

Abstract
Background and Purpose
Statin treatment may ameliorate viral infection-induced exacerbations of chronic obstructive pulmonary disease (COPD), which exhibit Th2-type bronchial inflammation. Thymic stromal lymphopoietin (TSLP), a hub cytokine switching on Th2 inflammation, is overproduced in viral and dsRNA-stimulated bronchial epithelial cells from COPD donors. Hence, TSLP may be causally involved in exacerbations. This study tests the hypothesis that simvastatin inhibits dsRNA-induced TSLP.
Experimental Approach
Epithelial cells, obtained by bronchoscopy from COPD (n = 7) and smoker control (n = 8) donors, were grown and stimulated with a viral infection and danger signal surrogate, dsRNA (10 μg·mL−1). Cells were treated with simvastatin (0.2–5 μg·mL−1), with or without mevalonate (13–26 μg·mL−1), or dexamethasone (1 μg·mL−1) before dsRNA. Cytokine expression and production, and transcription factor (IRF3 and NF-κB) activation were determined.
Key Results
dsRNA induced TSLP, TNF-α, CXCL8 and IFN-β. TSLP was overproduced in dsRNA-exposed COPD cells compared with control. Simvastatin, but not dexamethasone, concentration-dependently inhibited dsRNA-induced TSLP. Unexpectedly, simvastatin acted independently of mevalonate and did not affect dsRNA-induced NF-κB activation nor did it reduce production of TNF-α and CXCL8. Instead, simvastatin inhibited dsRNA-induced IRF3 phosphorylation and generation of IFN-β.
Conclusions and Implications
Independent of mevalonate and NF-κB, previously acknowledged anti-inflammatory mechanisms of pleiotropic statins, simvastatin selectively inhibited dsRNA-induced IRF3 activation and production of TSLP and IFN-β in COPD epithelium. These data provide novel insight into epithelial generation of TSLP and suggest paths to be exploited in drug discovery aimed at inhibiting TSLP-induced pulmonary immunopathology.
Keywords: TSLP, COPD, bronchial brushing epithelium, dsRNA, simvastatin, mevalonate independent, IRF3 phosphorylation, IFN-β
Introduction
Exacerbations of chronic obstructive pulmonary disease (COPD) are severe events that may also aggravate the natural development of the disease (Mallia et al., 2007; Hurst et al., 2010). These conditions lack efficient treatment options. COPD is also one of the world's leading causes of morbidity and mortality (Warren, 2009). Exacerbations of COPD are characterized by bronchial occurrence of Th2-type CD8+ T cells and Th2-type cytokines and chemokines (Papi et al., 2006b; Makris et al., 2008; Singh et al., 2010) in addition to the recognized Th1 features of this disease (Hogg et al., 2004; Hurst et al., 2010; Singh et al., 2010). Although the involved mechanisms have not been fully elucidated, rhinoviral infections, targeting bronchial epithelium, are considered to play a major causative role in exacerbations of COPD (Mallia and Johnston, 2006). Using primary bronchial epithelial cells from asthmatic and COPD donors, we have previously demonstrated that rhinoviral infection and its surrogate, dsRNA, induce overproduction of thymic stromal lymphopoietin (TSLP) (Uller et al., 2010; Calven et al., 2012). TSLP is an epithelial-derived, upstream hub cytokine linking innate and adaptive immunology (Soumelis et al., 2002; Takai, 2012) and is considered to be a compelling target for therapeutic intervention (Kopf et al., 2010). The current view is that NF-κB signalling is essential for epithelial production of TSLP (Lee and Ziegler, 2007; Brandelius et al., 2010; Takai, 2012). Based in part on mouse studies in which TSLP has been overproduced or its gene knocked out, respectively, TSLP has emerged as a ‘master switch’ of Th2-type inflammation (Liu, 2006; Takai, 2012). Studies involving human cells have generated additional features compatible with involvement of TSLP in further aspects of COPD immunopathogenesis. Thus, given an appropriate micro milieu, TSLP itself, or TSLP through priming of dendritic cells, may expand and proliferate cytotoxic T cells (Gilliet et al., 2003), which may be important in causing tissue destruction in COPD (Hogg et al., 2004; Wedzicha and Seemungal, 2007). TSLP may also induce generation of cytokines and cells potentially involved in development of bronchi-associated lymphatic tissue and autoimmunity (Hogg et al., 2004; Allman and Northrup, 2007; Tanaka et al., 2009), which are topical facets of severe COPD (Hogg et al., 2004; Duncan, 2010). These intriguing actions of TSLP, together with its increased occurrence in COPD lungs (Ying et al., 2008) and its overproduction by virally stimulated bronchial epithelial cells from asthmatic and COPD donors, underpin our hypothesis that TSLP may contribute to exacerbations and development of severe asthma and COPD (Uller et al., 2010; Calven et al., 2012). For that reason, we are also searching for small molecular inhibitors of viral and dsRNA-induced epithelial generation of TSLP (Mahmutovic-Persson et al., 2012).
During many years, novel statins have been developed based on their capacity of inhibiting the HMG-CoA (3-hydroxy-3-methylglutaryl-coenzyme A) reductase that produces mevalonic acid. Mevalonic acid is involved both in cholesterol synthesis and in the synthesis of isoprenoid intermediates, notably farnesyl and geranyl pyrophosphates, which can be linked to inflammation through Ras and Rho small G-protein activation (Jain and Ridker, 2005; Abeles and Pillinger, 2006). Rho-GTPases are known to activate NF-κB and thus be involved in generation of inflammatory cytokines. Indeed, most previously demonstrated pleiotropic anti-inflammatory effects of statins have been mevalonate-dependent and have been reported to reflect inhibition of NF-κB (Jain and Ridker, 2005; Abeles and Pillinger, 2006). Statins are also of interest in the context of COPD treatment. Retrospective studies have thus suggested that the use of these drugs, primarily indicated as cholesterol-lowering agents, is associated with reduced mortality and hospitalizations from COPD (Blamoun et al., 2008). One prospective study has recently been carried out supporting the possibility that statins have beneficial effects on COPD exacerbations (Bartziokas et al., 2011).
Cell death and epithelial injury repair are pronounced in severe COPD (Hogg et al., 2004). dsRNA, formed at rhinoviral replication, not only mimics some biological effects of rhinoviral infection (Vercammen et al., 2008; Calven et al., 2012) but dsRNA-evoked actions may additionally represent major pathogenic effects of necrotic cell-derived, danger-associated molecular patterns (DAMP), independent of infections (Cavassani et al., 2008; Vercammen et al., 2008; Lim and Wang, 2011). Hence, in this study, we have used dsRNA as a challenge to reflect effects of both viral infection and DAMP. We demonstrate here that simvastatin inhibits dsRNA-induced expression and production of TSLP in bronchial epithelial cells obtained from COPD donors. Unexpectedly, the simvastatin-induced anti-TSLP effect was independent of the mevalonate pathways and with a significant degree of selectivity both at transcriptional and cytokine levels. These novel data indicate modes of TSLP production in human diseased airways and suggest pathways for drug-induced inhibition of this hub cytokine.
