

Abstract
Background and Purpose
The KCa3.1 channel is a potential target for therapy of immune disease. We identified a compound from a new chemical class of KCa3.1 inhibitors and assessed in vitro and in vivo inhibition of immune responses.
Experimental Approach
We characterized the benzothiazinone NS6180 (4-[[3-(trifluoromethyl)phenyl]methyl]-2H-1,4-benzothiazin-3(4H)-one) with respect to potency and molecular site of action on KCa3.1 channels, selectivity towards other targets, effects on T-cell activation as well as pharmacokinetics and inflammation control in colitis induced by 2,4-dinitrobenzene sulfonic acid, a rat model of inflammatory bowel disease (IBD).
Key Results
NS6180 inhibited cloned human KCa3.1 channels (IC50 = 9 nM) via T250 and V275, the same amino acid residues conferring sensitivity to triarylmethanes such as like TRAM-34. NS6180 inhibited endogenously expressed KCa3.1 channels in human, mouse and rat erythrocytes, with similar potencies (15–20 nM). NS6180 suppressed rat and mouse splenocyte proliferation at submicrolar concentrations and potently inhibited IL-2 and IFN-γ production, while exerting smaller effects on IL-4 and TNF-α and no effect on IL-17 production. Antibody staining showed KCa3.1 channels in healthy colon and strong up-regulation in association with infiltrating immune cells after induction of colitis. Despite poor plasma exposure, NS6180 (3 and 10 mg·kg−1 b.i.d.) dampened colon inflammation and improved body weight gain as effectively as the standard IBD drug sulfasalazine (300 mg·kg−1 q.d.).
Conclusions and Implications
NS6180 represents a novel class of KCa3.1 channel inhibitors which inhibited experimental colitis, suggesting KCa3.1 channels as targets for pharmacological control of intestinal inflammation.
Keywords: IK channel, KCNN4, KCa3.1, Gárdos channel, TRAM-34, IBD, DNBS-induced colitis, autoimmune disease, NS6180, T-cell activation
Introduction
Following the activation of T cells, B cells, mast cells, macrophages and microglia, a highly regulated and sustained increase in cytosolic [Ca2+] is critical for the controlled expression of essential genes orchestrating proliferation, migration and cytokine production. The Ca2+-activated K+ channel, KCa3.1 (encoded by the KCNN4 gene), plays an important role in these processes by participating in the regulation of calcium entry (Ghanshani et al., 2000; Shumilina et al., 2008; Gao et al., 2010: channel nomenclature follows Alexander et al., 2011). A key supplemental route for Ca2+ release from intracellular stores is influx of extracellular Ca2+ primarily via Ca2+-release activated Ca2+ (CRAC) channels, which – due to the strong inward rectification of this channel – is effective only with a maintained hyperpolarizing drive provided by voltage-dependent (primarily Kv1.3) and Ca2+-activated K+-channels (KCa3.1 in normal T lymphocytes). In keeping with the role of KCa3.1 channels in immune cell activation, pharmacological inhibition of the channel has been shown to have beneficial effects in animal models of several autoimmune and inflammatory disorders, such as multiple sclerosis (Reich et al., 2005), traumatic brain injury (Mauler et al., 2004), ischemic stroke (Chen et al., 2011), inflammation related to atherosclerosis (Toyama et al., 2008) as well as inflammatory bowel disease (IBD) (Di et al., 2010). Confirmatory evidence from KCa3.1 knock-out animals has also been obtained in several cases (Toyama et al., 2008; Di et al., 2010).
With the exception of some rarely used dihydropyridine-derived phenylpyranes and cyclohexadienes from Bayer Pharmaceuticals (Urbahns et al., 2003; 2005; Mauler et al., 2004), the inhibitor pharmacology of KCa3.1 channels is dominated by analogues of the triarylmethane clotrimazole (Alvarez et al., 1992). A well-known example is ICA-17043 (Senicapoc), which was developed for sickle cell anaemia but failed in clinical phase 3 (lack of efficacy on ischaemic crises), despite clear improvements of haematology parameters related to in vivo inhibition of the erythrocyte KCa3.1 channel (the Gárdos channel) (Ataga et al., 2011). Clinical reprofiling in two smaller phase 2 studies gave promising results in allergen-induced, but not exercise-induced, asthma; and development was eventually stopped (Wulff et al., 2009). Another important compound in the triarylmethane class is TRAM-34 (Wulff et al., 2000), which is almost the only regularly used KCa3.1 channel inhibitor in scientific publications. TRAM-34 is a high-potency (10–20 nM) inhibitor of KCa3.1 channels with a 100- to 1000-fold selectivity towards most other ion channels, including the closely related small-conductance Ca2+-activated K+ channels KCa2.1–2.3. Furthermore, it is devoid of the potent P450 inhibition, which prohibits longer duration systemic administration of clotrimazole (Brugnara et al., 1996). However, despite TRAM-34's usefulness in many experimental situations, unknown off-target effects are always a concern with any tool compound, and it is therefore highly desirable to identify KCa3.1 blockers from a different chemical class, which are unlikely to have the same off-target effects as the triarylmethanes. This is particularly important for KCa3.1, which unlike many Kv channels or KCa1.1, currently has no specific peptidic blocker because charybdotoxin also affects KCa1.1 and Kv1.3, while maurotoxin potently blocks Kv1.2 (Wulff et al., 2007).
In this study we have characterized the benzothiazinone NS6180 with respect to potency on recombinant and endogenously expressed KCa3.1 channels, selectivity and molecular site of action. We further describe the effect of NS6180 on the activity of T-lymphocytes from wild-type and KCa3.1–/– knock-out mice, characterize its pharmacokinetics and show that it inhibits inflammation in an animal model of IBD.
Methods
Chemistry
All starting materials and reagents were obtained from commercial suppliers and used without further purification. The synthesis of NS6180 (4-[[3-(trifluoromethyl)phenyl]methyl]-2H-1,4-benzothiazin-3(4H)-one; CAS Registry Number 353262-04-1) is summarized in Figure 1 (upper panel, description in Supplementary Methods). TRAM-34 was synthesized in the Wulff laboratory (Wulff et al., 2000). All inorganic chemicals were from commercial suppliers and of the purest grade available. Sulfasalazine, cremophor; PEG-400, CCCP, A23187, ConA, PMA and ionomycin were from Sigma-Aldrich (St. Louis, MO, USA or Copenhagen, Denmark). 2,4-dinitrobenzene sulfonic acid (DNBS) was purchased from TCI (Tokyo, Japan).
Figure 1.
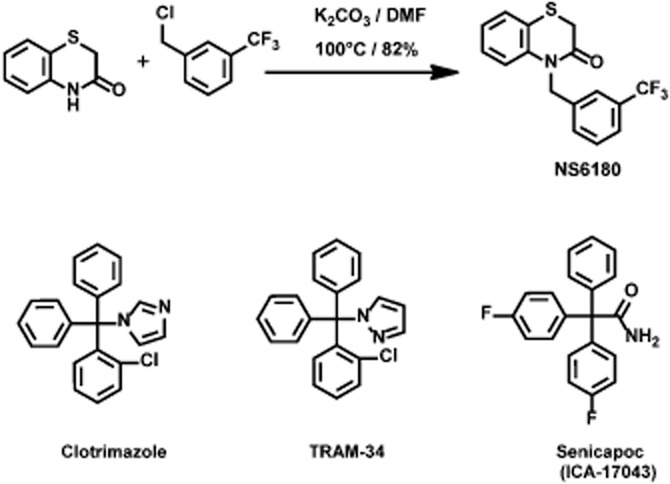
Chemical structure and reaction scheme for the synthesis of NS6180 (4-[[3-(trifluoromethyl)phenyl]methyl]-2H-1,4-benzothiazin-3(4H)-one) (see also Supplementary Methods). The structures of the triarylmethane inhibitors clotrimazole, TRAM-34 and senicapoc (ICA-17043) are shown for comparison.
