

Abstract
Background and Purpose
The success of antibody-drug conjugates (ADCs) depends on the therapeutic window rendered by the differential expression between normal and pathological tissues. The ability to identify and visualize target expression in normal tissues could reveal causes for target-mediated clearance observed in pharmacokinetic characterization. TENB2 is a prostate cancer target associated with the progression of poorly differentiated and androgen-independent tumour types, and ADCs specific for TENB2 are candidate therapeutics. The objective of this study was to locate antigen expression of TENB2 in normal tissues, thereby elucidating the underlying causes of target-mediated clearance.
Experimental Approach
A series of pharmacokinetics, tissue distribution and mass balance studies were conducted in mice using a radiolabelled anti-TENB2 ADC. These data were complemented by non-invasive single photon emission computed tomography – X-ray computed tomography imaging and immunohistochemistry.
Key Results
The intestines were identified as a saturable and specific antigen sink that contributes, at least in part, to the rapid target-mediated clearance of the anti-TENB2 antibody and its drug conjugate in rodents. As a proof of concept, we also demonstrated the selective disposition of the ADC in a tumoural environment in vivo using the LuCaP 77 transplant mouse model. High tumour uptake was observed despite the presence of the antigen sink, and antigen specificity was confirmed by antigen blockade.
Conclusions and Implications
Our findings provide the anatomical location and biological interpretation of target-mediated clearance of anti-TENB2 antibodies and corresponding drug conjugates. Further investigations may be beneficial in addressing the relative contributions to ADC disposition from antigen expression in both normal and pathological tissues.
Keywords: TENB2, antibody-drug conjugates, prostate cancer, indium-111-DOTA, tissue distribution, ELISA
Introduction
Antibody-drug conjugates (ADCs) combine the antigen targeting specificity of monoclonal antibodies (Chames et al., 2009) with the cytotoxic potency of chemotherapeutic drugs (Doronina et al., 2003; Carter and Senter, 2008; Alley et al., 2009; Senter, 2009; Teicher, 2009; Ducry and Stump, 2010). ADCs have gained renewed interest due to advances in antibody engineering, improvements in linker stability and recent clinical successes (Burris, 2011; Foyil and Bartlett, 2011). The effectiveness of targeted chemotherapy delivered via an antibody is contingent upon the therapeutic window rendered by differential antigen expression between the tumour and normal organs.
Antibody-based therapies, including ADCs, are currently being developed against specific antigens that are expressed in prostate tumours. Prostate cancer represents one quarter of newly diagnosed cancer cases in men and is associated with a high mortality rate surpassed only by lung cancer (Jemal et al., 2010). Despite significant efforts towards improving the diagnosis (Hricak et al., 2007; Ravizzini et al., 2009; Regino et al., 2009) and therapy (Feldman and Feldman, 2001; Denmeade and Isaacs, 2002; Michaelson et al., 2008; Lin GA et al., 2009a; Shepard and Raghavan, 2009; Rosenthal and Sandler, 2010) for this disease, few treatment options exist for metastatic prostate cancer, and developing effective treatments remains a critical priority.
One promising molecular target for prostate cancer therapy is a chondroitin sulphate proteoglycan known as TENB2 (Glynne-Jones et al., 2001), a transmembrane protein containing an EGF-like motif and two follistatin-like domains. Also known as TMEFF2 or tomoregulin (Afar et al., 2004; Zhao et al., 2005; 2008), TENB2 is overexpressed in both hormone-naïve and hormone-resistant prostate cancer, and is associated with the progression of poorly differentiated and androgen-independent tumour types (Afar et al., 2004). A humanized anti-TENB2 IgG1 antibody (Afar et al., 2004) was conjugated to a potent anti-mitotic auristatin drug via a protease-labile linker (Bai et al., 1990; Doronina et al., 2003; Francisco et al., 2003). This anti-TENB2 ADC was shown to have anti-tumour activity in explant and xenograft models and is being investigated for the treatment of prostate cancer (Afar et al., 2004).
The ability to identify and visualize antigen expression in normal tissues with an antibody provides a direct early assessment of the targeting specificity. This is particularly true for ADC targets, which exhibit therapeutic windows that are dependent upon the level and distribution of target in normal tissue, thus warranting rigorous safety assessment. In this report, we present pharmacokinetic (PK) data for a ThioMab-drug conjugate (Junutula et al., 2008) that specifically targets TENB2. Importantly, mouse and human TENB2 proteins share 98.9% sequence homology, with only four amino acid differences (Supporting Information Fig. S1). Furthermore, anti-TENB2 exhibited comparable binding affinity to both human and mouse TENB2 (Supporting Information Figure S2). The dose-dependent PK profile was consistent with rapid target-mediated clearance in mice, suggesting the presence of an antigenic sink in tissues. An integrated approach was subsequently pursued to provide a mechanistic basis for this rapid target-mediated clearance and, furthermore, to establish the biological basis for exploring alternative dosing regimens.
Methods
ThioMab production and monomethyl auristatin E conjugation
In these studies, we used a ThioMab version of the anti-TENB2 antibody, which contains a single cysteine residue engineered into an IgG heavy chain. These two cysteines serve as exclusive sites for conjugation to monomethyl auristatin E (MMAE) through a linker containing a thiol-reactive maleimide and the protease cleavable val-cit peptide (Figure 1). Methods for construction and production of the ThioMab variant and its drug conjugate were reported previously (Junutula et al., 2008). Briefly, a cysteine residue was engineered at Ala118 position of the anti-TENB2 heavy chain to produce its ThioMab variant from which the ADC was produced. Partial reduction and reoxidation yielded two free thiol groups per antibody, which were conjugated to the cytotoxic drug MMAE via the protease-labile maleimidocaproyl-valine-citrulline-para-aminobenzyloxycarbonyl linker (Figure 1). The drug to antibody ratio of ADC was determined to be approximately 2 as described earlier (Junutula et al., 2008).
Figure 1.
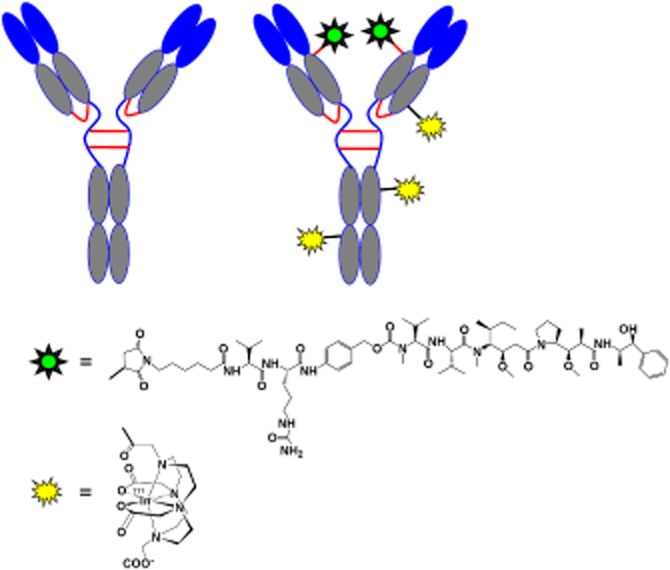
Structural depiction of the unconjugated, unlabelled mAb used for pre-dosing (anti-TENB2, left) and the radiolabelled anti-TENB2-MMAE under evaluation ([111In]-DOTA-anti-TENB2-MMAE, right). The auristatin drug, MMAE (green), was conjugated site-specifically to exactly two engineered thiols via the protease-labile maleimido-caproyl-valine-citrulline-para-amino-benzyloxy carbonyl (MC-vc-PAB) linker, while the radiometal chelate, DOTA (yellow), was randomly conjugated to lysine residues through amide bonds.
DOTA conjugation and characterization
The anti-TENB2 ADC was labelled with 1,4,7,10-tetraazacyclododecane-N,N′,N″,N′″-tetraacetic acid (DOTA) for indium-111 complexation by random modification of lysine residues (Figure 1) as previously described (Boswell et al., 2011b). Importantly, the DOTA conjugate showed acceptable (> 74%) retention of antigen binding, measured as % recovery in a TENB2-specific ELISA (see below). Because excessive chelate conjugation can influence electrostatics and, ultimately, PK (Boswell et al., 2010b), determination of the average number of attached chelates for each DOTA conjugate by radiometric assay was performed by modification of previously reported procedures (Meares et al., 1984; Lewis et al., 2001). Radiometric measurement of the average number of DOTA chelates attached per antibody molecule gave a value of 3.7 for DOTA-anti-TENB2-MMAE. This value was in good agreement with the estimate based on shifts of mass peaks before and after DOTA conjugation, which was 2.9 and 3.1 DOTA units per mAb using reduced and intact LC-MS analysis respectively (Supporting Information Fig. S3).
