

Abstract
Background and Purpose
Bone cancer pain is chronic and often difficult to control with opioids. However, recent studies have shown that several opioids have distinct analgesic profiles in chronic pain.
Experimental Approach
To clarify the mechanisms underlying these distinct analgesic profiles, functional changes in the μ-opioid receptor were examined using a mouse femur bone cancer (FBC) model.
Key Results
In the FBC model, the Bmax of [3H]-DAMGO binding was reduced by 15–45% in the periaqueductal grey matter (PAG), region ventral to the PAG (vPAG), mediodorsal thalamus (mTH), ventral thalamus and spinal cord. Oxycodone (10−8–10−5 M) and morphine (10−8–10−5 M) activated [35S]-GTPγS binding, but the activation was significantly attenuated in the PAG, vPAG, mTH and spinal cord in the FBC model. Interestingly, the attenuation of oxycodone-induced [35S]-GTPγS binding was quite limited (9–26%) in comparison with that of morphine (46–65%) in the PAG, vPAG and mTH, but not in the spinal cord. Furthermore, i.c.v. oxycodone at doses of 0.02–1.0 μg per mouse clearly inhibited pain-related behaviours, such as guarding, limb-use abnormalities and allodynia-like behaviour in the FBC model mice, while i.c.v. morphine (0.05–2.0 μg per mouse) had only partial or little analgesic effect on limb-use abnormalities and allodynia-like behaviour.
Conclusion and Implications
These results show that μ-opioid receptor functions are attenuated in several pain-related regions in bone cancer in an agonist-dependent manner, and suggest that modification of the μ-opioid receptor is responsible for the distinct analgesic effect of oxycodone and morphine.
Keywords: analgesic effect, bone cancer pain, oxycodone, morphine, G protein, μ-opioid receptor
Introduction
Cancer pain is treated using various therapeutic modalities, including non-steroidal anti-inflammatory drugs, opioids, radiation and surgical intervention (Levy, 1996; Cherny, 2000). For advanced cancer pain, opioids are often the mainstay of analgesic therapy (Koshy et al., 1998; Portenoy et al., 1999; Cherny, 2000); however, opioids are often insufficient in treating bone cancer pain (Mercadante and Arcuri, 1998; Portenoy et al., 1999). Recent studies have shown that morphine has different effects on bone cancer pain and inflammatory pain in animal models (Luger et al., 2002; Yamamoto et al., 2008), and that higher doses of morphine are required to obtain significant analgesic effects in a bone cancer pain model than in an inflammatory pain model. In the clinical setting, oxycodone effectively relieves bone cancer pain (Heiskanen and Kalso, 1997; Watson and Babul, 1998; Becker et al., 2000; Gimbel et al., 2003; Watson et al., 2003; Kalso, 2005; Bercovitch and Adunsky, 2006; Silvestri et al., 2008). Previously, we reported that oxycodone inhibited different pain-related behaviours in femur bone cancer (FBC) and nociceptive pain models over a similar dose range, whereas morphine was less effective in the FBC models (Minami et al., 2009). These results suggested that oxycodone had a unique, distinct analgesic profile in bone cancer pain.
Most strong opioids, including morphine and oxycodone, are μ-opioid receptor agonists, and several studies have addressed the pharmacological mechanism underlying the actions of opioids. μ-Opioid receptors are expressed in several brain regions that modulate ascending pain signal transmission from peripheral nociceptive sensory neurons to the CNS, including the spinal cord, periaqueductal grey matter (PAG), lateral thalamus and mediodorsal thalamus (mTH) (Cohen and Melzack, 1985; Basbaum and Jessell, 2000; Narita et al., 2008). The μ-opioid receptor belongs to the large GPCR superfamily (Cox and Weinstock, 1964; Veatch et al., 1964; Pert and Snyder, 1973; Martin et al., 1976; Chen et al., 1993; Min et al., 1994; Narita et al., 2008). Binding of a μ-opioid receptor agonist changes the receptor conformation allosterically, leading to the activation of G-proteins. Since a μ-opioid receptor agonist induces the binding of [35S]-GTPγS to G proteins, measuring [35S]-GTPγS binding to the μ-opioid receptor in pain-related brain regions is valuable for assessing functional changes in this receptor induced by opioid agonists (Lazareno, 1997; Narita et al., 1999).
In this study we investigated the mechanism underlying the unique analgesic profile of oxycodone by comparing it with morphine in a bone cancer pain model. One potential mechanism – differential activation of the μ-opioid receptor by oxycodone and morphine under the bone cancer pain – was observed in this model.
Methods
Animals
Two hundred and sixty four C3H/HeN mice (body weight 18–23 g) (CLEA Japan, Tokyo, Japan) were used in this study. The animals were housed in a room maintained at 23 ± 1°C under a 12-h light/12-h dark cycle and allowed access to water and food ad libitum. All procedures for the animal experiments were approved by the Animal Care and Use Committee of Shionogi Research Laboratories, Osaka, Japan in agreement with the internal guidelines for animal experiments and in adherence to the ethics policy of Shionogi & Co. The results of all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Drugs
Oxycodone (oxycodone hydrochloride) and morphine (morphine hydrochloride) were from Shionogi & Co. (Osaka, Japan). β-Funaltrexamine hydrochloride (β-FNA), [d-Ala2, N-Me-Phe4, Gly5-ol] enkephalin (DAMGO) and nor-binaltorphimine dihydrochloride (nor-BNI) were purchased from Tocris Bioscience (Bristol, UK). All drugs were dissolved in 0.9% physiological saline (Otsuka Pharmaceutical, Tokyo, Japan) for in vivo experiments and dissolved in the appropriate assay buffer for in vitro experiments.
The FBC model
The NCTC2472 tumour cells (American Type Culture Collection, Manassas, VA, USA) were maintained in DMEM (Invitrogen, Carlsbad, CA, USA), supplemented with 10% FBS (Invitrogen), 100 units·mL−1 penicillin and 100 μg·mL−1 streptomycin (Invitrogen) and cultured at 37 ± 0.2°C in a humidified atmosphere of 5% CO2. To prepare the FBC model, the NCTC2472 tumour cells were injected following a previously described protocol (Honore et al., 2000; Minami et al., 2009). In brief, C3H/HeN mice were anaesthetized with 3% isoflurane, and a left knee arthrotomy was performed; the level of anaesthesia was monitored by measuring their reaction to tail pinch. Tumour cells [1 × 105 cells in 5 μL of Hank's balanced salt solution (Invitrogen)] were injected directly into the medullary cavity of the distal femur, and the drill hole in the bone was closed with resin cement (ADFA; Shofu, Kyoto, Japan). In the sham group, 5 μL of Hank's balanced salt solution were injected instead of the tumour cells in the same manner. Fourteen days after tumour implantation, pain-related behaviour(s) was evaluated to confirm the phenotypic behaviour of the FBC model using previously described protocols (Minami et al., 2009). Mice showing guarding times that changed from 0–2 s (before tumour implantation) to 8–16 s were used for the in vitro experiments, and mice showing the above change in the guarding times and limb-use abnormality score that produced more than 3 on the ipsilateral side (14 days after tumour implantation) were used for the in vivo experiment.
Measurements of Bmax and Kd for μ-opioid receptors using [3H]-DAMGO
Tissue samples were prepared from sham-operated mice and from FBC model mice showing guarding behaviour for 8–16 s 14 days after the surgery. The PAG, region ventral to the PAG including red nucleus (vPAG; which does not contain the PAG.), mTH, ventral thalamus (vTH) and ipsilateral spinal cord were removed quickly after decapitation. Briefly, referring to the mouse brain atlas (Paxinos and Franklin, 2008), a coronal block of the PAG and vPAG was obtained from 2.7 to 3.7 mm posterior to the bregma, and a coronal block of the mTH and vTH was obtained from 1.0 to 2.0 mm posterior to the bregma. The collected tissues were then transferred to a tube filled with ice-cold sucrose (320 mM). The tissues were homogenized by the high-velocity revolution homogenizer. The homogenate was centrifuged at 4°C for 20 min at 2000× g, and the supernatant was centrifuged at 4°C for 20 min at 48 000× g. The pellet was resuspended in ice-cold assay buffer containing 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2 and 1 mM EDTA as the membrane fraction. The membrane proteins were quantified using a bicinchoninic acid compatible protein assay kit (Pierce, Rockford, IL, USA) with BSA as the standard. The amount of protein used in each assay was adjusted to within the range 100–150 μg depending on the region.