Electrophysiology
All experiments were performed in the whole-cell version of the patch-clamp technique using either an EPC-9 or EPC-10 amplifier controlled by Pulse software (HEKA Electronics, Lambrecht, Germany). HEK293 cells stably expressing human wild-type KCa3.1 channels or transiently transfected with the wild type or TRAM-34-insensitive double-mutant KCa3.1T250S,V275A (Jenkins et al., 2011) were used. Cells were seeded on 3.5 mm coverslips on the day of the experiments. Coverslips were transferred to a custom-made 15 μL microscope-mounted chamber and superfused with extracellular saline (in mM: 144 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES; pH adjusted to 7.4 with NaOH) at a rate of 1 mL·min−1. Upon establishment of the whole-cell configuration, a voltage ramp protocol (–120 to 30 mV in 150 ms) was applied every 5 s, and the current at −30 mV was followed online versus time. Equilibration of the Ca2+-buffered (400 nM free) pipette solution (in mM: 154 KCl, 8.1 CaCl2, 1.2 MgCl2, 10 EGTA, 10 HEPES; pH 7.2) with the cytoplasm fully activated the KCa3.1 currents typically within 2–5 min. The experiments on transiently transfected cells were performed in a saline with 150 mM KCl with voltage ramps from −80 to +80 mV. NS6180 and TRAM-34 were dissolved in DMSO and diluted at least 1000 times to achieve the final experimental concentration. IC50 values for blocking KCa3.1 channels were estimated either from concentration–response curves by fitting to the Hill equation or from a fit to the inhibition kinetics assuming that IC50 = Kd as detailed previously (Strobaek et al., 2006). Selectivity experiments using other ion channels were performed as described previously (Sankaranarayanan et al., 2009) or as detailed in the legend to Figure 5 or Supplementary Figure S3.
Figure 5.
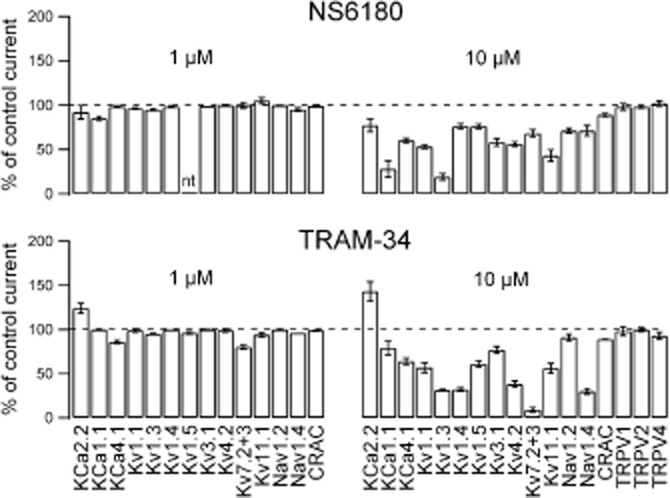
NS6180 and TRAM-34 exhibit different selectivity profiles when tested on other ion channels. NS6180 (upper panels) and TRAM-34 (lower panels) were tested in whole-cell experiments on recombinant channels expressed in mammalian cells using standard protocols and experimental solutions. Results obtained upon application of 1 μM compound are depicted on the left and those obtained at 10 μM depicted on the right. Results are listed as the % of control current. Data are averages of one to six experiments (±SEM for n > 3) (a few 1 μM testings were performed only once since no effects were seen at 10 μM). Slack (KCa4.1) channels were stably expressed in HEK cells and activated by 5 mM Na+ in the pipette solution as described (Yang et al., 2006). KCa1.1, KCa2.2, Kv7.2 + 7.3, Kv11.1 and Nav1.2 channels were expressed in HEK293 cells; and currents were measured from voltage ramps (KCa channels), from a depolarizing voltage step (Nav channels: peak current at 0 mV; Kv7.2 + Kv7.3 steady-state current at −30 mV) or as a tail current (Kv11.1). Kv1.1, Kv1.3, Kv1.4, Kv1.5, Kv3.1, Kv4.2 and Nav1.4 channels were stably expressed in various cell lines and recorded as described (Sankaranarayanan et al., 2009). CRAC and TRPV channel experiments are described in detail in Supplementary Figure S3.
Erythrocyte KCa3.1 (Gárdos channel) assay
Human blood was drawn from healthy human volunteers in a standard heparinized blood sampling vial (Vacutainer, Li/heparin, BD Bioscience, Plymouth, UK). The erythrocytes were packed by centrifugation, and the plasma and buffy coat were removed by aspiration. Erythrocytes were washed three times in the experimental salt solution and then stored at 0°C until use. Blood samples from NMRI mice or from Wistar rats were treated similarly. The methodological principle is outlined in Macey et al. (1978). In essence, activation of the erythrocyte KCa3.1 channels by addition of the Ca2+ ionophore A23187 causes synchronized hyperpolarization of the erythrocytes, which is reported as a CCCP-mediated shift in the unbuffered extracellular pH in a suspension of erythrocytes. Standard procedure: 3 mL unbuffered experimental salt solution (in mM: 2 KCl, 154 NaCl, 0.05 CaCl2) was heated to 37°C with stirring. Packed erythrocytes were added (50 μL, final cytocrit 1.5%), and the extracellular pH (pHo) followed with a glass/calomel (pHG200-8/REF200, Radiometer, Denmark) electrode pair. CCCP (3 μL, final concentration 20 μM) was added followed by varying concentrations of NS6180 or TRAM-34 (DMSO concentration constant). After pH stabilization at ∼7.2, A23187 (3 μL, final concentration 0.33 μM) was added to initiate the experiment. After the peak hyperpolarization was attained, the intracellular pH (pHi constant during the experiment) was found by haemolysing the erythrocytes via addition of 100 μL of Triton-X100.
The erythrocyte membrane potential, Vm, was calculated according to:
and the fractional remaining Ca2+-activated K+-conductance at the concentration C of blocker, fGK(C), was calculated from
where the K+ equilibrium potential EK = −107 mV, the Cl– equilibrium potential ECl = −12 mV and the Vm(0) and Vm(C) are the peak hyperpolarizations in the control and in the presence of a concentration of C of blocker respectively.
IC50 values were calculated from a plot of fGK(C) versus C by a fit to the Hill equation, where n represents the Hill coefficient:
using IGOR-Pro software (WaveMetrics, Lake Oswego, OR, USA).
Proliferation and cytokine assays
Mononuclear cells were isolated from rat spleens, and proliferation assays were performed as previously described (Beeton et al., 2001) (see also Supplementary Methods).
Amounts of secreted cytokines were determined by removing 50 μL per well of supernatant from plates after 40 h of incubation (before the [3H]-thymidine pulse) and frozen at −80°C pending analysis. Cytokines (IL-2, IL-4, IL-6, IL-10, IL-12, IL-17, IFN-γ, TNF-α, MCP-1) were then analysed with a Millipore Milliplex MAP rat cytokine/chemokine kit and Luminex 200™ reader according to the manufacturer's instructions.