Radiochemistry
Anti-TENB2-MMAE was radioiodinated with 125I at a 61.4% yield using the indirect iodogen method and purified using reported methods (Boswell et al., 2010a). Radiolabelling and characterization of DOTA-anti-TENB2-MMAE were conducted as reported earlier (Boswell et al., 2011b). A radiochemical yield of 84% was obtained for incorporation of 111In into DOTA-anti-TENB2-MMAE for the tissue distribution study. Specific activities for invasive biodistribution and non-invasive imaging studies in non-tumour-bearing mice were 0.70 and 0.74 MBq·μg−1, respectively, corresponding to dosing levels of 0.01 and 0.2 mg·kg−1. Specific activities for the dual (125I/111In) tracers in tumour-bearing mice were 0.075 and 0.15 MBq·μg−1 at tracer (0.1 mg·kg−1) level for 125I and 111In, respectively, after adjusting no-carrier-added preparations to 0.1 mg·kg−1 with unlabelled ADC. Size-exclusion HPLC characterization of all radioimmunoconjugates demonstrated radiochemically pure (≥ 98%) dosing material with no evidence of unconjugated 111In (Supporting Information Fig. S4).
PK studies in non-tumour-bearing mice
All experimental animal studies were conducted according to protocols that were reviewed and approved by the Institutional Animal Care and Use Committees of Genentech Laboratory Animal Resources. Animals were housed on 12 h light/dark cycles and were given ad libitum access to food and water. The total number of animals used was 150. Male severe combined immunodeficient (SCID) beige mice (CB-17.Cg-PrkdcscidLystbg/Crl; Charles River, Wilmington, MA, USA) ranging from 8 to 9 weeks old and weighing approximately 21–28 g at the initiation of the study received an i.v. bolus of unconjugated antibody at 5 mg·kg−1 or its respective drug conjugates at 0.3, 4 or 10 mg·kg−1. Blood samples were collected from each animal (three time points per animal; three animals per time point) via retroorbital bleed for up to 28 days and were used to derive plasma for total anti-TENB2 antibody concentration determination using an ELISA (see below). Plasma concentration−time data were obtained to estimate relevant PK parameters using WinNonlin® software (Version 5.2.1 Pharsight Corporation, Mountain View, CA, USA). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Total antibody ELISA
The plasma concentrations of either anti-TENB2 antibody or anti-TENB2-MMAE were measured with an ELISA that used TENB2-FLAG extracellular domain (Genentech, South San Francisco, CA, USA) antibody for capture and a goat anti-human IgG Fc detection antibody conjugated to horseradish peroxidase for detection. The assay has lower limits of quantification of 8.2 ng·mL−1 with a minimum dilution of 1:100.
Tissue distribution in non-tumour-bearing mice
Male SCID beige mice (CB-17.Cg-PrkdcscidLystbg/Crl) ranging from 6 to 8 weeks old and weighing approximately 20–30 g received a single bolus i.v. injection of 111In-labelled conjugates. Dosing solutions were prepared by using the 111In-anti-TENB2 conjugate alone (tracer only) or by addition of anti-TENB2 (conjugated to neither DOTA nor MMAE) to achieve a total protein dose of 1 or 10 mg·kg−1 and radioactive dose of approximately 14.8 MBq·kg−1 in no more than 100 μL of PBS. Administration of the tracer with an isotype-matched control antibody (unconjugated anti-STEAP1, an antibody against the six-transmembrane epithelial antigen of prostate 1) was also performed as a negative control, as the control antibody should not block specific uptake of radiolabelled anti-TENB2-MMAE. A 125 μL aliquot of whole blood was collected by retro-orbital bleed, after the mice had been anaesthetized by inhalation of isoflurane (2–4% v v-1), at 4 h and terminally at 1 and 3 days in lithium-heparin-treated tubes via cardiac puncture after administration of ketamine/xylazine. The mice were subsequently killed by thoracotomy after i.p. administration of ketamine (75–80 mg·kg−1) and xylazine (7.5–15 mg·kg−1) anaesthesia, and the following tissues were subsequently harvested: lungs, liver, kidneys, heart, spleen, gastrocnemius muscle, stomach, small intestine, large intestine, white body fat, prostate and dorsal skin. The contents of the stomach and gut were removed before the analysis. All tissues were rinsed with PBS, blot dried, weighed, frozen on dry ice and analysed as previously reported (Bumbaca et al., 2011; Pastuskovas et al., 2012). Statistical significance (P < 0.05) was determined using GraphPad Prism version 5.01 (La Jolla, CA, USA) by unpaired t-test when comparing two groups or by one-way anova followed by Tukey post-test for groups of three or more.
Mass balance and radioimmunoconjugate stability in non-tumour-bearing mice
Identical methods were used as for the tissue distribution studies (vide supra), except for the use of metabolic cages, to allow collection of urine and faeces throughout the duration of the study. Inhaled isoflurane was used for all blood collections and the mice were killed by cervical dislocation. Following tissue harvest, carcasses were sectioned into top and bottom sections and counted for radioactivity along with tissues, urine and faeces.
Radioimmunoconjugate stability in plasma and rate of elimination in urine were investigated as an adjunct to the mass balance study. Plasma and urine samples were obtained from separate mice used for single photon emission CT – X-ray CT (SPECT-CT) (see below), also housed in metabolic cages, because the higher levels of radioactivity used in imaging studies greatly facilitated radioanalytical detection. At 24 h, 48 h and at killing (72 h), a 150 μL volume of whole blood was collected into EDTA-coated tubes via retro-orbital or cardiac puncture, accordingly, under inhaled sevoflurane (3.5% v v-1) anaesthesia. A 100 μL portion was centrifuged for plasma preparation. Urine was collected in metabolic cages of individually housed mice and harvested daily. Both plasma and urine samples were stored at 4°C and promptly assessed by the same size-exclusion radio-HPLC conditions described earlier.
SPECT-CT imaging of non-tumour-bearing mice
SPECT-CT imaging of mice with 111In-anti-TENB2-MMAE was performed as an adjunct to the in vivo biodistribution study as previously reported (Bumbaca et al., 2011; Pastuskovas et al., 2012). One mouse received a co-dose (10 mg·kg−1) of cold non-drug-conjugated anti-TENB2 ThioMab, while the tracer-only mouse was dosed at 0.2 mg·kg−1. Both mice received an injection of 3.26–4.07 MBq 111In-anti-TENB2-MMAE. The higher dose level in SPECT-CT imaging compared with tissue distribution studies (0.2 vs. 0.01 mg·kg−1) is a consequence of the requirement for higher levels of radioactivity needed to acquire acceptable signal-to-noise ratios for gamma camera (SPECT) detection relative to gamma counting. SPECT acquisition time was 45 min for both mice. Light anaesthesia was maintained during the imaging for restraint only at the minimum concentration required to ensure the mice would not wake up during the scan, about 3.5% v v-1 sevoflurane. The eyes were lubricated with ophthalmic ointment, and body temperature was regulated at 37°C in a feedback system based on warm airflows. Mice were subsequently killed under inhaled sevoflurane sedation and tissues were collected for gamma counting in a manner identical to that used for the non-imaging arm of the study. SPECT quantification was accomplished using Amira software (TGS, San Diego, CA, USA).
Immunohistochemistry
Immunohistochemistry (IHC) of tissues from naïve, non-tumour-bearing mice was performed on 4-μm-thick formalin-fixed paraffin-embedded tissue sections mounted on glass slides. Staining for TENB2 was performed on the Discovery XT autostainer (Ventana Medical Systems, Tucson, AZ, USA). Tissues analysed included the following: brain, liver, kidney, small intestine and large intestine. In addition, a TENB2-positive tumour explant, LuCaP 77, was used as a positive control and exhibited the expected high level of staining. Cell conditioner 1 was utilized as the pretreatment. Primary antibody, humanized TENB2, clone PR-1, was diluted to 2.5 μg·mL−1, slides were incubated for 1 h at 37°C. Naïve human IgG (Jackson, West Grove, PA, USA) was used as a negative control antibody. Goat anti-human, biotin-conjugated (Vector Laboratories, Burlingame, CA, USA), secondary antibody was diluted to 7.5 μg·mL−1 and incubated for 30 min at room temperature. Detection was performed utilizing Ventana DabMAP.
