

Abstract
Background and Purpose
Anti-complement therapies have not been advanced for treating the inflammatory bowel diseases (IBDs) despite a growing body of evidence that blocking C5a protects against induced colitis in rodents. The purpose of this study was to further build on this evidence by examining the efficacy, mechanism and specificity of a potent, non-competitive and orally active C5a receptor (CD88) antagonist, PMX205, in the dextran sulphate sodium (DSS) model of murine innate colitis.
Experimental Approach
Mice with DSS added to their drinking water were orally administered 100 or 200 μg day−1 PMX205 in prophylactic and therapeutic regimens. Clinical illness, colon histology and local generation of inflammatory mediators were measured to evaluate the impact of PMX205 on disease.
Key Results
PMX205 significantly prevented DSS-induced colon inflammation in both regimens, associated with lower pro-inflammatory cytokine production and nitrotyrosine staining in colon sections. Additionally, the levels of anti-inflammatory cytokines IL-4 and IL-10 were increased. PMX205 had no significant effect on C5a levels. The beneficial effect of PMX205 was seen in two strains of mice of differing sensitivities to DSS inflammation, but was inactive in mice lacking CD88.
Conclusions and Implications
Pharmacological inhibition of C5a activity by PMX205 is efficacious in preventing DSS-induced colitis, providing further evidence that targeting CD88 in IBD patients could be a valuable therapeutic option.
Keywords: CD88, complement, anaphylatoxin, C5a, PMX205, DSS, IBD
Introduction
The inflammatory bowel diseases (IBDs), ulcerative colitis (UC) and Crohn's disease (CD) are disorders of the gastrointestinal tract characterized by chronic relapsing/remitting inflammation and mucosal damage. UC affects the large intestine and has contiguous mucosal lesions proceeding proximally from the rectum whereas CD can affect any part of the gastrointestinal tract and can include discontinuous but transmural lesions with granulomas. Various environmental factors combined with a genetic predisposition have been implicated in the pathogenesis of IBD (Satsangi et al., 2003; Hanauer, 2006; Molodecky and Kaplan, 2010). No single cure for either disease is available. While the first choice of therapies remains steroids and immune modulators, research into cytokine and chemokine networks continues to identify candidate therapeutic targets. One example of a target derived from the cytokine network is TNF, and the anti-TNF drugs have proven effective in a significant fraction of patients that fail the available choices of therapies; however, due to diversity in disease presentation, infectious risks and reactions to the drugs, still not all the patients benefit (Stallmach et al., 2010). Thus, the need to develop further targets remains, and importantly, pharmaceuticals that can be administered conveniently, for example, orally, in a chronic fashion.
One innate inflammatory mechanism that has not been well characterized during IBD is the complement cascade. This is despite evidence of increased mucosal and submucosal deposition of complement components and elevated expression of anaphylatoxins in IBD patients (Halstensen and Brandtzaeg, 1991; Chen et al., 2011). Patient studies are not yet available that determine whether split complement components might exacerbate the disease. Instead, recent investigations have focused on a possible link between UC and mutations that result in low concentrations of mannan binding lectin and a consequential risk of failing to respond to microbes (Sivaram et al., 2011).