Methods
Primary bronchial epithelial cells from COPD and healthy controls
Primary bronchial epithelial cells were established from seven COPD patients with smoke-induced COPD classified as GOLD stage II (Global Initiative for Obstructive Lung Disease) and eight healthy control smokers characterised in Table 1. Ethical approval was obtained from regional ethical review board at Lund University, Sweden. Epithelial brushings were obtained by bronchoscopy using a fiberoptic bronchoscope (Olympus, IT160, Tokyo, Japan), and a standard sterile single-sheathed nylon cytology brush was used to sample epithelial cells from the bronchi in accordance with standard published guidelines. On average, four consecutive brushings were sampled from the bronchial mucosa of the second- and third-generation bronchi. Cells were transferred to 5 mL sterile PBS after each brushing; 5 mL RPMI with 20% FBS was added and the sample centrifuged at 150× g for 5 min to harvest the cells. Epithelial cell purity was assessed by differential cell counts of the harvested cell suspension as previously described (Uller et al., 2010). Primary cultures of human bronchial epithelial cells were established by seeding bronchial brushings from COPD and healthy smoker subjects into collagen coated tissue culture flasks containing 3 mL of serum-free, hormonally supplemented bronchial epithelium growth medium (Clonetics, San Diego, CA), as previously described (Uller et al., 2010). Cultures were routinely tested for mycoplasma infection.
Table 1.
Study subjects and characteristics
| Characteristics | Controls | COPD | ||
|---|---|---|---|---|
| Number of subjects | 8 | 7 | ||
| Age (range) | 60 | (43–70) | 62 | (53–66) |
| Pack years (range) | 20 | (12–48) | 41 | (14–47) |
| Gender, M/F in % | 50/50 | 58/42 | ||
| FEV1% predicted | 98 | (89–132) | 61 | (41–70) |
Data in the Table are median values, with range given in parentheses. FEV1; % predicted = forced expiratory volume in 1 s expressed as a percentage of the predicted value. COPD patients were on maintenance treatment with inhaled glucocorticosteroids and long-acting β-agonist treatment, Symbicort®.
Stimulation of primary bronchial epithelial cells with dsRNA and treatment with simvastatin, dexamethasone and BX795
Bronchial epithelial cells were seeded into 12-well plates (Nunc; Life Technologies, Stockholm, Sweden), and when 80–90% confluent, the growth medium was replaced with bronchial epithelial basal medium (Clonetics, San Diego, CA) containing 1% insulin-transferrin-selenium and 0.1% BSA; and the cells were rendered quiescent for 24 h before start of the experiment. For all experiments, cells were used at passages 2–3 and stimulated with a synthetic dsRNA analogue, polyinosine-polycytidylic acid, (Invitrogen Ltd, Paisley, UK) and used at an optimal concentration, 10 μg·mL−1, as previously described (Brandelius et al., 2010). Simvastatin (Sigma-Aldrich, Gillingham, UK), 0.2–5 μg·mL−1, was added to the cells 18 h prior to stimulation with dsRNA. Dexamethasone 1. 0 μg·mL−1 (Sigma-Aldrich) was added to the cells 1 h prior to stimulation with 10 μg·mL−1 dsRNA. In preliminary experiments, BX795 (Sigma-Aldrich), an inhibitor of IFN regulatory factor 3 (IRF3), was similarly tested for effects on dsRNA-induced TSLP mRNA expression. All the compounds were dissolved in DMSO. Vehicle control was prepared as having the highest concentration of DMSO present. After treatment with drugs and control solution, the cells were stimulated for either 3 or 24 h by adding dsRNA to the wells. Cell supernatant was removed for protein analysis, and cells were lysed and harvested for either mRNA analysis or Western blot, as described below.
Effects of mevalonate-pathway intermediates on dsRNA-induced TSLP
Simvastatin was added with or without the mevalonic acid pathway intermediate mevalonate (Sigma-Aldrich); 13 μg·mL−1 has been a standard concentration in previous work demonstrating mevalonate dependence of anti-inflammatory effects of statins (Bu et al., 2010; Zhang et al., 2011). Two further intermediates were also tested: farnesyl pyrophosphate (Sigma-Aldrich) (4.3 μg·mL−1) or geranyl pyrophosphate (Sigma-Aldrich) (2.25 μg·mL−1). These intermediates were added, as was simvastatin, 18 h prior to stimulation with dsRNA.
RNA extraction and quantification of TSLP, TNF-α, CXCL8 and IFN-β gene expression with qRT-PCR
Total RNA was extracted from primary bronchial epithelial cells using a RNA extraction kit, Nucleospin RNA II (Machery-Nagel, Düren, Germany), according to the manufacturer's instructions; 1 μg of mRNA was reverse transcribed to cDNA using an RT-kit (Primerdesign, Southampton, UK), and quantitative PCR was performed as previously described (Uller et al., 2010). Briefly, thermocycling and real-time detection of PCR products were performed on an IcyclerIQ sequence detection system (Stratagene, Mx3000P, La Jolla, CA) with standard cycling parameters. The following primer sequences were used for TSLP, TNF-α, CXCL8 and IFN-β:
TSLP: AATCCAGAGCCTAACCTTCAATC (forward) and GTAGCATTTATCTGAGTTTCCGAATA (reverse). TNF-α: AGGTTCTCTTCCTCTCACATAC (forward) and ATCATGCTTTCAGTGCTCATG (reverse). CXCL8: CAGAGACAGCAGAGCACAC (forward) and AGCTTGGAAGTCATGTTTACAC (reverse). IFN-β: TTACTTCATTAACAGACTTACAGGT (forward) and TACATTAGCCATCAGTCACTTAAAC (reverse). Genes of interest were normalized to the geometric means of two housekeeping genes, ubiquitin C (UBC) and GAPDH, using the ΔΔCt method as previously described (Uller et al., 2010).
Cytokine measurement by elisa
Release of TSLP, CXCL8, TNF-α (R&D Systems, Abingdon, UK) and IFN-β (PBL Biosource, CA) into culture supernatants of the bronchial epithelial cells was measured at 24 h after stimulation with dsRNA, using elisa kits according to the manufacturer's instructions.