LuCaP 77 explant mouse model
The explant model of prostate cancer, LuCaP 77, was obtained from University of Washington (Seattle, WA, USA). This tumour model was derived from femur metastases at autopsy of a patient with hormone-refractory prostate cancer (Corey et al., 2003; Koochekpour et al., 2005; Corey and Vessella, 2007). The LuCaP 77 explant model was maintained by serial implantations (Boswell et al., 2012a). When donor mice had tumours of between 800 and 1000 mm3, tumour tissue was aseptically removed and dissected into small implantable-sized pieces (approximately 20−30 mm3), which were implanted s.c. into the right flanks of male C.B-17 SCID beige mice (CB17.Cg-PrkdcscidLystbg/Crl) followed by skin closure using wound clips.
PK and tumour uptake in LuCap 77 explant-bearing mice
To prevent thyroid sequestration of 125I, 100 μL of 30 mg·mL−1 of sodium iodide was administered i.p. 1 and 24 h prior to dosing. Dosing solution was prepared by mixing 125I- and [111In]-anti-TENB2-MMAE in PBS and administered i.v. to mice that had not received a pretreatment or had been pretreated with unconjugated anti-TENB2 24 h previously. Under ketamine/xylazine anaesthesia, whole blood was collected in lithium/heparin tubes at 4 h, 1, 2 and 3 days. Killing was performed under ketamine/xylazine anaesthesia at 24 h and 3 days post-dose (three mice per group) for tissue harvest and analysis as previously described (Bumbaca et al., 2011; Pastuskovas et al., 2012).
Results
Anti-TENB2 antibody and anti-TENB2-MMAE (ADC) exhibit non-linear PK and undergo significant target-mediated clearance in non-tumour-bearing mice
ELISA-derived plasma PK data are presented in Figure 2 as scatter plots of mean total anti-TENB2 antibody concentrations over time after i.v. administration of the unconjugated antibody at 5 mg·kg−1 or its respective MMAE-conjugated anti-TENB2 antibody at 0.3, 4 or 10 mg·kg−1 in mice. A summary of the PK parameters for total anti-TENB2 antibody in plasma is presented in Table 1. For all groups, total anti-TENB2 antibody plasma concentration–time profiles displayed multiphasic PK. Although the mean dose-normalized maximum-observed plasma concentration (Cmax/D; range: 17.8–21.6 kg*μg·mL−1·mg−1) appeared similar across anti-TENB2-MMAE (ADC) groups, a dose-dependent decrease in the mean CL (32.4, 13.1 and 9.13 mL·day−1·kg−1, respectively) was observed with increasing dose, indicating the presence of target-mediated clearance. The mean terminal half-lives (t1/2, β) ranged from 1.35 to 7.97 days. The PK profiles between 5 mg·kg−1 unconjugated antibody and 4 mg·kg−1 ADC were comparable, with clearance values of 14.4 and 13.1 mL·day−1·kg−1, suggesting MMAE conjugation had no effect on antibody clearance. The volume of distribution (V1) for all groups approximated to plasma volume in mice [50 mL·kg−1 for a 20 g mouse (Davies and Morris, 1993)], ranging from 44 to 62 mL·kg−1. The observed dose-dependent and saturable clearance with mouse reactive anti-TENB2 antibody and its conjugates suggests the presence of an antigen sink in mouse.
Figure 2.

Mean (±SD) total antibody plasma concentration–time (by ELISA) profile of anti-TENB2-MMAE (ADC) at various doses in non-tumour-bearing SCID mice. PK data for the corresponding unconjugated anti-TENB2 mAb at 5 mg·kg−1 are included for comparison.
Table 1.
Summary of total anti-TENB2 antibody (mAb) and anti-TENB2-MMAE (ADC) pharmacokinetic parameters in non-tumour-bearing SCID beige mice
| Dose (mg·kg−1) | Cmax/D (kg*μg·mL−1·mg−1) | AUCinf/D (day*kg*μg·mL−1·mg−1) | CL (mL·day−1·kg−1) | V1 (mL·kg−1) | t1/2 (day) |
|---|---|---|---|---|---|
| 5 (mAb) | 16.1 | 69.6 | 14.4 | 61.8 | 3.63 |
| 0.3 (ADC) | 17.8 | 30.9 | 32.4 | 55.9 | 1.35 |
| 4 (ADC) | 18.3 | 76.3 | 13.1 | 56.0 | 4.60 |
| 10 (ADC) | 21.6 | 110 | 9.13 | 43.8 | 7.97 |
AUCinf/D, dose-normalized area under the plasma concentration–time curve from the time of dosing extrapolated to infinity; CL, clearance; Cmax/D, dose-normalized maximum observed plasma concentration; t1/2, β, half-life of the terminal (elimination) phase; V1, volume of distribution of the central compartment (i.e. volume of initial dilution compartment).
Co-administration of anti-TENB2 antibody specifically increases systemic exposure of anti-TENB2-MMAE and blocks uptake in selected organs
To identify the location of the antigen sink in mice and evaluate the binding specificity with antigen, we prepared the radiolabelled 111In-anti-TENB2-MMAE tracer as shown in Figure 1. The choice of [111In]-DOTA as a probe was influenced by a desire to increase the likelihood of detecting any antigen-specific uptake in tissue sinks. Antibodies labelled with metal radionuclides via polyaminopolycarboxylate chelators (e.g. DOTA, diethylenetriaminepentaacetic acid) tend to accumulate in antigen-expressing tissues (Perera et al., 2007) following receptor-mediated endocytosis due to the residualizing properties of this charged, highly polar probe (Boswell and Brechbiel, 2007; Boswell et al., 2012b). The tissue distribution of the tracer was studied in SCID beige mice, and compared with profiles after co-administration with the corresponding non-DOTA-conjugated, non-drug-conjugated anti-TENB2 antibody or isotype control-humanized anti-STEAP1 antibody. This scenario allowed us to test for antigen-specific uptake, following competitive binding inhibition. The plasma clearance of the tracer administered at 0.01 mg·kg−1 was dramatically retarded by co-administration of 1 and 10 mg·kg−1 anti-TENB2 but not by control antibody to STEAP1 (Figure 3A). When co-administered with 1 mg·kg−1 anti-TENB2 antibody, the blood exposure measured by AUC increased by 1.8-fold, indicating blockage of antigen sites at peripheral tissues and concurrent congregation of anti-TENB2-MMAE in systemic circulation. Similar patterns between 1 and 10 mg·kg−1 anti-TENB2 dose levels indicated that the saturation of antigen sites was reached by 1 mg·kg−1. The blocking was antigen specific, as demonstrated by the lack of an effect induced by co-administering anti-STEAP1 antibody at 10 mg·kg−1. No evidence of binding to blood cells was observed by blood fractionation (Figure 3B, inset).
Figure 3.
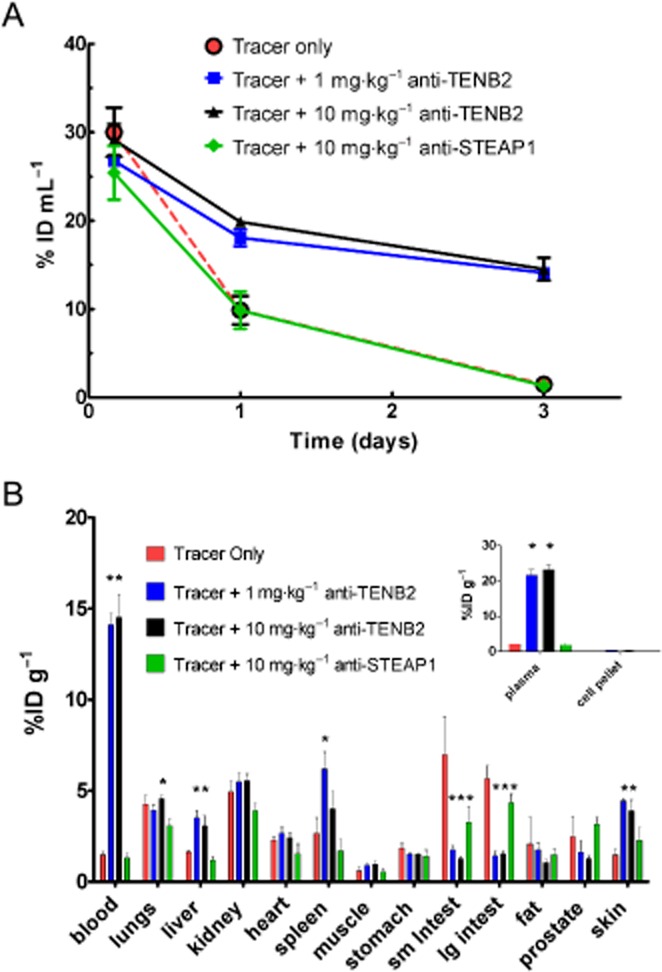
Dose-dependent and antigen-specific blood concentration–time profile and tissue distribution of [111In]-anti-TENB2-MMAE (ADC) in non-tumour-bearing SCID mice at 72 h after i.v. injection. The tracer was administered alone (red), in combination with unconjugated anti-TENB2 (ThioMab) at 1 (blue) or 10 (black) mg·kg−1, or in combination with 10 mg·kg−1 of an isotype-matched control antibody, anti-STEAP1 (green). (A) Whole blood PK data, plotted on a linear scale in a dose-normalized manner as % of injected dose mL-1 of blood, derived by gamma counting in tissue distribution studies. (B) Tissue distribution of [111In]-anti-TENB2-MMAE (ADC) in male SCID mice at 72 h expressed as mean %ID g−1 ± SEM for three mice per group. Asterisks indicate statistical significance (P < 0.05) by one-way anova followed by Tukey post-test.