Complement activation results in the generation of two major anaphylatoxins: C3a and C5a. C5a in particular is a potent inflammatory mediator and signalling through CD88 mediates chemotaxis of leukocytes, increased vascular permeability, inflammatory mediator release from mast cells and smooth muscle cell contraction, among other activities, but all of which contribute to inflammation and possibly tissue damage (Monk et al., 2007). Consequently, CD88 has been targeted as a treatment for various inflammatory conditions. To this end, two CD88 antagonists, PMX53 [AcF-(OPdChaWR)] and PMX205 [hydrocinnamate-(OPdChaWR)], have proven beneficial in both rat and murine models of acute and chronic inflammation (Woodruff et al., 2006; 2008; Fonseca et al., 2009). The PMX compounds have been shown to be protective in delayed-type hypersensitivity (trinitrobenzene sulfonic acid or TNBS) colitis in rats (Woodruff et al., 2003; 2005), as has a C5a blocking antibody used in mice (Chen et al., 2011). This was predictable considering that C5a has been directly implicated in the delayed-type hypersensitivity response (Tsuji et al., 2000). Yet these positive findings contrast a report using CD88 gene-deficient mice which, despite reduced clinical disease, had similar levels of colon pathology as wild-type mice after 8 days of dextran sulphate sodium (DSS)-induced colitis (Johswich et al., 2009). The TNBS model resembles CD whereas DSS colitis, which will occur in T cell-deficient animals (Dieleman et al., 1994), more closely resembles UC and responds to drugs similar to the response seen in UC (Melgar et al., 2008; Solomon et al., 2010). Therefore, there is uncertainty over whether blocking CD88 will have a beneficial effect in the DSS model of UC. To address this gap, we evaluated the effect of PMX205 in the DSS model. Our results indicate that PMX205 is indeed efficacious in a strain-independent manner in DSS colitis, through the specific inhibition of CD88, and includes a shift in the cytokine balance that favours anti-inflammatory IL-4 and IL-10.
Methods
Mice
Six- to eight-week-old mice were used for all the experiments. Specific pathogen-free male BALB/c and C57BL/6 (wild-type) strains were obtained from Charles River (St Constant, QC, Canada). They were acclimatized in the Health Centre animal facility for 1 week before experimentation. CD88 gene-deficient mice (CD88–/–, BALB/c background) were a generous gift from Dr Craig Gerard (Harvard University, Boston, MA, USA). (The receptor names conform to the standards identified in the Guide to Receptors and Channels (Alexander et al., 2011). These mice were bred and experiments were undertaken in the IWK Health Centre under the approval of the University Committee on Laboratory Animals, Dalhousie University. All the animals were housed in rooms maintained at 21°C in a 12 h dark and light cycles, and had free access to food and water. One hundred three animals were used in the study.
The results of all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010).
Induction of colitis and PMX205 treatment
Colitis was induced by administering DSS [36 000–50 000 mw from MP Biomedicals (Solon, OH, USA), 5% w/v for BALB/c, CD88–/– mice and 3% w/v for C57BL/6 mice] in their drinking water ad libitum from day 1 to day 6, followed by facility drinking water for an additional day. DSS was replaced every other day. While alert, mice underwent oral feeding of distilled water (control) or PMX205 [hydrocinnamate-(OPdChaWR)] at a dose of 100 μg per mouse (∼4–6 mg kg−1) or 200 μg per mouse (∼8–10 mg kg−1) dissolved in distilled water, daily, either starting 24 h before the addition of DSS (e.g. day 0) or after 48 h of DSS (day 3) and continuing until day 6. Untreated mice were kept on facility water from day 1 to day 7.
Assessment of clinical illness in DSS-induced colitis
Daily assessment of clinical disease for each mouse included measurements of body weight, stool consistency and the presence of blood in stool using Hemoccult developer (Beckman Coulter, Brea, CA, USA), as reported elsewhere (Stillie and Stadnyk, 2009). Clinical scores range from 0 (no weight loss, normal stool) to 10 (>20% weight loss, diarrhoea with blood in stool).
Blood collection and microscopic colon assessment
At the end of the treatment period, mice were anaesthetized using isoflurane and blood was collected by cardiac puncture, then the mice were killed by cervical dislocation while still under anaesthesia. The blood was left to clot for 30 min at room temperature, then serum was collected by centrifugation and stored at −20°C until further use. The colon was excised post-mortem and the length was measured. The colons were flushed with cold PBS and opened longitudinally and divided into three longitudinal parts. One full-length part was utilized for histology whereas the others were used for explant cultures and homogenates.
The colon part reserved for histology was prepared in a Swiss roll and fixed in 10% neutral buffered formalin overnight then dehydrated in ethanol and processed for paraffin embedding and sectioning. Four micron thick sections were stained with haematoxylin and eosin. Colon inflammation scoring was graded by an investigator, blinded to the treatments, based on the following parameters: extent of oedema (present, 1 or absent, 0), crypt damage (0–5), infiltration of cells (0–5) and ulcers (0–3) as described in detail elsewhere (Stillie and Stadnyk, 2009). The score for each parameter was subsequently added to obtain the total inflammation score, with a maximum of 14.