Western blot analysis for NF-κB and IRF3 activation
Western blots was performed for the quantification of IκBα degradation andt also for NF-κB p65 and NF-κB p105 release, induced by dsRNA in the primary epithelial cells. Experiments were performed as described above, but cells were stimulated with dsRNA for 2 h and then lysed using a sample buffer for Western blot containing 250 mM Tris–HCl, 6% SDS, 100 mM DTT, 10% glycerol and 25 mM NaF. Protein content for each sample was determined by Coomassie blue, and equal amount of each sample was loaded and analysed by electrophoresis on a 10% polyacrylamide gel. The fractionated protein was blotted onto a PVDF membrane. The membrane was blocked in blocking buffer (5% non-fat dry milk, TBST) and then incubated in primary rabbit polyclonal antibodies, anti-IκBα, phospho-NF-κB p105 (Ser933), phospho-NF-κB p65 (Ser536), anti-IRF3 or phospho-IRF3 (Ser396) antibody (Cell Signaling; Danvers, MA) diluted 1:1000, according to manufacturer's instructions. The secondary antibody was a HRP-conjugate, anti-rabbit IgG antibody, dilution 1:3000 (concentration 0.24 μg·mL−1) from (Fisher Scientific AB, Gothenburg, Sweden). As loading control, GAPDH protein was blotted, using mouse polyclonal primary antibody, diluted according to manufacturer's instructions (Cell Signaling). The secondary antibody was a HRP-conjugate, anti-mouse IgG antibody, dilution 1:10 000 (Cell Signaling). The immunoreactive proteins were detected with chemiluminescent system (ECL kit, GE Healthcare; Buckinghamshire, UK). After exposure to an X-ray film, the band density was calculated from the optical density using image analyser Quantity One (Bio-Rad, Hercules, CA).
Cell viability
The morphology of the bronchial epithelial cells was regularly recorded by light microscopy at different time points after dsRNA and simvastatin treatment. We also measured LDH levels in the cell supernatants. Our data, including LDH levels (all of which were below detection limit) and light microscopy, confirmed that the concentrations of simvastatin used here did not induce epithelial cell death.
Statistical analysis
Data are expressed as median ± IQR, and all data were analysed using non-parametric tests. Significant variation in the data within groups (COPD and healthy smokers) was investigated using Friedman's one-way anova using the software GraphPadPrism version 5.0 (GraphPad Software, San Diego, CA, http://www.graphpad.com). The two-tailed Mann–Whitney test was used to compare the difference between the groups healthy control smokers and COPD. The two-tailed Wilcoxon test was used to compare variance between groups. P-values of less than 0.05 were considered statistically significant.
Results
Clinical characterization of study subjects
Fifteen study subjects participated in the study, eight healthy control smoking individuals and seven patients with smoke-induced COPD classified as GOLD stage II. COPD patients were on maintenance treatment with inhaled glucocorticosteroids (Budesonide 160 μg per inhalation) and long-acting β-agonist (Formoterol 4.5 μg per inhalation) treatment, Symbicort® Turbuhaler®. There was no statistically significant difference in pack-years between the smoking control group and the COPD patients. The clinical characteristics of the subjects are shown in Table 1.
dsRNA-induced expression and production of TSLP
Baseline expression of TSLP mRNA as well as protein release were very low in cells from both COPD and control donors (Figure 1A). Based on previous studies (Brandelius et al., 2010; Uller et al., 2010) and our initial experiments, a concentration of 10 μg·mL−1 of dsRNA was chosen for optimal production of TSLP in this study. dsRNA markedly increased the mRNA expression of TSLP at 3 h in cells from healthy control smokers and patients with COPD (Figure 1A). TSLP mRNA was back to baseline values (data not shown) at 24 h when a significant increase in TSLP protein release was observed in both groups in response to dsRNA (Figure 1B). dsRNA induced greater TSLP mRNA expression (P < 0.05) and protein release (P < 0.05) in the COPD group compared with the smoker control group (Figure 1).
Figure 1.
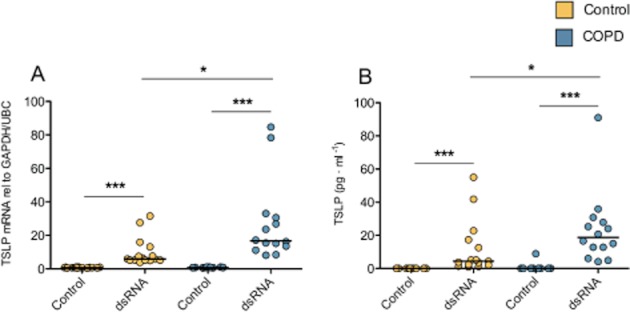
dsRNA induced greater expression and production of TSLP in primary human bronchial epithelial cells from COPD donors compared with smoking control donors. dsRNA 10 μg·mL−1 induced a significant increase in TSLP mRNA and protein in both smoking control and COPD cells compared with un-stimulated control cells. TSLP mRNA and protein release were overproduced in the COPD group. Data are presented as individual values in duplicate, and median values are shown. Healthy smoking controls (n = 8) and COPD (n = 7). *P ≤ 0.05, and ***P ≤ 0.001.
Simvastatin inhibited dsRNA-induced expression and production of TSLP
Simvastatin concentration-dependently inhibited the dsRNA-induced TSLP mRNA expression and protein release in both control and COPD cells (Figure 2). Simvastatin alone was without effect on the low baseline levels of TSLP mRNA or protein (Figure 2). Dexamethasone given at a concentration proven to produce maximum steroid anti-inflammatory effects in primary bronchial epithelial cells in previous studies (Brandelius et al., 2010) reduced dsRNA-induced TSLP mRNA in control cells (Figure 2A), but this effect was not significant in COPD cells nor was it associated with attenuation of dsRNA-induced TSLP protein release (Figure 2B, D).
Figure 2.
