolonging the NFAT-signal (Shibasaki et al., 1996). Indeed, nuclear translocation of Cn is required for the response to ET-1 in NRVM (Hallhuber et al., 2006; Higazi et al., 2009).
Cn-NFAT activity within the cardiomyocyte is undoubtedly important for the hypertrophic effects of ET-1, and there is a wealth of further evidence that convincingly demonstrates the pathological role of Cn-NFAT activity within the heart. Failing human myocardium exhibits increased expression and activity of Cn and translocation of NFATc4 to the nuclear compartment (Lim and Molkentin, 1999; Diedrichs et al., 2004). These observations have been further substantiated by in vitro and in vivo studies. Overexpression of a constitutively-active mutant of Cn in the mouse heart induces severe left ventricular hypertrophy, cardiomyocyte dysfunction and heart failure (Molkentin et al., 1998). To further confirm these conclusions, other studies have probed the role of Cn-NFAT downstream of hypertrophic stimuli such as ET-1 in vitro. ET-1 increases Cn enzymatic activity in NRVM, inhibition of which prevents the response to hypertrophic stimulation (Zhu et al., 2000). Moreover, multiple studies have verified that the deleterious effect of Cn on cardiac function is mediated through Cn-dependent regulation of one or more NFAT isoforms. Constitutively nuclear, constitutively active NFATc4 induces a phenotype of pathological left ventricular hypertrophy, confirming the damaging effect of increased NFAT-transcriptional activity in vivo (Molkentin et al., 1998). Moreover, cardio-specific deletion of either NFATc3 (Wilkins et al., 2002) or NFATc2 (Bourajjaj et al., 2008) impairs the hypertrophic response to increased Cn activity. Although these mouse models do not directly interrogate the role of ET-1-induced Cn-NFAT activity during hypertrophy, when combined with the evidence for ET-1-dependent NFAT translocation in isolated myocytes and the requirement for Cn in the hypertrophic response to ET-1, this pathway can be considered to be an important mediator of ET-1 induced hypertrophic responses.
Signalling to CaMKII, HDACs and MEF2 to induce hypertrophy
Although Cn-NFAT are strongly implicated in the hypertrophic response to ET-1, other studies have shown that ET-1-induced nuclear IICR elicits its pro-hypertrophic effect by engaging co-localized CaMKII and eliciting gene expression dependent upon the pro-hypertrophic transcription factor MEF2 (Wu et al., 2006). Basally, MEF2 activity is restrained in the myocyte as a consequence of its association with class II histone deacetylases (HDACs 4,5,7 and 9), which negatively regulate transcription by deacetylating histone proteins, resulting in condensation of chromatin and impeding the access of transcriptional activators. In response to peri-nuclear IICR, CaMKII phosphorylates HDAC4 creating a docking site for 14-3-3 proteins. Association of HDAC4 with 14-3-3 induces conformational changes that mask NLS and expose a nuclear export sequence, facilitating HDAC4 nuclear export and depressing MEF2-dependent transcriptional responses (Lu et al., 2000; Zhang et al., 2002; Wu et al., 2006). ET-1 induces nuclear export of both HDAC4 and HDAC5 in rat ventricular myocytes (Wu et al., 2006; Peng et al., 2009), and ET-1 induced activation of MEF2-dependent transcription is likely to form a key component of the cellular response to the peptide. Deletion of MEF2D abrogates the hypertrophic response induced by hypertension-induced hypertrophy (Chang et al., 2004; Kim et al., 2008), whilst overexpression of MEF2A, C or D in vivo is sufficient to induce pathological cardiac remodelling (Kim et al., 2008).
Recent reports indicate that the spatial pattern of ET-1-induced signalling is important in the regulation of HDAC phosphorylation. ET-1 activates CaMKII kinase activity, and this activity is necessary for the hypertrophic response to the peptide in NRVM (Zhu et al., 2000). CaMKII itself is a key inducer of HDAC nuclear export, with cardiomyocytes predominantly expressing the CaMKIIδ isoform, localized to either the nucleus (CaMKIIδB) or cytosol (CaMKIIδC) (Anderson, 2005). CaMKIIδB is therefore present in the vicinity of its activating signal (ET-1-induced nuclear calcium transients) and its target (HDAC4). Although CaMKII directly targets HDAC4, it does not directly phosphorylate HDAC5. HDAC5 is rendered sensitive to CaMKII by forming oligomers with HDAC4 (Backs et al., 2008), indicating that ET-1-induced IICR could lead to nuclear export of both of the key MEF2 repressors. More recently, the novel calcium independent isoforms of PKC and the PKC target PKD have also been shown to phosphorylate HDAC5 and induce its nuclear export (Phan et al., 2011). ET-1 activates both PKC and PKD in cardiomyocytes (Phan et al., 2011); however, this is unlikely to form part of the mechanism whereby ET-1 depresses MEF2. ET-1-induced HDAC5 nuclear export is PKC-independent and although ET-1 activates subsarcolemmal PKD, it does not stimulate PKD translocation to the nucleus, preventing access of the kinase to its nuclear targets (Bossuyt et al., 2011). Therefore, peri-nuclear IICR and the CaMKII activity it elicits are considered to be the key signals for ET-1-induced MEF2 depression.
ET-1 activation of PKC in hypertrophy
ET-1 activates the novel isoforms of PKC in cardiomyocytes. PKC isoform expression is species, tissue and cell-type-specific, with cardiomyocytes from murine species and humans expressing multiple differentially regulated PKC isoenzymes (Salazar et al., 2007; Palaniyandi et al., 2009). NRVM express PKCs α, δ, ε and η (Clerk et al., 1995; Erdbrugger et al., 1997; Markou et al., 2006), whilst human cardiac tissue also expresses PKC βI and βII (Bowling et al., 1999). In NRVM, ET-1 induces membrane association and activation of the DAG-sensitive, calcium-insensitive PKC δ and ε isoforms within 5 min of stimulation (Clerk et al., 1994; Clerk and Sugden, 1997), and PKC ε is the major ET-1-activated isoform in AVM (Bogoyevitch et al., 1993). The role of PKC isoenzymes in hypertrophy has been intensively investigated (Braz et al., 2002; Vega et al., 2004; Vijayan et al., 2004). This has lead to the identification of many downstream targets of PKC relevant to hypertrophy in cardiomyocytes, such as the small G-protein Ras (Montessuit and Thorburn, 1999), mTOR, ribosomal S6 kinase (Moschella et al., 2007), GSK3β (Zhai et al., 2007), GATA4 (Wang et al., 2005) and PKD (Vega et al., 2004). Although PKC regulates multiple targets, activation of Ras and downstream MAPK cascades by the DAG-sensitive isoforms of PKC is considered to have a predominant role in cardiac remodelling (Braz et al., 2002).
MAPK signalling pathways in the hypertrophic response
Upon ET-1 binding to the ETA receptor, the classical small g protein Ras is converted into an active, GTP-bound form. Multiple mechanisms have been suggested to explain this effect, relying on the activity of PKC as described above and/or ET-1-induced production of intracellular reactive oxygen species (ROS) (Sugden and Clerk, 2006). Clinically, activating mutations in Ras and its target Raf result in Costello and Noonen syndromes respectively, both of which induce cardiomyopathy amongst other abnormalities (Lin et al., 2011; Wu et al., 2011). Induction of hypertrophy by activated Ras and Raf is ERK1/2 dependent, suggesting that this kinase is responsible for the hypertrophic response to Ras and Raf activation (Harris et al., 2004; Wu et al., 2011). However, although it has been intensively investigated, the precise role and function of activated ERK1/2 in the heart remains controversial (Bueno and Molkentin, 2002; Kehat and Molkentin, 2010). It has been appreciated for many years that ERK1/2 is activated in NRVM in response to hypertrophic agonists including ET-1 (Bogoyevitch et al., 1994). Moreover, inhibition of ERK1/2 activation using either pharmacological inhibitors of its activating kinases MEK1/2 or adenoviral-mediated overexpression of dominant-negative MEK prevents the hypertrophic response to ET-1 in NRVM (Ueyama et al., 2000; Yue et al., 2000). Indeed, microarray studies demonstrated the reliance of ET-1-induced hypertrophic gene expression on ERK1/2 activity (Marshall et al., 2010). However, these observations have been called into question following in vivo studies of transgenic mouse models. Cardiac-specific overexpression of constitutively-active MEK1 induces ERK1/2 phosphorylation and compensated concentric hypertrophy (Bueno et al., 2000). Therefore, activation of ERK1/2 above the baseline level, as occurs in vitro in response to ET-1, can induce cardiac remodelling. However, in vivo loss-of-function studies of ERK1/2 have demonstrated that under certain conditions the hypertrophic response is preserved, calling into question the necessity of these kinases during pathology. Echoing the current thinking regarding calcium-regulated signalling, interesting observations suggest that spatial and contextual aspects of ERK1/2 activation may govern whether it exhibits pro-hypertrophic activity. Interestingly, in vascular smooth muscle cells and fibroblasts, ET-1 induces nuclear translocation of ERK1/2 (Lenormand et al., 1993; El-Mowafy and White, 1999) where it can phosphorylate targets involved in regulation of gene transcription. These include transcription factors (e.g. GATA4 and Elk), nuclear kinases (e.g. MSK1) and chromatin modifying enzymes (e.g. the p300 histone acetylase). In cardiomyocytes, a novel phosphorylated form of ERK1/2 (p-Thr188-ERK1/2) accumulates within the nucleus over the duration of the hypertrophic response in vivo, an effect that could also be simulated in vitro by agonists of Gαq-coupled receptors (Lorenz et al., 2009). Hence, it is conceivable that ERK1/2 activity becomes more important in the later stages of hypertrophy, once sufficient nuclear accumulation has occurred. Furthermore, the basal and stimulus-induced levels of ERK1/2 activity may have different functional effects within the myocyte (Kehat et al., 2011). In the context of cellular stimulation with agonists such as ET-1, mechanisms may be activated that permit ERK1/2 to perform functions other than those active under non-stimulated conditions, changing the cellular response to the kinase. Discriminating whether, when and how ERK1/2 regulates hypertrophy is an interesting topic of future research.
Although the requirement for ERK1/2 during remodelling remains controversial, many of the targets of the kinase have defined pro-hypertrophic roles. Genome-wide studies of myocyte gene expression profiles have shown that ET-1 induces distinct waves of gene expression, with the earlier phases of the response being largely ERK1/2 dependent (Cullingford et al., 2008; Marshall et al., 2010). Demonstrating the importance of these acute events, inhibiting activity of the activator protein-1 (AP1) complex of the immediate-early genes c-fos and c-jun prevents ET-1-induced hypertrophic remodelling (Omura et al., 2002). Other transcription factors also exhibit ET-1-induced ERK1/2-dependent activity. For example, GATA4 shows ET-1-dependent DNA-binding activity in response to increased myocardial stretch (Hautala et al., 2001; 2002; Liang et al., 2001) and is regulated by ERK1/2-dependent phosphorylation (Liang et al., 2001). Expression of GATA4 harbouring phospho-mimetic activating mutations is pro-hypertrophic both in vitro and in vivo, whilst replacing endogenous GATA4 with a mutant that cannot be phosphorylated on its activating sites impedes hypertrophic responses (van Berlo et al., 2011). Interestingly, GATA4 can form higher order complexes with other pro-hypertrophic transcription factors such as NFAT and AP1, providing a mechanism whereby pro-hypertrophic signalling pathways can be integrated (Sanna et al., 2005).
Are JNK and p38-MAPK important for ET-1-induced remodelling?