A saturation binding experiment was performed in duplicate with increasing concentrations of [3H]-DAMGO (0.5–16 nM; specific activity, 59.0 Ci·mmol−1; PerkinElmer Life Science, Arlington Heights, IL, USA) in a final volume of 0.5 mL. The binding assay preparation was incubated in the assay buffer (50 mM Tris-HCl (pH 7.4), 5 mM MgCl2 and 1 mM EDTA) for 1 h at 25°C, and non-specific binding was determined in the presence of 10 μM DAMGO. The binding was terminated by rapid filtration through Whatman GF/C glass filters (Brandel, Gaithersburg, MD, USA). The filters were then washed three times with 50 mM Tris-HCl, pH 7.4 and transferred to scintillation vials. Next, 5 mL of Clear-sol 2 (Nacalai Tesque, Kyoto, Japan) were added to the vials. Radioactivity in the samples was determined using a liquid scintillation analyser.
[35S]-GTPγS binding assay
Tissue samples were prepared from sham-operated mice and from FBC model mice showing guarding behaviour of 8–16 s 14 days after the operation. The PAG, vPAG, mTH, vTH and ipsilateral spinal cord were removed quickly after decapitation, as described earlier. The collected tissues were then transferred to tubes filled with ice-cold 50 mM Tris-HCl buffer, pH 7.4, 5 mM MgCl2 and 1 mM EDTA. The membrane homogenate (5–10 μg per assay) was prepared as described previously (Narita et al., 2001) and incubated at 25°C for 2 h in 0.5 mL of assay buffer (50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 1 mM EDTA and 100 mM NaCl) with various concentrations of the opioid agonist, 30 μM GDP and 50 pM [35S]-GTPγS (specific activity, 1000 Ci mmol−1; PerkinElmer Life Science). Agonist (oxycodone or morphine) and antagonist (β-FNA or nor-BNI) were applied simultaneously and co-incubated (Figures 2E, F, 3E, F, and 4C). It has been reported that the GTP activity of morphine is not inhibited by a δ-opioid receptor antagonist or a κ-opioid receptor antagonist but is completely antagonized by β-FNA, a μ-opioid receptor antagonist under the similar co-incubation method (Narita et al., 2008), suggesting that the β-FNA treatment in the present study selectively antagonized the μ-opioid receptor activity. The reaction was terminated by filtration using Whatman GF/C glass filters (Brandel) that had been pre-soaked in 50 mM Tris-HCl, pH 7.4 and 12.5 mM MgCl2 at 4°C for 2 h. The filters were washed three times with 5 mL of ice-cold 50 mM Tris-HCl buffer, pH 7.4, and then transferred to scintillation-counting vials. Then, 5 mL of Clear-sol 2 (Nacalai Tesque) were added to the vials. Radioactivity in the samples was determined using a liquid scintillation analyser. Non-specific binding was measured in the presence of 10 μM unlabelled GTPγS. G-protein activation by each agonist was expressed as % stimulation, which was calculated as (T1 − T0)/(T2 − T0) × 100, where T0 is the non-specific activity, T1 is the [35S]-GTPγS activity in the presence of various concentrations of oxycodone or morphine and T2 is the [35S]-GTPγS activity in the absence of the agonist. The inhibition ratio of G-protein activation between samples from the tumour-implanted and sham-operated mice was reported as % inhibition for each opioid, which was calculated as 100 − (T3/T4) × 100, where T3 is % stimulation of the tumour-implanted mice samples treated with either 10−5 M oxycodone or morphine and T4 is % stimulation of the sham-operated mice samples treated with 10−5 M of the respective opioid.
Figure 2.
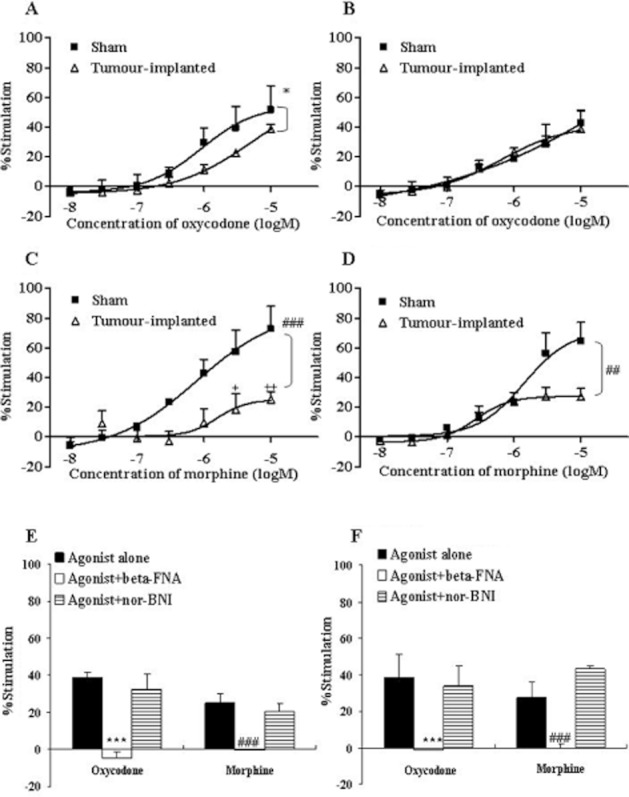
Concentration–response curves of oxycodone and morphine for [35S]-GTPγS binding to cell membranes of PAG and vPAG. The cell membranes of PAG (A, C) and vPAG (B, D) were prepared 14 days after the sham operation (Sham) or tumour implantation (tumour-implanted) and incubated in the presence of different concentrations of oxycodone (10−8–10−5 M) (A, B) or morphine (10−8–10−5 M) (C, D). The membrane-bound [35S]-GTPγS was measured and expressed as % stimulation relative to the basal level. Each symbol represents the mean ± SEM of three independent samples (four mice per sample). The effects of the μ-opioid receptor antagonist β-FNA (10−6 M) and κ-opioid receptor antagonist nor-BNI (10−6 M) on oxycodone- (10−5 M) or morphine- (10−5 M) induced [35S]-GTPγS binding in the PAG (E) and vPAG (F) of tumour-implanted mice are also shown. Each column represents the mean ± SEM of three independent samples (four mice per sample). In each graph, the y-axis indicates % of G-protein activation by each agonist. F(1,28) = 4.822; *P < 0.05 versus sham oxycodone group, F(1,28) = 9.203; ##P < 0.01 or F(1,28) = 18.94; ###P < 0.001 versus sham morphine group (two-way ANOVA; A, C or D). +P < 0.05 or ++P < 0.01 versus sham morphine group (Bonferroni multiple comparison post test; C). ***P < 0.001 versus oxycodone alone group, ###P < 0.001 versus morphine alone group (two-way ANOVA, Dunnett's multiple comparison test; E or F).
Figure 3.