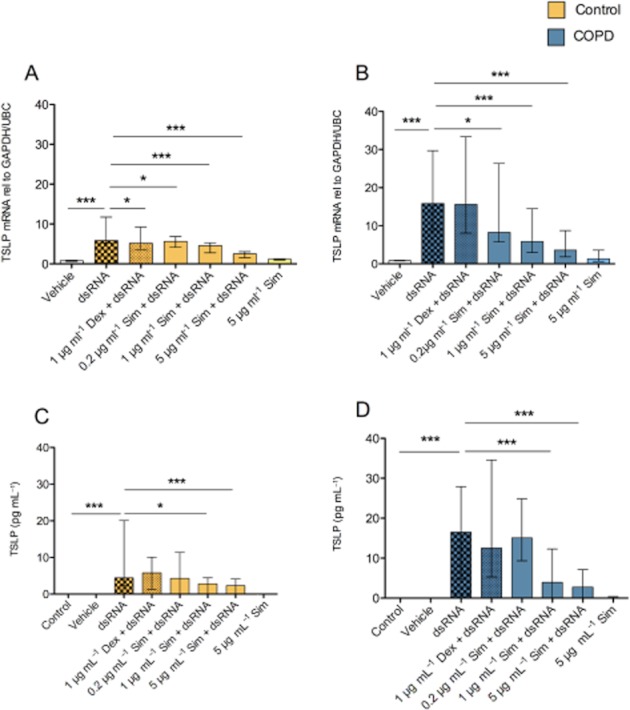
Effects of simvastatin (Sim) and dexamethasone (Dex) on dsRNA-induced TSLP gene expression (A,B) and protein production (C,D). The dsRNA-induced gene expression and protein production were inhibited concentration-dependently by simvastatin, whereas dexamethasone did not produce a significant anti-TSLP effect. Data are presented as median ± IQR. Healthy smoking controls (n = 8) and COPD (n = 7). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
Simvastatin inhibited TSLP gene expression and protein production independent of the mevalonic pathway
Adding mevalonate to the employed test systems is commonly used to determine mevalonate dependency of statin-induced anti-inflammatory effects. To investigate if the mevalonate pathway had an influence on the inhibition of TSLP, we added simvastatin with or without the presence of mevalonate. Mevalonate at a concentration (13 μg·mL−1) that commonly is used to demonstrate mevalonate dependence of anti-inflammatory statin actions in cell cultures (Bu et al., 2010; Zhang et al., 2011) did not reduce the simvastatin-induced inhibitory effect on dsRNA-induced TSLP gene expression and protein production nor did a doubling of the mevalonate concentration show any tendency of reducing the effect of simvastatin. The experiments with both concentrations of mevalonate are, therefore, grouped together (Figure 3A, B). In accord, neither the addition of geranyl pyrophosphate nor farnesyl pyrophosphate reduced the inhibitory effect of simvastatin (data not shown).
Figure 3.
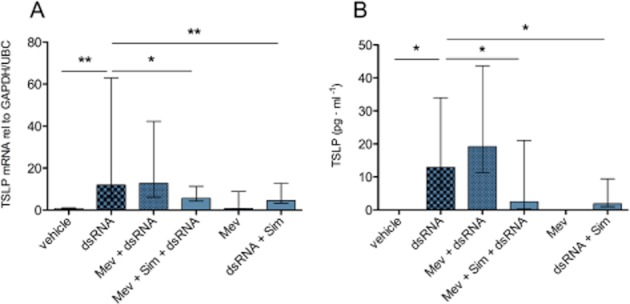
Simvastatin inhibited TSLP gene expression and protein production independent of the mevalonic pathway. Simvastatin (Sim) was added to the COPD bronchial epithelial cells with or without mevalonate (Mev) using two concentrations 13 or 26 μg·mL−1. Mevalonate (13 μg·mL−1) that commonly is used to demonstrate mevalonate dependence of anti-inflammatory statin actions in cell cultures did not reduce the simvastatin-induced inhibitory effect on dsRNA-induced TSLP gene expression and protein production nor did a doubling of the mevalonate concentration show any tendency of reducing the effect of simvastatin. The experiments with both concentrations of mevalonate are, therefore, grouped together. Data are presented as median ± IQR (n = 8). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001. n = 8 includes six COPD donors and two healthy smoking control.
dsRNA-induced epithelial TSLP generation was inhibited by simvastatin without effects on NF-κB
We next analysed the effects of simvastatin on three different regulatory molecules involved in NF-κB signalling. dsRNA reduced protein expression of the NF-κB repressor IκBα and increased the NF-κB subunits p105 and p65 (Figure 4). Simvastatin was without significant effect on any of these three molecules (Figure 4).
Figure 4.
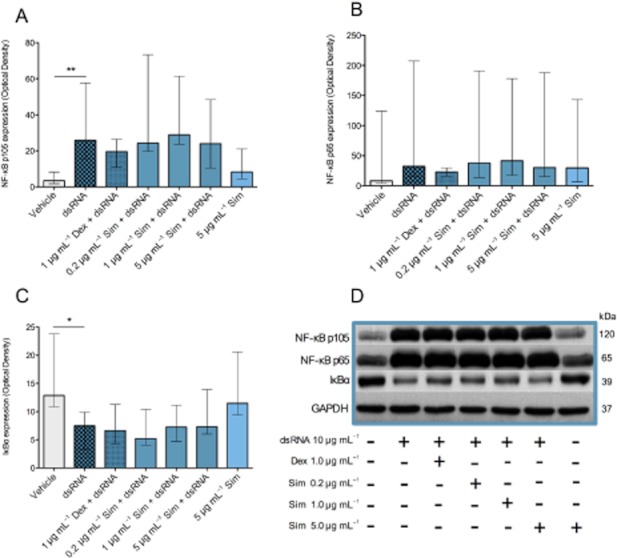
Effects of simvastatin (Sim) on NF-κB signalling molecules IκBα, p65 and P105 in bronchial epithelial cells from COPD donors. dsRNA reduced the NF-κB repressor IκBα (C, D) and increased the NF-κB subunits p65 and p105 (A,B and D). Simvastatin did not attenuate these effects on NF-κB signalling nor did dexamethasone produce any significant effects. Data are presented as median ± IQR (n = 6). *P ≤ 0.05, **P ≤ 0.01 (A, B and C). A representative Western blot micrograph (D) illustrates activation of NF-κB signalling by dsRNA and lack of effect by simvastatin or dexamethasone.
Simvastatin did not inhibit dsRNA-induced generation of TNF-α and CXCL8
dsRNA induced marked TNF-α and CXCL8 gene expression and protein production with no significant difference (P > 0.05) between healthy and COPD epithelial cells (Figures 5 and 6). Simvastatin did not inhibit dsRNA-induced gene expression and protein production of CXCL8 and TNF-α, whereas dexamethasone slightly reduced TNF-α production in healthy cells (Figure 5C).
Figure 5.
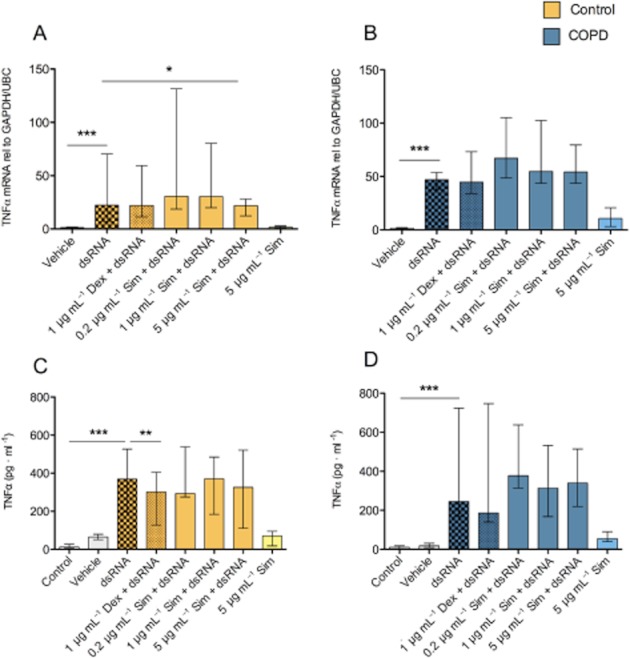
Effects of simvastatin (Sim) and dexamethasone (Dex) on dsRNA-induced TNF-α gene expression (A,B) and protein production (C,D). Simvastatin did not inhibit dsRNA-induced TNF-α protein production in smoking control or COPD epithelial cells (C,D). Dexamethasone slightly attenuated TNF-α production in smoking control epithelial cells (C). Data are presented as median ± IQR, smoking controls (n = 8) and COPD (n = 7). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
Figure 6.