Immunohistochemistry
Following macroscopic scoring, pieces of the colon taken at 2.5, 5 and 7 cm from the anus were fixed in 10% buffer formalin, embedded in paraffin and sectioned at 5 μm. Sections were dewaxed with xylene, rehydrated through an alcohol gradient and heated with 10 mM Na+ citrate (pH 6) in a microwave for 15 min to retrieve antigenic determinants. After treatment with 1% H2O2 to inactivate endogenous peroxidase activity and blocking with 5% goat serum in PBS, the sections were incubated overnight at 4°C with the primary antibody in PBS containing 2% goat serum. The following primary antibodies were used: KCa3.1 channel (1:3000; AV35098, Sigma), CD68 (ED1, 1:1000; Serotec, Raleigh, NC, USA) and CD43 (1:2500; Serotec). Bound primary antibodies were detected with a biotinylated donkey anti-mouse IgG secondary antibody for CD68 and CD43 or with a biotinylated goat anti-rabbit IgG secondary antibody (both 1:500, Jackson ImmunoResearch, West Grove, PA, USA) for KCa3.1 channels followed by a HRP-conjugated avidin complex (Vectastain Elite ABC Kit, Vector Laboratories, Burlingame, CA, USA). Peroxidase activity was visualized with 3,3′-diaminobenzidine (DAB Substrate Kit for Peroxidase, Vector Laboratories). Sections were counterstained with haematoxylin.
IBD model
All animal care and experimental procedures were in accordance with the Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, D.C., 1996). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). We used a total of 78 animals in these experiments. Groups of eight Wistar-derived male rats weighing 205 ± 5 g were fasted for 24 h before distal colitis was induced by intra-colonic instillation of DNBS (2,4-dinitrobenzene sulfonic acid, 30 mg in 0.5 mL 30% ethanol/0.9% NaCl) with a 10 cm catheter, followed by gentle injection of air (2 mL) through the catheter to ensure that the solution remained in the colon. Test substances were solubilized in a CremophoreEL/PEG400/water (10%/10%/80%) vehicle (0.3 and 1 mg·mL−1) and administered orally (10 mL·kg−1) twice daily (b.i.d) for seven consecutive days. The first dose was given 1 day before DNBS instillation. The positive control treatment, sulfasalazine at 300 mg·kg−1, was given 24 h and 2 h before DNBS instillation and then once daily (q.d.) for five consecutive days. One normal control group was treated daily with vehicle without DNBS challenge (0.9% NaCl replaced DNBS). During the experiment, fecal occult blood and stool consistency were monitored daily and scored (see Supplementary Figure S4). Animals were killed 12 h (b.i.d. dose) or 24 h (sulfasalazine-treated group) after the final dosing, and the colon was removed and weighed. Adhesions between the colon and other organs or the presence of colonic ulcerations were recorded (see Supplementary Figure S4).
Results
Figure 1 shows the synthesis and chemical structure of NS6180 and, for comparison, the structures of the triarylmethanes TRAM-34, clotrimazole and ICA-17043 (senicapoc). NS6180 is a benzothiazinone and as such constitutes the first member of a new chemical scaffold of KCa3.1 channel blockers. Like TRAM-34, it is a small and compact molecule without significant acid/base properties, but with fewer freely rotatable bonds.
Effect on cloned hKCa3.1 and delineation of molecular site of action
NS6180 was identified as a KCa3.1 channel blocker in a high-throughput screening campaign (Fluorescence Image Plate Reader technology) based on Ca2+-activated Tl+ influx in HEK293 cells stably expressing hKCa3.1 channels (Susanne Jørgensen, pers. comm.). Figure 2 shows inhibition of human KCa3.1 currents by NS6180 and TRAM-34 as evaluated by whole-cell voltage clamp. The IV curves in the upper panel were obtained from HEK293 cells expressing KCa3.1 channels in the absence or presence of 10 nM of NS6180 (A) or TRAM-34 (B). The middle panels show the time courses of inhibition at various concentrations. Equilibrium inhibition is obtained faster at higher concentrations; and it should be noted that at 10 nM, which is close to the half-maximal inhibition concentration, it takes ∼4 min to reach equilibrium. The lower panel summarizes the results for NS6180 and TRAM-34 with the curves representing fits to the Hill equation, revealing IC50 values of 9.4 nM for NS6180 and 8.4 nM for TRAM-34. Exploiting the kinetic information (middle panel) on experiments with compounds applied in concentrations from 3 to 300 nM gave similar potencies [Kd values of 11 ± 1.7 nM (n = 20) for NS6180 and 13 ± 1.4 nM (n = 19) for TRAM-34]. The high-affinity interaction of triarylmethanes like TRAM-34 and clotrimazole with KCa3.1 channels is mediated via the amino acid residues T250 and V275, positioned below the selectivity filter in the inner pore, and substitutions with the corresponding residues from KCa2.3 channels reduced blocker potency by more than 10 000-fold. We tested whether the benzothiazinone NS6180 might share a common binding site with the triarylmethanes by using the double mutant channel, KCa3.1T250S,V275A. Surprisingly, at physiological concentrations of extracellular K+, this construct exhibited irreversible rundown within in a few minutes when expressed in HEK293 cells (results not shown). We therefore performed the following series of experiments with high extracellular K+, since currents from the double mutant remained stable under these conditions. Figure 3A shows ramp currents obtained with the wildtype KCa3.1 channels in the absence and presence of 30 nM NS6180 (Kd = 10 ± 6 nM, n = 4), whereas Figure 3B shows that 10 μM NS6180 (n = 9) fails to inhibit the KCa3.1T250S,V275A channel (>1000 times loss of potency). Charybdotoxin (Figure 3) and nitrendipine (results not shown) were tested in the same experiments. A 20-fold reduction in potency was observed for nitrendipine (WT: Kd = 0.3 ± 0.03 μM, n = 9; Double mutant: Kd = 6 ± 1 μM, n = 8), while charybdotoxin was only slightly less potent (46% inhibition at 100 nM instead of 72%). In conclusion, the inhibitory effect of NS6180 depends on the same amino acid residues as TRAM-34, possibly indicating identical or overlapping interaction sites on KCa3.1 channels for triarylmethanes and benzothiazinones.
Figure 2.
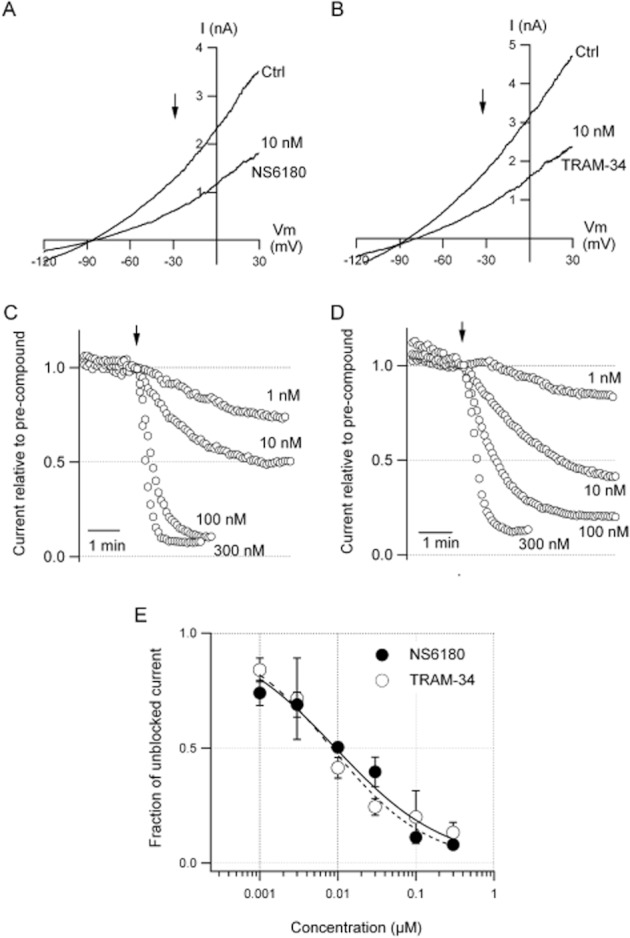
Potent inhibition of KCa3.1 channels by NS6180 and TRAM-34. Whole-cell voltage clamp experiments were performed on HEK293 cells stably expressing human KCa3.1 channels. Currents were activated with a pipette solution containing free Ca2+ buffered at 400 nM. (A, B) Current traces elicited by voltage ramps ranging from −120 to 30 mV. Each panel has a trace obtained before (Ctrl) and after addition of 10 nM NS6180 (A) or TRAM-34 (B) to the bath solution. The arrows at −30 mV indicate where the current versus time analyses depicted in panel (C) for NS6180 and (D) for TRAM-34 were performed. The currents were normalized to the amplitude just before compound application and traces are averages of three to six experiments at the concentrations indicated in the panels. Data points were acquired every 5 s. (E) The fraction of unblocked current measured at the end of compound application as a function of the test concentration. The lines are the curves resulting from fits of the data to the Hill equation described by IC50 values and Hill coefficients of 9.4 nM and 0.62 for NS6180 and 8.4 nM and 0.70 for TRAM-34 respectively.