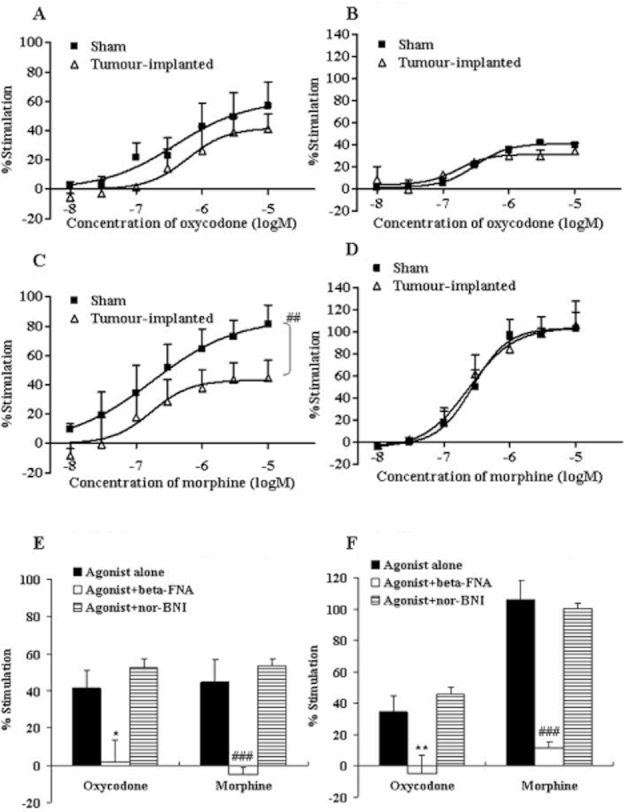
Concentration–response curves of oxycodone and morphine for [35S]-GTPγS binding to cell membranes from the mTH (A, C) and vTH (B, D). The membranes were prepared 14 days after the sham operation (Sham) or tumour implantation (tumour-implanted) and incubated in the presence of different concentrations of oxycodone (10−8–10−5 M) (A, B) or morphine (10−8–10−5 M) (C, D). The membrane-bound [35S]-GTPγS was measured and expressed as % stimulation relative to the basal level. Each symbol represents the mean ± SEM of three independent samples (four mice per sample). The effects of β-FNA (10−6 M) and nor-BNI (10−6 M) on oxycodone- (10−5 M) or morphine- (10−5 M) induced [35S]-GTPγS binding in the mTH (E) and vTH (F) of tumour-implanted mice are also shown. Each column represents the mean ± SEM of three independent samples (four mice per sample). In each graph, the y-axis indicates % of G-protein activation by each agonist. F(1,28) = 10.71; ##P < 0.01 versus sham morphine group (two-way ANOVA; C). *P < 0.05 or **P < 0.01 versus oxycodone alone group, ###P < 0.001 versus morphine alone group (two-way ANOVA, Dunnett's multiple comparison test; E or F).
Figure 4.
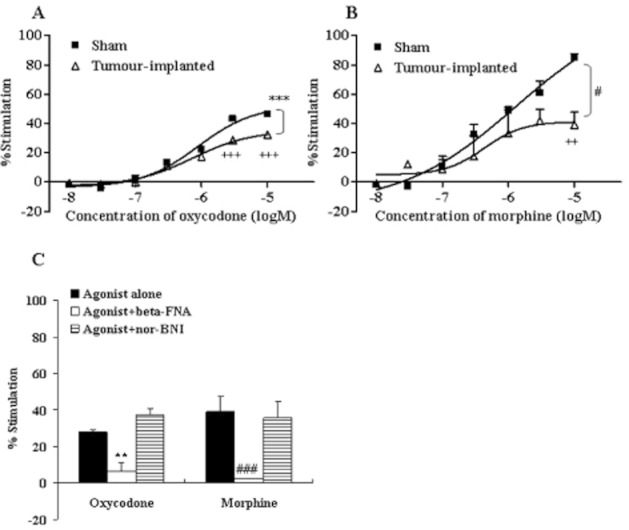
Concentration–response curves of oxycodone and morphine for [35S]-GTPγS binding to cell membranes of the ipsilateral spinal cord. Spinal cord cell membranes (A, B) were prepared 14 days after the sham operation (Sham) or tumour implantation (tumour-implanted) and incubated in the presence of different concentrations of oxycodone (10−8–10−5 M) (A) or morphine (10−8–10−5 M) (B). The membrane-bound [35S]-GTPγS was measured and expressed as % stimulation from the basal level. Each symbol represents the mean ± SEM of three independent samples (four mice per sample). The effects of β-FNA (10−6 M) and nor-BNI (10−6 M) on oxycodone (10−5 M)- or morphine (10−5 M)-induced [35S]-GTPγS binding in the spinal cord (C) of tumour-implanted mice are also shown. Each column represents the mean ± SEM of three independent samples (four mice per sample). In each graph, the y-axis indicates % G-protein activation by each agonist. F(1,28) = 34.41; ***P < 0.001 versus sham oxycodone group, F(1,28) = 8.252; #P < 0.05 versus sham morphine group (two-way ANOVA; A or B). +++P < 0.001 versus sham oxycodone group or ++P < 0.01 versus sham morphine group (Bonferroni multiple comparison post test; A or B). **P < 0.01 versus oxycodone alone group, ###P < 0.001 versus morphine alone group (two-way ANOVA, Dunnett's multiple comparison test; C).
Measurement of μ-opioid receptor binding affinities of oxycodone and morphine using [3H]-DAMGO
The μ-opioid receptor binding affinities were measured by displacement of [3H]-DAMGO. Sample preparation, the amount of membrane proteins, composition of assay buffer, incubation time and filtration method were as described earlier (Measurements of Bmax and Kd for μ-opioid receptor using the [3H]-DAMGO). The μ-opioid receptor binding of each agonist was measured by displacement of [3H]-DAMGO (2 nM) binding by different concentrations of oxycodone (10−10–10−6 M) or morphine (10−10–10−6 M), and was expressed as % of [3H]-DAMGO binding, in which the [3H]-DAMGO binding in the absence of oxycodone or morphine was defined as 100%. The value of each sample was calculated as: (T1 − T0)/(T2 − T0) × 100, where T0 is the non-specific binding, T1 is the [3H]-DAMGO binding in the presence of various concentrations of oxycodone or morphine and T2 is the [3H]-DAMGO binding in the absence of respective agonist.
Measuring pain-related behaviour in the FBC model
Before evaluation, the mouse was placed in a clear plastic observation box and allowed to habituate for 15 min. Spontaneous guarding behaviour, that is the amount of time the hind paw on the ipsilateral side was lifted during ambulation, was assessed for a 2-min observation period. Limb-use abnormality on the ipsilateral side during spontaneous ambulation was scored on a scale of 0 to 4: 0, normal limb use; 1, slight limp; 2, obvious limp; 3, partial non-use of the limb; and 4, complete non-use of the limb.
Allodynia-like behaviour was recognized as paw withdrawal in response to tactile stimuli using a series of von Frey monofilaments (pressure: 0.008, 0.02, 0.04, 0.07, 0.16, 0.4, 0.6 and 1 g). The up–down method of the von Frey monofilament test (Zhao et al., 2007) was used, as described previously (Minami et al., 2009).
The effects on guarding time and limb-use abnormality are expressed as % maximum possible effect (MPE), which was calculated as ([T0 − T1] × 100/T0), where T0 and T1 are the values (time or score) before and after administering the agonist in each animal, respectively. The inhibition of allodynia-like behaviour is expressed as % MPE, which was calculated as ([T2 − T1] × 100/[T0 − T1]), where T0 is the threshold stimulation (g) before tumour implantation, and T1 and T2 are the respective threshold stimulations (g) before and after administering the agonist in each animal.
Tail-flick test
The anti-nociceptive effects of oxycodone and morphine were determined by the tail-flick test (UGO-BASILE, Comerio, VA, Italy). The intensity of the heat stimulus was adjusted so that the animal flicked its tail within 2–4 s after the application of the stimulus. Anti-nociceptive effect is expressed as % MPE and was calculated by (T1 − T0) × 100/(T2 − T0), where T0 and T1 are the tail-flick latencies before and after the administration of opioid agonist, respectively, and T2 is the cut-off time (set at 10 s) in the tests to avoid damage to the tail.
I.c.v. administration
I.c.v. drug administration was performed as described previously, with some modifications (Narita et al., 2008). The day before i.c.v. administration, the mice were anaesthetized briefly with 3% isoflurane, and a 2 mm double needle (tip: 27G, 2 mm; base: 22G, 10 mm, Natsume Seisakusyo, Tokyo, Japan) attached to a 25 μL Hamilton microsyringe was inserted into a unilateral injection site in order to make a hole in the skull for injection. The unilateral injection site was approximately 2 mm caudal and 2 mm lateral from the bregma, and the needle was inserted perpendicular to the skull. When drugs were administered, the injection volume was 2 μL for each mouse. Each solution was injected without injection cannulae.