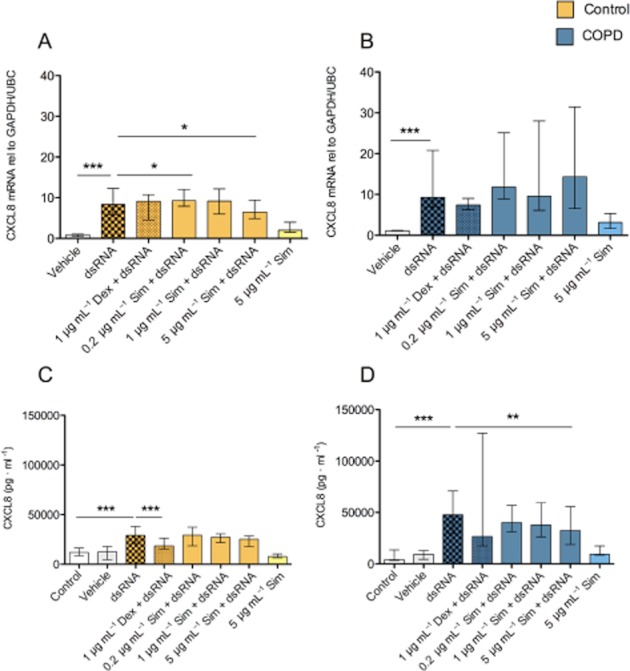
Effects of simvastatin (Sim) and dexamethasone (Dex) on dsRNA-induced CXCL8 gene expression (A,B) and protein production (C,D). Simvastatin only had marginal inhibitory effects on dsRNA-induced CXCL8 gene expression and protein release in epithelial cells from COPD and control donors. Dexamethasone reduced CXCL8 production in control cells. Data are presented as median ± IQR, smoking controls (n = 8) and COPD (n = 7). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
dsRNA-induced IRF3 phosphorylation and inhibition by simvastatin
In preliminary experiments involving different exposure times of the bronchial epithelial cells to dsRNA (0.5–24 h), a 2 h exposure was selected for these experiments. Significant IRF3 phosphorylation was thus induced in epithelial cells exposed to dsRNA for 2 h. This effect was inhibited by simvastatin (Figure 7). The total levels of IRF3 were not affected by either dsRNA alone or dsRNA together with simvastatin.
Figure 7.
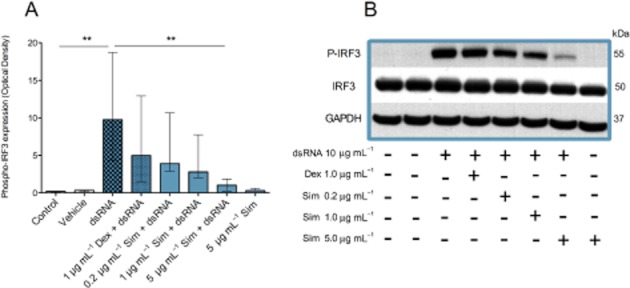
Effects of simvastatin (Sim) and dexamethasone (Dex) on IRF3 phosphorylation in bronchial epithelial cells from COPD donors. dsRNA increased phosphorylation of IRF3 (A,B) without changing IRF3 levels (B). Dexamethasone was without significant effects (A,B) whereas simvastatin inhibited phosphorylation of IRF3 (A,B). Data are presented as median ± IQR (n = 6). **P < 0.01. A representative Western blot is shown (B).
Effects of simvastatin treatment on dsRNA-induced IFN-β
IRF3 phosphorylation is known to involve anti-viral IFN-β gene expression. We therefore analysed effects of dsRNA and simvastatin on IFN-β expression and protein production. IFN-β expression and protein were induced by dsRNA. Although IFN-β protein release in dsRNA-exposed COPD cells was slightly less than in control, this response did not differ significantly (P > 0.05) between control and COPD cells (Figure 8). Similar to its effect on TSLP production, simvastatin inhibited the dsRNA-induced expression and production of IFN-β (Figure 8).
Figure 8.
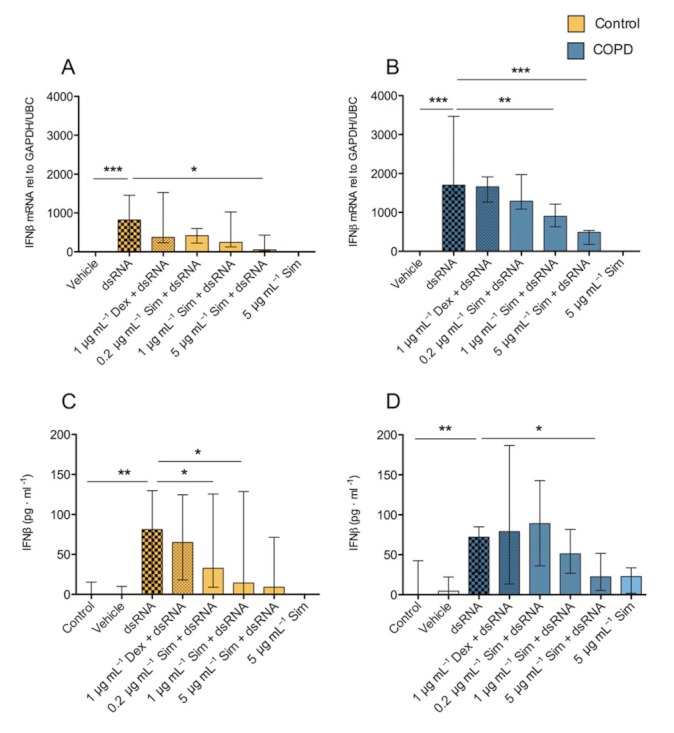
Effects of simvastatin (Sim) and dexamethasone (Dex) on dsRNA-induced IFN-β gene expression and protein production. dsRNA induced IFN-β gene expression (A,B) and protein release (C,D) were inhibited by simvastatin but not by dexamethasone. Data are presented as median ± IQR, smoking controls (n = 8) and COPD (n = 7). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
Effects of a known inhibitor of IRF3 and IFN-β on dsRNA-induced TSLP
In preliminary experiments, we observed that a relatively specific inhibitor of TANK-binding kinase 1 (TBK1) and IκB kinase ε (IKKε), BX795, that is known to inhibit activation of IRF3 and IFN-β production (Clark et al., 2009), inhibited dsRNA-induced TSLP (Supporting Information Figure S1).