Another longitudinal part of colon was weighed and washed with PBS containing penicillin (100 U mL−1) and streptomycin (100 μg mL−1), then placed in a well of a 12-well tissue culture plate (Costar: 3513, Corning, NY, USA) containing 1 mL of high glucose DMEM (Invitrogen, Burlington, ON, Canada) supplemented with penicillin (100 U mL−1) and streptomycin (100 μg mL−1), 0.5% FBS, 10 mM HEPES, 2 mM L-glutamine and 50 μM 2-mercaptoethanol. Organ cultures were incubated at 37°C for 24 h. The supernatants were then collected and stored at −20°C until further use.
Another longitudinal part of the colon was homogenized in 50 mM HEPES buffer (4 μL mg−1), supplemented with soybean trypsin inhibitor (100 μg mL−1) and centrifuged at 16000× g for 30 min at 4°C. Supernatants were stored at −20°C for later estimation of anaphylatoxins. The cell pellet was again homogenized in 0.5% cetyltrimethylammonium chloride (4 μL mg−1) and centrifuged at 16000× g for 30 min at 4°C and the supernatant was collected for measuring myeloperoxidase (MPO).
In some experiments, the third colon part was homogenized in RIPA buffer containing a cocktail of protease and phosphatase inhibitors (Sigma, St. Louis, MO, USA). Total protein concentration was estimated using the Bradford assay and the homogenates were utilized for Western blot analysis.
Measurement of pro-inflammatory mediators
IL-6, CXCL2, IL-12, IL-1β, TNF and IL-4 were measured in the explant culture supernatants using elisa kits from Peprotech (Dollard, QC, Canada). An elisa kit from eBiosciences (San Diego, CA, USA) was used for IL-17A. C3a and C5a concentrations were determined utilizing elisa reagents from BD Pharmingen (Mississauga, ON, Canada). For IL-10 detection, capture antibody (clone JES5-16E3) and biotinylated detection antibody (clone JES5-2A5) were purchased from eBiosciences. All the protocols were performed according to the manufacturers' instructions.
MPO assay
MPO was estimated as described elsewhere (Yang et al., 2011). Briefly, equal volumes (75 μL) of samples and substrate (3 mM 3,3′,5,5′-tetramethylbenzidine dihydrochloride, 120 μmol L−1 resorcinol and 2.2 mmol L−1 H2O2) were mixed and incubated at room temperature for 2 min. The reaction was stopped by adding 2 M sulphuric acid (150 μL) and optical density was measured at 450 nm.
Immunohistochemistry
Four micron thick tissue sections were deparaffinized and incubated with either rat anti-mouse Ly6G antibody (1:4000; BD Pharmingen), F4/80 (1:100; Serotec, Raleigh, NC, USA) or anti-nitrotyrosine (NT) antibody (1:500; Millipore, Billerica, MA, USA) overnight at 4°C. For arginase I, mouse anti-mouse arginase I antibody (1:250; BD Biosciences, San Jose, CA, USA) was used and slides were stained utilizing a mouse on mouse detection kit, according to the manufacturer's instructions (Vector Laboratories, Burlington, ON, Canada). The tissue sections were then incubated with biotinylated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and then with a peroxidase conjugate (Vector Laboratories). Staining was developed using diaminobenzidine and counterstained with haematoxylin. Cells were counted in the mucosa and submucosa in 20 different high power fields (400×) per section and averaged among all mice per group. The results are expressed as number of cells per 10 high power fields. For NT, the whole length of the colon was examined and scored as follows: 0 = less than 25% stained, 1 = 25–50% stained, 2 = 50–75% stained and 3 = more than 75% stained (Mañé et al., 2009).