Early studies in NRVM demonstrated that both cellular osmotic stress and ET-1 induce phosphorylation and activation of JNK1/2. However, the response to stress-associated stimuli was much greater than the response to ETA receptor activation. Subsequently, the role of JNK1/2 in hypertrophy has been extensively examined. Overexpression of dominant-negative MKK4, the kinase upstream of JNK1/2, attenuates the hypertrophic response to ET-1 stimulation of NRVM (Choukroun et al., 1998). These and other data suggest that JNK1/2 mediates a pro-hypertrophic effect in vitro. However, this observation has been questioned by other studies showing that activation of JNK1/2 in vitro inhibits the hypertrophic response to ET-1 (Nemoto et al., 1998). To attempt to address the conflicting data sets, in vivo studies have been performed. Currently, the prevailing consensus suggests that although ectopic activation of JNK1/2 can induce cardiac remodelling and eventual heart failure (Choukroun et al., 1999), these changes occur in the absence of left ventricular hypertrophy. Instead, myocardial stiffening has been observed as a consequence of extracellular matrix remodelling (Petrich et al., 2004). Intriguingly JNK1−/− knock-out mice exhibited a hastened early deterioration in cardiac performance during hypertension-induced hypertrophy, suggesting that the basal level of activity of this isoform may be important for maintaining cardiac function (Tachibana et al., 2006). The cardio-protective actions of JNK have been attributed to JNK-dependent phosphorylation and subsequent nuclear export of the highly pro-hypertrophic transcription factor NFAT (Chow et al., 1997) and/or JNK1/2-dependent positive regulation of the anti-hypertrophic protein JunD. The importance of JNK activity within the hypertrophic response to ET-1 is likely to be governed by the balance of stimuli within the ET-1-regulated signalling network.
Similarly to JNK, the exact role of p38-MAPK within the myocardium has proven to be a matter for debate, with data generated in vitro and in vivo producing somewhat conflicting conclusions. Both ET-1 and cellular stress induce p38-MAPK phosphorylation and activation. However, the kinase has been demonstrated to either be necessary or dispensable for the ET-1-induced hypertrophic response. Furthermore, the different isoforms of p38-MAPK may have variable pathological roles, with p38-MAPKα implicated in the induction of apoptosis whilst p38-MAPKβ is involved in hypertrophy. The weight of evidence suggests that p38-MAPK activity is not required for the initial development of left ventricular hypertrophy (Liao et al., 2001; Braz et al., 2003; Zhang et al., 2003; Nishida et al., 2004). Indeed, akin to the action of JNK1/2, p38-MAPK isoforms may be anti-hypertrophic as a consequence of their NFAT-kinase activity during the early hypertrophic response (Gomez del Arco et al., 2000; Braz et al., 2003). In the latter stages of cardiac decompensation and heart failure, pathological functions for p38-MAPK have been described. The kinase promotes ventricular dilation, stiffening and fibrosis thereby impairing contractility (Liao et al., 2001; Zhang et al., 2003). Consequently, the effect of ET-1 stimulated p38-MAPK activity may not be important for the initial response to the peptide, but become more significant over time. A possible reason for this is that the remodelled physiology and signalling pathways of the hypertrophic heart creates a different profile of substrates or cellular context for the action of p38-MAPK.
ET-1 signalling – a question of location and timing?
When considered in their entirety, the importance of ET-1-induced signalling pathways within the hypertrophic response appears to be determined by two governing rules. First, as described above, as a consequence of the remodelling of gene expression that occurs during hypertrophy, the necessity of the various ET-1-regulated mediators for the induction of pathology may be altered over time. Secondly, subcellular localization of ET-1-dependent signals is a critical determinant of their pro-hypertrophic role. This is elegantly demonstrated by the specific pro-hypertrophic effect of peri-nuclear release of calcium in response to IP3RII activation. Interestingly, the ETA receptor itself may also be subject to regulated localization, a mechanism that could be important for its cellular effects. The localization of GPCRs has been well studied and several GPCRs are localized at the nuclear membrane of cardiac myocytes and other cells types (Tadevosyan et al., 2010; Vaniotis et al., 2011a; 2011b; Wright et al., 2012). Although ETA receptors are largely found within the sarcolemma, several studies have demonstrated functional ETA receptor and ETB receptor at the nuclear envelope of ventricular cardiomyocytes (Boivin et al., 2003; Bkaily et al., 2011). This may result from translocation of ET receptors from the cell surface or by de novo receptor synthesis. In order for nuclear receptors to be functional, a source of intracellular ligand must exist. It has been hypothesized that ET-1 produced intracellularly in the myocyte can signal in an intracrine manner and that ET-1 is taken into the cell from an extracellular source to act on nuclear ET receptors (Boivin et al., 2003; Tadevosyan et al., 2012). Activation of nuclear ETA receptor has been postulated to stimulate similar downstream pathways as sarcolemmal ETAR, such as PKC activation and IP3 generation, thus potentially activating hypertrophic gene expression and regulating calcium homeostasis (Bkaily et al., 2011). As ET-1 production is increased in hypertrophic and failing myocytes, under hypertrophic conditions, there would be a larger stimulus to ET receptors located within the cell. Furthermore, if present in the nucleus specifically, the ETA receptor and the signalling pathways it initiates would be clustered in close proximity of their cellular effectors, facilitating ET-1-dependent cellular responses. Further studies to substantiate the localization of ETA receptors within the nucleus and their significance and relevance to the hypertrophic response are required to allow inclusion of sub-cellular ET-1 signalling within the accepted models of ET-1 action.
Non-myocyte effects of ET in the heart
ET-1 signalling within the heart regulates a wide spectrum of cardiovascular processes, ranging from development through to life-threatening cardiovascular diseases. In this review, we have focused upon ET signalling in cardiac myocytes. By inducing the activity of a complex web of signalling pathways, this important peptide can directly influence myocyte survival, contractility and remodelling. However, the heart is composed of multiple cell types and despite providing the greater part of cardiac mass, myocytes account for only ∼20% of the total number of cells in humans (Rubart and Field, 2006). The non-myocyte population is comprised of endothelial cells, smooth muscle cells and fibroblasts (Nag, 1980; Banerjee et al., 2007), all of which are responsive to ET-1 (Kedzierski and Yanagisawa, 2001). Cardiac endothelial and smooth muscle cells form the myocardial vasculature. ET-1 interacts with these cells to regulate vascular tone, influencing myocardial blood supply and metabolic activity (Wang et al., 1998). Cardiac fibroblasts are essential for secretion of extracellular matrix and for cardiac repair (Biernacka and Frangogiannis, 2011). In response to elevated levels of ET-1, cardiac fibroblasts proliferate, differentiate into myofibroblasts and increase their secretion of matrix proteins (Shi-Wen et al., 2004a; 2004b). These fibroblast responses underlie scar formation following cardiac damage and the myocardium-wide fibrosis associated with cardiac hypertrophy, where the increased abundance of cardiac fibroblasts replaces the myocytes lost as a result of pathology (Swynghedauw, 1999). The reactive response of the fibroblast is essential for the maintenance of cardiac function during disease, without which the integrity of the heart is compromised (Shimazaki et al., 2008). However, fibroblast infiltration alters the electrophysiological properties of the myocardium, affecting both action potential propagation and allowing arrhythmic events at the cellular level to propagate (Mayyas et al., 2010; Karagueuzian, 2011). Cardiac fibrosis also decreases elasticity of the heart, stiffening the myocardial wall and impeding cardiac output (Lakatta and Levy, 2003a). Extensive fibrosis of the myocardium is not only a characteristic of pathology but is also a prominent feature of the aged, senescent heart and in common with pathological situations, fibrosis of the aged heart arises due to hypertrophy and myocyte loss (Lakatta and Levy, 2003a; Biernacka and Frangogiannis, 2011). When combined with the decreased relaxation rate of the aged heart, these effects are manifest in diastolic dysfunction, impacting exercise tolerance, quality of life and contributing to future heart failure (Lakatta and Levy, 2003a). As ET-1 increases in concentration with age (Maeda et al., 2003) and induces fibrosis (Shi-Wen et al., 2004a), the importance of the ET-1 system for the decline in myocardial function associated with age should be considered paramount.
Role of ET-1 in PAH and right ventricular hypertrophy
The compelling evidence linking the ET-1 system to cardiovascular pathologies has prompted its targeting for therapeutic benefit. Currently, the combined ETA-ETB receptor antagonist bosentan and the selective ETA receptor antagonist ambrisentan are licensed and successfully used for the treatment of pulmonary arterial hypertension (PAH) (Kirkby et al., 2008), a condition which induces right ventricular hypertrophy, right heart failure and eventually death (Gomberg-Maitland et al., 2011). PAH increases pulmonary arterial pressure and vascular resistance, stretching and increasing the load on the right ventricle. Under these conditions, the type 1 angiotensin receptor is activated in response to myocardial stretch and stimulates ROS formation. Subsequently, increased intracellular ROS indirectly triggers the release of ET-1. ET-1-induced increases in intracellular calcium stimulate NFAT and CaMKII, culminating in hypertrophic remodelling (Cingolani et al., 2010). Interestingly, during PAH, pro-hypertrophic GATA4 is also active within the right ventricle (Park et al., 2010), suggesting that multiple ET-1 regulated signalling pathways may be important in this condition. Significantly, ETA receptor blockade prevents right ventricular remodelling in an animal model (Jasmin et al., 2003), demonstrating the importance of the link between PAH, right ventricular hypertrophy and ET-1. These effects may be responsible for the therapeutic benefit of ETA receptor antagonists for the treatment of PAH.
Why have ET receptor antagonists failed in the treatment of left ventricular hypertrophy and congestive heart failure and where may they provide benefit?
Despite the myriad of lines of evidence from both animal models and humans for a role of ET-1 and ET-1-dependent signalling pathways in left ventricular hypertrophy and congestive heart failure (CHF), it is disappointing to note that ET-1 receptor antagonists have not yet proven efficacious for the treatment of CHF (Kirkby et al., 2008). Although initial studies were promising (Sutsch et al., 1998), clinical trials of the combined ETA–ETB receptor antagonist bosentan for the treatment of CHF were halted owing to problems with liver toxicity. Furthermore, later studies were unable to demonstrate a reduction in the mortality or symptoms of patients suffering from CHF when a lower dose of bosentan was administered (Kalra et al., 2002; Kelland and Webb, 2006). A number of explanations for the failure of this approach have been proposed. Blockade of the ETB receptor impairs ET-1 clearance, increasing plasma ET-1 levels and allowing the effects of ETA receptor antagonism to be overcome (Cowburn et al., 2005). Furthermore, ETB receptor inhibition leads to systemic vasoconstriction, a deleterious response in patients with cardiovascular conditions (Cowburn et al., 2005). To avoid the complicating effects of ETB receptor inhibition, studies aiming to selectively block the ETA receptor have been carried out (Anand et al., 2004; Cowburn et al., 2005; Leslie et al., 2005). Although cardiac output was acutely improved, no long-term benefit of this treatment regimen was observed (Anand et al., 2004). However, these studies may have utilized a dose of drug at which antagonism of the ETB receptor cannot be completely excluded (Kelland and Webb, 2007). In view of such limitations, it would be informative if trials were performed using ETA receptor antagonists at a dose that more selectively targets the ETA receptor. In addition, the results of many of the clinical trials performed to date have not been fully published and analysed, meaning that a thorough interpretation of the data is lacking (Kelland and Webb, 2007). Further progress will only be made by a more detailed analysis of these trials. For example, there may be patient sub-populations where ET receptor inhibition is effective. It will also be important to gain a better understanding of the interaction of the ET-1 system with patient lifestyle and age. Indeed, ET-1 levels are elevated in aged and obese populations, which may suggest that ET receptor antagonists would provide greater benefit in these individuals. Interestingly, improved lifestyle benefits these populations, with weight loss and exercise reducing ET-1 levels in the obese and aged respectively (Maeda et al., 2003; 2006). Furthermore, our increased understanding of ET-1 biology and ET-1-induced cellular responses may henceforth guide more intelligent and targeted drug design. In view of the disparate effects of ETA and ETB receptor inhibition in humans, it will be important to consider the influence of dimers and heterodimers of the ET receptor subtypes upon downstream signalling and pharmacology (Evans and Walker, 2008a; 2008b). More knowledge regarding the persistence, localization and integration of these ET-1-induced responses is also required. Agents that allow selective control of the activation of ETA and ETB receptors and/or the individual limbs of the signalling pathways they activate downstream would be particularly informative. The development of biased agonists/antagonists akin to those for the angiotensin receptor would allow for the specific targeting of key signalling pathways (Watts, 2010) and perhaps provide a window of opportunity to manipulate ET-1-induced signalling for therapeutic benefit.