Discussion
In this study, we demonstrate that an accepted mimic of effects of viral infection and an emerging mimic of effects of necrotic cells, dsRNA, induced overproduction of TSLP in bronchial epithelial cells from donors with moderate COPD compared with a group of healthy control smokers. Furthermore, simvastatin concentration-dependently inhibited TSLP gene expression and protein production. Unexpectedly, this inhibitory effect was independent of acknowledged anti-inflammatory mechanisms of statins. Instead, simvastatin inhibited dsRNA-induced phosphorylation of IRF3 and production of IFN-β along with its inhibition of TSLP. These data provide novel aspects on generation of TSLP and identify molecular leads for targeting epithelial TSLP production in airway diseases.
Bronchial epithelial cells are central disease-driving cells in COPD and asthma (Holgate, 2007). Rhinoviral infections targeting bronchial epithelium may be responsible for about 50% of exacerbations in COPD and about 90% of exacerbations in asthma (Nicholson et al., 1993; Papi et al., 2006b). In exacerbations and severe disease, COPD and asthma may share several pathogenic mechanisms (Kraft, 2006). A Th2-type inflammation, considered typical of asthma, may also characterize the bronchi of patients with severe or exacerbating COPD (Papi et al., 2006a; Makris et al., 2008; Singh et al., 2010). Agreeing with potential similarities between exacerbation mechanisms in asthma and COPD, we have previously shown that TSLP is overproduced in virally stimulated epithelial cells from donors with asthma (Uller et al., 2010) and from donors with severe COPD (Calven et al., 2012). In this study, dsRNA induced overproduction of TSLP in cells from COPD patients having markedly milder disease than in our previous study (Calven et al., 2012). In COPD as well as in control cells, dexamethasone produced only marginal inhibitory effects on dsRNA-induced cytokines. It also appears that potent glucocorticoids have insufficient effects in COPD patients (Mallia et al., 2007; Hurst et al., 2010). The concentrations of simvastatin required for inhibition of TSLP production in this study was some 20-fold higher than blood levels obtained in patients on the drug (Ucar et al., 2004). Such supra-clinical drug levels have also been employed in previously demonstrated statin-induced anti-inflammatory effects in animal models and cell culture (Ucar et al., 2004; Abeles and Pillinger, 2006; Sakoda et al., 2006; Qi et al., 2009; Bu et al., 2010; Takai, 2012). Although little discussed, this concentration difference is likely to contribute to an apparently poor translation of consistently beneficial effects of statins in respiratory disease models to patients (Abeles and Pillinger, 2006; Rubin, #b1002). Until further proof becomes available, we suggest that the present anti-TSLP effect is of interest more as a molecular lead to development of novel drugs than as an effect that may explain the reported effects of statin treatment on COPD exacerbations (Bartziokas et al., 2011).
Anti-inflammatory effects of statins in different test systems have previously been shown to be mevalonate-dependent and associated with reduced NF-κB activation (Jain and Ridker, 2005; Abeles and Pillinger, 2006; Qi et al., 2009). The present independence of both mevalonate and NF-κB would thus rule out roles of several anti-inflammatory mechanisms currently attributed to statins. Although a role of IRF3 has not been excluded (Kato et al., #b1001), the current view is that NF-κB is essential for epithelial TSLP production (Lee and Ziegler, 2007; Takai, 2012). Our previous data support a significant involvement of the endosomal Toll-like receptors, TLR3, in dsRNA-induced human bronchial epithelial cell-induced TSLP production in health and disease (Calven et al., 2012). We now demonstrate that complete inhibition of dsRNA-induced TSLP gene expression and protein production can be achieved without attenuation of NF-κB (p65 and p105). Lee and Ziegler (2007) reported that inducible human and mouse TSLP mRNA in epithelial cell lines required activation of NF-κB and involved an NF-κB binding site upstream of the transcription initiation site. However, these authors also reported that dsRNA was without effect on TSLP mRNA in their test system of commercial epithelial cells obtained by lung digestion from only one individual. By contrast, in our study involving epithelial cells from bronchial brushings, dsRNA clearly increased TSLP expression and production and activated both NF-κB and IRF3, albeit with variations between the individual donors. Furthermore, we demonstrated practically complete inhibition of TSLP generation without any attenuation of dsRNA-induced NF-κB and without attenuation of typically NF-κB-dependent generation of TNF-α and CXCL8. Future work extending the number of experiments and including analyses of effects on phosphorylation, nuclear translocation and promoter binding is warranted to more clearly demonstrate any involvement of NF-κB in the present generation and inhibition of TSLP production. We also cannot exclude the possibility of cooperation between NF-κB and other transcription factors, such as IRF3 (Ieki et al., 2004), in generation of TSLP. However, our data suggest that effects on NF-κB were not significantly involved in the present, simvastatin-induced, inhibition of TSLP production.
It was lack of effects on NF-κB signalling that prompted the present analysis of IRF3, a key transcription factor for induction of antiviral IFNs (Fitzgerald et al., 2003; Chow et al., 2007). IRF3 is phosphorylated in response to viral infection or dsRNA (Fitzgerald et al., 2003; Chow et al., 2007) (this study). It subsequently dimerizes and translocates to the nucleus where IRF3 aids in gene transactivation (Chow et al., 2007). The best characterized of these genes is that for IFN-β and this was also increased in response to dsRNA in this study. The present study disclosed that inhibition of IRF3 activation was associated with inhibition of TSLP production. Hence, simvastatin was unlikely to have acted on mechanisms involved in the entry of dsRNA into the cells to reach the endosomal TLR3 (DeWitte-Orr et al., 2010). Fitzgerald et al. (2003) reported that IKKε and TBK-1 are essentially involved in dsRNA-induced activation of IRF3. The present observations may agree with a reported statin-induced inhibition of LPS-induced IRF3 phosphorylation in macrophages (RAW264) (Abe et al., 2008). However, there appears to be no known effect of statins on IKKε and TBK-1. It is relevant that a specific antagonist of IKKε and TBK-1, BX795 (Clark et al., 2009), blocked phosphorylation, nuclear translocation, transcriptional activity of IRF3 and production of IFN-β in response to dsRNA (50 μg·mL−1) in HEK293 cells (Clark et al., 2009). Moreover, similar to simvastatin in this study, BX795 selectively inhibited IRF3 without affecting NF-κB activation (Clark et al., 2009). The present preliminary experiments showed that relatively high concentrations of BX795 also inhibited dsRNA-induced TSLP expression in bronchial epithelial cells from COPD donors. Hence, studies of possible IKKε and TBK-1 antagonism by simvastatin seem warranted. It needs emphasizing that IFN-β is a major antiviral factor and that its mobilization in response to viral infection and dsRNA may be already compromised in diseased airways (Wark et al., 2005; Uller et al., 2010; Sykes et al., 2012). The inhibition of production of IFN-β is probably an undesirable component of the pulmonary actions of simvastatin.