Statistical analysis
Statistical analysis was performed with GraphPad Prism version 5 (La Jolla, CA, USA). Parametric data are shown as mean ± SEM. Colon lengths, inflammatory mediators and granulocyte count comparisons were analysed using either t-test (two groups) or one-way anova with Tukey's multiple comparison tests as a post hoc test. Body weights were compared using two-way anova with Bonferroni post-test. Clinical illness, total inflammation scores and NT quantifications are shown as median score and were compared using either Mann–Whitney (two groups) or Kruskal–Wallis test (more than two groups) with Dunn's multiple comparison post-test. A significant difference was defined as P < 0.05.
Results
The generation of C3a and C5a in DSS colitis and effect of PMX205
Johswich et al. reported that C3a levels in the blood of mice with DSS colitis were increased (Johswich et al., 2009). Considering that C5a levels have not been reported in DSS inflamed mice and in order to directly confirm that DSS leads to complement activation in the colon, levels of C3a and C5a were determined by elisa from colon explant supernatants and colon homogenates of healthy and inflamed mice. Anaphylatoxin levels were detectable above the threshold of the elisa assay in supernatants from healthy BALB/c mice, but were significantly increased in supernatants harvested from colons at the end of the DSS cycle (Figure 1). Being a selective CD88 antagonist, PMX205 presumably should not have any direct effect on complement activation nor on the local generation of C3a and C5a. As predicted, no difference was observed in the C3a (Figure 1A) or C5a (Figure 1B) levels in the colon supernatants between the DSS control and PMX205-treated groups. C3a and C5a concentrations showed similar patterns in colon homogenates (data not shown). Having established that anaphylatoxin levels are increased due to DSS and that PMX205 does not significantly alter this increase, we next examined mice for illness and inflammation.
Figure 1.
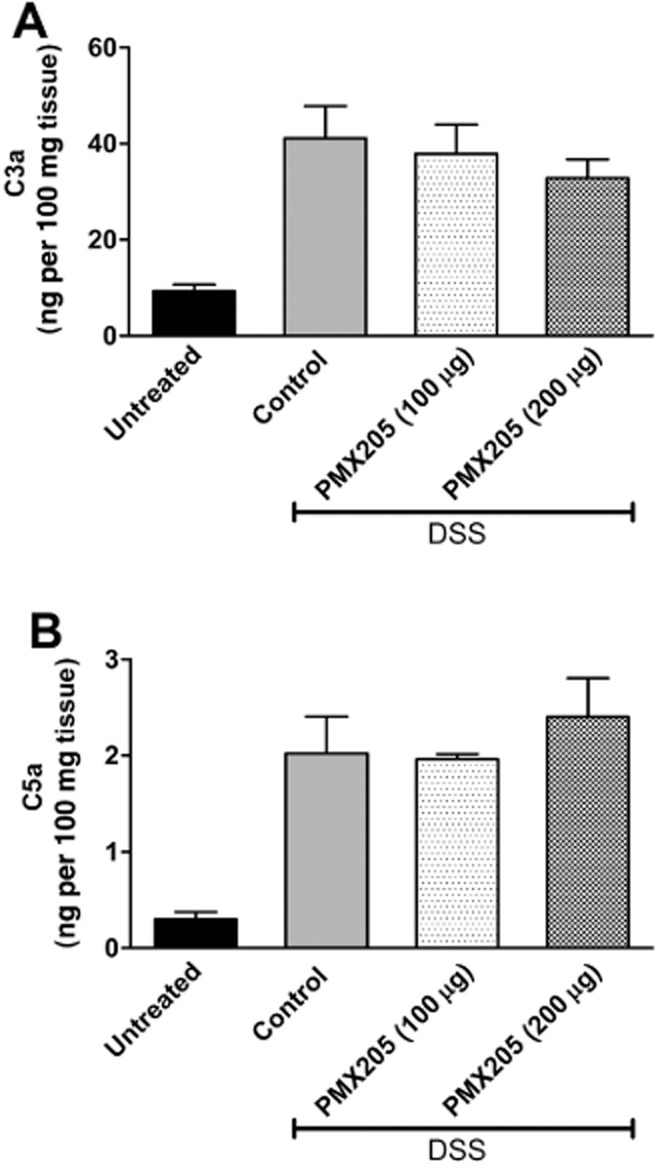
DSS-induced local generation of C3a and C5a is not altered by PMX205. BALB/c mice were fed 5% DSS in water and treated with either water (control) or PMX205 (100 or 200 μg per mouse) in a prophylactic regimen until day 6. Untreated mice were kept on facility water. On the day of killing, the colon was excised and explant supernatants were analysed for C3a (A) and C5a (B) by ELISA. Values are shown as mean concentrations ± SEM (n = 5–11 mice per group). All DSS-treated groups are significantly different from the untreated group.