Statistical analysis
The data are expressed as mean ± SEM. SAS (ver. 8; SAS Institute, Tokyo, Japan) and GraphPad Prism 4.0 (Graphad Software Inc., La Jolla, CA, USA) were used for the statistical analysis. The Kd and Bmax of saturation binding and IC50 of displacement binding were calculated using GraphPad Prism 4.0. The ED50 and IC50 values were determined from the regression analysis. The dose-dependent analgesic response, G-protein activation and receptor binding curves were fitted using GraphPad Prism 4.0. The statistical significance of differences among groups was assessed using two-way ANOVA followed by Dunnett's or Bonferroni multiple comparison tests. A probability value (P) of <0.05 was considered to be statistically significant.
Our drug/molecular target nomenclature conforms to British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2011).
Results
Reduction in μ-opioid receptors on cell membranes in supraspinal sites and the spinal cord in the FBC model
One of the possible mechanisms by which opioid potency in bone cancer pain is reduced is via a reduction in μ-opioid receptors. Therefore, we examined whether the total number of μ-opioid receptors was affected in chronic bone cancer pain in the FBC model by measuring the Bmax and Kd of the μ-opioid receptor (Figure 1).
Figure 1.

Saturation curves for the specific binding of [3H]-DAMGO on cell membranes of PAG, vPAG, mTH, vTH and spinal cord prepared from sham-operated and FBC model mice. Tissue samples were collected 14 days after the sham operation (Sham) or tumour implantation (tumour-implanted), and membranes prepared from the PAG (A), vPAG (B), mTH (C), vTH (D) and spinal cord (E) were used for the binding assay. [3H]-DAMGO binding was examined using concentrations from 0.5 to 16 nM. Specific binding was defined as the difference in binding observed in the absence and presence of 10 μM unlabelled DAMGO. Each value represents the mean ± SD of two independent experiments (eight mice per sample in each experiment). The Kd and Bmax of [3H]-DAMGO in those regions are shown in (F). The values were determined from the saturation curves and Scatchard plots analysis, and at least six concentrations were used for each analysis. ***P < 0.001 versus sham group (two-way ANOVA, Dunnett's multiple comparison test).
We evaluated [3H]-DAMGO saturation binding to cell membranes prepared from pain-related regions, including the PAG (Figure 1A), vPAG (Figure 1B), thalamus (mTH or vTH; Figure 1C, D) and spinal cord (Figure 1E) in sham-operated and tumour-implanted mice. High specific binding of [3H]-DAMGO was observed in membrane samples prepared from the mouse brain regions and spinal cord, and specific binding increased linearly with protein concentration up to 400 μg (data not shown). Figure 1 shows the saturation curves of [3H]-DAMGO (0.5–16 nM) binding to membrane samples from the PAG, vPAG, mTH, vTH and spinal cord in sham-operated and tumour-implanted mice. Scatchard plot analysis showed that the Bmax of [3H]-DAMGO decreased significantly by 15–45% in all regions in tumour-implanted mice as compared with sham-operated mice, while the Kd of [3H]-DAMGO binding did not differ significantly between sham-operated and tumour-implanted mice (Figure 1F). This showed that the total amount of μ-opioid receptors on tissue membranes was reduced in the brain regions and spinal cord in the FBC model mice.
Different activation of GTPγS binding by oxycodone and morphine in the supraspinal sites and spinal cord in the FBC model
In order to examine the mechanism underlying the distinct analgesic profiles of the opioids, we investigated the μ-opioid receptor agonist activities of oxycodone and morphine in several pain-related regions in the FBC model (Figures 5).
Figure 5.
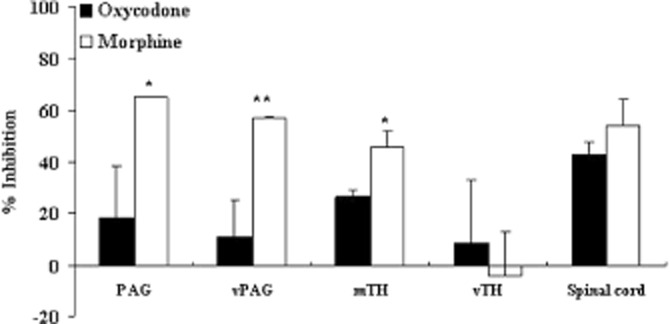
Change in the maximum activation of μ-opioid receptors by oxycodone and morphine in the FBC model. Activation of the μ-opioid receptor was determined by the [35S]-GTPγS assay, and the levels stimulated by oxycodone (10−5 M) or morphine (10−5 M) were compared between the sham and tumour-implanted groups. The y-axis represents the inhibition ratio of G-protein activation in the samples from the tumour-implanted mice as compared with those from the sham-operated mice. Each column represents the mean ± SEM of three independent experiments. The inhibition ratio of G-protein activation between samples from the tumour-implanted and sham-operated mice was calculated as % inhibition for each opioid as described in the text. The variability of the inhibition ratio was determined by the variability of the ratio of the GTP activation between the tumour-implanted and sham-operated group among the three experiments, and therefore, the SEM values in this figure are different from those in Figures 4. *P < 0.05, **P < 0.01 versus oxycodone group (two-way ANOVA, Dunnett's multiple comparison test).
In the PAG, oxycodone (10−8–10−5 M) increased [35S]-GTPγS binding in a concentration-dependent manner, but the maximum activation (Emax) of [35S]-GTPγS binding was reduced in tumour-implanted mice (Emax = 38.6%) as compared with control mice (Emax = 51.6%) (Figure 2A). Morphine (10−8–10−5 M)-induced G-protein activation was also attenuated in tumour-implanted mice (Emax: tumour-implanted mice = 25.2%, sham control mice = 72.6%) in the PAG (Figure 2C). In the vPAG, the difference between the two opioids was more apparent. Similarly, oxycodone (10−8–10−5 M) induced G-protein activation in both tumour-implanted (Emax = 38.8%) and sham-operated (Emax = 42.9%) mice (Figure 2B), while μ-opioid receptor activation by morphine (10−8–10−5 M) was significantly attenuated in the tumour-implanted mice (Emax = 27.3%) as compared with sham control mice (Emax = 64.5%) (Figure 2D).
A similar result was observed for the mTH; oxycodone (10−8–10−5 M)-induced G-protein activation was reduced in tumour-implanted mice (Emax: sham-operated mice = 57.0%, tumour-implanted mice = 41.3%), and the morphine effect was significantly and more severely attenuated in tumour-implanted mice (Emax: sham-operated mice = 81.5%, tumour-implanted mice = 44.6%) (Figure 3A, C). By contrast, the effects of oxycodone and morphine on G-protein activation in the vTH (Emax in sham-operated mice: oxycodone = 39.9%, morphine = 102.7%) were not altered in tumour-implanted mice (Emax in tumour-implanted mice: oxycodone = 34.1%, morphine = 106.2%) (Figure 3B, D). In the ipsilateral spinal cord of sham control mice, both oxycodone and morphine produced concentration-dependent G-protein activation (Emax: oxycodone = 46.6%, morphine = 85.1%) and Emax was attenuated in the tumour-implanted mice (Emax: oxycodone = 32.2%, morphine = 38.9%) (Figure 4).
GTP binding induced by oxycodone and morphine in the brain regions and spinal cord was inhibited completely by β-FNA (10−6 M), a μ-opioid receptor-selective antagonist, but not affected by nor-BNI (10−6 M), a κ-opioid receptor-selective antagonist. These results confirm that the observed GTP binding was predominantly due to μ-opioid receptor activation (Figures 2E, F, 3E, F and 4C).
The ratio of the inhibition of μ-opioid receptor activation in the tumour-implanted group to that in the sham operated group was compared between the two opioids at the maximum concentration (10−5 M) (Figure 5). In the PAG, vPAG and mTH, the activation of μ-opioid receptors was reduced more severely (i.e. % inhibition was increased significantly) in the morphine-treated group (PAG = 65.3%, vPAG = 57.5% and mTH = 46.0%) as compared with the oxycodone-treated group (PAG = 18.9%, vPAG = 11.5% and mTH = 26.8%). By contrast, the effects of the two opioids were reduced similarly in the spinal cord (oxycodone 43.2%, morphine 54.5%). Thus, the functional activation by oxycodone and morphine was attenuated in several regions related to pain signalling with tumour implantation; interestingly, the activation by morphine was attenuated more severely than that by oxycodone in the mTH, PAG and vPAG.