Figure 3.
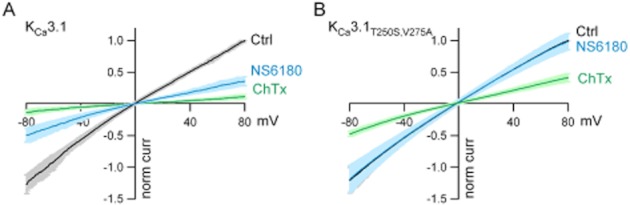
The inhibition by NS6180 is dependent on amino acid residues T250 and V275. Whole-cell voltage clamp experiments were performed using 150 mM K+ in the extracellular solution and voltage ramps from −80 to +80 mV during 200 ms were applied every 5 s. Currents were normalized to the control current measured at 80 mV (norm curr) and average I–V curves ± SD are shown. (A) Currents mediated by HEK293 cells transiently transfected with WT KCa3.1 channels (n = 4) in the absence (Ctrl) or presence of NS6180 (30 nM) and in the presence of charybdotoxin (100 nM, ChTx). (B) Same type of experiment but with cells transfected with the double mutant channel KCa3.1T250SV275A (n = 9) and using 10 μM NS6180.
Effect on erythrocyte KCa3.1 channels
In order to elucidate whether NS6180 exhibits similar potency on endogenous KCa3.1 channels from various species as on the cloned human channel, we performed experiments with human, mice and rat erythrocytes and activated the erythrocyte KCa3.1 channel (the ‘Gárdos channel’) by addition of the Ca2+ ionophore A23187. Figure 4A shows CCCP-reported hyperpolarizations of human erythrocytes in the presence of increasing concentrations of NS6180 (1, 10, 100 and 1000 nM). As seen in Figure 4B NS6180 and TRAM-34 blocked the erythrocyte KCa3.1 channels with nearly the same potency, Hill slopes and efficacy in all three species (Human: NS6180; IC50 = 14 nM, nH = 0.90; TRAM-34; IC50 = 18 nM, nH = 0.87; Mouse: NS6180; IC50 = 15 nM, nH = 1.2. TRAM-34; IC50 = 19 nM, nH = 1.0; Rat: NS6180; IC50 = 9 nM, nH = 0.79, TRAM-34, IC50 = 17 nM, nH = 0.90).
Figure 4.
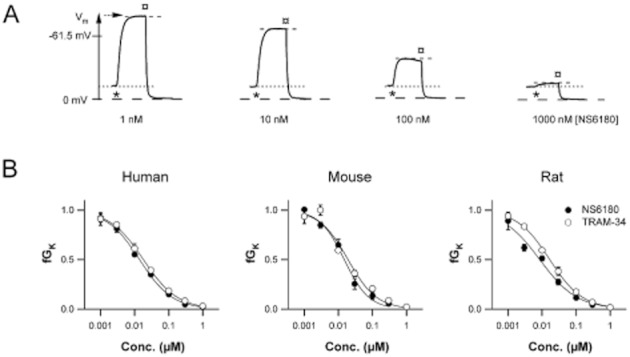
Nearly equipotent block by NS6180 and TRAM-34 of the endogenous Ca2+-activated K+ conductance from human, mouse and rat erythrocytes. (A) CCCP-reported hyperpolarizations from human erythrocytes in the presence of 1, 10, 100 and 1000 nM NS6180 incubated for 1 min before addition of the Ca2+ ionophore A23187 (0.33 μM), which initiates Ca2+ influx, activation of KCa3.1 channels and the ensuing hyperpolarization. Triton-X-100 was added after 1 min causing haemolysis and measurement of the intracellular pH, which defines Vm = 0 mV. The maximal hyperpolarization achieved in the absence of NS6180 is indicated. The erythrocyte resting membrane potential (= ECl = −12 mV) is indicated. (B) The fractional remaining Ca2+-activated K+ conductance, fGK(c), is calculated as detailed in the Methods section and plotted (mean ± SD, N = 3) versus the concentration of the compounds.
Selectivity
In order to determine whether NS6180 is selective for KCa3.1 channels, the compound was tested at 1 and 10 μM (concentrations 100- and 1000-fold above the IC50 for KCa3.1 inhibition) on the T-cell Ca2+ channel ICRAC consisting of Orai-1 and STIM1 and a range of Kv, KCa, Nav and TRP channels ( Figure 5 and Supplementary Figure S3). While both TRAM-34 and NS6180 exerted only minor effects on any of the tested channels at 1 μM, the two compounds clearly differed in their ‘off-target’ channel profiles at higher concentrations. At 10 μM, NS6180 inhibited KCa1.1 (BK), Kv1.3 and Kv11.1 (hERG) channels by more than 50%, while TRAM-34 blocked Kv1.3, Kv1.4, Kv7.2 + Kv7.3 and Nav1.4 channels. A notable difference was also the small but significant stimulation of KCa2 channels by TRAM-34. NS6180 was further tested for selectivity in a lead profiling binding assay screen (LeadProfilerScreen; internal reference #1107076, Ricerca Biosciences LLC, Taiwan) containing 69 receptors (both GPCRs and channels) covering the major transmitter and hormonal systems (adrenergic, GABAergic etc.). Only four targets reached the predefined significance level of 50% inhibition at 10 μM – the noradrenaline transporter (59%), the dopamine transporter (50%), L-type Ca2+ channel, dihydropyridine site (59%) and the melatonin receptor MT1 (70%). In conclusion, NS6180 and TRAM34 should both be cautiously used at concentrations above 1 μM, which is further accentuated by their lack of solubility in aqueous media, above 10 μM.
Inhibition of T-lymphocyte activation and cytokine release
Since NS6180 is as potent and selective as TRAM-34 we next evaluated its immunosuppressive effects in vitro. Similar to TRAM-34, which has previously been used to probe the role of KCa3.1 channels in T- and B-cell activation (Ghanshani et al., 2000; Wulff et al., 2004), NS6180 suppressed both concanavalin (ConA)- and (PMA + ionomycin)-stimulated [3H]-thymidine incorporation of rat splenocytes with IC50s of ∼1 μM and ∼200 nM (Figure 6A). Experiments with WT mouse splenocytes revealed similar inhibitions, whereas splenocytes from KCa3.1–/– mice were insensitive to both compounds (Supplementary Figure S1). We further examined the effects of N6180 and TRAM-34 on the secretion of IL-2, IL-4, IL-6, IL-10, IL-12, IL-17, IFN-γ, TNF-α and MCP-1. While levels of IL-6, IL-10, IL-12 and MCP-1 were below or close to the detection limit and therefore not analyzable, the cells produced copious amounts of IL-2 (∼50 000 pg·mL−1), IL-17 (∼23 000 pg·mL−1) and IFN-γ (∼9000 pg·mL−1) as well as detectable amounts of IL-4 and TNF-α (Figure 6B). Both KCa3.1 channel blockers potently inhibited production of the Th1 cytokines IL-2 and IFN-γ (IC50 ∼50 nM), but had less effect on IL-4 and TNF-α (Figure 6B). In keeping with previous reports that Th17 functions were normal in KCa3.1–/– mice (Di et al., 2010), both KCa3.1 channel blockers exerted no effect on the production of rat IL-17 in our hands (data not shown).