PMX205 prevents DSS-induced colon damage in BALB/c mice
Mice administered 5% DSS in drinking water for 5 days developed illness marked by weight loss, diarrhoea, and occult blood or visible blood in their stool. There was no mortality among the mice at this concentration of DSS. Mice were gavaged with either 100 or 200 μg of PMX205 daily, and it was observed that the dose of 200 μg per mouse more effectively prevented the clinical manifestations associated with DSS. The average maximum weight loss among control mice was 8% as compared with 4% in the PMX205 (200 μg day−1) group (Figure 2A). The clinical illness score was significantly lower in the PMX205-treated animals (200 μg day−1) (Figure 2B). Shortening colon lengths is an objective indicator directly associated with pathology during DSS colitis, and there was a difference between the two groups, with PMX205-treated mice (200 μg) having, on average, statistically significantly longer colons (Figure 2C).
Figure 2.
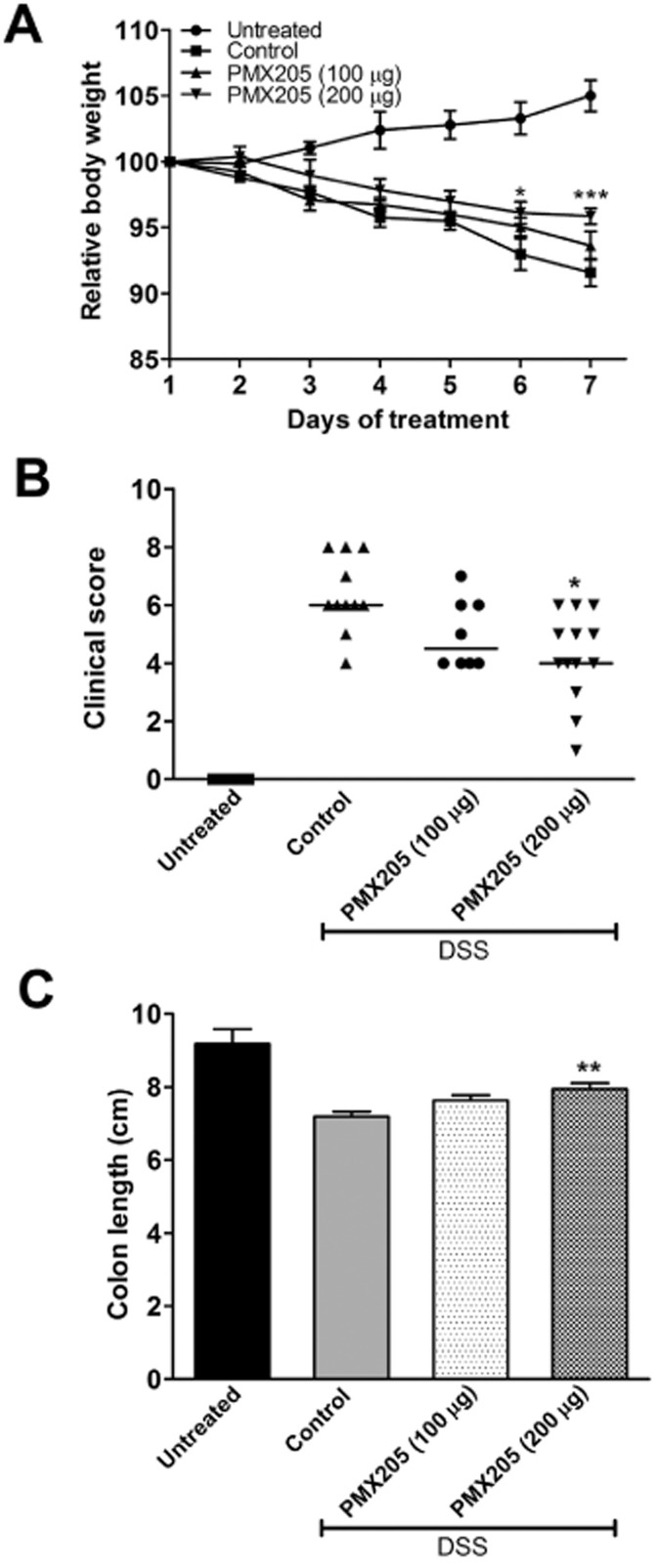
Effect of PMX205 on DSS-induced body weight loss, clinical disease and colon length in BALB/c mice. BALB/c mice had 5% DSS added to their water for 5 days. PMX205 (100 or 200 μg/mouse) or water (control) was administered orally beginning 1 day prior (Day 0) to the DSS start. The untreated group was kept on facility water until day 7. (A) Each animal's weight was measured daily and is expressed as the percentage of their weight on day 1. Shown are the averages ± SEM (n = 5–13 mice per group). (B) Clinical illness scores were derived from combinations of weight loss, stool consistency and presence of blood in stool. (C) Colon lengths from the groups were measured after killing. Results are expressed as mean colon length ± SEM (n = 5–13 per group). *P < 0.05, **P < 0.01 and ***P < 0.001 versus controls. Although different from the DSS control group, the PMX205 group also remained statistically different from the untreated group.