Cardiac failure is the dominant cause of mortality in the over 65 population. This population is likely to double in number in the next 25 years; hence, future therapies will need to be developed to tackle the increasing burden of cardiac disease. The prominent role that ET-1 performs in the pathological alterations in physiology and function of the multiple cell types of the heart, in particular during ageing, identify it as a key target for further development of therapeutic strategies.
Acknowledgments
Work in the authors' laboratory is funded by the BHF (grant #PG/11/12/28717) and the BBSRC. HLR is a Royal Society Research Fellow.
Glossary
- AngII
angiotensin II
- CICR
calcium-induced-calcium release
- Cn
calcineurin
- ECC
excitation-contraction-coupling
- IICR
IP3-induced calcium release
- IP3R
inositol triphosphate receptor
- MyBP-C
myosin binding protein C
- RyR
ryanodine receptor
- TnI
troponin I
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164:1–2. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BG, Phuong LL, Farhat H, Chevalier D. Both endothelin-A and endothelin-B receptors are present on adult rat cardiac ventricular myocytes. Can J Physiol Pharmacol. 2003;81:95–104. doi: 10.1139/y02-155. [DOI] [PubMed] [Google Scholar]
- Alvarez BV, Pérez NG, Ennis IL, Camilión de Hurtado MC, Cingolani HE. Mechanisms underlying the increase in force and Ca(2+) transient that follow stretch of cardiac muscle: a possible explanation of the Anrep effect. Circ Res. 1999;85:716–722. doi: 10.1161/01.res.85.8.716. [DOI] [PubMed] [Google Scholar]
- Anand I, McMurray J, Cohn JN, Konstam MA, Notter T, Quitzau K, et al. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the EndothelinA Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364:347–354. doi: 10.1016/S0140-6736(04)16723-8. [DOI] [PubMed] [Google Scholar]
- Anderson ME. Calmodulin kinase signaling in heart: an intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol Ther. 2005;106:39–55. doi: 10.1016/j.pharmthera.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Arai H, Hori S, Aramori I, Ohkubo H, Nakanishi S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature. 1990;348:730–732. doi: 10.1038/348730a0. [DOI] [PubMed] [Google Scholar]
- Arai M, Yoguchi A, Iso T, Takahashi T, Imai S, Murata K, et al. Endothelin-1 and its binding sites are upregulated in pressure overload cardiac hypertrophy. Am J Physiol. 1995;268:H2084–H2091. doi: 10.1152/ajpheart.1995.268.5.H2084. [DOI] [PubMed] [Google Scholar]
- Aramori I, Nakanishi S. Coupling of two endothelin receptor subtypes to differing signal transduction in transfected Chinese hamster ovary cells. J Biol Chem. 1992;267:12468–12474. [PubMed] [Google Scholar]
- Attinà T, Camidge R, Newby DE, Webb DJ. Endothelin antagonism in pulmonary hypertension, heart failure, and beyond. Heart. 2005;91:825–831. doi: 10.1136/hrt.2004.053991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backs J, Backs T, Bezprozvannaya S, McKinsey TA, Olson EN. Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol Cell Biol. 2008;28:3437–3445. doi: 10.1128/MCB.01611-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard C, Schaffer S. Stimulation of the Na+/Ca2+ exchanger by phenylephrine, angiotensin II and endothelin 1. J Mol Cell Cardiol. 1996;28:11–17. doi: 10.1006/jmcc.1996.0002. [DOI] [PubMed] [Google Scholar]
- Baltogiannis GG, Tsalikakis DG, Mitsi AC, Hatzistergos KE, Elaiopoulos D, Fotiadis DI, et al. Endothelin receptor – a blockade decreases ventricular arrhythmias after myocardial infarction in rats. Cardiovasc Res. 2005;67:647–654. doi: 10.1016/j.cardiores.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol. 2007;293:H1883–H1891. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- van Berlo JH, Elrod JW, Aronow BJ, Pu WT, Molkentin JD. Serine 105 phosphorylation of transcription factor GATA4 is necessary for stress-induced cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2011;108:12331–12336. doi: 10.1073/pnas.1104499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology. 2006;21:380–387. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- Bianchini L, L'Allemain G, Pouyssegur J. The p42/p44 mitogen-activated protein kinase cascade is determinant in mediating activation of the Na+/H+ exchanger (NHE1 isoform) in response to growth factors. J Biol Chem. 1997;272:271–279. doi: 10.1074/jbc.272.1.271. [DOI] [PubMed] [Google Scholar]
- Biernacka A, Frangogiannis NG. Aging and cardiac fibrosis. Aging Dis. 2011;2:158–173. [PMC free article] [PubMed] [Google Scholar]
- Bkaily G, Avedanian L, Al-Khoury J, Provost C, Nader M, D'Orléans-Juste P, et al. Nuclear membrane receptors for ET-1 in cardiovascular function. Am J Physiol Regul Integr Comp Physiol. 2011;300:R251–R263. doi: 10.1152/ajpregu.00736.2009. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch MA, Parker PJ, Sugden PH. Characterization of protein kinase C isotype expression in adult rat heart. Protein kinase C-epsilon is a major isotype present, and it is activated by phorbol esters, epinephrine, and endothelin. Circ Res. 1993;72:757–767. doi: 10.1161/01.res.72.4.757. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch MA, Glennon PE, Andersson MB, Clerk A, Lazou A, Marshall CJ, et al. Endothelin-1 and fibroblast growth factors stimulate the mitogen-activated protein kinase signaling cascade in cardiac myocytes. The potential role of the cascade in the integration of two signaling pathways leading to myocyte hypertrophy. J Biol Chem. 1994;269:1110–1119. [PubMed] [Google Scholar]
- Boivin B, Chevalier D, Villeneuve LR, Rousseau E, Allen BG. Functional endothelin receptors are present on nuclei in cardiac ventricular myocytes. J Biol Chem. 2003;278:29153–29163. doi: 10.1074/jbc.M301738200. [DOI] [PubMed] [Google Scholar]
- Boixel C, Dinanian S, Lang-Lazdunski L, Mercadier JJ, Hatem SN. Characterization of effects of endothelin-1 on the L-type Ca2+ current in human atrial myocytes. Am J Physiol. 2001;281:H764–H773. doi: 10.1152/ajpheart.2001.281.2.H764. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Harzheim D, Smyrnias I, Conway SJ, Roderick HL. Temporal changes in atrial EC-coupling during prolonged stimulation with endothelin-1. Cell Calcium. 2007;42:489–501. doi: 10.1016/j.ceca.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Bossuyt J, Chang C-W, Helmstadter K, Kunkel MT, Newton AC, Campbell KS, et al. Spatiotemporally distinct protein kinase D activation in adult cardiomyocytes in response to phenylephrine and endothelin. J Biol Chem. 2011;286:33390–33400. doi: 10.1074/jbc.M111.246447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourajjaj M, Armand A-S, da Costa Martins PA, Weijts B, van der Nagel R, Heeneman S, et al. NFATc2 is a necessary mediator of calcineurin-dependent cardiac hypertrophy and heart failure. J Biol Chem. 2008;283:22295–22303. doi: 10.1074/jbc.M801296200. [DOI] [PubMed] [Google Scholar]
- Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, et al. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- Boyden PA, Dun W, Barbhaiya C, Ter Keurs HE. 2APB- and JTV519(K201)-sensitive micro Ca2+ waves in arrhythmogenic Purkinje cells that survive in infarcted canine heart. Heart Rhythm. 2004;1:218–226. doi: 10.1016/j.hrthm.2004.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz JC, Bueno OF, De Windt LJ, Molkentin JD. PKC alpha regulates the hypertrophic growth of cardiomyocytes through extracellular signal-regulated kinase1/2 (ERK1/2) J Cell Biol. 2002;156:905–919. doi: 10.1083/jcb.200108062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, et al. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno OF, Molkentin JD. Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res. 2002;91:776–781. doi: 10.1161/01.res.0000038488.38975.1a. [DOI] [PubMed] [Google Scholar]
- Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24:8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PP, Patel JR, Rybakova IN, Walker JW, Moss RL. Protein kinase A-induced myofilament desensitization to Ca(2+) as a result of phosphorylation of cardiac myosin-binding protein C. J Gen Physiol. 2010;136:615–627. doi: 10.1085/jgp.201010448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166:193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng TH, Chang CY, Wei J, Lin CI. Effects of endothelin 1 on calcium and sodium currents in isolated human cardiac myocytes. Can J Physiol Pharmacol. 1995;73:1774–1783. doi: 10.1139/y95-242. [DOI] [PubMed] [Google Scholar]
- Cheng TH, Shih NL, Chen SY, Wang DL, Chen JJ. Reactive oxygen species modulate endothelin-I-induced c-fos gene expression in cardiomyocytes. Cardiovasc Res. 1999;41:654–662. doi: 10.1016/s0008-6363(98)00275-2. [DOI] [PubMed] [Google Scholar]
- Choukroun G, Hajjar R, Kyriakis JM, Bonventre JV, Rosenzweig A, Force T. Role of the stress-activated protein kinases in endothelin-induced cardiomyocyte hypertrophy. J Clin Invest. 1998;102:1311–1320. doi: 10.1172/JCI3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choukroun G, Hajjar R, Fry S, del Monte F, Haq S, Guerrero JL, et al. Regulation of cardiac hypertrophy in vivo by the stress-activated protein kinases/c-Jun NH(2)-terminal kinases. J Clin Invest. 1999;104:391–398. doi: 10.1172/JCI6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CW, Rincon M, Cavanagh J, Dickens M, Davis RJ. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science. 1997;278:1638–1641. doi: 10.1126/science.278.5343.1638. [DOI] [PubMed] [Google Scholar]
- Chun M, Liyanage UK, Lisanti MP, Lodish HF. Signal transduction of a G protein-coupled receptor in caveolae: colocalization of endothelin and its receptor with caveolin. Proc Natl Acad Sci USA. 1994;91:11728–11732. doi: 10.1073/pnas.91.24.11728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun M, Lin HY, Henis YI, Lodish HF. Endothelin-induced endocytosis of cell surface ETA receptors. Endothelin remains intact and bound to the ETA receptor. J Biol Chem. 1995;270:10855–10860. doi: 10.1074/jbc.270.18.10855. [DOI] [PubMed] [Google Scholar]
- Chung KY, Kang M, Walker JW. Contractile regulation by overexpressed ETA requires intact T tubules in adult rat ventricular myocytes. Am J Physiol. 2008;294:H2391–H2399. doi: 10.1152/ajpheart.00011.2008. [DOI] [PubMed] [Google Scholar]
- Cingolani H, Ennis I, Aiello E, Pérez N. Role of autocrine/paracrine mechanisms in response to myocardial strain. Pflugers Arch. 2011;462:29–38. doi: 10.1007/s00424-011-0930-9. [DOI] [PubMed] [Google Scholar]
- Cingolani HE, Ennis IL. Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation. 2007;115:1090–1100. doi: 10.1161/CIRCULATIONAHA.106.626929. [DOI] [PubMed] [Google Scholar]
- Cingolani HE, Pérez NG, Caldiz CI, Garciarena CD, Giusti VC, Correa MV. Early hypertrophic signals after myocardial stretch. Role of reactive oxygen species and the sodium/hydrogen exchanger. In: Kamkin A, Kiseleva I, et al., editors. Mechanosensitivity of the Heart. Houten: Springer Netherlands; 2010. pp. 327–371. [Google Scholar]