Figure 6.
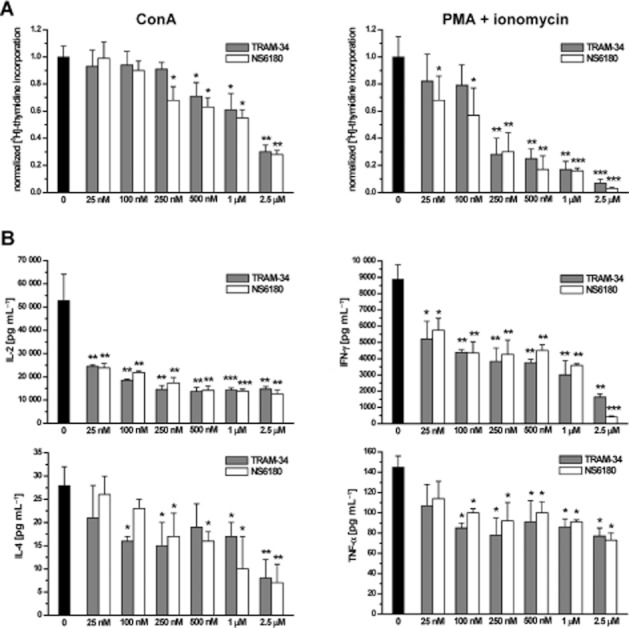
NS6180 and TRAM-34 inhibit rat lymphocyte proliferation and cytokine secretion. (A) Suppression of [3H]-thymidine incorporation by rat splenocytes stimulated for 48 h with either 5 μg·mL−1 ConA (left) or 10 nM PMA plus 175 nM ionomycin (right). Control counts (42 000 cpm for ConA and 120 000 cpm for PMA + ionomycin) were normalized to 1 and fractional [3H]-thymidine incorporations from triplicate wells from one representative experiment are shown as mean ± SD. (B) Effect of N6180 and TRAM-34 on IL-2, IFN-γ, IL-4 and TNF-α production. Data shown are concentrations of cytokines (means ± SD from triplicate wells).
In vivo pharmacokinetics of NS6180
The basic pharmacokinetics of NS6180 in rats is outlined in Supplementary Figure S2 and Supplementary Methods. To summarize, NS6180 had a plasma half-life of 3.8 h and was best fitted with a three-compartment model. Oral or i.p. administration gave low plasma exposure (Cmax: 186 nM and 33 nM, respectively, after administration of 10 mg·kg−1) indicating extremely low bioavailability of NS6180.
KCa3.1 channels as targets for IBD treatment
In order to evaluate its efficacy in blocking KCa3.1 channels in a disease model, we tested NS6180 in DNBS-induced colitis in rats, a model of human IBD. In keeping with the known expression of KCa3.1 channels in intestinal epithelium (Rufo et al., 1997), immunohistochemical staining for these channels in colon sections from normal rats revealed KCa3.1 channels on the epithelial cells lining the well-aligned parallel crypts (Figure 7, upper panels) and on occasional ED1+ macrophages or CD43+ T cells in the muscularis mucosae and lamina propria (Figure 7, upper panels). Sections of colons from rats treated with DNBS exhibited crypt deformation, cryptitis, extensive inflammatory cell infiltration, oedema and vasodilation, accompanied by increased staining for KCa3.1 channels, which was particularly strong on crypt, mucosa and submucosa infiltrating ED1+ macrophages and CD43+ T cells (Figure 7, lower panels).
Figure 7.

Photomicrographs of sections of colons from control or DNBS treated rats. The upper panels show immunlocalization of KCa3.1 channels to crypt-lining epithelial cells and to occasional macrophages (ED1+) and T cells (CD43+) in the lamina propria and muscularis mucosae of a normal colon. The three smaller images show a 400× magnification of the boxed area A (KCa3.1 stain) or 400× magnifications of serial sections from the boxed area A (ED1, macrophages) or B (CD43, T cells). The three lower panels show serial sections from the colon of a DNBS-treated rat stained for KCa3.1, ED1 or CD43. The smaller images are 400× magnifications of the boxed areas A, B or C.
In a separate series of experiments, groups of DNBS challenged rats were treated with two doses (3 and 10 mg·kg−1 b.i.d.) of NS6180 for 7 days in direct comparison with the IBD drug sulfasalazine (300 mg·kg−1 q.d.). The disease symptoms were followed, and Figure 8 shows the relative colon (Figure 8A) and body weight development (Figure 8B) of the different groups after 8 days of treatment. Both doses of NS6180 significantly improved weight gain and decreased inflammation induced swelling of the colon as determined by relative colon weight. The positive control sulfasalazine also significantly affected both body weight and colon inflammation (Figure 8). The effects of NS6180 and sulfasalazine did not differ significantly in magnitude. For complementary effect parameters from this study (macroscopic colonic damage, stool consistency and stool blood scores), see Supplementary Figure S4.
Figure 8.
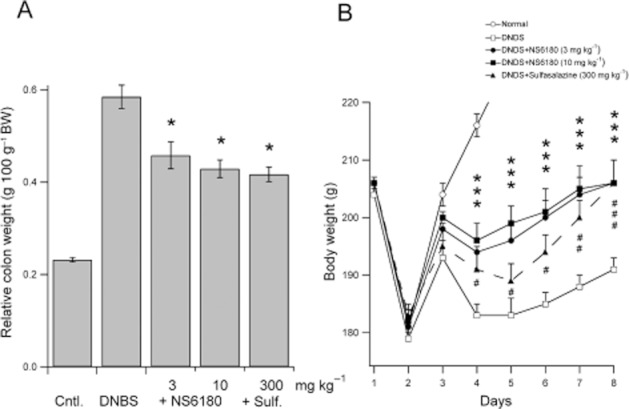
NS6180 reduces DNBS-induced experimental colitis in rats. (A) Relative colon weights (g * 100 g–1 bodyweight) 8 days after rectal instillation of physiological saline (Control animals, Cntl.) or DNBS (DNBS, +NS6180 and +Sulf.) at day 1. Test animals were dosed twice daily with 3 or 10 mg·kg−1 NS6180 or once daily with 300 mg·kg−1 sulfasalazine (Sulf) for 7 consecutive days. Data shown are means ± SEM (n = 8). *P < 0.05, significant difference between treatment groups; one-way anova, followed by Dunnett's post hoc test. (B) Body weight gain during the treatment period. The acute weight loss observed at day 2 is due to dosing-related diarrhoea and invariable between the groups. The Y-axis has been truncated at 220 g in order to highlight the differences between DNBS and the treatment groups. The control group reaches 258 ± 1 g at day 8. A two-way anova (followed by Holm–Sidak's post hoc test) comparison between groups revealed highly significant effects of treatment compared to the DNBS group after day 3. *P < 0.001 for both the 3 and 10 mg·kg−1 dosing groups at all days; #P < 0.05 for sulfasalazine day 4–6, P < 0.01 at day 7, and P < 0.001 at day 8. No statistical differences were detected between the sulfasalazine group and either of the NS6180 groups or between the two NS6180 groups. Additional data from this experiment is found in Supplementary Figure S1.