Post-mortem microscopic examination of the BALB/c colons revealed DSS-induced damage including mucosal ulcerations with a highly infiltrated mucosa, dilatation of crypts and oedema of the submucosa. In contrast, BALB/c mice given oral treatment with PMX205 (200 μg day−1) resulted in significant attenuation in inflammation characterized by less submucosal oedema, cellular infiltrate, and protection against crypt damage and epithelial loss (Figure 3A). Fewer ulcers were present along the length of the colon in PMX205-treated animals. Indeed, the total inflammation score of the PMX205-treated group (200 μg) was statistically significantly lower than the score of control mice (Figure 3B). Although administration of 100 μg per mouse PMX205 showed less illness, statistical significance was not reached when compared with controls. Hence, for all further experiments, PMX205 was used at 200 μg day−1 per mouse.
Figure 3.
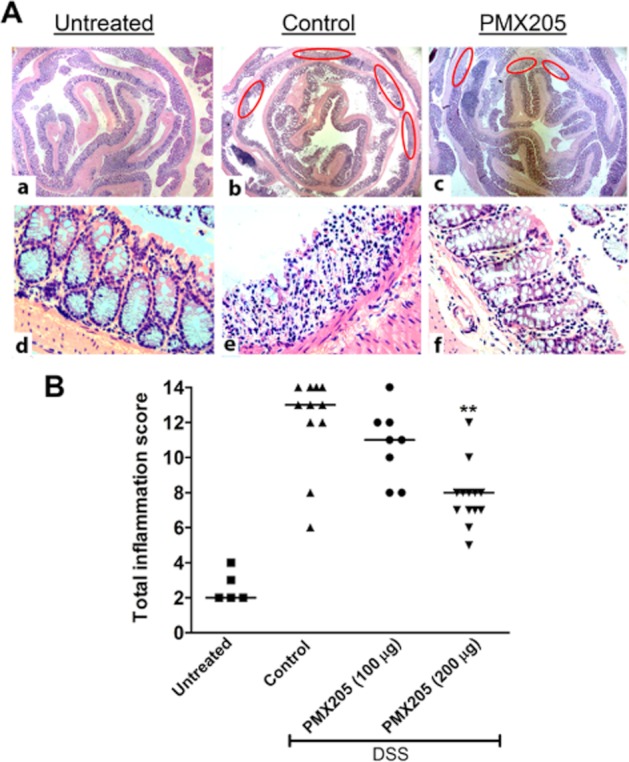
PMX205 attenuates DSS-induced histopathological features in BALB/c mice. (A) Representative figures of the colon prepared from (a,d) untreated, (b,e) control and (c,f) PMX205 groups. In (b,c) ovals identify the ulcers along the length of the colon. Compared with the control group, PMX205-treated mice typically showed fewer and shorter ulcers, less crypt damage, fewer mice with oedema and less of the colon was infiltrated. (B) The inflammation score was determined from these criteria by an investigator blinded to the treatment. Each dot is the data from a single mouse and the line represents the median of the inflammation scores. **P < 0.01 versus controls.