- Clerk A, Sugden PH. Regulation of phospholipases C and D in rat ventricular myocytes: stimulation by endothelin-1, bradykinin and phenylephrine. J Mol Cell Cardiol. 1997;29:1593–1604. doi: 10.1006/jmcc.1997.0395. [DOI] [PubMed] [Google Scholar]
- Clerk A, Bogoyevitch MA, Anderson MB, Sugden PH. Differential activation of protein kinase C isoforms by endothelin-1 and phenylephrine and subsequent stimulation of p42 and p44 mitogen-activated protein kinases in ventricular myocytes cultured from neonatal rat hearts. J Biol Chem. 1994;269:32848–32857. [PubMed] [Google Scholar]
- Clerk A, Bogoyevitch MA, Fuller SJ, Lazou A, Parker PJ, Sugden PH. Expression of protein kinase C isoforms during cardiac ventricular development. Am J Physiol. 1995;269:H1087–H1097. doi: 10.1152/ajpheart.1995.269.3.H1087. [DOI] [PubMed] [Google Scholar]
- Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357:695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- Coura JR, Borges-Pereira J. Chagas disease: 100 years after its discovery. A systemic review. Acta Trop. 2010;115:5–13. doi: 10.1016/j.actatropica.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Coura JR, Viñas PA. Chagas disease: a new worldwide challenge. Nature. 2010;465:S6–S7. doi: 10.1038/nature09221. [DOI] [PubMed] [Google Scholar]
- Cowburn PJ, Cleland JGF, McDonagh TA, McArthur JD, Dargie HJ, Morton JJ. Comparison of selective ET(A) and ET(B) receptor antagonists in patients with chronic heart failure. Eur J Heart Fail. 2005;7:37–42. doi: 10.1016/j.ejheart.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Cuello F, Bardswell SC, Haworth RS, Yin X, Lutz S, Wieland T, et al. Protein kinase D selectively targets cardiac troponin I and regulates myofilament Ca2+ sensitivity in ventricular myocytes. Circ Res. 2007a;100:864–873. doi: 10.1161/01.RES.0000260809.15393.fa. [DOI] [PubMed] [Google Scholar]
- Cuello F, Snabaitis AK, Cohen MS, Taunton J, Avkiran M. Evidence for direct regulation of myocardial Na+/H+ exchanger isoform 1 phosphorylation and activity by 90-kDa ribosomal S6 kinase (RSK): effects of the novel and specific RSK inhibitor fmk on responses to alpha1-adrenergic stimulation. Mol Pharmacol. 2007b;71:799–806. doi: 10.1124/mol.106.029900. [DOI] [PubMed] [Google Scholar]
- Cuello F, Bardswell SC, Haworth RS, Ehler E, Sadayappan S, Kentish JC, et al. Novel role for p90 ribosomal S6 kinase in the regulation of cardiac myofilament phosphorylation. J Biol Chem. 2011;286:5300–5310. doi: 10.1074/jbc.M110.202713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullingford TE, Markou T, Fuller SJ, Giraldo A, Pikkarainen S, Zoumpoulidou G, et al. Temporal regulation of expression of immediate early and second phase transcripts by endothelin-1 in cardiomyocytes. Genome Biol. 2008;9:R32. doi: 10.1186/gb-2008-9-2-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Giusti VC, Correa MV, Villa-Abrille MC, Beltrano C, Yeves AM, de Cingolani GEC, et al. The positive inotropic effect of endothelin-1 is mediated by mitochondrial reactive oxygen species. Life Sci. 2008;83:264–271. doi: 10.1016/j.lfs.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Diedrichs H, Chi M, Boelck B, Mehlhorn U, Schwinger RH. Increased regulatory activity of the calcineurin/NFAT pathway in human heart failure. Eur J Heart Fail. 2004;6:3–9. doi: 10.1016/j.ejheart.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Diwan A, Dorn GW. Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology. 2007;22:56–64. doi: 10.1152/physiol.00033.2006. [DOI] [PubMed] [Google Scholar]
- Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA, Blatter LA. IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am J Physiol. 2008;294:H596–H604. doi: 10.1152/ajpheart.01155.2007. [DOI] [PubMed] [Google Scholar]
- Dupuis J, Goresky CA, Fournier A. Pulmonary clearance of circulating endothelin-1 in dogs in vivo: exclusive role of ETB receptors. J Appl Physiol. 1996;81:1510–1515. doi: 10.1152/jappl.1996.81.4.1510. [DOI] [PubMed] [Google Scholar]
- Eguchi S, Hirata Y, Marumo F. Endothelin subtype B receptors are coupled to adenylate cyclase via inhibitory G protein in cultured bovine endothelial cells. J Cardiovasc Pharmacol. 1993a;22(Suppl. 8):S161–S163. doi: 10.1097/00005344-199322008-00043. [DOI] [PubMed] [Google Scholar]
- Eguchi S, Hirata Y, Imai T, Marumo F. Endothelin receptor subtypes are coupled to adenylate cyclase via different guanyl nucleotide-binding proteins in vasculature. Endocrinology. 1993b;132:524–529. doi: 10.1210/endo.132.2.7678793. [DOI] [PubMed] [Google Scholar]
- El-Mowafy AM, White RE. Resveratrol inhibits MAPK activity and nuclear translocation in coronary artery smooth muscle: reversal of endothelin-1 stimulatory effects. FEBS Lett. 1999;451:63–67. doi: 10.1016/s0014-5793(99)00541-4. [DOI] [PubMed] [Google Scholar]
- Erdbrugger W, Keffel J, Knocks M, Otto T, Philipp T, Michel MC. Protein kinase C isoenzymes in rat and human cardiovascular tissues. Br J Pharmacol. 1997;120:177–186. doi: 10.1038/sj.bjp.0700877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergul A, Grubbs AL, Zhang Y, Spinale FG. Selective upregulation of endothelin converting enzyme-1a in the human failing heart. J Card Fail. 2000a;6:314–320. doi: 10.1054/jcaf.2000.19227. [DOI] [PubMed] [Google Scholar]
- Ergul A, Walker CA, Goldberg A, Baicu SC, Hendrick JW, King MK, et al. ET-1 in the myocardial interstitium: relation to myocyte ECE activity and expression. Am J Physiol. 2000b;278:H2050–H2056. doi: 10.1152/ajpheart.2000.278.6.H2050. [DOI] [PubMed] [Google Scholar]
- Evans NJ, Walker JW. Endothelin receptor dimers evaluated by FRET, ligand binding, and calcium mobilization. Biophys J. 2008a;95:483–492. doi: 10.1529/biophysj.107.119206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans NJ, Walker JW. Sustained Ca2+ signaling and delayed internalization associated with endothelin receptor heterodimers linked through a PDZ finger. Can J Physiol Pharmacol. 2008b;86:526–535. doi: 10.1139/Y08-050. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Fearnley CJ, Roderick HL, Bootman MD. Calcium signaling in cardiac myocytes. Cold Spring Harb Perspect Biol. 2011;3:a004242. doi: 10.1101/cshperspect.a004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filtz TM, Grubb DR, McLeod-Dryden TJ, Luo J, Woodcock EA. Gq-initiated cardiomyocyte hypertrophy is mediated by phospholipase Cbeta1b. FASEB J. 2009;23:3564–3570. doi: 10.1096/fj.09-133983. [DOI] [PubMed] [Google Scholar]
- Firth JD, Ratcliffe PJ. Organ distribution of the three rat endothelin messenger RNAs and the effects of ischemia on renal gene expression. J Clin Invest. 1992;90:1023–1031. doi: 10.1172/JCI115915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegel L, Karmazyn M. The cardiac Na-H exchanger: a key downstream mediator for the cellular hypertrophic effects of paracrine, autocrine and hormonal factors. Biochem Cell Biol. 2004;82:626–635. doi: 10.1139/o04-129. [DOI] [PubMed] [Google Scholar]
- Francis G, Benedict C, Johnstone D, Kirlin P, Nicklas J, Liang C, et al. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD) Circulation. 1990;82:1724–1729. doi: 10.1161/01.cir.82.5.1724. [DOI] [PubMed] [Google Scholar]
- Freedman NJ, Ament AS, Oppermann M, Stoffel RH, Exum ST, Lefkowitz RJ. Phosphorylation and desensitization of human endothelin A and B receptors. Evidence for G protein-coupled receptor kinase specificity. J Biol Chem. 1997;272:17734–17743. doi: 10.1074/jbc.272.28.17734. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci USA. 1992;89:3686–3690. doi: 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisaki H, Ito H, Hirata Y, Tanaka M, Hata M, Lin M, et al. Natriuretic peptides inhibit angiotensin II-induced proliferation of rat cardiac fibroblasts by blocking endothelin-1 gene expression. J Clin Invest. 1995;96:1059–1065. doi: 10.1172/JCI118092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariepy CE, Ohuchi T, Williams SC, Richardson JA, Yanagisawa M. Salt-sensitive hypertension in endothelin-B receptor-deficient rats. J Clin Invest. 2000;105:925–933. doi: 10.1172/JCI8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JC, Shirayama T, Pappano AJ. Inositol trisphosphate promotes Na-Ca exchange current by releasing calcium from sarcoplasmic reticulum in cardiac myocytes. Circ Res. 1991;69:1632–1639. doi: 10.1161/01.res.69.6.1632. [DOI] [PubMed] [Google Scholar]
- Go LO, Moschella MC, Watras J, Handa KK, Fyfe BS, Marks AR. Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. J Clin Invest. 1995;95:888–894. doi: 10.1172/JCI117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohla A, Offermanns S, Wilkie TM, Schultz G. Differential involvement of Galpha12 and Galpha13 in receptor-mediated stress fiber formation. J Biol Chem. 1999;274:17901–17907. doi: 10.1074/jbc.274.25.17901. [DOI] [PubMed] [Google Scholar]
- Gomberg-Maitland M, Dufton C, Oudiz RJ, Benza RL. Compelling evidence of long-term outcomes in pulmonary arterial hypertension? A clinical perspective. J Am Coll Cardiol. 2011;57:1053–1061. doi: 10.1016/j.jacc.2010.11.020. [DOI] [PubMed] [Google Scholar]
- Gomez del Arco P, Martinez-Martinez S, Maldonado JL, Ortega-Perez I, Redondo JM. A role for the p38 MAP kinase pathway in the nuclear shuttling of NFATp. J Biol Chem. 2000;275:13872–13878. doi: 10.1074/jbc.275.18.13872. [DOI] [PubMed] [Google Scholar]
- Goonasekera SA, Molkentin JD. Unraveling the secrets of a double life: contractile versus signaling Ca2+ in a cardiac myocyte. J Mol Cell Cardiol. 2012;52:317–322. doi: 10.1016/j.yjmcc.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Gorza L, Schiaffino S, Volpe P. Inositol 1,4,5-trisphosphate receptor in heart: evidence for its concentration in Purkinje myocytes of the conduction system. J Cell Biol. 1993;121:345–353. doi: 10.1083/jcb.121.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griendling KK, Tsuda T, Alexander RW. Endothelin stimulates diacylglycerol accumulation and activates protein kinase C in cultured vascular smooth muscle cells. J Biol Chem. 1989;264:8237–8240. [PubMed] [Google Scholar]
- Hallhuber M, Burkard N, Wu R, Buch MH, Engelhardt S, Hein L, et al. Inhibition of nuclear import of calcineurin prevents myocardial hypertrophy. Circ Res. 2006;99:626–635. doi: 10.1161/01.RES.0000243208.59795.d8. [DOI] [PubMed] [Google Scholar]
- Harris IS, Zhang S, Treskov I, Kovacs A, Weinheimer C, Muslin AJ. Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation. 2004;110:718–723. doi: 10.1161/01.CIR.0000138190.50127.6A. [DOI] [PubMed] [Google Scholar]
- Harzheim D, Movassagh M, Foo RS, Ritter O, Tashfeen A, Conway SJ, et al. Increased InsP3Rs in the junctional sarcoplasmic reticulum augment Ca2+ transients and arrhythmias associated with cardiac hypertrophy. Proc Natl Acad Sci USA. 2009;106:11406–11411. doi: 10.1073/pnas.0905485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harzheim D, Talasila A, Movassagh M, Foo RS, Figg N, Bootman MD, et al. Elevated InsP3R expression underlies enhanced calcium fluxes and spontaneous extra-systolic calcium release events in hypertrophic cardiac myocytes. Channels (Austin) 2010;4:67–71. doi: 10.4161/chan.4.1.10531. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Fujiwara H, Koshiji M, Inada T, Ohtani S, Doyama K, et al. Endothelin-1 and its receptor in hypertrophic cardiomyopathy. Hypertension. 1996;27:259–264. doi: 10.1161/01.hyp.27.2.259. [DOI] [PubMed] [Google Scholar]