In conclusion, simvastatin inhibited dsRNA-induced TSLP in bronchial epithelial cells from COPD donors without reducing dsRNA-induced TNF-α and CXCL8. This inhibitory effect was independent of the mevalonate pathway and of NF-κB signalling. Instead, simvastatin inhibited dsRNA-induced IRF3 activation and IFN-β production. Thus, simvastatin has been an instrumental small molecule in the present unravelling of novel aspects of TSLP generation and inhibition. These data advance our understanding of mechanisms and drug targets involved in epithelial generation of a hub pathogenic cytokine in asthma and COPD.
Acknowledgments
The authors are grateful for the technical assistance by Brita Sunden-Andersson. This work was supported by Vetenskapsrådet, Vinnova, Swedish Heart and Lung foundation, and the Crafoord Foundation, Sweden.
Glossary
- COPD
chronic obstructive pulmonary disease
- DAMP
danger-associated molecular patterns
- dsRNA
double stranded RNA
- GOLD
global initiative for obstructive lung disease
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- IKK
IκB kinase
- IRF3
IFN regulatory factor 3
- RV
rhinoviral
- TANK
TRAF family member-associated NF-κB activator
- TBK-1
TANK-binding kinase-1
- Th
T helper cells
- TLR3
Toll receptor 3
- TRPV1
transient receptor potential cation channel subfamily V member 1
- TSLP
thymic stromal lymphopoietin
- UBC
Ubiquitin C
Conflicts of interest
The authors declare that they have no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effects of BX795 on dsRNA-induced TSLP mRNA. BX795 is known to inhibit activation of IRF3 and IFN production inhibited dsRNA-induced TSLP in COPD bronchial epithelial cells (n = 3). Data are presented as median ± IQR.
References
- Abe M, Matsuda M, Kobayashi H, Miyata Y, Nakayama Y, Komuro R, et al. Effects of statins on adipose tissue inflammation: their inhibitory effect on MyD88-independent IRF3/IFN-beta pathway in macrophages. Arterioscler Thromb Vasc Biol. 2008;28:871–877. doi: 10.1161/ATVBAHA.107.160663. [DOI] [PubMed] [Google Scholar]
- Abeles AM, Pillinger MH. Statins as antiinflammatory and immunomodulatory agents: a future in rheumatologic therapy? Arthritis Rheum. 2006;54:393–407. doi: 10.1002/art.21521. [DOI] [PubMed] [Google Scholar]
- Allman D, Northrup D. Giving B cell tolerance the ‘TSLiP’. Nat Immunol. 2007;8:481–483. doi: 10.1038/ni0507-481. [DOI] [PubMed] [Google Scholar]
- Bartziokas K, Papaioannou AI, Minas M, Kostikas K, Banya W, Daniil ZD, et al. Statins and outcome after hospitalization for COPD exacerbation: a prospective study. Pulm Pharmacol Ther. 2011;24:625–631. doi: 10.1016/j.pupt.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Blamoun AI, Batty GN, Debari VA, Rashid AO, Sheikh M, Khan MA. Statins may reduce episodes of exacerbation and the requirement for intubation in patients with COPD: evidence from a retrospective cohort study. Int J Clin Pract. 2008;62:1373–1378. doi: 10.1111/j.1742-1241.2008.01731.x. [DOI] [PubMed] [Google Scholar]
- Brandelius A, Yudina Y, Calven J, Bjermer L, Andersson M, Persson C, et al. dsRNA-induced expression of thymic stromal lymphopoietin (TSLP) in asthmatic epithelial cells is inhibited by a small airway relaxant. Pulm Pharmacol Ther. 2010;24:59–66. doi: 10.1016/j.pupt.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Bu DX, Tarrio M, Grabie N, Zhang Y, Yamazaki H, Stavrakis G, et al. Statin-induced Kruppel-like factor 2 expression in human and mouse T cells reduces inflammatory and pathogenic responses. J Clin Invest. 2010;120:1961–1970. doi: 10.1172/JCI41384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calven J, Yudina Y, Hallgren O, Westergren-Thorsson G, Davies DE, Brandelius A, et al. Viral stimuli trigger exaggerated thymic stromal lymphopoietin expression by chronic obstructive pulmonary disease epithelium: role of endosomal TLR3 and cytosolic RIG-I-like helicases. J Innate Immun. 2012;4:86–89. doi: 10.1159/000329131. [DOI] [PubMed] [Google Scholar]
- Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow EK, Razani B, Cheng G. Innate immune system regulation of nuclear hormone receptors in metabolic diseases. J Leukoc Biol. 2007;82:187–195. doi: 10.1189/jlb.1206741. [DOI] [PubMed] [Google Scholar]
- Clark K, Plater L, Peggie M, Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem. 2009;284:14136–14146. doi: 10.1074/jbc.M109.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewitte-Orr SJ, Collins SE, Bauer CM, Bowdish DM, Mossman KL. An accessory to the ‘Trinity’: SR-As are essential pathogen sensors of extracellular dsRNA, mediating entry and leading to subsequent type I IFN responses. Plos Pathog. 2010;6:e1000829. doi: 10.1371/journal.ppat.1000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SR. What is autoimmunity and why is it likely to be important in chronic lung disease? Am J Respir Crit Care Med. 2010;181:4–5. doi: 10.1164/rccm.200910-1488ED. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, Mcwhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Gilliet M, Soumelis V, Watanabe N, Hanabuchi S, Antonenko S, De Waal-Malefyt R, et al. Human dendritic cells activated by TSLP and CD40L induce proallergic cytotoxic T cells. J Exp Med. 2003;197:1059–1063. doi: 10.1084/jem.20030240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- Holgate ST. The epithelium takes centre stage in asthma and atopic dermatitis. Trends Immunol. 2007;28:248–251. doi: 10.1016/j.it.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Hurst JR, Vestbo J, Anzueto A, Locantore N, Mullerova H, Tal-Singer R, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363:1128–1138. doi: 10.1056/NEJMoa0909883. [DOI] [PubMed] [Google Scholar]
- Ieki K, Matsukura S, Kokubu F, Kimura T, Kuga H, Kawaguchi M, et al. Double-stranded RNA activates RANTES gene transcription through co-operation of nuclear factor-kappaB and interferon regulatory factors in human airway epithelial cells. Clin Exp Allergy. 2004;34:745–752. doi: 10.1111/j.1365-2222.2004.1941.x. [DOI] [PubMed] [Google Scholar]
- Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. 2005;4:977–987. doi: 10.1038/nrd1901. [DOI] [PubMed] [Google Scholar]