Discussion and conclusions
We here identified a novel KCa3.1 channel inhibitor (NS6180), a benzothiazinone that bears no structural resemblance to previous KCa3.1 channel inhibitors based on the TRAM or cyclohexadiene/phenylpyrane scaffolds (Urbahns et al., 2003; 2005; Mauler et al., 2004). NS6180 was compared with TRAM-34 (Wulff et al., 2000) in a number of classic in vitro experiments in order to qualify the compound as an alternative tool compound for KCa3.1 channels. NS6180 was equipotent (10–20 nM) with TRAM-34 on both cloned and endogeneous human KCa3.1 channels and showed no species variation for inhibiting KCa3.1-mediated hyperpolarizations from human, rat and mice erythrocytes (Gárdos responses). The identical potencies found in the patch clamp (single cell under flow conditions) and erythrocyte assays (population ∼5 × 108 cells under stirred conditions) reflect lack of competing high-affinity/high-capacity sites for NS6180 (and TRAM-34) in erythrocytes. Furthermore, the potency of NS6180 for the erythrocyte KCa3.1 channel is close to the reported potency of senicapoc (Stocker et al., 2003), a TRAM compound that has been in clinical phase 3 for sickle cell anaemia (Ataga et al., 2011). Despite its non-TRAM structure, the effect of NS6180 depends critically on the residues T250 and V275, which confer TRAM sensitivity on KCa3.1 channels (Wulff et al., 2001), suggesting that NS6180 interacts with the same binding site in the inner pore vestibule as TRAM-34. NS6180 was selective for KCa3.1 channels with a concentration 100 times the IC50 value giving maximally 15% inhibition of any other channel tested. On detailed inspection, however, NS6180 exhibited a different selectivity profile from that of TRAM-34 at 10 μM, with a general trend towards less pronounced inhibition. The selectivity against Kv7.2 + Kv7.3 channels (NS6180 better than TRAM-34) is noteworthy, since that particular channel has the critical threonine and valine positioned at the nominally same positions as in KCa3.1. Obviously, these amino acids do not define a high-affinity interaction in the KCNQ channel family as they do in the KCNN family.
To evaluate its immunosuppressive activity, we next tested NS6180 in a series of in vitro assays with rat splenocytes: NS6180 inhibited proliferation stimulated by the lectin ConA, which cross-links T-cell receptors, as well as proliferation driven by the combination of the PKC activator PMA and the calcium ionophore ionomycin, which together stimulate T-cell activation downstream of the T-cell receptor. Consistent with previous (Wulff et al., 2000) and current results with TRAM-34 the anti-proliferative effect of NS6180 (IC50 ∼100–250 nM) is more pronounced using the (PMA + ionomycin) than ConA stimulation (IC50 ∼1 μM) reflecting a particularly important role of KCa3.1 channels when proliferation is driven by the Ca2+-dependent NFAT and NF-kB pathways. Analysis of cytokine production revealed equipotent inhibitory effects of the two compounds on the production of IL-2 (IC50 < 25 nM) and IFN-γ (IC50 ∼100 nM) in the (PMA + ionomycin) assay, as well as the quantitatively minor cytokines IL-4 and TNF-α; whereas IL-17 was not affected. As expected, assuming that the effects of TRAM-34 and NS6180 are solely due to inhibition of KCa3.1 channels, both compounds showed no effect on the proliferation of KCa3.1–/– mouse splenocytes. However, as previously reported, these cells are not anergenic, possibly due to a partial compensatory up-regulation of Kv1.3 channels, but show impaired IL-2, TNF-α, and INF-γ production (Di et al., 2010). The identical cytokine profiles obtained with KCa3.1–/– splenocytes and KCa3.1 channel inhibitors strongly support their pharmacological specificity in these assays.
Finally, we tested NS6180 in DNBS induced rat colitis, a widely used animal model for human IBD and showed that it was nearly as effective and much more potent than sulfasalazine, the current first-line treatment for IBD (Nielsen and Munck, 2007), in treating macroscopic indices of the disease. Immunohistochemistry showed a strong positive staining for KCa3.1 channels on crypts, mucosa and submucosa infiltrating macrophages and T cells, both of which we consider the most likely targets of KCa3.1 blockers in colitis. This notion is supported by observations made in an adoptive transfer model of IBD (injection of a subset of CD4+ T lymphocytes into rag2–/– mice), where T cells taken from KCa3.1–/– mice – in contrast to wild-type mice – failed to induce colitis. The same study also demonstrated that systemic treatment with TRAM-34 ameliorated hapten-induced colitis in mice (Di et al., 2010). Furthermore, clotrimazole has also been reported to inhibit experimental colitis and accompanying angiogenesis via a NF-κB-dependent mechanism (Thapa et al., 2008). Interestingly, however, a recent clinical study reported reduced basolateral expression of KCa3.1 channels in the colonic epithelium from ulcerative colitis patients (but not patients with Crohn's disease) concurrent with increased KCa3.1 immunoreactivity on infiltrating cells (Al-Hazza et al., 2012). The authors hypothesize that the reduced epithelial expression might be impairing salt and water absorption across the inflamed mucosa, thereby contributing to diarrhoea in colitis. However, since KCa3.1 channel blockade with clotrimazole inhibits chloride secretion in normal rabbit colonic mucosal sheets and effectively treats diarrhoea in mouse model of cholera (Rufo et al., 1997), the reduced epithelial expression of KCa3.1 channels could alternatively be interpreted as a pathophysiological counter-regulatory attempt to reduce chloride and fluid secretion across the colon epithelium.
Recent drug development for IBD has concentrated on biological agents, such as antibodies against inflammatory cytokines and receptors (see Caprioli et al., 2012 and Bouguen et al., 2011) and several anti-TNF-α drugs have been approved for Crohn's disease and ulcerative colitis. Furthermore, drugs targeting IL-6, IL-12/IL-23, IFN-γ and IL-2 have been in clinical trials with varying results. Despite their promising and versatile treatment opportunities, anti-cytokine therapies remain expensive, are not orally applicable, may cause serious side effects such as allergic reactions and increased incidence of infections and lymphomas and may also have therapeutic weaknesses, such as loss of efficacy as demonstrated for anti-TNF-α therapy (Ben-Horin and Chowers, 2011), which may be due to neutralizing antibodies raised against the biological drug, or changes in the inflammation process, for example cytokine switching. Thus, the highly specific interaction of biological drugs with their target molecule may eventually be suboptimal for maintenance treatment.
Modulation of immune responses via block of ion channels on immune cells may be an advantageous alternative to anti-cytokine therapy. As shown, inhibition of KCa3.1 channels induced a profiled inhibition of specific T-cell populations and reduced the production of both TH1 and TH2 cytokines, while leaving others unaffected. The clear anti-inflammatory action of NS6180 in the DNBS model supports KCa3.1 channels as a target for the treatment of IBD. Furthermore, the novel chemical scaffold of NS6180 clearly illustrates the amount of unexploited chemical space for development of KCa3.1 channel inhibitors, which is particularly important since the original patents for TRAM and phenylpyranes/cyclohexadienes are expiring soon, precluding their clinical development.
Acknowledgments
Vibeke Meyland-Smith is greatly acknowledged for her expert handling of the erythrocyte assay and patch-clamp experiments. We further would like to thank LeeLee Zhu for technical assistance with the UPLC/MS operation. The human Orai-1 and STIM-1 clones were kindly provided by Richard Lewis (Department of Molecular and Cellular Physiology, Stanford). We thank Jie Zheng (Department of Physiology and Membrane Biology, UC Davis) for providing the TRPV clones.