- Hautala N, Tokola H, Luodonpaa M, Puhakka J, Romppanen H, Vuolteenaho O, et al. Pressure overload increases GATA4 binding activity via endothelin-1. Circulation. 2001;103:730–735. doi: 10.1161/01.cir.103.5.730. [DOI] [PubMed] [Google Scholar]
- Hautala N, Tenhunen O, Szokodi I, Ruskoaho H. Direct left ventricular wall stretch activates GATA4 binding in perfused rat heart: involvement of autocrine/paracrine pathways. Pflugers Arch. 2002;443:362–369. doi: 10.1007/s004240100699. [DOI] [PubMed] [Google Scholar]
- He JQ, Pi Y, Walker JW, Kamp TJ. Endothelin-1 and photoreleased diacylglycerol increase L-type Ca2+ current by activation of protein kinase C in rat ventricular myocytes. J Physiol (Lond) 2000;524(Pt 3):807–820. doi: 10.1111/j.1469-7793.2000.00807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel FR, MacQuaide N, Biesmans L, Sipido K. Dyssynchrony of Ca2+ release from the sarcoplasmic reticulum as subcellular mechanism of cardiac contractile dysfunction. J Mol Cell Cardiol. 2011;50:390–400. doi: 10.1016/j.yjmcc.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Higazi DR, Fearnley CJ, Drawnel FM, Talasila A, Corps EM, Ritter O, et al. Endothelin-1-stimulated InsP3-induced Ca2+ release is a nexus for hypertrophic signaling in cardiac myocytes. Mol Cell. 2009;33:472–482. doi: 10.1016/j.molcel.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Hilal-Dandan R, Ramirez MT, Villegas S, Gonzalez A, Endo-Mochizuki Y, Brown JH, et al. Endothelin ETA receptor regulates signaling and ANF gene expression via multiple G protein-linked pathways. Am J Physiol. 1997;272:H130–H137. doi: 10.1152/ajpheart.1997.272.1.H130. [DOI] [PubMed] [Google Scholar]
- Hirata Y, Fukuda Y, Yoshimi H, Emori T, Shichiri M, Marumo F. Specific receptor for endothelin in cultured rat cardiocytes. Biochem Biophys Res Commun. 1989;160:1438–1444. doi: 10.1016/s0006-291x(89)80165-2. [DOI] [PubMed] [Google Scholar]
- Hirose M, Stuyvers B, Dun W, Ter Keurs HE, Boyden PA. Wide long lasting perinuclear Ca2+ release events generated by an interaction between ryanodine and IP3 receptors in canine Purkinje cells. J Mol Cell Cardiol. 2008;45:176–184. doi: 10.1016/j.yjmcc.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Hosoda K, Nakao K, Arai H, Suga S, Ogawa Y, Mukoyama M, et al. Cloning and expression of human endothelin-1 receptor cDNA. FEBS Lett. 1991;287:23–26. doi: 10.1016/0014-5793(91)80007-p. [DOI] [PubMed] [Google Scholar]
- Inoue A, Yanagisawa M, Kimura S, Kasuya Y, Miyauchi T, Goto K, et al. The human endothelin family: three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc Natl Acad Sci USA. 1989;86:2863–2867. doi: 10.1073/pnas.86.8.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Yanagisawa M, Kimura S, Goto K, Masaki T. Positive chronotropic effects of endothelin, a novel endothelium-derived vasoconstrictor peptide. Pflugers Arch. 1988a;413:108–110. doi: 10.1007/BF00581239. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Yanagisawa M, Kimura S, Goto K, Masaki T. Positive inotropic action of novel vasoconstrictor peptide endothelin on guinea pig atria. Am J Physiol. 1988b;255:H970–H973. doi: 10.1152/ajpheart.1988.255.4.H970. [DOI] [PubMed] [Google Scholar]
- Ito H, Hirata Y, Hiroe M, Tsujino M, Adachi S, Takamoto T, et al. Endothelin-1 induces hypertrophy with enhanced expression of muscle- specific genes in cultured neonatal rat cardiomyocytes. Circ Res. 1991;69:209–215. doi: 10.1161/01.res.69.1.209. [DOI] [PubMed] [Google Scholar]
- Ito H, Hirata Y, Adachi S, Tanaka M, Tsujino M, Koike A, et al. Endothelin-1 is an autocrine/paracrine factor in the mechanism of angiotensin II-induced hypertrophy in cultured rat cardiomyocytes. J Clin Invest. 1993;92:398–403. doi: 10.1172/JCI116579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Hiroe M, Hirata Y, Fujisaki H, Adachi S, Akimoto H, et al. Endothelin ET(A) receptor antagonist blocks cardiac hypertrophy provoked by hemodynamic overload. Circulation. 1994;89:2198–2203. doi: 10.1161/01.cir.89.5.2198. [DOI] [PubMed] [Google Scholar]
- Jain J, Miner Z, Rao A. Analysis of the preexisting and nuclear forms of nuclear factor of activated T cells. J Immunol. 1993;151:837–848. [PubMed] [Google Scholar]
- Jasmin JF, Cernacek P, Dupuis J. Activation of the right ventricular endothelin (ET) system in the monocrotaline model of pulmonary hypertension: response to chronic ETA receptor blockade. Clin Sci (Lond) 2003;105:647–653. doi: 10.1042/CS20030139. [DOI] [PubMed] [Google Scholar]
- Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348:2007–2018. doi: 10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- Ju Y-K, Liu J, Lee BH, Lai D, Woodcock EA, Lei M, et al. Distribution and functional role of inositol 1,4,5-trisphosphate receptors in mouse sinoatrial node. Circ Res. 2011;109:848–857. doi: 10.1161/CIRCRESAHA.111.243824. [DOI] [PubMed] [Google Scholar]
- Kakita T, Hasegawa K, Iwai-Kanai E, Adachi S, Morimoto T, Wada H, et al. Calcineurin pathway is required for endothelin-1-mediated protection against oxidant stress-induced apoptosis in cardiac myocytes. Circ Res. 2001;88:1239–1246. doi: 10.1161/hh1201.091794. [DOI] [PubMed] [Google Scholar]
- Kalra PR, Moon JC, Coats AJ. Do results of the ENABLE (Endothelin Antagonist Bosentan for Lowering Cardiac Events in Heart Failure) study spell the end for non-selective endothelin antagonism in heart failure? Int J Cardiol. 2002;85:195–197. doi: 10.1016/s0167-5273(02)00182-1. [DOI] [PubMed] [Google Scholar]
- Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]
- Kapur N, Banach K. Inositol-1,4,5-trisphosphate-mediated spontaneous activity in mouse embryonic stem cell-derived cardiomyocytes. J Physiol (Lond) 2007;581:1113–1127. doi: 10.1113/jphysiol.2006.125955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagueuzian HS. Targeting cardiac fibrosis: a new frontier in antiarrhythmic therapy? Am J Cardiovasc Dis. 2011;1:101–109. [PMC free article] [PubMed] [Google Scholar]
- Kawamura T, Ono K, Morimoto T, Akao M, Iwai-Kanai E, Wada H, et al. Endothelin-1-dependent nuclear factor of activated T lymphocyte signaling associates with transcriptional coactivator p300 in the activation of the B cell leukemia-2 promoter in cardiac myocytes. Circ Res. 2004;94:1492–1499. doi: 10.1161/01.RES.0000129701.14494.52. [DOI] [PubMed] [Google Scholar]
- Kedzierski RM, Yanagisawa M. Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol. 2001;41:851–876. doi: 10.1146/annurev.pharmtox.41.1.851. [DOI] [PubMed] [Google Scholar]
- Kehat I, Molkentin JD. Extracellular signal-regulated kinase 1/2 (ERK1/2) signaling in cardiac hypertrophy. Ann N Y Acad Sci. 2010;1188:96–102. doi: 10.1111/j.1749-6632.2009.05088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehat I, Davis J, Tiburcy M, Accornero F, Saba-El-Leil MK, Maillet M, et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ Res. 2011;108:176–183. doi: 10.1161/CIRCRESAHA.110.231514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelland NF, Webb DJ. Clinical trials of endothelin antagonists in heart failure: a question of dose? Exp Biol Med. 2006;231:696–699. [PubMed] [Google Scholar]
- Kelland NF, Webb DJ. Clinical trials of endothelin antagonists in heart failure: publication is good for the public health. Heart. 2007;93:2–4. doi: 10.1136/hrt.2006.089250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelland NF, Kuc RE, McLean DL, Azfer A, Bagnall AJ, Gray GA, et al. Endothelial cell-specific ETB receptor knockout: autoradiographic and histological characterisation and crucial role in the clearance of endothelin-1. Can J Physiol Pharmacol. 2010;88:644–651. doi: 10.1139/Y10-041. [DOI] [PubMed] [Google Scholar]
- Kim Y, Phan D, van Rooij E, Wang DZ, McAnally J, Qi X, et al. The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J Clin Invest. 2008;118:124–132. doi: 10.1172/JCI33255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby NS, Hadoke PWF, Bagnall AJ, Webb DJ. The endothelin system as a therapeutic target in cardiovascular disease: great expectations or bleak house? Br J Pharmacol. 2008;153:1105–1119. doi: 10.1038/sj.bjp.0707516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Miyauchi T, Sakai S, Kobayashi M, Yamaguchi I, Goto K, et al. Expression of endothelin-1, ETA and ETB receptors, and ECE and distribution of endothelin-1 in failing rat heart. Am J Physiol. 1999;276:H1197–H1206. doi: 10.1152/ajpheart.1999.276.4.H1197. [DOI] [PubMed] [Google Scholar]
- Kockskamper J, Seidlmayer L, Walther S, Hellenkamp K, Maier LS, Pieske B. Endothelin-1 enhances nuclear Ca2+ transients in atrial myocytes through Ins(1,4,5)P3-dependent Ca2+ release from perinuclear Ca2+ stores. J Cell Sci. 2008;121:186–195. doi: 10.1242/jcs.021386. [DOI] [PubMed] [Google Scholar]
- Kockskämper J, Lewinski D, Khafaga M, Elgner A, Grimm M, Eschenhagen T, et al. The slow force response to stretch in atrial and ventricular myocardium from human heart: functional relevance and subcellular mechanisms. Prog Biophys Mol Biol. 2008a;97:250–267. doi: 10.1016/j.pbiomolbio.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockskämper J, Zima AV, Roderick HL, Pieske B, Blatter LA, Bootman MD. Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J Mol Cell Cardiol. 2008b;45:128–147. doi: 10.1016/j.yjmcc.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolettis TM, Baltogiannis GG, Tsalikakis DG, Tzallas AT, Agelaki MG, Fotopoulos A, et al. Effects of dual endothelin receptor blockade on sympathetic activation and arrhythmogenesis during acute myocardial infarction in rats. Eur J Pharmacol. 2008;580:241–249. doi: 10.1016/j.ejphar.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Komukai K, O-Uchi J, Morimoto S, Kawai M, Hongo K, Yoshimura M, et al. Role of Ca(2+)/calmodulin-dependent protein kinase II in the regulation of the cardiac L-type Ca(2+) current during endothelin-1 stimulation. Am J Physiol. 2010;298:H1902–H1907. doi: 10.1152/ajpheart.01141.2009. [DOI] [PubMed] [Google Scholar]
- Kuhn C, Frank D, Will R, Jaschinski C, Frauen R, Katus HA, et al. DYRK1A is a novel negative regulator of cardiomyocyte hypertrophy. J Biol Chem. 2009;284:17320–17327. doi: 10.1074/jbc.M109.006759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y, Kurihara H, Oda H, Maemura K, Nagai R, Ishikawa T, et al. Aortic arch malformations and ventricular septal defect in mice deficient in endothelin-1. J Clin Invest. 1995;96:293–300. doi: 10.1172/JCI118033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a ‘set up’ for vascular disease. Circulation. 2003a;107:139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation. 2003b;107:346–354. doi: 10.1161/01.cir.0000048893.62841.f7. [DOI] [PubMed] [Google Scholar]