At 72 h post-injection, the tissue distribution of tracer only and tracer plus 10 mg·kg−1 anti-STEAP1 was vastly different from both tracer plus anti-TENB2 groups (Figure 3B). Among the various organs that we collected, uptake trends in liver, spleen and skin roughly mirrored that of the blood, while uptake in lungs, kidney, heart, muscle and stomach was relatively flat across the four dose groups. For instance, tracer uptake levels in liver expressed as % of injected dose g-1 tissue (%ID g−1) were 1.6 ± 0.1, 3.5 ± 0.4, 3.1 ± 0.6 and 1.2 ± 0.2 for tracer only, 1 and 10 mg·kg−1 anti-TENB2, and 10 mg·kg−1 anti-STEAP respectively. In contrast, the same respective levels in muscle were 0.6 ± 0.2, 0.9 ± 0.1, 0.9 ± 0.2 and 0.6 ± 0.2 %ID g−1. Interestingly, mice receiving tracer only or tracer plus anti-STEAP1 showed very low levels of radioactivity in blood, but elevated levels in small and large intestines relative to groups administered tracer plus 1 and 10 mg·kg−1 anti-TENB2. Small and large intestines showed antigen-specific blockage when co-administered anti-TENB2. For instance, tracer uptake levels in small intestine decreased from 7 ± 2 to 1.8 ± 0.2 and 1.2 ± 0.1 %ID g−1 after co-administration of 1 and 10 mg·kg−1 anti-TENB2 mAb. A similar scenario was evident in large intestine, tracer uptake levels decreased from 5.7 ± 0.7 to 1.4 ± 0.2 and 1.5 ± 0.1 %ID g−1 but to a lesser extent with anti-STEAP1 mAb (4.3 ± 0.5 %ID g−1). Even after correcting for tissue blood volume using published values for murine tissues (Boswell et al., 2011a), the small and large intestines still emerge as the most probable underlying tissues responsible for the rapid clearance (data not shown).
Mass balance study re-affirms gut uptake and further implicates antibody catabolism and subsequent urinary excretion as the major route of whole-body clearance
To account for the rapid decline in radioactivity in blood and to further assess the contribution of gut uptake in overall distribution, we conducted a mass balance study using [111In]-anti-TENB2-MMAE alone (tracer only) or in combination with anti-TENB2 antibody. At 72 h post-injection, much lower levels of radioactivity in blood and higher levels of radioactivity in gut were observed for mice receiving tracer only relative to those receiving tracer plus 10 mg·kg−1 anti-TENB2 (Figure 4A). For instance, tracer uptake levels in blood in the tracer only and 10 mg·kg−1 groups were 2.3 ± 0.2 and 21 ± 2 %ID mL−1 respectively. Tracer levels for the same respective groups were 7.7 ± 0.4 and 2.5 ± 0.4 %ID g−1 in small intestine and 7 ± 3 and 1.9 ± 0.3 %ID g−1 in large intestine. Note that these values are consistent with the data shown in Figure 3B from a separate study. In addition, a 10-fold higher concentration of radioactivity was detected in the urine of mice receiving tracer only (9 ± 2 %ID mL−1) than at 10 mg·kg−1 (0.9 ± 0.1 %ID mL−1). Expression of these data in terms of percentage of injected dose (ID) for mass balance analysis highlights that the amount of radioactivity excreted in urine at the tracer only dose (22% ID) is of much greater magnitude than the amount bound or resident in the gut (5 and 2% ID for small and large intestines, respectively), consistent with whole-body urinary clearance of 111In-containing catabolites (Figure 4B). In contrast, no such discrepancy existed in faecal excretion of tracer between tracer only (4 ± 1 %ID) and 10 mg·kg−1 (3.5 ± 0.8 %ID) groups.
Figure 4.
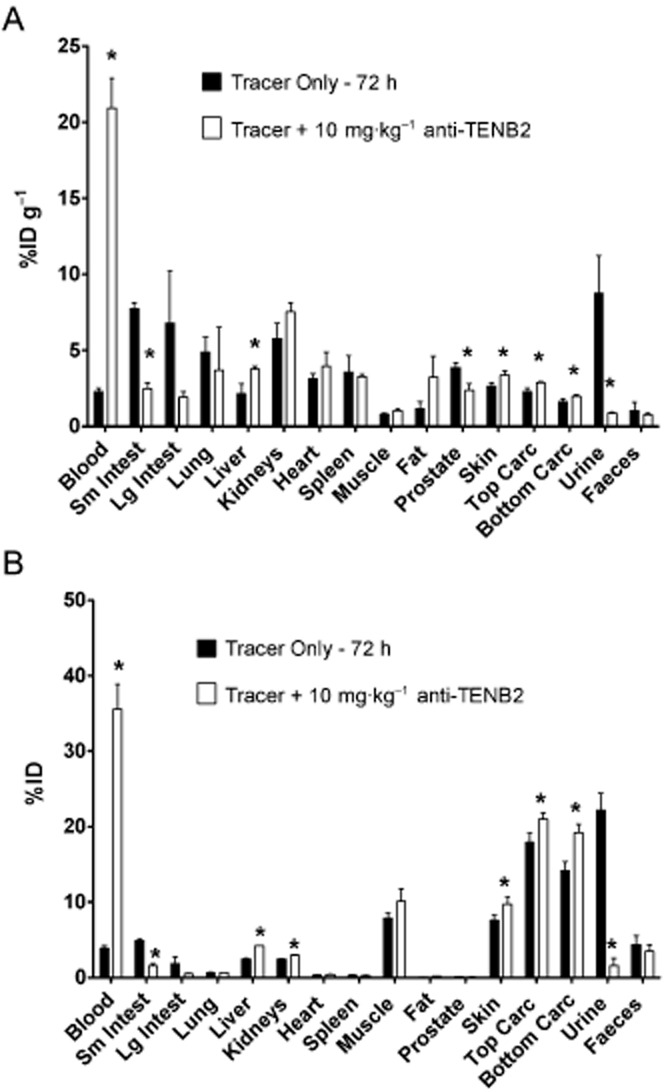
Comprehensive tissue distribution (A) and mass balance analysis (B) of [111In]-anti-TENB2-MMAE (ADC) in non-tumour-bearing SCID mice at 72 h after i.v. injection. The tracer was administered alone or in combination with unconjugated anti-TENB2 (ThioMab) at 10 mg·kg−1. Data are presented as % of injected dose g-1 tissue (A) or % of injected dose (B) as mean ± SD for three mice per group. ‘Carc’ denotes carcass remaining after tissue harvest. Asterisks indicate statistical significance (P < 0.05) by unpaired t-test.
As part of the mass balance study, we also characterized the 111In-labelled species present in plasma, the majority of which was consistent with intact ADC (Figure 5). Size-exclusion HPLC analysis shows the consistent presence of the radioimmunoconjugate [retention time (RT) ∼17 min] in plasma of mice receiving [111In]-anti-TENB2-MMAE tracer only (A) or a dose of 10 mg·kg−1 (B). In contrast, no intact protein and only radiolabelled catabolites (RT ∼24 min) were excreted in urine from mice receiving tracer only (D) or a dose of 10 mg·kg−1 (D). Given that radioimmunoconjugate stability should not depend on dose (or blood concentration), the presence of only intact protein (Figure 5B) and very low levels of excreted radioactivity (Figure 5D) at 10 mg·kg−1 demonstrates the in vivo stability of our radioimmunoconjugate.
Figure 5.