- Kato A, Favoreto S, Avila PC, Schleimer RP. TLR3- and Th2 Cytokine-Dependent Production of Thymic Stromal Lymphopoietin in Human Airway Epithelial Cells. J Immunol. 2007;179:1080–1087. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf M, Bachmann MF, Marsland BJ. Averting inflammation by targeting the cytokine environment. Nat Rev Drug Discov. 2010;9:703–718. doi: 10.1038/nrd2805. [DOI] [PubMed] [Google Scholar]
- Kraft M. Asthma and chronic obstructive pulmonary disease exhibit common origins in any country! Am J Respir Crit Care Med. 2006;174:238–240. doi: 10.1164/rccm.2604007. [DOI] [PubMed] [Google Scholar]
- Lee HC, Ziegler SF. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proc Natl Acad Sci U S A. 2007;104:914–919. doi: 10.1073/pnas.0607305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DM, Wang ML. Toll-like receptor 3 signaling enables human esophageal epithelial cells to sense endogenous danger signals released by necrotic cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:91–99. doi: 10.1152/ajpgi.00471.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YJ. Thymic stromal lymphopoietin: master switch for allergic inflammation. J Exp Med. 2006;203:269–273. doi: 10.1084/jem.20051745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmutovic-Persson I, Johansson M, Brandelius A, Calven J, Bjermer L, Yudina Y, et al. Capacity of capsazepinoids to relax human small airways and inhibit TLR3-induced TSLP and IFNbeta production in diseased bronchial epithelial cells. Int Immunopharmacol. 2012;13:292–300. doi: 10.1016/j.intimp.2012.04.007. [DOI] [PubMed] [Google Scholar]
- Makris D, Lazarou S, Alexandrakis M, Kourelis TV, Tzanakis N, Kyriakou D, et al. Tc2 response at the onset of COPD exacerbations. Chest. 2008;134:483–488. doi: 10.1378/chest.07-2626. [DOI] [PubMed] [Google Scholar]
- Mallia P, Johnston SL. How viral infections cause exacerbation of airway diseases. Chest. 2006;130:1203–1210. doi: 10.1378/chest.130.4.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallia P, Contoli M, Caramori G, Pandit A, Johnston SL, Papi A. Exacerbations of asthma and chronic obstructive pulmonary disease (COPD): focus on virus induced exacerbations. Curr Pharm Des. 2007;13:73–97. doi: 10.2174/138161207779313777. [DOI] [PubMed] [Google Scholar]
- Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ. 1993;307:982–986. doi: 10.1136/bmj.307.6910.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papi A, Bellettato CM, Braccioni F, Romagnoli M, Casolari P, Caramori G, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006a;173:1114–1121. doi: 10.1164/rccm.200506-859OC. [DOI] [PubMed] [Google Scholar]
- Papi A, Luppi F, Franco F, Fabbri LM. Pathophysiology of exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006b;3:245–251. doi: 10.1513/pats.200512-125SF. [DOI] [PubMed] [Google Scholar]
- Qi XF, Kim DH, Yoon YS, Li JH, Jin D, Teng YC, et al. Fluvastatin inhibits expression of the chemokine MDC/CCL22 induced by interferon-gamma in HaCaT cells, a human keratinocyte cell line. Br J Pharmacol. 2009;157:1441–1450. doi: 10.1111/j.1476-5381.2009.00311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin BK. Statins for the treatment of asthma: a discovery well, dry hole or just snake oil. Thorax. 2009;64:4–5. doi: 10.1136/thx.2008.106757. [DOI] [PubMed] [Google Scholar]
- Sakoda K, Yamamoto M, Negishi Y, Liao JK, Node K, Izumi Y. Simvastatin decreases IL-6 and IL-8 production in epithelial cells. J Dent Res. 2006;85:520–523. doi: 10.1177/154405910608500608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Lee SH, Porter P, Xu C, Ohno A, Atmar RL, et al. Human rhinovirus proteinase 2A induces TH1 and TH2 immunity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2010;125:1369–1378. doi: 10.1016/j.jaci.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- Sykes A, Edwards MR, Macintyre J, Del Rosario A, Bakhsoliani E, Trujillo-Torralbo MB, et al. Rhinovirus 16-induced IFN-alpha and IFN-beta are deficient in bronchoalveolar lavage cells in asthmatic patients. J Allergy Clin Immunol. 2012;129:1506–1514. doi: 10.1016/j.jaci.2012.03.044. [DOI] [PubMed] [Google Scholar]
- Takai T. TSLP expression: cellular sources, triggers, and regulatory mechanisms. Allergol Int. 2012;1:3–17. doi: 10.2332/allergolint.11-RAI-0395. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Watanabe N, Kido M, Saga K, Akamatsu T, Nishio A, et al. Human TSLP and TLR3 ligands promote differentiation of Th17 cells with a central memory phenotype under Th2-polarizing conditions. Clin Exp Allergy. 2009;39:89–100. doi: 10.1111/j.1365-2222.2008.03151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ucar M, Neuvonen M, Luurila H, Dahlqvist R, Neuvonen PJ, Mjorndal T. Carbamazepine markedly reduces serum concentrations of simvastatin and simvastatin acid. Eur J Clin Pharmacol. 2004;59:879–882. doi: 10.1007/s00228-003-0700-5. [DOI] [PubMed] [Google Scholar]
- Uller L, Leino M, Bedke N, Sammut D, Green B, Lau L, et al. Double-stranded RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon-beta in bronchial epithelial cells from donors with asthma. Thorax. 2010;65:626–632. doi: 10.1136/thx.2009.125930. [DOI] [PubMed] [Google Scholar]
- Vercammen E, Staal J, Beyaert R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin Microbiol Rev. 2008;21:13–25. doi: 10.1128/CMR.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CP. The nature and causes of chronic obstructive pulmonary disease: a historical perspective. The Christie Lecture 2007, Chicago, USA. Can Respir J. 2009;16:13–20. doi: 10.1155/2009/540527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedzicha JA, Seemungal TA. COPD exacerbations: defining their cause and prevention. Lancet. 2007;370:786–796. doi: 10.1016/S0140-6736(07)61382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, O'connor B, Ratoff J, Meng Q, Fang C, Cousins D, et al. Expression and cellular provenance of thymic stromal lymphopoietin and chemokines in patients with severe asthma and chronic obstructive pulmonary disease. J Immunol. 2008;181:2790–2798. doi: 10.4049/jimmunol.181.4.2790. [DOI] [PubMed] [Google Scholar]
- Zhang X, Tao Y, Troiani L, Markovic-Plese S. Simvastatin inhibits IFN regulatory factor 4 expression and Th17 cell differentiation in CD4+ T cells derived from patients with multiple sclerosis. J Immunol. 2011;187:3431–3437. doi: 10.4049/jimmunol.1100580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.