No change in binding affinity of oxycodone or morphine to μ-opioid receptors in the supraspinal sites and spinal cord in the FBC model
We next investigated whether the μ-opioid receptor binding affinity was reduced depending on the agonists under the bone cancer pain condition. We performed competitive displacement-binding assay of [3H]-DAMGO with different concentrations (10−10–10−6 M) of unlabelled opioid agonist using membrane samples from sham-operated and tumour-implanted mice (Figure 6). The [3H]-DAMGO binding was displaced by oxycodone or morphine in a concentration-dependent manner in the membrane samples from all regions (Figure 6A–E). When the levels of [3H]-DAMGO displacement were compared between the sham-treated group and the tumour-implanted group in the respective opioids, displacement curves were similar between two groups, suggesting that affinities of these two opioids to μ-opioid receptor were not altered in the FBC model (Figure 6A–E). The IC50 value of either oxycodone or morphine did not differ significantly between sham-operated and tumour-implanted mice (Figure 6F). These results suggest that the change in the μ-opioid receptor binding affinity is not likely to be the mechanism for the agonist-dependent attenuation of μ-opioid receptor activation under the bone cancer pain condition.
Figure 6.
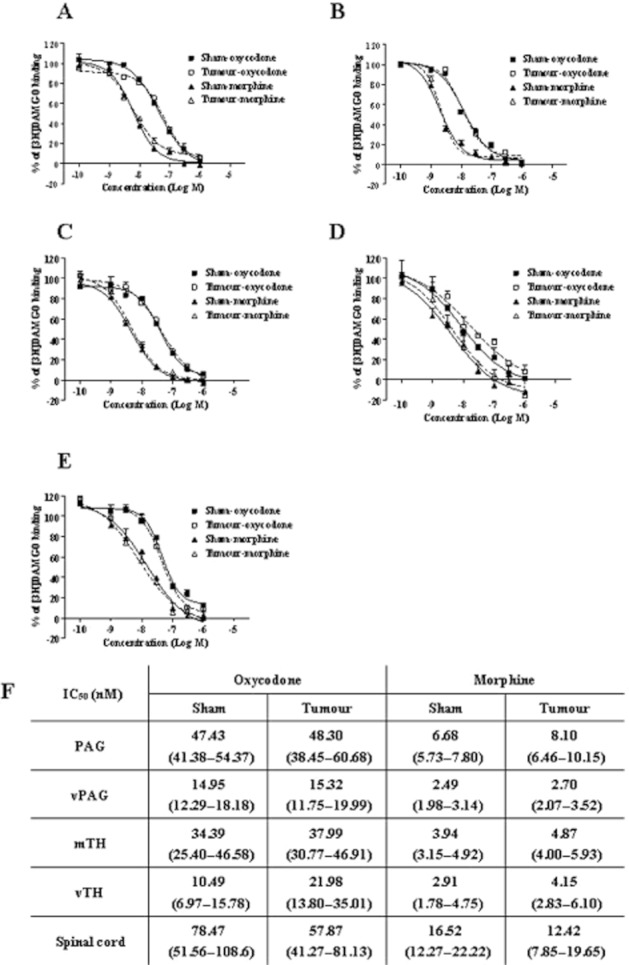
Displacement of [3H]-DAMGO binding on membranes of PAG, vPAG, mTH, vTH and spinal cord by oxycodone or morphine. Tissue samples were collected 14 days after sham operation (sham) or tumour implantation (tumour-implanted), and membranes prepared from PAG (A), vPAG (B), mTH (C), vTH (D) and spinal cord (E) were used for the displacement assay. Experiments were performed in the presence of [3H]-DAMGO (2 nM) and increasing concentrations of oxycodone (10−10–10−6 M) or morphine (10−10–10−6 M). The specific binding was defined as the difference in bindings observed in the absence and presence of 10 μM unlabelled DAMGO. Each column represents the mean ± SEM of three independent samples (eight mice per sample). The IC50 values for oxycodone and morphine are shown in (F). The IC50 values were determined by ANOVA and linear regression techniques, and at least nine concentrations were used for each analysis. Values in parentheses indicate the 95% confidence range.
Analgesic effects induced by the i.c.v. administration of oxycodone and morphine in the FBC model
Since the proportionate reduction of GTPγS stimulation was distinct for morphine compared to oxycodone in the pain-related supraspinal sites, we next examined whether the analgesic effects of supraspinal oxycodone and morphine were affected in the FBC model (Figure 7).
Figure 7.
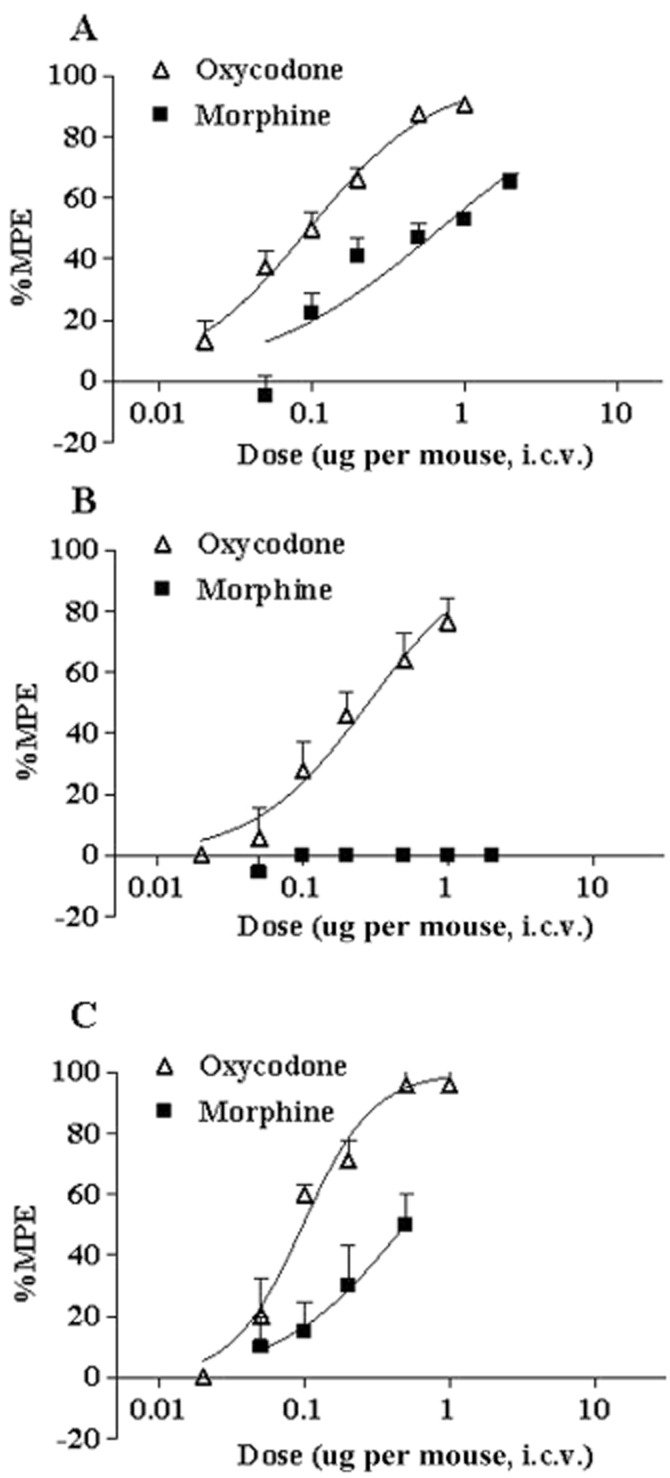
Dose–response curves for the analgesic effects induced by i.c.v. oxycodone and morphine in the FBC model. The analgesic effects of oxycodone (0.02–1 μg per mouse, i.c.v.) and morphine (0.05–2 μg per mouse, i.c.v.) on ongoing pain, ambulatory pain and the allodynia-like response were evaluated based on guarding behaviour (A), limb-use abnormalities (B) and the von Frey test (C), respectively. FBC model mice were used 14 days after tumour implantation, and the analgesic effects were determined 10 min after opioid administration. The y-axes represent % MPE of the analgesic effect. Data plotted are the mean ± SEM of 6–8 mice.