- Lauer MR, Gunn MD, Clusin WT. Endothelin activates voltage-dependent Ca2+ current by a G protein-dependent mechanism in rabbit cardiac myocytes. J Physiol (Lond) 1992;448:729–747. doi: 10.1113/jphysiol.1992.sp019067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc N, Hume JR. Sodium current-induced release of calcium from cardiac sarcoplasmic reticulum. Science. 1990;248:372–376. doi: 10.1126/science.2158146. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Lenormand P, Sardet C, Pagès G, L'Allemain G, Brunet A, Pouyssegur J. Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J Cell Biol. 1993;122:1079–1088. doi: 10.1083/jcb.122.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie SJ, Spratt JCS, McKee SP, Strachan FE, Newby DE, Northridge DB, et al. Direct comparison of selective endothelin A and non-selective endothelin A/B receptor blockade in chronic heart failure. Heart. 2005;91:914–919. doi: 10.1136/hrt.2004.040386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zima AV, Sheikh F, Blatter LA, Chen J. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res. 2005;96:1274–1281. doi: 10.1161/01.RES.0000172556.05576.4c. [DOI] [PubMed] [Google Scholar]
- Liang Q, Wiese RJ, Bueno OF, Dai YS, Markham BE, Molkentin JD. The transcription factor GATA4 is activated by extracellular signal-regulated kinase 1- and 2-mediated phosphorylation of serine 105 in cardiomyocytes. Mol Cell Biol. 2001;21:7460–7469. doi: 10.1128/MCB.21.21.7460-7469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci USA. 2001;98:12283–12288. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HW, Molkentin JD. Calcineurin and human heart failure. Nat Med. 1999;5:246–247. doi: 10.1038/6430. [DOI] [PubMed] [Google Scholar]
- Lin AE, Alexander ME, Colan SD, Kerr B, Rauen KA, Noonan J, et al. Clinical, pathological, and molecular analyses of cardiovascular abnormalities in Costello syndrome: a Ras/MAPK pathway syndrome. Am J Med Genet. 2011;155A:486–507. doi: 10.1002/ajmg.a.33857. [DOI] [PubMed] [Google Scholar]
- Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W, et al. Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr Biol. 2000;10:939–942. doi: 10.1016/s0960-9822(00)00624-2. [DOI] [PubMed] [Google Scholar]
- Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat Med. 2009;15:75–83. doi: 10.1038/nm.1893. [DOI] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Nicol RL, Olson EN. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci USA. 2000;97:4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie L, Bootman MD, Berridge MJ, Lipp P. Predetermined recruitment of calcium release sites underlies excitation-contraction coupling in rat atrial myocytes. J Physiol. 2001;530:417–429. doi: 10.1111/j.1469-7793.2001.0417k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie L, Bootman MD, Laine M, Berridge MJ, Thuring J, Holmes A, et al. The role of inositol 1,4,5-trisphosphate receptors in Ca(2+) signalling and the generation of arrhythmias in rat atrial myocytes. J Physiol (Lond) 2002;541:395–409. doi: 10.1113/jphysiol.2001.013411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie L, Roderick HL, Berridge MJ, Conway SJ, Bootman MD. The spatial pattern of atrial cardiomyocyte calcium signalling modulates contraction. J Cell Sci. 2004;117:6327–6337. doi: 10.1242/jcs.01559. [DOI] [PubMed] [Google Scholar]
- McMullen JR, Jennings GL. Differences between pathological and physiological cardiac hypertrophy: novel therapeutic strategies to treat heart failure. Clin Exp Pharmacol Physiol. 2007;34:255–262. doi: 10.1111/j.1440-1681.2007.04585.x. [DOI] [PubMed] [Google Scholar]
- Maeda S, Tanabe T, Miyauchi T, Otsuki T, Sugawara J, Iemitsu M, et al. Aerobic exercise training reduces plasma endothelin-1 concentration in older women. J Appl Physiol. 2003;95:336–341. doi: 10.1152/japplphysiol.01016.2002. [DOI] [PubMed] [Google Scholar]
- Maeda S, Jesmin S, Iemitsu M, Otsuki T, Matsuo T, Ohkawara K, et al. Weight loss reduces plasma endothelin-1 concentration in obese men. Exp Biol Med (Maywood) 2006;231:1044–1047. [PubMed] [Google Scholar]
- Maekawa N, Abe J-I, Shishido T, Itoh S, Ding B, Sharma VK, et al. Inhibiting p90 ribosomal S6 kinase prevents (Na+)-H+ exchanger-mediated cardiac ischemia-reperfusion injury. Circulation. 2006;113:2516–2523. doi: 10.1161/CIRCULATIONAHA.105.563486. [DOI] [PubMed] [Google Scholar]
- Markou T, Yong CS, Sugden PH, Clerk A. Regulation of protein kinase C delta by phorbol ester, endothelin-1, and platelet-derived growth factor in cardiac myocytes. J Biol Chem. 2006;281:8321–8331. doi: 10.1074/jbc.M508398200. [DOI] [PubMed] [Google Scholar]
- Marshall AK, Barrett OP, Cullingford TE, Shanmugasundram A, Sugden PH, Clerk A. ERK1/2 signaling dominates over RhoA signaling in regulating early changes in RNA expression induced by endothelin-1 in neonatal rat cardiomyocytes. PLoS ONE. 2010;5:e10027. doi: 10.1371/journal.pone.0010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayyas F, Niebauer M, Zurick A, Barnard J, Gillinov AM, Chung MK, et al. Association of left atrial endothelin-1 with atrial rhythm, size, and fibrosis in patients with structural heart disease. Circ Arrhythm Electrophysiol. 2010;3:369–379. doi: 10.1161/CIRCEP.109.924985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Lehnart S, Pieske B, Schlottauer K, Munk S, Holubarsch C, et al. Influence of endothelin 1 on human atrial myocardium – myocardial function and subcellular pathways. Basic Res Cardiol. 1996;91:86–93. doi: 10.1007/BF00788869. [DOI] [PubMed] [Google Scholar]
- Molenaar P, O'Reilly G, Sharkey A, Kuc RE, Harding DP, Plumpton C, et al. Characterization and localization of endothelin receptor subtypes in the human atrioventricular conducting system and myocardium. Circ Res. 1993;72:526–538. doi: 10.1161/01.res.72.3.526. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montessuit C, Thorburn A. Activation of Ras by phorbol esters in cardiac myocytes. Role of guanine nucleotide exchange factors. FEBS Lett. 1999;460:57–60. doi: 10.1016/s0014-5793(99)01223-5. [DOI] [PubMed] [Google Scholar]
- Moravec CS, Reynolds EE, Stewart RW, Bond M. Endothelin is a positive inotropic agent in human and rat heart in vitro. Biochem Biophys Res Commun. 1989;159:14–18. doi: 10.1016/0006-291x(89)92397-8. [DOI] [PubMed] [Google Scholar]
- Moschella MC, Marks AR. Inositol 1,4,5-trisphosphate receptor expression in cardiac myocytes. J Cell Biol. 1993;120:1137–1146. doi: 10.1083/jcb.120.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschella PC, Rao VU, McDermott PJ, Kuppuswamy D. Regulation of mTOR and S6K1 activation by the nPKC isoforms, PKCε and PKCδ, in adult cardiac muscle cells. J Mol Cell Cardiol. 2007;43:754–766. doi: 10.1016/j.yjmcc.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motte S, van Beneden R, Mottet J, Rondelet B, Mathieu M, Havaux X, et al. Early activation of cardiac and renal endothelin systems in experimental heart failure. Am J Physiol. 2003;285:H2482–H2491. doi: 10.1152/ajpheart.00419.2003. [DOI] [PubMed] [Google Scholar]
- Munzel F, Muhlhauser U, Zimmermann W-H, Didié M, Schneiderbanger K, Schubert P, et al. Endothelin-1 and isoprenaline co-stimulation causes contractile failure which is partially reversed by MEK inhibition. Cardiovasc Res. 2005;68:464–474. doi: 10.1016/j.cardiores.2005.06.020. [DOI] [PubMed] [Google Scholar]
- Nag AC. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28:41–61. [PubMed] [Google Scholar]
- Nagasaka T, Izumi M, Hori M, Ozaki H, Karaki H. Positive inotropic effect of endothelin-1 in the neonatal mouse right ventricle. Eur J Pharmacol. 2003;472:197–204. doi: 10.1016/s0014-2999(03)01936-8. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Bodi I, Maillet M, DeSantiago J, Domeier TL, Mikoshiba K, et al. The IP3 receptor regulates cardiac hypertrophy in response to select stimuli/novelty and significance. Circ Res. 2010;107:659–666. doi: 10.1161/CIRCRESAHA.110.220038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JW, Clipstone NA. Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J Biol Chem. 2001;276:3666–3673. doi: 10.1074/jbc.M004888200. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Sheng Z, Lin A. Opposing effects of Jun kinase and p38 mitogen-activated protein kinases on cardiomyocyte hypertrophy. Mol Cell Biol. 1998;18:3518–3526. doi: 10.1128/mcb.18.6.3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, et al. p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol Cell Biol. 2004;24:10611–10620. doi: 10.1128/MCB.24.24.10611-10620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimaru K, Miura Y, Endoh M. Mechanisms of endothelin-1-induced decrease in contractility in adult mouse ventricular myocytes. Br J Pharmacol. 2007;152:456–463. doi: 10.1038/sj.bjp.0707392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez DJ, Brown MJ, Davenport AP, Neylon CB, Schofield JP, Wyse RK. Endothelin-1 mRNA is widely expressed in porcine and human tissues. J Clin Invest. 1990;85:1537–1541. doi: 10.1172/JCI114601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Yoshiyama M, Yoshida K, Nakamura Y, Kim S, Iwao H, et al. Dominant negative mutant of c-Jun inhibits cardiomyocyte hypertrophy induced by endothelin 1 and phenylephrine. Hypertension. 2002;39:81–86. doi: 10.1161/hy0102.100783. [DOI] [PubMed] [Google Scholar]
- Ono K, Tsujimoto G, Sakamoto A, Eto K, Masaki T, Ozaki Y, et al. Endothelin-A receptor mediates cardiac inhibition by regulating calcium and potassium currents. Nature. 1994;370:301–304. doi: 10.1038/370301a0. [DOI] [PubMed] [Google Scholar]
- Ono K, Masumiya H, Sakamoto A, Christé G, Shijuku T, Tanaka H, et al. Electrophysiological analysis of the negative chronotropic effect of endothelin-1 in rabbit sinoatrial node cells. J Physiol (Lond) 2001;537:467–488. doi: 10.1111/j.1469-7793.2001.00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaniyandi SS, Sun L, Ferreira JC, Mochly-Rosen D. Protein kinase C in heart failure: a therapeutic target? Cardiovasc Res. 2009;82:229–239. doi: 10.1093/cvr/cvp001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A-M, Wong C-M, Jelinkova L, Liu L, Nagase H, Suzuki YJ. Pulmonary hypertension-induced GATA4 activation in the right ventricle. Hypertension. 2010;56:1145–1151. doi: 10.1161/HYPERTENSIONAHA.110.160515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Lambert AA, Papst P, Pitts KR. Agonist-induced nuclear export of GFP-HDAC5 in isolated adult rat ventricular myocytes. J Pharmacol Toxicol Methods. 2009;59:135–140. doi: 10.1016/j.vascn.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Perez PJ, Ramos-Franco J, Fill M, Mignery GA. Identification and functional reconstitution of the type 2 inositol 1,4,5-trisphosphate receptor from ventricular cardiac myocytes. J Biol Chem. 1997;272:23961–23969. doi: 10.1074/jbc.272.38.23961. [DOI] [PubMed] [Google Scholar]
- Petkova SB, Tanowitz HB, Magazine HI, Factor SM, Chan J, Pestell RG, et al. Myocardial expression of endothelin-1 in murine Trypanosoma cruzi infection. Cardiovasc Pathol. 2000;9:257–265. doi: 10.1016/s1054-8807(00)00045-4. [DOI] [PubMed] [Google Scholar]