Glossary
- TRAM-34
(5-[(2-chlorophenyl)(diphenyl)methyl]-1H-pyrazole)
- CRAC
Ca2+ release-activated channel
- CCCP
carbonylcyanide-m-chloro-phenyl-hydrazone
- ConA
concanavalin A
- IBD
inflammatory bowel disease
- KCa3.1
intermediate-conductance Ca2+-activated K+ channel
- q.d
once a day dosing
- PMA
phorbol 12-myristate 13-acetate
- TRAM
triarylmethane
- b.i.d
twice a day dosing
- A23187
5-(methylamino)-2-(methyl)-1,3-benzoxazole-4-carboxylic acid
- NS6180
(4-[[3-(trifluoromethyl)phenyl]methyl]-2H-1,4-benzothiazin-3(4H)-one)
- DNBS
2,4-dinitrobenzene sulfonic acid
Conflicts of interests
None of the authors have any conflict of interests.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 NS6180 and TRAM-34 inhibit ConA-stimulated proliferation of splenocytes from wild-type mice but not from KCa3.1−/− mice. Cells were stimulated for 48 h with 5 μg·mL−1 ConA in the presence and absence of TRAM-34 or NS6180. Control counts were normalized to 1, and fractional [3H]-thymidine incorporations from triplicate wells from one representative experiment are shown as mean ± SD. Splenocytes (frozen MNC) from KCa3.1−/− mice (Si et al., 2006) and C57Bl6 littermates were a generous gift from Dr Ralf Köhler, University of Southern Denmark, Odense. The cells were thawed, allowed to recover for 3 h and then seeded at 1 × 105 cells into flat-bottom 96-well plates and stimulated with 5 μg·mL−1 ConA.
{kind=link}
Figure S2 Pharmacokinetics of NS6180 in rats. Total NS6180 plasma concentrations following i.v., i.p. and oral administration at 10 mg·kg−1 (n = 3 per route of application). Following i.v. administration, plasma concentrations fell from a peak of 9 ± 2 μM after 5 min to 90 nM at 24 h (n = 3). Similar to TRAM-34 (Chen et al., 2011), the i.v. data were best fitted triexponentially reflecting a three-compartment model with rapid distribution from blood into tissue (t1/2 distribution = 0.15 h) followed by elimination (t1/2 = 3.8 h), and slow repartitioning from body fat acting as a deep compartment back into plasma (t1/2 = 18 h). Bioavailability by i.p. and p.o. routes were low and resulted in Cmax concentrations of 186 ± 14 nM and 33 ± 4 nM only. Inset: Same data with rescaled X- and Y-axes to better visualize the i.p. and oral data. All values are means ± SD.
{kind=link}
Figure S3 Effect of NS6180 and TRAM-34 on CRAC-, TRPV1-, TRPV2- and TRPV4-mediated currents expressed in HEK293 cells. (A) CRAC currents from the co-expression of human GFP-Myc-Orail1 and human mCherry-STIM1. Left: I–V relation during voltage ramps (−120 to +40 mV) following application of 1 and 10 μM NS6180 (not leak-subtracted). Right: Time course of currents measured at −110 mV during application of NS6180 (1 and 10 μM) and 2-APB (30 μM). Ca2+ currents were elicited by passive store depletion with 2 mM external Ca2+. Pipette solution contained (in mM): CsAsp 133, CsCl 2, MgCl2 8, HEPES 15 and BAPTA 11 (pH 7.2 adjusted with CsOH; 309 mOsm). (B) Human TRPV1 currents: Left: Normalized I–V plots under control conditions, after application of 250 nM capsaicin, 10 μM NS6180 plus 250 nM capsaicin or 10 μM TRAM-34 plus 250 nM capsaicin. Currents at each voltage were generated by 200 ms depolarizing steps (−120 to +70 mV in 10 mV increments; 1 s delay between steps). Right: Voltage ramps (−120 to +80 mV) under control conditions, after application of 250 nM capsaicin, and 10 μM NS6180 plus 250 nM capsaicin or 10 μM TRAM-34 plus 250 nM capsaicin. (C) Human TRPV2 currents. Voltage ramps (−120 to +80 mV) under control conditions, after application of 200 μM 2-aminoethoxydiphenylborane (2-APB), 10 μM NS6180 plus 200 μM 2-APB or 10 μM TRAM-34 plus 200 μM 2-APB. (D) Human TRPV4 currents. Voltage ramps (−120 to +80 mV) under control conditions, after application of 500 nM 4α-phorbol 12,13-didecanoate (4α-PDD), and 10 μM NS6180 plus 500 nM 4α-PDD (top) or 10 μM TRAM-34 plus 500 nM 4α-PDD (bottom). For all recordings from TRP channels pipette solutions contained (in mM): NaF 160, MgCl2 2, HEPES 10, EGTA 10 (pH 7.2 adjusted with NaOH; 301 mOsm). Na+-Ringer was used as an external solution for all recordings (in mM); NaCl 160, KCl 4.5, MgCl2 1, CaCl2 2, HEPES 10 (pH 7.4 adjusted with NaOH; 301 mOsm).
{kind=link}
Figure S4 Additional data from the rat colitis experiment illustrated in Figure 8. (A) Colonic macroscopic damage score after 8 days of treatment. The individual colons were scored for adhesions (0–2), strictures (0–2), ulcers/inflammation (0–5) and wall thickness (1–2) and summarized. The bar graph depicts the averaged values from all animals (n = 8). Sulfasalazine reduced the total score significantly (one-way anova followed by Dunnett's post hoc test), whereas the NS6180 trends did not reach statistical significance. (B) Cumulative daily scorings for stool appearance comprising rectal bleeding (left column) and stool consistency (right column). Both parameters were scored (0, 1, 2), and the results from all animals are averaged daily. Both doses of NS6126 as well as sulfasalazine significantly (one-way anova followed by Dunnett's post hoc test) reduced the scores on days 6, 7 and 8.