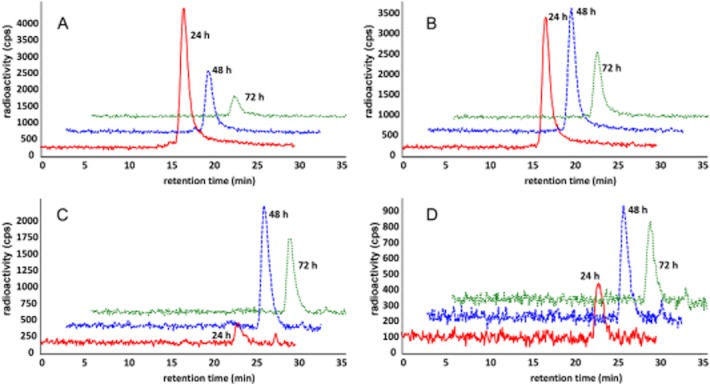
Radioactive anti-TENB2 was stable in plasma over the course of the study, and its catabolites were excreted in urine. No plasma protein complex formation was evident by HPLC analysis at 24 (red), 48 (blue) and 72 (green) h post-injection. Consistent presence of the radioimmunoconjugate [retention time (RT) ∼17 min, see Supporting Information Fig. S4] was observed in plasma of mice receiving [111In]-anti-TENB2-MMAE (ADC) at tracer only (A) or 10 mg·kg−1 (B) dose levels. In contrast, no intact protein and only radiolabelled catabolites (RT ∼24 min) were observed in urine from mice receiving tracer only (C) or 10 mg·kg−1 (D) dose. Note the difference in y-axis scale, particularly between (C) and (D).
SPECT-CT imaging confirms the elevated gut uptake with the tracer-only dose and highlights clearance through the urinary bladder
SPECT-CT fusion images (Figure 6) of SCID beige mice demonstrated both increased gut uptake and accelerated blood pool clearance in mice receiving tracer only compared with tracer plus unlabelled antibody. Imaging of mice administered [111In]-anti-TENB2-MMAE at tracer only (Figure 6A, C, E) or 10 mg·kg−1 (Figure 6B, D, F) dose levels at 3, 48 and 72 h after i.v. injection (A–B, C–D and E–F, respectively) showed distinct, time-dependent differences in liver, heart and gut intensity. Three-dimensional volume rendering images from sagittal (left) and coronal (centre) perspectives and coronal tomographic images (right) are shown in each panel. At the early 3 h time point, a noticeably stronger signal in the liver was present with the tracer only than at 10 mg·kg−1 (Figure 6A and B). A slightly stronger gut signal was evident at 48 h for the tracer-only subject than for the mouse administered 10 mg·kg−1 (Figure 6C and D). By 72 h, the whole-body clearance had already occurred at the tracer-only dose (Figure 6E) whereas significant heart and blood pool activity was still apparent following co-administration of 10 mg·kg−1 unlabelled antibody (Figure 6F).
Figure 6.
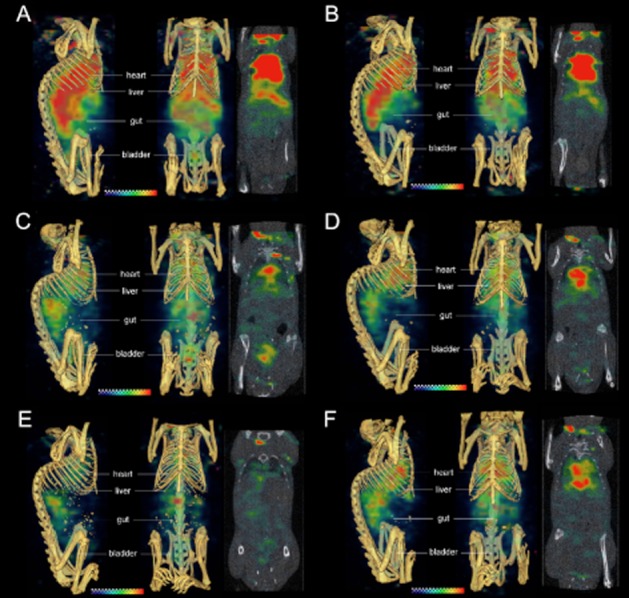
SPECT-CT fusion images of non-tumour-bearing SCID mice receiving [111In]-anti-TENB2-MMAE (ADC) at tracer only (A, C, E) or 10 mg·kg−1 (B, D, F) dose levels at 3, 48 and 72 h after i.v. injection (A–B, C–D and E–F respectively). Three-dimensional volume rendering images from sagittal (left) and coronal (centre) perspectives and coronal tomographic images (right) are shown in each panel. Note that the high signals above the thorax and below the animals' heads are both caused by reconstruction edge artefacts that are more severe at early time points when higher levels of radioactivity remain in the blood pool.
Immunohistochemistry
To determine the tissue expression of TENB2 in the mouse, we performed an immunohistochemical stain of normal mouse tissues using an anti-TENB2 antibody. TENB2 was found to be expressed at moderate to strong levels in the enterocytes of large intestines (Figure 7A), with low expression levels also present in the small intestines. Control antibody showed no staining in the same section (Figure 7B). Little to no specific TENB2 expression was detected in bone, kidney, liver and brain. These results are consistent with the gut acting as an antigen sink for TENB2.
Figure 7.
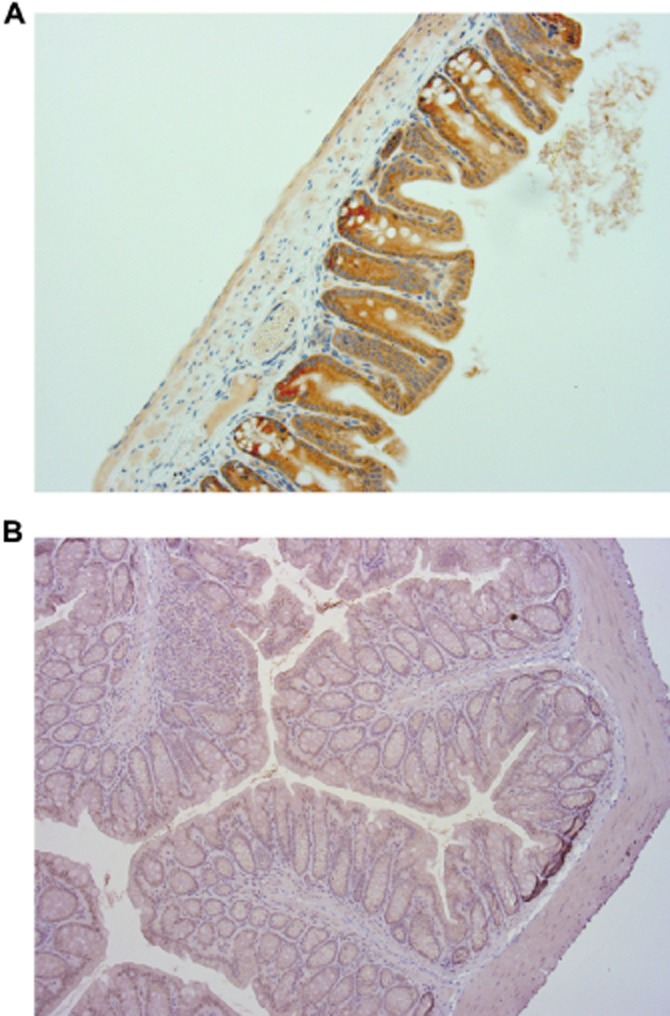
Representative immunohistochemical staining of TENB2 expression in the large intestine of a naïve, non-tumour-bearing SCID mouse. Large intestine exhibited moderate to strong staining by a TENB2-specific antibody (A) in a cytoplasmic distribution pattern. In contrast, a negative control antibody (B) did not result in staining of large intestine.
Antigen blockade with anti-TENB2 increases systemic exposure of radiolabelled anti-TENB2-MMAE in LuCaP 77 tumour-bearing mice
To demonstrate the selective tumour disposition of the ADC in vivo, we tested anti-TENB2-MMAE in the LuCaP 77 model. The radiolabelled ADC showed extremely rapid clearance and low overall exposure at the tracer level, similar to that observed in normal mice (Figure 8A and B). The dose-normalized blood exposures of tracer in the presence of low amounts (∼0.1 mg·kg−1) of anti-TENB2-MMAE, expressed as area under the concentration–time curve for 3 days (AUC0–3), were only 25 and 21 %ID mL−1 × days (in terms of 125I and 111In, respectively; data calculated from Figure 8A and B). For comparison, the analogous AUC0–3 value for trastuzumab (10 mg·kg−1), a typical non-binding humanized IgG1 with linear PK in mice, was 68 %ID g−1 × days (Boswell et al., 2010a). The rapid clearance of the cross-reactive anti-TENB2-MMAE reflects its dynamic interaction with both human and murine TENB2 in the LuCaP 77 tumour-bearing mouse model, resulting in extensive tissue and tumour uptake.
Figure 8.