Effect of PMX205 on cellular infiltration
DSS colitis is characterized by massive infiltration by neutrophils and macrophages (Stevceva et al., 2001), and C5a is a known chemoattractant for these cell types (Snyderman et al., 1970; 1975). Consequently, we sought to determine whether protection by PMX205 was associated with changes in the numbers of either leukocyte type. Colon sections were stained for Ly6G (neutrophils, Figure 4A) and F4/80 (macrophages, Figure 4B). Enumerating Ly6G-positive neutrophils showed a significant difference between DSS-treated groups in the mucosa but not in the submucosa (Figure 4C). No significant difference was found in F4/80 stained cell numbers between the two groups (Figure 4D). In addition, an MPO assay was performed. As shown in Figure 4E, the MPO concentration was significantly higher in both DSS groups compared with untreated mice, but no significant difference was observed between control and PMX205 groups.
Figure 4.
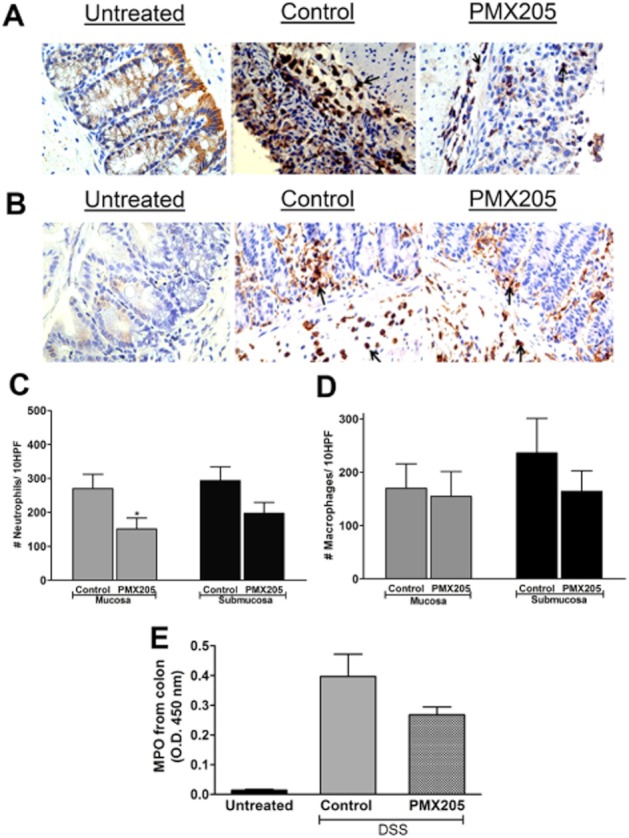
Effect of PMX205 on DSS-induced granulocyte infiltration and myeloperoxidase: Representative examples of immunohistochemical staining for (A) Ly6G and (B) F4/80. Quantification of stained cell numbers of (C) neutrophils and (D) macrophages in the mucosa and submucosa of DSS groups. Data are shown as mean ± SEM (n = 5–8/group). Original magnification: 400×. (E) MPO levels were not significantly different between the DSS groups although both groups were different from untreated values. Values shown are mean O.D. ± SEM (n = 5/group). *P < 0.05. An isotype control antibody did not show any visible staining (data not shown).