{kind=link}
References
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hazza A, Linley JE, Aziz Q, Maclennan KA, Hunter M, Sandle GI. Potential role of reduced basolateral potassium (IKCa3.1) channel expression in the pathogenesis of diarrhoea in ulcerative colitis. J Pathol. 2012;226:463–470. doi: 10.1002/path.2994. [DOI] [PubMed] [Google Scholar]
- Alvarez J, Montero M, Garcia-Sancho J. High affinity inhibition of Ca(2+)-dependent K+ channels by cytochrome P-450 inhibitors. J Biol Chem. 1992;267:11789–11793. [PubMed] [Google Scholar]
- Ataga KI, Reid M, Ballas SK, Yasin Z, Bigelow C, James LS, et al. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: a phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapoc (ICA-17043) Br J Haematol. 2011;153:92–104. doi: 10.1111/j.1365-2141.2010.08520.x. [DOI] [PubMed] [Google Scholar]
- Beeton C, Wulff H, Barbaria J, Clot-Faybesse O, Pennington M, Bernard D, et al. Selective blockade of T lymphocyte K(+) channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc Natl Acad Sci U S A. 2001;98:13942–13947. doi: 10.1073/pnas.241497298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Horin S, Chowers Y. Review article: loss of response to anti-TNF treatments in Crohn's disease. Aliment Pharmacol Ther. 2011;33:987–995. doi: 10.1111/j.1365-2036.2011.04612.x. [DOI] [PubMed] [Google Scholar]
- Bouguen G, Chevaux JB, Peyrin-Biroulet L. Recent advances in cytokines: therapeutic implications for inflammatory bowel diseases. World J Gastroenterol. 2011;17:547–556. doi: 10.3748/wjg.v17.i5.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugnara C, Gee B, Armsby CC, Kurth S, Sakamoto M, Rifai N, et al. Therapy with oral clotrimazole induces inhibition of the Gardos channel and reduction of erythrocyte dehydration in patients with sickle cell disease. J Clin Invest. 1996;97:1227–1234. doi: 10.1172/JCI118537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprioli F, Caruso R, Sarra M, Pallone F, Monteleone G. Disruption of inflammatory signals by cytokine-targeted therapies for inflammatory bowel diseases. Br J Pharmacol. 2012;165:820–828. doi: 10.1111/j.1476-5381.2011.01614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Raman G, Bodendiek S, O'Donnell ME, Wulff H. The KCa3.1 blocker TRAM-34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J Cereb Blood Flow Metab. 2011;12:2363–2374. doi: 10.1038/jcbfm.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L, Srivastava S, Zhdanova O, Ding Y, Li Z, Wulff H, et al. Inhibition of the K+ channel KCa3.1 ameliorates T cell-mediated colitis. Proc Natl Acad Sci U S A. 2010;107:1541–1546. doi: 10.1073/pnas.0910133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YD, Hanley PJ, Rinne S, Zuzarte M, Daut J. Calcium-activated K(+) channel (K(Ca)3.1) activity during Ca(2+) store depletion and store-operated Ca(2+) entry in human macrophages. Cell Calcium. 2010;48:19–27. doi: 10.1016/j.ceca.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, et al. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J Biol Chem. 2000;275:37137–37149. doi: 10.1074/jbc.M003941200. [DOI] [PubMed] [Google Scholar]
- Jenkins DP, Strobaek D, Hougaard C, Jensen ML, Hummel R, Sorensen US, et al. Negative gating modulation by (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphthylamine (NS8593) depends on residues in the inner pore vestibule: pharmacological evidence of deep-pore gating of K(Ca)2 channels. Mol Pharmacol. 2011;79:899–909. doi: 10.1124/mol.110.069807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macey RI, Adorante JS, Orme FW. Erythrocyte membrane potentials determined by hydrogen ion distribution. Biochim Biophys Acta. 1978;512:284–295. doi: 10.1016/0005-2736(78)90253-5. [DOI] [PubMed] [Google Scholar]
- Mauler F, Hinz V, Horvath E, Schuhmacher J, Hofmann HA, et al. Selective intermediate-/small-conductance calcium-activated potassium channel (KCNN4) blockers are potent and effective therapeutics in experimental brain oedema and traumatic brain injury caused by acute subdural haematoma. Eur J Neurosci. 2004;20:1761–1768. doi: 10.1111/j.1460-9568.2004.03615.x. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reportingexperiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen OH, Munck LK. Drug insight: aminosalicylates for the treatment of IBD. Nat Clin Pract Gastroenterol Hepatol. 2007;4:160–170. doi: 10.1038/ncpgasthep0696. [DOI] [PubMed] [Google Scholar]
- Reich EP, Cui L, Yang L, Pugliese-Sivo C, Golovko A, Petro M, et al. Blocking ion channel KCNN4 alleviates the symptoms of experimental autoimmune encephalomyelitis in mice. Eur J Immunol. 2005;35:1027–1036. doi: 10.1002/eji.200425954. [DOI] [PubMed] [Google Scholar]
- Rufo PA, Merlin D, Riegler M, Ferguson-Maltzman MH, Dickinson BL, Brugnara C, et al. The antifungal antibiotic, clotrimazole, inhibits chloride secretion by human intestinal T84 cells via blockade of distinct basolateral K+ conductances. Demonstration of efficacy in intact rabbit colon and in an in vivo mouse model of cholera. J Clin Invest. 1997;100:3111–3120. doi: 10.1172/JCI119866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan A, Raman G, Busch C, Schultz T, Zimin PI, Hoyer J, et al. Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol. 2009;75:281–295. doi: 10.1124/mol.108.051425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumilina E, Lam RS, Wolbing F, Matzner N, Zemtsova IM, Sobiesiak M, et al. Blunted IgE-mediated activation of mast cells in mice lacking the Ca2+-activated K+ channel KCa3.1. J Immunol. 2008;180:8040–8047. doi: 10.4049/jimmunol.180.12.8040. [DOI] [PubMed] [Google Scholar]
- Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, Grgic I, et al. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ Res. 2006;99:537–544. doi: 10.1161/01.RES.0000238377.08219.0c. [DOI] [PubMed] [Google Scholar]
- Stocker JW, De Franceschi L, McNaughton-Smith GA, Corrocher R, Beuzard Y, Brugnara C. ICA-17043, a novel Gardos channel blocker, prevents sickled red blood cell dehydration in vitro and in vivo in SAD mice. Blood. 2003;101:2412–2418. doi: 10.1182/blood-2002-05-1433. [DOI] [PubMed] [Google Scholar]
- Strobaek D, Hougaard C, Johansen TH, Sorensen US, Nielsen EO, Nielsen KS, et al. Inhibitory gating modulation of small conductance Ca2+-activated K+ channels by the synthetic compound (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphtylamine (NS8593) reduces afterhyperpolarizing current in hippocampal CA1 neurons. Mol Pharmacol. 2006;70:1771–1782. doi: 10.1124/mol.106.027110. [DOI] [PubMed] [Google Scholar]
- Thapa D, Lee JS, Park SY, Bae YH, Bae SK, Kwon JB, et al. Clotrimazole ameliorates intestinal inflammation and abnormal angiogenesis by inhibiting interleukin-8 expression through a nuclear factor-kappaB-dependent manner. J Pharmacol Exp Ther. 2008;327:353–364. doi: 10.1124/jpet.108.141887. [DOI] [PubMed] [Google Scholar]
- Toyama K, Wulff H, Chandy KG, Azam P, Raman G, Saito T, et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest. 2008;118:3025–3037. doi: 10.1172/JCI30836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbahns K, Horvath E, Stasch JP, Mauler F. 4-Phenyl-4H-pyrans as IK(Ca) channel blockers. Bioorg Med Chem Lett. 2003;13:2637–2639. doi: 10.1016/s0960-894x(03)00560-2. [DOI] [PubMed] [Google Scholar]
- Urbahns K, Goldmann S, Kruger J, Horvath E, Schuhmacher J, Grosser R, et al. IKCa-channel blockers. Part 2: discovery of cyclohexadienes. Bioorg Med Chem Lett. 2005;15:401–404. doi: 10.1016/j.bmcl.2004.10.063. [DOI] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci U S A. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Gutman GA, Cahalan MD, Chandy KG. Delineation of the clotrimazole/TRAM-34 binding site on the intermediate conductance calcium-activated potassium channel, IKCa1. J Biol Chem. 2001;276:32040–32045. doi: 10.1074/jbc.M105231200. [DOI] [PubMed] [Google Scholar]
- Wulff H, Knaus HG, Pennington M, Chandy KG. K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity. J Immunol. 2004;173:776–786. doi: 10.4049/jimmunol.173.2.776. [DOI] [PubMed] [Google Scholar]
- Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier JM, Shakkottai V. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem. 2007;14:1437–1457. doi: 10.2174/092986707780831186. [DOI] [PubMed] [Google Scholar]
- Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8:982–1001. doi: 10.1038/nrd2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Gribkoff VK, Pan J, Damagnez V, Dworetzky SI, Boissard CG, et al. Pharmacological activation and inhibition of Slack (Slo2.2) channels. Neuropharmacology. 2006;51:896–906. doi: 10.1016/j.neuropharm.2006.06.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.