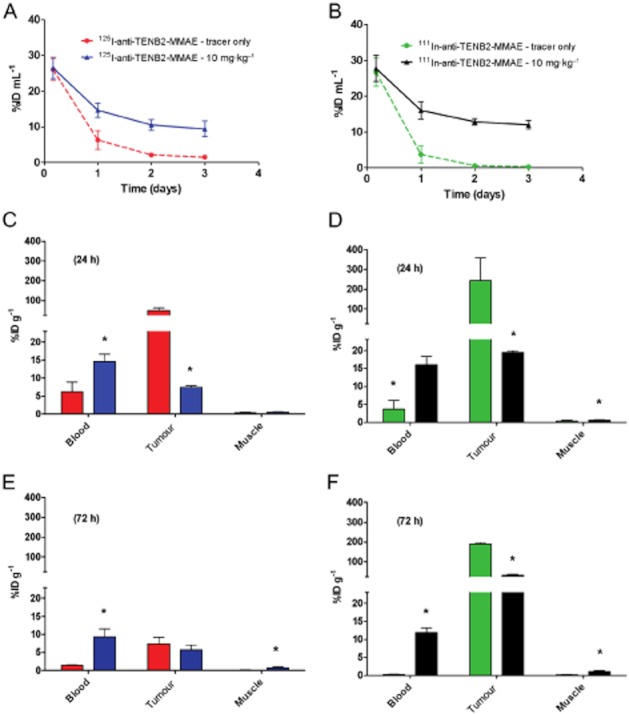
Comparative disposition of 125I- and 111In-labelled anti-TENB2-MMAE in the LuCaP 77 tumour explant model with and without a blocking dose of anti-TENB2. Dual-label tracers were co-administered at 0.1 mg·kg−1. Blood concentration–time profiles of anti-TENB2-MMAE labelled with 125I (A) and 111In (B) were obtained in mice (n = 3) that received no block (red and green, respectively) or an i.v. anti-TENB2 blocking dose of 10 mg·kg−1 (blue and black, respectively) 24 h before administration of the tracer. Tissue distribution (n = 3) of anti-TENB2-MMAE labelled with 125I (C, E) and 111In (D, F) in mice from the same four treatment groups was also performed at 24 (C, D) and 72 (E, F) h after radiotracer administration. *P < 0.05 by unpaired t-test.
At 4 h, blood concentrations from all dose groups with both tracers were comparable at ∼26 %ID mL−1 in both the 125I and the 111In energy window (Figure 8A and B) in mice that were blocked with 0 or 10 mg·kg−1 anti-TENB2 respectively. The modulation of systemic exposure from the antigen blockade emerged as time elapsed. At 48 h, in the blocked group, blood concentrations increased by ∼5-fold (11 ± 2 vs. 2 ± 0.2 %ID mL−1) and ∼20-fold (13 ± 0.9 vs. 0.6 ± 0.1 %ID mL−1) in the 125I and 111In energy windows respectively (Figure 8A and B). In addition, blood PK were largely independent of labelling method, indicating similar exposures of the two radioprobes.
Dual radiotracer study with non-residualizing (125I) and residualizing (111In) demonstrates the effect of antigen blockade on LuCaP 77 tumour localization and cellular internalization
Tumour uptake was strongly affected by both antigen blockade and labelling method. Pretreating with a saturating 10 mg·kg−1 dose of anti-TENB2 significantly inhibited the tumour uptake of radiolabelled ADC at 24 h (Figure 8C and D). Measuring 125I levels, tumour uptake at 24 h was reduced from 50 ± 14 to 7 ± 0.4 %ID g−1 in mice by blocking tumour uptake of 111In at 24 h was 243 ± 116 without block relative to 19 ± 0.5 %ID g−1 with block. However, by 72 h, 125I levels had decreased to 7 ± 2 with block relative to 6 ± 1 %ID g−1 without block, while 111In values were 189 ± 7 and 32 ± 4 %ID g−1 respectively. Higher tumour uptake was observed for 111In than 125I at all time points with or without a block. This reflects the residualizing nature of the radiometal 111In and the gradual accumulation of ADC in the tumour. In comparison to the tumour, uptake in muscle, a representative non-tumour antigen-expressing tissue, was low and unaffected by pre-dosing or the labelling method (Figure 8C–F).
Discussion and conclusions
Considerable efforts have been placed on understanding the PK (Stephan et al., 2008; Lin K et al., 2009b), tissue distribution (Mandler et al., 2004), metabolism (Austin et al., 2005; Perera et al., 2007; Doronina et al., 2008) and pharmacological effects (Alley et al., 2008; Dornan et al., 2009; Zheng et al., 2009) of ADCs. There is a limited understanding of interactions between antigen expression and ADC disposition, and their effect on efficacy and toxicity. The present work demonstrates a unique strategy for determining antigen-specific disposition of a potent drug conjugate, anti-TENB2-MMAE (Figure 1).
A significantly non-linear PK profile was observed for anti-TENB2-MMAE in mice, which presents an experimental model of an ADC target with an antigen sink. Tissue distribution studies were executed to explore the location(s) of the antigenic sink, the doses at which they were saturated, and the kinetics of tissue PK with respect to circulatory exposure. Non-invasive imaging further corroborated the identity of the tissue sink revealed by tissue distribution. Furthermore, a mass balance study was performed to characterize the rate of elimination of the radioimmunoconjugate under scrutiny to give further evidence for the site and mechanism of antigen-mediated clearance. The non-linear blood clearance of the ADC tracer was remarkably similar in either the presence (Figure 8A and B) or absence (Figure 3A) of a tumour, suggesting that the gut's role as an antigen sink has a greater influence than the tumour on systemic PK. While this may seem counterintuitive due to much higher TENB2 expression levels in tumour than in gut, one must consider that the small and large intestines are much larger in size than the tumours.
Both the mouse cross-reactive humanized anti-TENB2 mAb and its ADC demonstrated pronounced target-mediated disposition in mice, and indicated the presence of an antigen sink (Figure 2). Co-administration of 10 mg·kg−1 of the unconjugated anti-TENB2 antibody successfully blocked TENB2-specific interactions in the small and large intestines, with a less pronounced but significant (P < 0.05) change observed for the isotype-matched control IgG, anti-STEAP1 (Figure 3). This key finding implicated gut as the antigenic sink responsible, at least in part, for the target-mediated clearance observed in PK studies. In addition, dose-dependent hepatic uptake was observed at 3 h by SPECT-CT. However, liver uptake was not detected by imaging at 48 h and beyond or in the tissue distribution study at 72 h. Overall, these observations raise the possibility that at least some of the intestinal uptake may result from re-absorption of catabolites originating from hepatic metabolism. To address this concern, we repeated the study in rats and found that dose-dependent intestinal uptake occurred whether or not the animals were bile duct cannulated (data not shown); this finding suggests that the ADC is directly binding to antigen expressed in gut, rather than being a downstream consequence of hepatic uptake and subsequent metabolism. Concordantly, the positive IHC staining of murine intestine (Figure 7) combined with negative staining in the liver (data not shown) provides strong evidence for direct binding of anti-TENB2 to antigen on gut cells. Furthermore, a rigorous mass balance study utilizing metabolic cages demonstrated that urinary clearance of catabolites was an additional mechanism in accounting for the discrepancy between tracer and 10 mg·kg−1 dose levels (Figure 4). It is conceivable that the catabolites cleared through urinary excretion originated from the uptake and subsequent catabolism in gut tissues.
Collection and radiochromatographic analysis of plasma and urine samples demonstrated that, while the radioimmunoconjugate remained largely intact in blood, only radiolabelled catabolites were present in the urine (Figure 5), suggesting that lysosomal degradation of the antibody probably occurred within the cells of the intestines. Non-invasive SPECT-CT imaging was useful in (i) re-affirming the gut as a tissue sink; (ii) further indicating transient (3 h) hepatic uptake at the tracer-only dose; and (iii) delineating bladder uptake that was consistent with urinary excretion of the tracer-only dose (Figure 6). Finally, a positive immunohistochemical staining of gut tissue (Figure 7) validated the results from the tracer studies.
Our study design involved selective radiolabelling of the drug conjugate, anti-TENB2-MMAE, with indium to exploit the radiolabel as a surrogate marker for tissue-specific delivery of the potent small molecule drug. If cellular catabolism of radioimmunoconjugates parallels that of ADCs, then MMAE or other small molecule drug catabolites may have similar metabolic fates as compared with the expected radiolabelled catabolite, [111In]-DOTA-lysine (Rogers et al., 1995). If this is the case, then the uptake of radioindium may serve as a surrogate marker of MMAE uptake and/or retention in tissues. This would be particularly important for the current example, as unexpected gut uptake of 111In, and therefore, MMAE, could potentially have negative safety implications.