The i.c.v. administration of oxycodone (0.02–1.0 μg per mouse) reduced the guarding time and limb-use abnormality score and reversed the lowered paw withdrawal threshold in the FBC model (Figure 7), indicating that oxycodone dose-dependently ameliorated all pain-related responses. The highest dose (1.0 μg per mouse) had a 91% MPE for guarding behaviour, 76% MPE for limb-use abnormality and 96% MPE for allodynia-like behaviour. The i.c.v. morphine produced dose-dependent analgesic effects on guarding (0.05–2.0 μg per mouse) (Figure 7A) and allodynia-like (0.05–0.5 μg per mouse) behaviours (Figure 7C), but did not improve limb-use abnormalities in the dose range used (0.05–2.0 μg per mouse) (Figure 7B). The highest dose of morphine exhibited 65% MPE for guarding behaviour and 50% MPE for allodynia-like behaviour.
Table 1 shows the ED50 values of the analgesic effects of i.c.v. oxycodone and morphine on the pain-related behaviours calculated using regression equations. The ED50 of i.c.v. oxycodone for guarding, limb-use and allodynia-like behaviours was 0.096, 0.28 and 0.097 μg per mouse, respectively. The ED50 of morphine for guarding and allodynia-like behaviours was 0.69 and 0.50 μg per mouse, respectively, while the ED50 for the limb-use abnormality could not be calculated because morphine did not improve limb-use abnormalities in the dose range used (0.05–2.0 μg per mouse). This shows that oxycodone had approximately 5–7 times greater potency for the inhibition of guarding and allodynia-like behaviours than morphine, and that overall analgesic potency was greater than morphine after i.c.v. administration in the FBC model.
Table 1.
ED50 values for the analgesic effects of oxycodone and morphine
| ED50 (μg per mouse) | ||
|---|---|---|
| i.c.v. administration | Oxycodone | Morphine |
| Tumour-implanted | ||
| Guarding behaviour | 0.096 (0.080–0.12) | 0.69 (0.48–0.99) |
| Limb-use abnormality | 0.28 (0.20–0.40) | N.D. |
| Allodynia-like behaviour | 0.097 (0.075–0.12) | 0.5 (0.22–1.10) |
| Sham-operated | ||
| Tail-flick test | 0.28 (0.21–0.38) | 0.23 (0.16–0.33) |
The ED50 was determined by ANOVA and linear regression using data from at least four doses for each opioid. Values in parentheses indicate the 95% confidence range.
In order to compare the analgesic potency of oxycodone and morphine under the sham-operated condition, we compared antinociceptive potency of i.c.v. administered oxycodone and morphine in sham-operated mice using the tail-flick test. The i.c.v. administration of both oxycodone (0.3–10 μg per mouse) and morphine (0.3–10 μg per mouse) dose-dependently increased the tail-flick latency in sham-operated mice with 100% MPE at the highest dose (10 μg per mouse) (data not shown). The ED50 values were 0.28 μg per mouse in the oxycodone-treated groups and 0.23 μg per mouse in the morphine-treated groups (Table 1), showing that both oxycodone and morphine possess similar antinociceptive potencies in the sham-operated mice.
Discussion
In this study, we found that μ-opioid receptor activation by oxycodone and morphine was attenuated in the PAG, vPAG, mTH and spinal cord in the FBC model. In addition, the attenuation of μ-opioid receptor activation by oxycodone was less than that of morphine in supraspinal regions, such as the PAG, vPAG and mTH. Therefore, in bone cancer pain, μ-opioid receptor activation in the brain regions related to pain signalling was modified in an agonist-dependent manner. Consistent with this, when the analgesic effects of i.c.v. oxycodone and morphine were examined using three different pain-related behaviours in the FBC model, the overall analgesic potency of oxycodone was greater than that of morphine.
Bone cancer pain is often insufficiently controlled by opioids (Mercadante and Arcuri, 1998; Portenoy et al., 1999). The FBC model is a good animal model for investigating the mechanisms of bone cancer pain (Honore et al., 2000; Minami et al., 2009), because this model mimics some clinical features of human bone cancer pain (Komiya et al., 1999; Pandit-Taskar et al., 2004). For example, pathological changes, such as bone destruction and nerve compression, appear within a few weeks after the implantation of tumour cells (Luger et al., 2002; Minami et al., 2009). Since we have reported that oxycodone has a unique analgesic profile as compared with other opioids in this model (Minami et al., 2009), the mechanism underlying the distinct analgesic effects of oxycodone in bone cancer pain can be investigated.
In the receptor binding assay, FBC mice exhibited a marked decrease in the Bmax of [3H]-DAMGO binding without affecting the Kd in pain-related regions, indicating that the number of μ-opioid receptors on cell membranes was reduced in bone cancer pain. Yamamoto et al. (2008) reported that μ-opioid receptor levels were reduced in the spinal cord and dorsal root ganglion in bone cancer pain model mice. In this study, we also found a reduction in μ-opioid receptors, not only in the spinal site but also in the supraspinal region. The reduction in μ-opioid receptors may result from their phosphorylation and/or internalization with the continuous release of endogenous opioids. It has been reported that pain and electrical stimulation induces the release of endogenous opioids in the brain (Zangen et al., 1998; Zubieta et al., 2001), and the endogenous opioids rectify pain by stimulating the μ-opioid receptor as an internal compensation system. Although we did not measure endogenous opioid release in this model, perhaps such a change in the endogenous opioid level plays a role in the reduction in μ-opioid receptors on cell membranes in pain-related regions.
Our results showed that reduction in the μ-opioid receptor levels depended on the brain regions. When we analysed the relationship between changes in the μ-opioid receptor levels and changes in the GTP activities by oxycodone and morphine, a consistent correlation was not observed. For example, while both the reduction in the number of μ-opioid receptors and that in the opioid-activated GTP level were relatively small at the vTH compared with other regions, the reduction in the μ-opioid receptor level was only 25% in PAG, although the morphine-activated GTP level was more severely decreased (as about 60% reduction) at the PAG. These results showed that the magnitude of the change in the μ-opioid receptor level does not simply reflect the magnitude of the change in the agonist activity level.
One of our main findings was the limited attenuation of μ-opioid receptor activation (measured by GTPγS binding activity) induced by oxycodone as compared with morphine in the pain-related regions in bone cancer pain. There are at least three possibile mechanisms that may explain the agonist-dependent attenuation of μ-opioid receptor activation; (i) a change in the number of μ-opioid receptors; (ii) a change in the μ-opioid receptor binding affinity; and (iii) a change in regulatory mechanism of G-protein activation. It has been reported that agonist activity of a partial agonist tends to be more affected by the change in the number of receptors than that of a full agonist (Cordeaux et al., 2000; McDonald et al., 2003). Since our data and the previous studies by others (Lemberg et al., 2006; Narita et al., 2008) showed that oxycodone is a more nearly partial agonist than morphine, a reduction of μ-opioid receptors in the FBC model mouse would be expected to affect the agonist activity of oxycodone more than that of morphine. However, our results do not corroborate this prediction and, therefore, this possibility is unlikely. The second possibility is that μ-opioid receptor binding affinity was reduced depending on the agonists under the bone cancer pain condition. However, when we examined whether the μ-opioid receptor binding affinities of oxycodone and morphine were affected in the FBC model, no significant change was observed in the displacement levels of [3H]-DAMGO binding by either oxycodone or morphine between the tumour-implanted mice and sham-operated mice. This suggests that changes in μ-opioid receptor binding affinity are an unlikely mechanism for the attenuation of μ-opioid receptor activation. The third possibility is that a regulatory mechanism of GDP-GTP exchange is responsible for the the agonist-dependent modulation in the FBC model. Since we used a non-hydrolysable analogue of GTP ([35S]-GTPγS) in this study, the processes involved in GTP hydrolysation cannot account for the differences in GTP stimulation, and the distinct μ-opioid receptor activation caused by oxycodone and morphine is probably due to the cellular processes following ligand binding to the receptors, before Gα-GDP is exchanged for Gα-GTP. Thus, the differential activation of the μ-opioid receptor by oxycodone and morphine might be due to the mechanism that regulates the binding of GTP to G proteins in the FBC model. Our hypothesis is supported by previous findings showing that the GDP-GTP exchange factor modulates agonist-induced pharmacological effects (Birukova et al., 2006). However, little is known about the importance of the GDP-GTP exchange mechanism in the regulation of GTP activation by opioid agonists; further studies are required to identify the underlying mechanism of the observed agonist-dependent effect.