- Petrich BG, Eloff BC, Lerner DL, Kovacs A, Saffitz JE, Rosenbaum DS, et al. Targeted activation of c-Jun N-terminal kinase in vivo induces restrictive cardiomyopathy and conduction defects. J Biol Chem. 2004;279:15330–15338. doi: 10.1074/jbc.M314142200. [DOI] [PubMed] [Google Scholar]
- Phan D, Stratton MS, Huynh QK, McKinsey TA. A novel protein kinase C target site in protein kinase D is phosphorylated in response to signals for cardiac hypertrophy. Biochem Biophys Res Commun. 2011;411:335–341. doi: 10.1016/j.bbrc.2011.06.143. [DOI] [PubMed] [Google Scholar]
- Philipson KD, Nicoll DA. Sodium-calcium exchange: a molecular perspective. Annu Rev Physiol. 2000;62:111–133. doi: 10.1146/annurev.physiol.62.1.111. [DOI] [PubMed] [Google Scholar]
- Pi Y, Zhang D, Kemnitz KR, Wang H, Walker JW. Protein kinase C and A sites on troponin I regulate myofilament Ca2+ sensitivity and ATPase activity in the mouse myocardium. J Physiol (Lond) 2003;552:845–857. doi: 10.1113/jphysiol.2003.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieske B, Schlotthauer K, Schattmann J, Beyersdorf F, Martin J, Just H, et al. Ca(2+)-dependent and Ca(2+)-independent regulation of contractility in isolated human myocardium. Basic Res Cardiol. 1997;92(Suppl. 1):75–86. doi: 10.1007/BF00794071. [DOI] [PubMed] [Google Scholar]
- Pieske B, Beyermann B, Breu V, Loffler BM, Schlotthauer K, Maier LS, et al. Functional effects of endothelin and regulation of endothelin receptors in isolated human nonfailing and failing myocardium. Circulation. 1999;99:1802–1809. doi: 10.1161/01.cir.99.14.1802. [DOI] [PubMed] [Google Scholar]
- Pikkarainen S, Tokola H, Kerkela R, Ilves M, Mäkinen M, Orzechowski H-D, et al. Inverse regulation of preproendothelin-1 and endothelin-converting enzyme-1beta genes in cardiac cells by mechanical load. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1639–R1645. doi: 10.1152/ajpregu.00559.2005. [DOI] [PubMed] [Google Scholar]
- Ping P, Anzai T, Gao M, Hammond HK. Adenylyl cyclase and G protein receptor kinase expression during development of heart failure. Am J Physiol. 1997;273:H707–H717. doi: 10.1152/ajpheart.1997.273.2.H707. [DOI] [PubMed] [Google Scholar]
- Proven A, Roderick HL, Conway SJ, Berridge MJ, Horton JK, Capper SJ, et al. Inositol 1,4,5-trisphosphate supports the arrhythmogenic action of endothelin-1 on ventricular cardiac myocytes. J Cell Sci. 2006;119:3363–3375. doi: 10.1242/jcs.03073. [DOI] [PubMed] [Google Scholar]
- Puri TS, Gerhardstein BL, Zhao XL, Ladner MB, Hosey MM. Differential effects of subunit interactions on protein kinase A- and C-mediated phosphorylation of L-type calcium channels. Biochemistry. 1997;36:9605–9615. doi: 10.1021/bi970500d. [DOI] [PubMed] [Google Scholar]
- Remus TP, Zima AV, Bossuyt J, Bare DJ, Martin JL, Blatter LA, et al. Biosensors to measure inositol 1,4,5-trisphosphate concentration in living cells with spatiotemporal resolution. J Biol Chem. 2006;281:608–616. doi: 10.1074/jbc.M509645200. [DOI] [PubMed] [Google Scholar]
- Rinne A, Blatter LA. Activation of NFATc1 is directly mediated by IP3 in adult cardiac myocytes. Am J Physiol. 2010;299:H1701–H1707. doi: 10.1152/ajpheart.00470.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne A, Kapur N, Molkentin JD, Pogwizd SM, Bers DM, Banach K, et al. Isoform- and tissue-specific regulation of the Ca2+-sensitive transcription factor NFAT in cardiac myocytes and heart failure. Am J Physiol. 2010;298:H2001–H2009. doi: 10.1152/ajpheart.01072.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robu VG, Pfeiffer ES, Robia SL, Balijepalli RC, Pi Y, Kamp TJ, et al. Localization of functional endothelin receptor signaling complexes in cardiac transverse tubules. J Biol Chem. 2003;278:48154–48161. doi: 10.1074/jbc.M304396200. [DOI] [PubMed] [Google Scholar]
- Roderick HL, Berridge MJ, Bootman MD. Calcium-induced calcium release. Curr Biol. 2003;13:R425. doi: 10.1016/s0960-9822(03)00358-0. [DOI] [PubMed] [Google Scholar]
- Rubart M, Field LJ. Cardiac regeneration: repopulating the heart. Annu Rev Physiol. 2006;68:29–49. doi: 10.1146/annurev.physiol.68.040104.124530. [DOI] [PubMed] [Google Scholar]
- Russell FD, Davenport AP. Characterization of the binding of endothelin ETB selective ligands in human and rat heart. Br J Pharmacol. 1996;119:631–636. doi: 10.1111/j.1476-5381.1996.tb15720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell FD, Molenaar P. The human heart endothelin system: ET-1 synthesis, storage, release and effect. Trends Pharmacol Sci. 2000;21:353–359. doi: 10.1016/s0165-6147(00)01524-8. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- Sakai S, Miyauchi T, Kobayashi M, Yamaguchi I, Goto K, Sugishita Y. Inhibition of myocardial endothelin pathway improves long-term survival in heart failure. Nature. 1996;384:353–355. doi: 10.1038/384353a0. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Yanagisawa M, Takuwa Y, Miyazaki H, Kimura S, Goto K, et al. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature. 1990;348:732–735. doi: 10.1038/348732a0. [DOI] [PubMed] [Google Scholar]
- Salazar NC, Chen J, Rockman HA. Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim Biophys Acta. 2007;1768:1006–1018. doi: 10.1016/j.bbamem.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomone OA, Caeiro TF, Madoery RJ, Amuchástegui M, Omelinauk M, Juri D, et al. High plasma immunoreactive endothelin levels in patients with Chagas' cardiomyopathy. Am J Cardiol. 2001;87:1217–1220. doi: 10.1016/s0002-9149(01)01502-8. A7. [DOI] [PubMed] [Google Scholar]
- Sanna B, Bueno OF, Dai YS, Wilkins BJ, Molkentin JD. Direct and indirect interactions between calcineurin-NFAT and MEK1-extracellular signal-regulated kinase 1/2 signaling pathways regulate cardiac gene expression and cellular growth. Mol Cell Biol. 2005;25:865–878. doi: 10.1128/MCB.25.3.865-878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvais PL, Robert A, Ahn S, van Linden F, Ketelslegers JM, Pouleur H, et al. Direct comparison between endothelin-1, N-terminal proatrial natriuretic factor, and brain natriuretic peptide as prognostic markers of survival in congestive heart failure. J Card Fail. 2000;6:201–207. doi: 10.1054/jcaf.2000.8833. [DOI] [PubMed] [Google Scholar]
- Serneri GGN, Cecioni I, Vanni S, Paniccia R, Bandinelli B, Vetere A, et al. Selective upregulation of cardiac endothelin system in patients with ischemic but not idiopathic dilated cardiomyopathy: endothelin-1 system in the human failing heart. Circ Res. 2000;86:377–385. doi: 10.1161/01.res.86.4.377. [DOI] [PubMed] [Google Scholar]
- Shah AM, Lewis MJ, Henderson AH. Inotropic effects of endothelin in ferret ventricular myocardium. Eur J Pharmacol. 1989;163:365–367. doi: 10.1016/0014-2999(89)90208-2. [DOI] [PubMed] [Google Scholar]
- Shattock MJ, Bers DM. Rat vs. rabbit ventricle: Ca flux and intracellular Na assessed by ion-selective microelectrodes. Am J Physiol. 1989;256:C813–C822. doi: 10.1152/ajpcell.1989.256.4.C813. [DOI] [PubMed] [Google Scholar]
- Shaw KT, Ho AM, Raghavan A, Kim J, Jain J, Park J, et al. Immunosuppressive drugs prevent a rapid dephosphorylation of transcription factor NFAT1 in stimulated immune cells. Proc Natl Acad Sci USA. 1995;92:11205–11209. doi: 10.1073/pnas.92.24.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki F, Price ER, Milan D, McKeon F. Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF-AT4. Nature. 1996;382:370–373. doi: 10.1038/382370a0. [DOI] [PubMed] [Google Scholar]
- Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, et al. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med. 2008;205:295–303. doi: 10.1084/jem.20071297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-Wen X, Chen Y, Denton CP, Eastwood M, Renzoni EA, Bou-Gharios G, et al. Endothelin-1 promotes myofibroblast induction through the ETA receptor via a rac/phosphoinositide 3-kinase/Akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol Biol Cell. 2004a;15:2707–2719. doi: 10.1091/mbc.E03-12-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-Wen X, Howat SL, Renzoni EA, Holmes A, Pearson JD, Dashwood MR, et al. Endothelin-1 induces expression of matrix-associated genes in lung fibroblasts through MEK/ERK. J Biol Chem. 2004b;279:23098–23103. doi: 10.1074/jbc.M311430200. [DOI] [PubMed] [Google Scholar]
- Shohet RV, Kisanuki YY, Zhao XS, Siddiquee Z, Franco F, Yanagisawa M. Mice with cardiomyocyte-specific disruption of the endothelin-1 gene are resistant to hyperthyroid cardiac hypertrophy. Proc Natl Acad Sci USA. 2004;101:2088–2093. doi: 10.1073/pnas.0307159101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyrnias I, Mair W, Harzheim D, Walker SA, Roderick HL, Bootman MD. Comparison of the T-tubule system in adult rat ventricular and atrial myocytes, and its role in excitation-contraction coupling and inotropic stimulation. Cell Calcium. 2010;47:210–223. doi: 10.1016/j.ceca.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Snabaitis AK, Muntendorf A, Wieland T, Avkiran M. Regulation of the extracellular signal-regulated kinase pathway in adult myocardium: differential roles of G(q/11), Gi and G(12/13) proteins in signalling by alpha1-adrenergic, endothelin-1 and thrombin-sensitive protease-activated receptors. Cell Signal. 2005;17:655–664. doi: 10.1016/j.cellsig.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Snabaitis AK, Cuello F, Avkiran M. Protein kinase B/Akt phosphorylates and inhibits the cardiac Na+/H+ exchanger NHE1. Circ Res. 2008;103:881–890. doi: 10.1161/CIRCRESAHA.108.175877. [DOI] [PubMed] [Google Scholar]
- Song L-S, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci USA. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden PH. An overview of endothelin signaling in the cardiac myocyte. J Mol Cell Cardiol. 2003;35:871–886. doi: 10.1016/s0022-2828(03)00153-6. [DOI] [PubMed] [Google Scholar]
- Sugden PH, Clerk A. Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxid Redox Signal. 2006;8:2111–2124. doi: 10.1089/ars.2006.8.2111. [DOI] [PubMed] [Google Scholar]
- Sutsch G, Kiowski W, Yan XW, Hunziker P, Christen S, Strobel W, et al. Short-term oral endothelin-receptor antagonist therapy in conventionally treated patients with symptomatic severe chronic heart failure. Circulation. 1998;98:2262–2268. doi: 10.1161/01.cir.98.21.2262. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Kumazaki T, Mitsui Y. Endothelin-1 is produced and secreted by neonatal rat cardiac myocytes in vitro. Biochem Biophys Res Commun. 1993;191:823–830. doi: 10.1006/bbrc.1993.1291. [DOI] [PubMed] [Google Scholar]
- Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- Tachibana H, Perrino C, Takaoka H, Davis RJ, Naga Prasad SV, Rockman HA. JNK1 is required to preserve cardiac function in the early response to pressure overload. Biochem Biophys Res Commun. 2006;343:1060–1066. doi: 10.1016/j.bbrc.2006.03.065. [DOI] [PubMed] [Google Scholar]