The LuCap 77 tumour explant model provided a platform to assess ADC uptake in both targets expressing normal tissue and tumour. As a proof of concept, we evaluated the disposition of anti-TENB2 ADC in this TENB2-expressing model and confirmed the modulation of exposure and tumour uptake upon blocking with a saturating dose of the unconjugated antibody. A potentially useful approach warranting further investigation is the idea of deliberately pre-dosing with selected amounts of unconjugated antibody to reduce uptake in non-target tissues. In our separate but related preclinical proof-of-concept study, pre-administration of 1 mg·kg−1 anti-TENB2 24 h before the radiolabelled ADC significantly increased blood exposure and reduced intestinal, hepatic and splenic uptake in mice while not affecting tumour accretion (Boswell et al., 2012b). However, we did not attempt to optimize the time window between unlabelled antibody and radiolabelled ADC. Furthermore, translating such a strategy to the clinic would require careful consideration of the relative amounts of off-target antigen expression and tumour burden in patients. Similar dosing strategies have been successfully developed for the radioimmunotherapeutic agents Zevalin and Bexxar (Boswell and Brechbiel, 2007).
In conclusion, we have utilized a combination of PK, tissue distribution, non-invasive imaging and immunohistochemical studies to characterize tissue-specific antigen expression of the prostate cancer target TENB2, thereby elucidating the underlying causes of target-mediated clearance. Concentration–time profiles of TENB2 in mice showed considerable non-linearity, indicating the presence of a target sink in normal murine tissue. For ADCs, target-mediated clearance is not only an important mechanistic component of antibody clearance, but also of critical importance for the therapeutic window and toxicity in normal tissue. Quantitative localization and characterization of drug uptake across relevant species is important for target validation, toxicity mitigation and therapeutic window projection. Given the sensitivity of ADC uptake in tissue to unlabelled unconjugated antibody and potency difference between them, this provides an opportunity to improve systemic exposure, to mask peripheral tissue uptake of potent and toxic ADC with benign antibody, and to improve the therapeutic window.
Acknowledgments
The authors would like to thank Shang-Fan Yu and Jennifer A. Lacap for tumour model expertise. We also thank Michelle Schweiger, Kirsten Messick, Jose Imperio, Bernadette Johnstone, Misia Bruski, Michael Reich and Jason Ho for animal studies support.
Glossary
- %ID g−1
percentage of injected dose per gram of tissue
- ADC
antibody-drug conjugate
- CL
clearance
- Cmax
maximum concentration
- DOTA
1,4,7,10-tetraazacyclododecane-N,N′,N″,N′″-tetraacetic acid
- IHC
immunohistochemistry
- mAb
monoclonal antibody
- MMAE
monomethyl auristatin E
- PK
pharmacokinetics
- SCID
severe combined immunodeficient
- SPECT-CT
single photon emission CT – X-ray CT
- STEAP1
six-transmembrane epithelial antigen of prostate 1
- TENB2 (i.e. TMEFF2 or tomoregulin)
transmembrane protein with EGF-like and two follistatin-like domains
Conflict of interest
All authors are employees of Genentech, Inc., a member of the Roche Group, and hold financial interest in Roche.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Sequence alignments of human and mouse TENB2. Human (NM_016192.2) and mouse (NM_019790.4) TENB2 (TmeFF2) protein sequences from the NCBI database were aligned using GSeqweb (Genentech, Inc.) for human and mouse TENB2 similarity analysis.
Figure S2 Flow cytometric analysis of anti-TENB2 binding to HEK cells (HEK293, obtained from ATCC) that were stably transfected with human or mouse TENB2. Cross-reactivity of anti-TENB2 to human and mouse TENB2 was evaluated in HEK cells (HEK293, obtained from ATCC) that were stably transfected with human or mouse TENB2. Cells were selected and maintained using G418 antibiotic solution (Invitrogen) at 400 μg·mL−1. Cells were grown to 90% confluence and removed from plates using Cell Dissociation Buffer (Invitrogen). Cells were washed and resuspended in fluorescence-activated cell-sorting (FACS) buffer (PBS with 1% BSA) and incubated for 45 min with humanized anti-TENB2 (ranging from 0.008 to 18 μg·mL−1) followed by a 30 min incubation with anti-human secondary antibody conjugated to phycoerythrin (anti-PE). Analysis was performed with a FACS Caliber flow cytometer (BD Biosciences).
Figure S3 Mass spectrometry data for anti-TENB2-MMAE (ADC) before (top) and after (bottom) DOTA conjugation. An aliquot of each protein sample was injected on a PLRP-S column [1000 Å, 50 × 2.1 mm, Varian (formerly Polymer Laboratories), Amherst, MA] maintained at 75°C with a linear gradient from 34–45% acetonitrile in 0.01% formic acid and water. The column eluent was directed by electrospray ionization into an Agilent 6220 time-of-flight mass spectrometer (Agilent Technologies, Santa Clara, CA) with fragmentor, capillary, skimmer and octupole RF voltages, respectively, at 415, 5500, 120 and 300 V. Data analysis was performed using the maximum entropy deconvolution algorithm within the Agilent Mass Hunter Qualitative Analysis software.
Figure S4 Size-exclusion HPLC radiochromatogram of an aliquot of the radioimmunoconjugate, 111In-anti-TENB2-MMAE (ADC), used for a tracer in tissue distribution and imaging studies. Size-exclusion high performance liquid chromatography (HPLC) characterization of all radioimmunoconjugates was carried out on a Phenomenex™ BioSep-SEC-S 3000, 300 × 7.8 mm, 5 μm column.
References
- Afar DE, Bhaskar V, Ibsen E, Breinberg D, Henshall SM, Kench JG, et al. Preclinical validation of anti-TMEFF2-auristatin E-conjugated antibodies in the treatment of prostate cancer. Mol Cancer Ther. 2004;3:921–932. [PubMed] [Google Scholar]
- Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, et al. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem. 2008;19:759–765. doi: 10.1021/bc7004329. [DOI] [PubMed] [Google Scholar]
- Alley SC, Zhang X, Okeley NM, Anderson M, Law CL, Senter PD, et al. The pharmacologic basis for antibody-auristatin conjugate activity. J Pharmacol Exp Ther. 2009;330:932–938. doi: 10.1124/jpet.109.155549. [DOI] [PubMed] [Google Scholar]
- Austin CD, Wen X, Gazzard L, Nelson C, Scheller RH, Scales SJ. Oxidizing potential of endosomes and lysosomes limits intracellular cleavage of disulfide-based antibody-drug conjugates. Proc Natl Acad Sci U S A. 2005;102:17987–17992. doi: 10.1073/pnas.0509035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai RL, Pettit GR, Hamel E. Binding of dolastatin 10 to tubulin at a distinct site for peptide antimitotic agents near the exchangeable nucleotide and vinca alkaloid sites. J Biol Chem. 1990;265:17141–17149. [PubMed] [Google Scholar]
- Boswell CA, Brechbiel MW. Development of radioimmunotherapeutic and diagnostic antibodies: an inside-out view. Nucl Med Biol. 2007;34:757–778. doi: 10.1016/j.nucmedbio.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boswell CA, Ferl GZ, Mundo EE, Schweiger MG, Marik J, Reich MP, et al. Development and evaluation of a novel method for preclinical measurement of tissue vascular volume. Mol Pharmaceutics. 2010a;7:1848–1857. doi: 10.1021/mp100183k. [DOI] [PubMed] [Google Scholar]
- Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem. 2010b;21:2153–2163. doi: 10.1021/bc100261d. [DOI] [PubMed] [Google Scholar]
- Boswell CA, Ferl GZ, Mundo EE, Bumbaca D, Schweiger MG, Theil FP, et al. Effects of anti-VEGF on predicted antibody biodistribution: roles of vascular volume, interstitial volume, and blood flow. PLoS ONE. 2011a;6:e17874. doi: 10.1371/journal.pone.0017874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boswell CA, Mundo EE, Zhang C, Bumbaca D, Valle N, Kozak K, et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjug Chem. 2011b;22:1994–2004. doi: 10.1021/bc200212a. [DOI] [PubMed] [Google Scholar]