Since oxycodone and morphine activate μ-opioid receptors differently in pain-related brain regions, the supraspinal administration of oxycodone and morphine leads to distinct analgesic effects in the FBC model. While i.c.v. oxycodone and morphine had equipotent antinociceptive effects in the sham-operated mouse of FBC (C3H/HeN mouse; Table 1) and the naïve mouse (Narita et al., 2008), i.c.v. oxycodone had 5∼7 times greater analgesic potency than morphine in the evaluation of guarding and allodynia-like behaviours in the FBC model. Consequently, the analgesic potencies of oxycodone and morphine are consistent with their profiles of μ-opioid receptor activation in supraspinal sites in the FBC model.
Of the regions examined in this study, the PAG and mTH are thought to play important roles in the analgesic effects of opioids. The PAG is part of the descending analgesic pathway, and morphine suppresses the release of the neurotransmitter GABA from neurons in the PAG (Basbaum and Fields, 1984). In the mTH, the stimulation of μ-opioid receptors by opioids results in an increase of the inwardly rectifying potassium channel current, which hyperpolarizes the cell and changes its firing pattern on the postsynaptic membrane (Brunton and Charpak, 1998). The unique μ-opioid receptor activation profiles in the PAG and mTH with bone cancer pain may result in the distinct in vivo analgesic potency of oxycodone.
A previous study using a neuropathic pain model reported that morphine-induced μ-opioid receptor activation in the PAG, thalamus and spinal cord did not differ significantly between sham-operated and sciatic nerve-ligated mice (Narita et al., 2008). In our study, on the other hand, the Emax of morphine for μ-opioid receptors was attenuated markedly in several regions in the FBC model. These results suggest that the function of μ-opioid receptors is differentially modulated depending on the type of pain.
There are reports that the antinociceptive effect of oxycodone is antagonized by nor-BNI, a κ-opioid receptor antagonist, at a dose that did not affect the antinociceptive effect of morphine in two rat neuropathic pain models (Ross and Smith, 1997; Nielsen et al., 2007). This suggests that oxycodone produces its analgesic effect by acting on the κ-opioid receptor. Conversely, several different groups have reported that oxycodone-induced antinociception is mediated by the μ-opioid receptor, and that oxycodone binds selectively to μ-opioid receptors (Lemberg et al., 2006; Peckham and Traynor, 2006; Narita et al., 2008). There is a possibility that the discrepancies of the receptors responsible for the oxycodone effect might be due to the different routes of administration. Since oxycodone is metabolized to oxymorphone, an active metabolite, by the peripheral administration, the oxycodone-induced pharmacological effect may be the result of both oxycodone and oxymorphone. It has been reported, however, that both oxycodone- and oxymorphone-induced antinociception are mediated by μ-opioid receptors rather than κ-opioid receptors (Lemberg et al., 2006). These studies suggest that the difference in the administration route does not simply account for the different roles of the μ- and κ-opioid receptor in the opioid analgesic effects. In the present study, we showed that increased GTPγS binding by oxycodone and morphine was antagonized by β-FNA, but not by nor-BNI. Our data are consistent with the idea that the pharmacological effects, for example analgesic effects, of both oxycodone and morphine are mediated via the μ-opioid receptor.
Finally, this study showed that the effects of oxycodone and morphine are modulated differently in bone cancer pain, and that μ-opioid receptor activation by oxycodone in brain regions related to pain signalling was attenuated less as compared with the effects of morphine. Consistent with this, the overall analgesic potency of oxycodone was stronger than that of morphine when they were administered i.c.v. in the FBC model. Therefore, the modulation of μ-opioid receptor function under bone cancer pain appears to be one of the mechanisms underlying the unique analgesic profile of oxycodone, and such modulation may determine analgesic efficacy of a particular opioid in chronic pain.
Glossary
- [3H]-DAMGO
[tylosil-3,5-(3)H(N)]-[D-Ala(2),N-Me-Phe(4),Gly-ol(5)]enkephalin
- DAMGO
[d-Ala2, N-Me-Phe4, Gly5-ol] enkephalin
- Emax
maximum activation
- FBC
femur bone cancer
- MPE
maximum possible effect
- mTH
mediodorsal thalamus
- nor-BNI
nor-binaltorphimine dihydrochloride
- PAG
periaqueductal grey matter
- vPAG
region ventral to PAG
- vTH
ventral thalamus
- β-FNA
β-funaltrexamine hydrochloride
Conflict of interest
None of the authors have any conflicts of interest to disclose relating to this submission. AN, MH, KM, TK, TT, AN and AK are employees of Shionogi Co., Ltd, the manufacturer of oxycodone and morphine.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Fields HL. Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu Rev Neurosci. 1984;7:309–338. doi: 10.1146/annurev.ne.07.030184.001521. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Jessell TM. The perception of pain. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principles of Neural Science. New York: McGraw-Hill; 2000. pp. 472–491. [Google Scholar]
- Becker R, Jakob D, Uhle EI, Riegel T, Bertalanffy H. The significance of intrathecal opioid therapy for the treatment of neuropathic cancer pain conditions. Stereotact Funct Neurosug. 2000;75:16–26. doi: 10.1159/000048379. [DOI] [PubMed] [Google Scholar]
- Bercovitch M, Adunsky A. High dose controlled-release oxycodone in hospice care. J Pain Palliat Care Pharmacother. 2006;20:33–39. [PubMed] [Google Scholar]
- Birukova AA, Adyshev D, Gorshkov B, Bokoch GM, Birukov KG, Verin AD. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2006;390:L540–L548. doi: 10.1152/ajplung.00259.2005. [DOI] [PubMed] [Google Scholar]
- Brunton J, Charpak S. mu-Opioid peptides inhibit thalamic neurons. J Neurosci. 1998;18:1671–1678. doi: 10.1523/JNEUROSCI.18-05-01671.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Mestek A, Liu J, Hurley JA, Yu L. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol Pharmacol. 1993;44:8–12. [PubMed] [Google Scholar]
- Cherny N. New strategies in opioid therapy for cancer pain. J Oncol Manage. 2000;9:8–15. [PubMed] [Google Scholar]
- Cohen SR, Melzack R. Morphine injected into the habenula and dorsal posteromedial thalamus produces analgesia in the formalin test. Brain Res. 1985;359:131–139. doi: 10.1016/0006-8993(85)91420-9. [DOI] [PubMed] [Google Scholar]
- Cordeaux Y, Briddon SJ, Megson AE, Mcdonnell J, Dickenson J. Influence of receptor number on functional responses elicited by agonists acting at the human adenosine A1 receptor: evidence for signalling pathway-dependent changes in agonist potency and relative intrinsic activity. Mol Pharmacol. 2000;58:1075–2000. doi: 10.1124/mol.58.5.1075. [DOI] [PubMed] [Google Scholar]
- Cox BM, Weinstock M. Quantitative studies of the antagonism by nalorphine of some of the actions of morphine-like analgesia drugs. Br J Pharmacol. 1964;22:289–300. doi: 10.1111/j.1476-5381.1964.tb02034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbel JS, Richards P, Portenoy RK. Controlled-release oxycodone for pain in diabetic neuropathy: a randomized controlled trial. Neurology. 2003;60:927–934. doi: 10.1212/01.wnl.0000057720.36503.2c. [DOI] [PubMed] [Google Scholar]