- Tadevosyan A, Maguy A, Villeneuve LR, Babin J, Bonnefoy A, Allen BG, et al. Nuclear-delimited angiotensin receptor-mediated signaling regulates cardiomyocyte gene expression. J Biol Chem. 2010;285:22338–22349. doi: 10.1074/jbc.M110.121749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadevosyan A, Vaniotis G, Allen BG, Hébert TE, Nattel S. G protein-coupled receptor signalling in the cardiac nuclear membrane: evidence and possible roles in physiological and pathophysiological function. J Physiol (Lond) 2012;590:1313–1330. doi: 10.1113/jphysiol.2011.222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanashi M, Endoh M. Characterization of positive inotropic effect of endothelin on mammalian ventricular myocardium. Am J Physiol. 1991;261:H611–H619. doi: 10.1152/ajpheart.1991.261.3.H611. [DOI] [PubMed] [Google Scholar]
- Takuwa Y, Kasuya Y, Takuwa N, Kudo M, Yanagisawa M, Goto K, et al. Endothelin receptor is coupled to phospholipase C via a pertussis toxin-insensitive guanine nucleotide-binding regulatory protein in vascular smooth muscle cells. J Clin Invest. 1990;85:653–658. doi: 10.1172/JCI114488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanowitz HB, Huang H, Jelicks LA, Chandra M, Loredo ML, Weiss LM, et al. Role of endothelin 1 in the pathogenesis of chronic chagasic heart disease. Infect Immun. 2005;73:2496–2503. doi: 10.1128/IAI.73.4.2496-2503.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuinenburg AE, Van Veldhuisen DJ, Boomsma F, Van Den Berg MP, De Kam PJ, Crijns HJ. Comparison of plasma neurohormones in congestive heart failure patients with atrial fibrillation versus patients with sinus rhythm. Am J Cardiol. 1998;81:1207–1210. doi: 10.1016/s0002-9149(98)00092-7. [DOI] [PubMed] [Google Scholar]
- Uchida K, Aramaki M, Nakazawa M, Yamagishi C, Makino S, Fukuda K, et al. Gene knock-outs of inositol 1,4,5-trisphosphate receptors types 1 and 2 result in perturbation of cardiogenesis. PLoS ONE. 2010;5:e12500. doi: 10.1371/journal.pone.0012500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueyama T, Kawashima S, Sakoda T, Rikitake Y, Ishida T, Kawai M, et al. Requirement of activation of the extracellular signal-regulated kinase cascade in myocardial cell hypertrophy. J Mol Cell Cardiol. 2000;32:947–960. doi: 10.1006/jmcc.2000.1135. [DOI] [PubMed] [Google Scholar]
- Vaniotis G, Allen BG, Hébert TE. Nuclear GPCRs in cardiomyocytes: an insider's view of β-adrenergic receptor signaling. Am J Physiol. 2011a;301:H1754–H1764. doi: 10.1152/ajpheart.00657.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaniotis G, Del Duca D, Trieu P, Rohlicek CV, Hébert TE, Allen BG. Nuclear β-adrenergic receptors modulate gene expression in adult rat heart. Cell Signal. 2011b;23:89–98. doi: 10.1016/j.cellsig.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, et al. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigne P, Lazdunski M, Frelin C. The inotropic effect of endothelin-1 on rat atria involves hydrolysis of phosphatidylinositol. FEBS Lett. 1989;249:143–146. doi: 10.1016/0014-5793(89)80611-8. [DOI] [PubMed] [Google Scholar]
- Vijayan K, Szotek EL, Martin JL, Samarel AM. Protein kinase C-alpha-induced hypertrophy of neonatal rat ventricular myocytes. Am J Physiol. 2004;287:H2777–H2789. doi: 10.1152/ajpheart.00171.2004. [DOI] [PubMed] [Google Scholar]
- Vinge LE, Øie E, Andersson Y, Grøgaard HK, Andersen G, Attramadal H. Myocardial distribution and regulation of GRK and beta-arrestin isoforms in congestive heart failure in rats. Am J Physiol. 2001;281:H2490–H2499. doi: 10.1152/ajpheart.2001.281.6.H2490. [DOI] [PubMed] [Google Scholar]
- Vinge LE, Andressen KW, Attramadal T, Andersen GO, Ahmed MS, Peppel K, et al. Substrate specificities of g protein-coupled receptor kinase-2 and -3 at cardiac myocyte receptors provide basis for distinct roles in regulation of myocardial function. Mol Pharmacol. 2007;72:582–591. doi: 10.1124/mol.107.035766. [DOI] [PubMed] [Google Scholar]
- Wang H, Grant JE, Doede CM, Sadayappan S, Robbins J, Walker JW. PKC-betaII sensitizes cardiac myofilaments to Ca2+ by phosphorylating troponin I on threonine-144. J Mol Cell Cardiol. 2006;41:823–833. doi: 10.1016/j.yjmcc.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Wang J, Paradis P, Aries A, Komati H, Lefebvre C, Wang H, et al. Convergence of protein kinase C and JAK-STAT signaling on transcription factor GATA-4. Mol Cell Biol. 2005;25:9829–9844. doi: 10.1128/MCB.25.22.9829-9844.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JX, Paik G, Morgan JP. Endothelin 1 enhances myofilament Ca2+ responsiveness in aequorin-loaded ferret myocardium. Circ Res. 1991;69:582–589. doi: 10.1161/01.res.69.3.582. [DOI] [PubMed] [Google Scholar]
- Wang QD, Gonon A, Shimizu M, Sjöquist PO, Pernow J. Contribution of endothelin to the coronary vasoconstriction in the isolated rat heart induced by nitric oxide synthase inhibition. Acta Physiol Scand. 1998;163:325–330. doi: 10.1046/j.1365-201X.1998.t01-1-00364.x. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Endoh M. Characterization of the endothelin-1-induced regulation of L-type Ca2+ current in rabbit ventricular myocytes. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:654–664. doi: 10.1007/s002109900130. [DOI] [PubMed] [Google Scholar]
- Watts SW. Endothelin receptors: what's new and what do we need to know. Am J Physiol Regul Integr Comp Physiol. 2010;298:R254–R260. doi: 10.1152/ajpregu.00584.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb DJ. Endothelin receptors cloned, endothelin converting enzyme characterized and pathophysiological roles for endothelin proposed. Trends Pharmacol Sci. 1991;12:43–46. doi: 10.1016/0165-6147(91)90492-b. [DOI] [PubMed] [Google Scholar]
- Westfall MV, Lee AM, Robinson DA. Differential contribution of troponin I phosphorylation sites to the endothelin-modulated contractile response. J Biol Chem. 2005;280:41324–41331. doi: 10.1074/jbc.M506043200. [DOI] [PubMed] [Google Scholar]
- Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, et al. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol Cell Biol. 2002;22:7603–7613. doi: 10.1128/MCB.22.21.7603-7613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CD, Wu SC, Dahl EF, Sazama AJ, O'Connell TD. Nuclear localization drives α1-adrenergic receptor oligomerization and signaling in cardiac myocytes. Cell Signal. 2012;24:794–802. doi: 10.1016/j.cellsig.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DQ, Lee CH, Rhee SG, Simon MI. Activation of phospholipase C by the alpha subunits of the Gq and G11 proteins in transfected Cos-7 cells. J Biol Chem. 1992;267:1811–1817. [PubMed] [Google Scholar]
- Wu ML, Tseng YZ. The modulatory effects of endothelin-1, carbachol and isoprenaline upon Na(+)-H+ exchange in dog cardiac Purkinje fibres. J Physiol (Lond) 1993;471:583–597. doi: 10.1113/jphysiol.1993.sp019917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, et al. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116:675–682. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Simpson J, Hong JH, Kim K-H, Thavarajah NK, Backx PH, et al. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1L613V mutation. J Clin Invest. 2011;121:1009–1025. doi: 10.1172/JCI44929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Zhou P, Xu SM, Liu Y, Feng X, Bai SH, et al. Intermolecular failure of L-type Ca2+ channel and ryanodine receptor signaling in hypertrophy. Plos Biol. 2007;5:e21. doi: 10.1371/journal.pbio.0050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Komuro I, Kudoh S, Zou Y, Shiojima I, Hiroi Y, et al. Endothelin-1 is involved in mechanical stress-induced cardiomyocyte hypertrophy. J Biol Chem. 1996;271:3221–3228. doi: 10.1074/jbc.271.6.3221. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- Yang HT, Sakurai K, Sugawara H, Watanabe T, Norota I, Endoh M. Role of Na+/Ca2+ exchange in endothelin-1-induced increases in Ca2+ transient and contractility in rabbit ventricular myocytes: pharmacological analysis with KB-R7943. Br J Pharmacol. 1999;126:1785–1795. doi: 10.1038/sj.bjp.0702454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LL, Gros R, Kabir MG, Sadi A, Gotlieb AI, Husain M, et al. Conditional cardiac overexpression of endothelin-1 induces inflammation and dilated cardiomyopathy in mice. Circulation. 2004;109:255–261. doi: 10.1161/01.CIR.0000105701.98663.D4. [DOI] [PubMed] [Google Scholar]
- Yorikane R, Sakai S, Miyauchi T, Sakurai T, Sugishita Y, Goto K. Increased production of endothelin-1 in the hypertrophied rat heart due to pressure overload. FEBS Lett. 1993;332:31–34. doi: 10.1016/0014-5793(93)80476-b. [DOI] [PubMed] [Google Scholar]
- Yue TL, Gu JL, Wang C, Reith AD, Lee JC, Mirabile RC, et al. Extracellular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275:37895–37901. doi: 10.1074/jbc.M007037200. [DOI] [PubMed] [Google Scholar]
- Zhai P, Gao S, Holle E, Yu X, Yatani A, Wagner T, et al. Glycogen synthase kinase-3alpha reduces cardiac growth and pressure overload-induced cardiac hypertrophy by inhibition of extracellular signal-regulated kinases. J Biol Chem. 2007;282:33181–33191. doi: 10.1074/jbc.M705133200. [DOI] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Weinheimer C, Courtois M, Kovacs A, Zhang CE, Cheng AM, et al. The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J Clin Invest. 2003;111:833–841. doi: 10.1172/JCI16290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Anger T, Su J, Hao J, Xu X, Zhu M, et al. Selective loss of fine tuning of Gq/11 signaling by RGS2 protein exacerbates cardiomyocyte hypertrophy. J Biol Chem. 2006;281:5811–5820. doi: 10.1074/jbc.M507871200. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Hancox JC. Regulation of cardiac Na+-Ca2+ exchanger activity by protein kinase phosphorylation – still a paradox? Cell Calcium. 2009;45:1–10. doi: 10.1016/j.ceca.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Zhao X-S, Pan W, Bekeredjian R, Shohet RV. Endogenous endothelin-1 is required for cardiomyocyte survival in vivo. Circulation. 2006;114:830–837. doi: 10.1161/CIRCULATIONAHA.105.577288. [DOI] [PubMed] [Google Scholar]
- Zhou P, Zhao Y-T, Guo Y-B, Xu S-M, Bai S-H, Lakatta EG, et al. Beta-adrenergic signaling accelerates and synchronizes cardiac ryanodine receptor response to a single L-type Ca2+ channel. Proc Natl Acad Sci USA. 2009;106:18028–18033. doi: 10.1073/pnas.0906560106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Zou Y, Shiojima I, Kudoh S, Aikawa R, Hayashi D, et al. Ca2+/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275:15239–15245. doi: 10.1074/jbc.275.20.15239. [DOI] [PubMed] [Google Scholar]
- Zima AV, Bare DJ, Mignery GA, Blatter LA. IP3-dependent nuclear Ca2+ signalling in the mammalian heart. J Physiol. 2007;584:601–611. doi: 10.1113/jphysiol.2007.140731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolk O, Quattek J, Seeland U, El-Armouche A, Eschenhagen T, Böhm M. Activation of the cardiac endothelin system in left ventricular hypertrophy before onset of heart failure in TG(mREN2)27 rats. Cardiovasc Res. 2002;53:363–371. doi: 10.1016/s0008-6363(01)00468-0. [DOI] [PubMed] [Google Scholar]