- Boswell CA, Bumbaca D, Fielder PJ, Khawli LA. Compartmental tissue distribution of antibody therapeutics: experimental approaches and interpretations. AAPS J. 2012a;14:612–618. doi: 10.1208/s12248-012-9374-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boswell CA, Mundo EE, Zhang C, Stainton SL, Yu SF, Lacap JA, et al. Differential effects of predosing on tumor and tissue uptake of an 111In-labeled anti-TENB2 antibody-drug conjugate. J Nucl Med. 2012b;53:1454–1461. doi: 10.2967/jnumed.112.103168. [DOI] [PubMed] [Google Scholar]
- Bumbaca D, Xiang H, Boswell CA, Port RE, Stainton SL, Mundo EE, et al. Maximizing anti-neuropilin-1 tumour exposure requires saturation of non-tumour tissue antigenic sinks in mice. Br J Pharmacol. 2011;10:1476–5381. doi: 10.1111/j.1476-5381.2011.01777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris HA. Trastuzumab emtansine: a novel antibody-drug conjugate for HER2-positive breast cancer. Expert Opin Biol Ther. 2011;11:807–819. doi: 10.1517/14712598.2011.580273. [DOI] [PubMed] [Google Scholar]
- Carter PJ, Senter PD. Antibody-drug conjugates for cancer therapy. Cancer J. 2008;14:154–169. doi: 10.1097/PPO.0b013e318172d704. [DOI] [PubMed] [Google Scholar]
- Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey E, Vessella RL. Xenograft models of human prostate cancer. In: Chung LWK, Isaacs WB, Simons JW, editors. Prostate Cancer: Biology, Genetics, and the New Therapeutics. 2nd edn. Totowa, NJ: Humana Press; 2007. pp. 3–31. [Google Scholar]
- Corey E, Quinn JE, Vessella RL. A novel method of generating prostate cancer metastases from orthotopic implants. Prostate. 2003;56:110–114. doi: 10.1002/pros.10235. [DOI] [PubMed] [Google Scholar]
- Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nat Rev Cancer. 2002;2:389–396. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornan D, Bennett F, Chen Y, Dennis M, Eaton D, Elkins K, et al. Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood. 2009;114:2721–2729. doi: 10.1182/blood-2009-02-205500. [DOI] [PubMed] [Google Scholar]
- Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21:778–784. doi: 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- Doronina SO, Bovee TD, Meyer DW, Miyamoto JB, Anderson ME, Morris-Tilden CA, et al. Novel peptide linkers for highly potent antibody-auristatin conjugate. Bioconjug Chem. 2008;19:1960–1963. doi: 10.1021/bc800289a. [DOI] [PubMed] [Google Scholar]
- Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21:5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- Foyil KV, Bartlett NL. Brentuximab vedotin for the treatment of CD30+ lymphomas. Immunotherapy. 2011;3:475–485. doi: 10.2217/imt.11.15. [DOI] [PubMed] [Google Scholar]
- Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102:1458–1465. doi: 10.1182/blood-2003-01-0039. [DOI] [PubMed] [Google Scholar]
- Glynne-Jones E, Harper ME, Seery LT, James R, Anglin I, Morgan HE, et al. TENB2, a proteoglycan identified in prostate cancer that is associated with disease progression and androgen independence. Int J Cancer. 2001;94:178–184. doi: 10.1002/ijc.1450. [DOI] [PubMed] [Google Scholar]
- Hricak H, Choyke PL, Eberhardt SC, Leibel SA, Scardino PT. Imaging prostate cancer: a multidisciplinary perspective. Radiology. 2007;243:28–53. doi: 10.1148/radiol.2431030580. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–932. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koochekpour S, Zhuang YJ, Beroukhim R, Hsieh CL, Hofer MD, Zhau HE, et al. Amplification and overexpression of prosaposin in prostate cancer. Genes Chromosomes Cancer. 2005;44:351–364. doi: 10.1002/gcc.20249. [DOI] [PubMed] [Google Scholar]
- Lewis MR, Kao JY, Anderson AL, Shively JE, Raubitschek A. An improved method for conjugating monoclonal antibodies with N-hydroxysulfosuccinimidyl DOTA. Bioconjug Chem. 2001;12:320–324. doi: 10.1021/bc0000886. [DOI] [PubMed] [Google Scholar]
- Lin GA, Aaronson DS, Knight SJ, Carroll PR, Dudley RA. Patient decision aids for prostate cancer treatment: a systematic review of the literature. CA Cancer J Clin. 2009a;59:379–390. doi: 10.3322/caac.20039. [DOI] [PubMed] [Google Scholar]
- Lin K, Lou T, Ferl G, Leipold D, Graham R, Kozak KR, et al. Cross-species pharmacokinetic characterization of antibody drug conjugate TenB2-vc-E to understand target biology. 2009b. AAPS Meeting Abstracts 2009.
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandler R, Kobayashi H, Hinson ER, Brechbiel MW, Waldmann TA. Herceptin-geldanamycin immunoconjugates: pharmacokinetics, biodistribution, and enhanced antitumor activity. Cancer Res. 2004;64:1460–1467. doi: 10.1158/0008-5472.can-03-2485. [DOI] [PubMed] [Google Scholar]
- Meares CF, McCall MJ, Reardan DT, Goodwin DA, Diamanti CI, McTigue M. Conjugation of antibodies with bifunctional chelating agents: isothiocyanate and bromoacetamide reagents, methods of analysis, and subsequent addition of metal ions. Anal Biochem. 1984;142:68–78. doi: 10.1016/0003-2697(84)90517-7. [DOI] [PubMed] [Google Scholar]
- Michaelson MD, Cotter SE, Gargollo PC, Zietman AL, Dahl DM, Smith MR. Management of complications of prostate cancer treatment. CA Cancer J Clin. 2008;58:196–213. doi: 10.3322/CA.2008.0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastuskovas CV, Mundo EE, Williams SP, Nayak TK, Ho J, Ulufatu S, et al. Effects of anti-VEGF on pharmacokinetics, biodistribution, and tumor penetration of trastuzumab in a preclinical breast cancer model. Mol Cancer Ther. 2012;11:752–762. doi: 10.1158/1535-7163.MCT-11-0742-T. [DOI] [PubMed] [Google Scholar]
- Perera RM, Zoncu R, Johns TG, Pypaert M, Lee FT, Mellman I, et al. Internalization, intracellular trafficking, and biodistribution of monoclonal antibody 806: a novel anti-epidermal growth factor receptor antibody. Neoplasia. 2007;9:1099–1110. doi: 10.1593/neo.07721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravizzini G, Turkbey B, Kurdziel K, Choyke PL. New horizons in prostate cancer imaging. Eur J Radiol. 2009;70:212–226. doi: 10.1016/j.ejrad.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regino CA, Wong KJ, Milenic DE, Holmes EH, Garmestani K, Choyke PL, et al. Preclinical evaluation of a monoclonal antibody (3C6) specific for prostate-specific membrane antigen. Curr Radiopharm. 2009;2:9–17. doi: 10.2174/1874471010902010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers BE, Franano FN, Duncan JR, Edwards WB, Anderson CJ, Connett JM, et al. Identification of metabolites of 111In-diethylenetriaminepentaacetic acid-monoclonal antibodies and antibody fragments in vivo. Cancer Res. 1995;55(Suppl. 23):5714s–5720s. [PubMed] [Google Scholar]
- Rosenthal SA, Sandler HM. Treatment strategies for high-risk locally advanced prostate cancer. Nat Rev Urol. 2010;7:31–38. doi: 10.1038/nrurol.2009.237. [DOI] [PubMed] [Google Scholar]
- Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13:235–244. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]
- Shepard DR, Raghavan D. Innovations in the systemic therapy of prostate cancer. Nat Rev Clin Oncol. 2009;7:13–21. doi: 10.1038/nrclinonc.2009.187. [DOI] [PubMed] [Google Scholar]
- Stephan JP, Chan P, Lee C, Nelson C, Elliott JM, Bechtel C, et al. Anti-CD22-MCC-DM1 and MC-MMAF conjugates: impact of assay format on pharmacokinetic parameters determination. Bioconjug Chem. 2008;19:1673–1683. doi: 10.1021/bc800059t. [DOI] [PubMed] [Google Scholar]
- Teicher BA. Antibody-drug conjugate targets. Curr Cancer Drug Targets. 2009;9:982–1004. doi: 10.2174/156800909790192365. [DOI] [PubMed] [Google Scholar]
- Zhao XY, Schneider D, Biroc SL, Parry R, Alicke B, Toy P, et al. Targeting tomoregulin for radioimmunotherapy of prostate cancer. Cancer Res. 2005;65:2846–2853. doi: 10.1158/0008-5472.CAN-04-4019. [DOI] [PubMed] [Google Scholar]
- Zhao XY, Liu HL, Liu B, Willuda J, Siemeister G, Mahmoudi M, et al. Tomoregulin internalization confers selective cytotoxicity of immunotoxins on prostate cancer cells. Transl Oncol. 2008;1:102–109. doi: 10.1593/tlo.08124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Fuji RN, Elkins K, Yu SF, Fuh FK, Chuh J, et al. In vivo effects of targeting CD79b with antibodies and antibody-drug conjugates. Mol Cancer Ther. 2009;8:2937–2946. doi: 10.1158/1535-7163.MCT-09-0369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.