- Heiskanen T, Kalso E. Controlled-release oxycodone and morphine in cancer related pain. Pain. 1997;73:37–45. doi: 10.1016/s0304-3959(97)00072-9. [DOI] [PubMed] [Google Scholar]
- Honore P, Lugar NM, Sabino MAC, Schwei MJ, Rogers SD, Mash DB, et al. Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-induced neurochemical reorganization of the spinal cord. Nat Med. 2000;6:521–528. doi: 10.1038/74999. [DOI] [PubMed] [Google Scholar]
- Kalso E. Oxycodone. J Pain Symptom Manage. 2005;29:S47–S56. doi: 10.1016/j.jpainsymman.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya S, Zenmyo M, Inoue A. Bone tumors in the pelvis presenting growth during pregnancy. Arch Orthop Trauma Surg. 1999;119:22–29. doi: 10.1007/s004020050349. [DOI] [PubMed] [Google Scholar]
- Koshy RC, Rhodes D, Devi A, Grossman SA. Cancer pain management in developing countries: a mosaic of complex issues resulting in inadequate analgesia. Support Care Cancer. 1998;6:430–437. doi: 10.1007/s005200050190. [DOI] [PubMed] [Google Scholar]
- Lazareno S. Measurement of agonist-stimulated [35S]GTPγS binding to cell membranes. Methods Mol Biol. 1997;83:107–116. doi: 10.1385/0-89603-495-X:107. [DOI] [PubMed] [Google Scholar]
- Lemberg KK, Kontinen VK, Siiskonen AO, Viljakka KM, Yli-Kauhaluoma JT, Korpi ER, et al. Antinociception by spinal and systemic oxycodone: why does the route make a difference? In vitro and in vivo studies in rats. Anesthesiology. 2006;105:801–812. doi: 10.1097/00000542-200610000-00027. [DOI] [PubMed] [Google Scholar]
- Levy MH. Pharmacologic treatment of cancer pain. N Engl J Med. 1996;10:1124–1132. doi: 10.1056/NEJM199610103351507. [DOI] [PubMed] [Google Scholar]
- Luger NM, Sabino MA, Schwei MJ, Mach DB, Pomonis JD, Keyser CP, et al. Efficacy of systemic morphine suggests a fundamental difference in the mechanisms that generate bone cancer vs inflammatory pain. Pain. 2002;99:397–406. doi: 10.1016/S0304-3959(02)00102-1. [DOI] [PubMed] [Google Scholar]
- McDonald J, Barnes TA, Okawa H, Williams J, Calo G, Rowbotham DJ, et al. Partial agonist behaviour depends upon the level of nociceptin/orphanin FQ receptor expression: studies using the ecdysone-inducuble mammalian expression system. Br J Pharmacol. 2003;140:61–70. doi: 10.1038/sj.bjp.0705401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine and nalorphine-like drugs in the nondependent and morphine dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197:517–532. [PubMed] [Google Scholar]
- Mercadante S, Arcuri E. Breakthrough pain in cancer patients: pathophysiology and treatment. Cancer Treat Rev. 1998;24:425–432. doi: 10.1016/s0305-7372(98)90005-6. [DOI] [PubMed] [Google Scholar]
- Min BH, Augustin LB, Felsheim RF, Fuchs JA, Loh HH. Genomic structure and analysis of promoter sequence of a mouse μ opioid receptor gene. Proc Natl Acad Sci U S A. 1994;91:9081–9085. doi: 10.1073/pnas.91.19.9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami K, Hasegawa M, Ito H, Nakamura A, Tomii T, Matsumoto M, et al. Morphine, oxycodone, and fentanyl exhibit different analgesic profiles in mouse pain models. J Pharmacol Sci. 2009;111:60–72. doi: 10.1254/jphs.09139fp. [DOI] [PubMed] [Google Scholar]
- Narita M, Mizoguchi H, Narita M, Sora I, Uhl GR, Tseng LF. Absence of G-protein activation by μ-opioid receptor knockout mice. Br J Pharmacol. 1999;123:451–456. doi: 10.1038/sj.bjp.0702330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Mizoguchi H, Narita M, Nagase H, Suzuki T, Tseng LF. Involvement of spinal protein kinase Cγ in the attenuation of opioid μ-receptor-mediated G-protein activation after chronic intrathecal administration of [D-Ala2, N-MePhe4, Gly-Ol5] enkephalin. J Neurosci. 2001;21:3715–3720. doi: 10.1523/JNEUROSCI.21-11-03715.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Nakamura A, Ozaki M, Imai S, Miyoshi K, Suzuki M, et al. Comparative pharmacological profiles of morphine and oxycodone under a neuropathic pain-like state in mice: evidence for less sensitivity to morphine. Neuropsychopharmacology. 2008;33:1097–1112. doi: 10.1038/sj.npp.1301471. [DOI] [PubMed] [Google Scholar]
- Nielsen CK, Ross FB, Lotfipour S, Saini KS, Edwards SR, Smith MT. Oxycodone and morphine have distinctly different pharmacological profiles: radioligand binding and behavioural studies in two rat models of neuropathic pain. Pain. 2007;132:289–300. doi: 10.1016/j.pain.2007.03.022. [DOI] [PubMed] [Google Scholar]
- Pandit-Taskar N, Batraki M, Divgi CR. Radiopharmaceutical therapy for palliation of bone pain from osseous metastases. J Nucl Med. 2004;45:1358–1365. [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. New York, NY: Academic Press; 2008. [Google Scholar]
- Peckham EM, Traynor JR. Comparison of the antinociceptive responses to morphine and morphine-like compounds in male and female Sprague-Dawley rats. J Pharmacol Exp Ther. 2006;316:1195–1201. doi: 10.1124/jpet.105.094276. [DOI] [PubMed] [Google Scholar]
- Pert CB, Snyder SH. Opiate receptor: demonstration in nervous tissue. Science. 1973;179:1011–1014. doi: 10.1126/science.179.4077.1011. [DOI] [PubMed] [Google Scholar]
- Portenoy RK, Payne D, Jacobsen P. Breakthrough pain: characteristics and impact in patients with cancer pain. Pain. 1999;81:129–134. doi: 10.1016/s0304-3959(99)00006-8. [DOI] [PubMed] [Google Scholar]
- Ross FB, Smith MT. The intrinsic antinociceptive effects of oxycodone appear to be κ-opioid receptor mediated. Pain. 1997;73:151–157. doi: 10.1016/S0304-3959(97)00093-6. [DOI] [PubMed] [Google Scholar]
- Silvestri B, Bandieri E, Del Prete S, Ianniello GP, Micheletto G, Dambrosio M, et al. Oxycodone controlled-release as first-choice therapy for moderate-to-severe cancer pain in Italian patients: results of an open-label, multicentre, observational study. Clin Drug Investig. 2008;28:399–407. doi: 10.2165/00044011-200828070-00001. [DOI] [PubMed] [Google Scholar]
- Veatch RM, Adler TK, Way EL. The importance of steric configuration in certain morphine-mimetics of synthetic analgetics. J Pharmacol Exp Ther. 1964;145:11–19. [PubMed] [Google Scholar]
- Watson CP, Babul N. Efficacy of oxycodone in neuropathic pain. A randomized trial in post herpetic neuralgia. Neurology. 1998;50:1837–1841. doi: 10.1212/wnl.50.6.1837. [DOI] [PubMed] [Google Scholar]
- Watson CP, Moulin D, Watt-Watson J, Gordon A, Eisenhoffer J. Controlled-release oxycodone relieves neuropathic pain: a randomized controlled trial in painful diabetic neuropathy. Pain. 2003;105:71–78. doi: 10.1016/s0304-3959(03)00160-x. [DOI] [PubMed] [Google Scholar]
- Yamamoto J, Kawamata T, Niiyama Y, Omote K, Namiki A. Down-regulation of mu opioid receptor expression within distinct subpopulations of dorsal root ganglion neurons in a murine model of bone cancer pain. Neuroscience. 2008;6:843–853. doi: 10.1016/j.neuroscience.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Zangen A, Herzberg U, Vogel Z, Yadid G. Nociceptive stimulus induced release of endogenous beta-endorphin in the rat brain. Neuroscience. 1998;85:659–662. doi: 10.1016/s0306-4522(98)00050-5. [DOI] [PubMed] [Google Scholar]
- Zhao C, Chen L, Tao YX, Tall JM, Borzan J, RingKamp M, et al. Lumbar sympathectomy attenuates cold allodynia but not mechanical allodynia and hyperalgesia in rats with spared nerve injury. J Pain. 2007;8:931–937. doi: 10.1016/j.jpain.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, et al. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311–315. doi: 10.1126/science.1060952. [DOI] [PubMed] [Google Scholar]