As C5a has been reported to delay as well as enhance cell apoptosis depending on the cell type (Perianayagam et al., 2002; Pavlovski et al., 2012), we explored whether blocking CD88 modified the number of cells undergoing apoptosis in the colon. No difference in colonic-activated caspase-3 between the DSS groups was detected using Western blot or immunohistochemistry (data not shown).
PMX205 prevents the production of pro-inflammatory cytokines
Because high production of inflammatory cytokines is associated with IBD, we next sought to determine the effect of PMX205 on the production of local pro-inflammatory markers. All pro-inflammatory cytokines measured were significantly up-regulated by DSS compared with the untreated group (statistics not shown). PMX205 treatment resulted in significantly less production of IL-1β, IL-6 and TNF compared with controls (Figure 5). Differences between the two DSS groups of all other cytokines did not achieve statistical significance. Colonic supernatants were also analysed for the production of IL-10 and IL-4, both anti-inflammatory cytokines. IL-4 and IL-10 (Figure 5G,H) were significantly elevated in the PMX205 group compared with control group concentrations.
Figure 5.
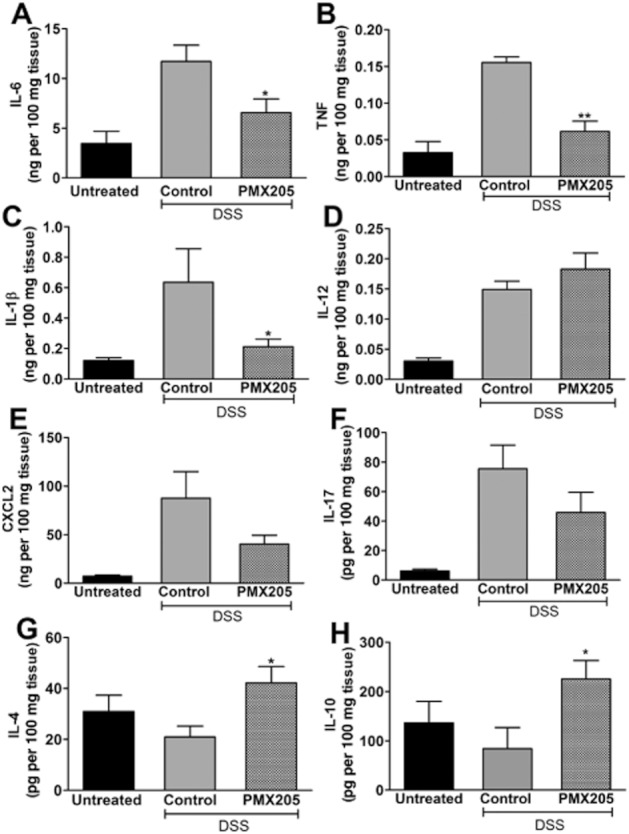
PMX205 prevents the production of pro-inflammatory cytokines and induces the production of IL-4 and IL-10. Colon culture supernatants from untreated, PMX205 or water-treated DSS inflamed mice were analysed for the production of (A) IL-6, (B) TNF, (C) IL-1β, (D) IL-12, (E) CXCL2, (F) IL-17, (G) IL-4 and (H) IL-10. Values shown are mean concentrations ± SEM (n = 5–10 mice per group). In panels A through F, the cytokine levels of the DSS control colons are significantly different from untreated levels but not in G and H, shown in the figure. *P < 0.05, **P < 0.01 PMX205 versus DSS controls.
Effect of PMX205 on NT and arginase I expression
C5a stimulation of murine macrophages polarizes the cells towards the M1 phenotype with increased IL-6, TNF and low IL-10 (Langer et al., 2010). Alternatively, M2 macrophages (high IL-0 producers) protect mice from colitis (Hunter et al., 2010). The reduction in IL-6 and TNF but increased IL-10 raised the possibility of a phenotype shift from M1 to M2 macrophages. We stained colon sections for NT (a marker of M1 and product of NO) (Figure 6A) and arginase I (marker of M2). The NT staining intensity was significantly less in the PMX205-treated group (Figure 6B). This observation of reduced NO activity was despite similar numbers of arginase positive cells in the two groups (data not shown).
Figure 6.
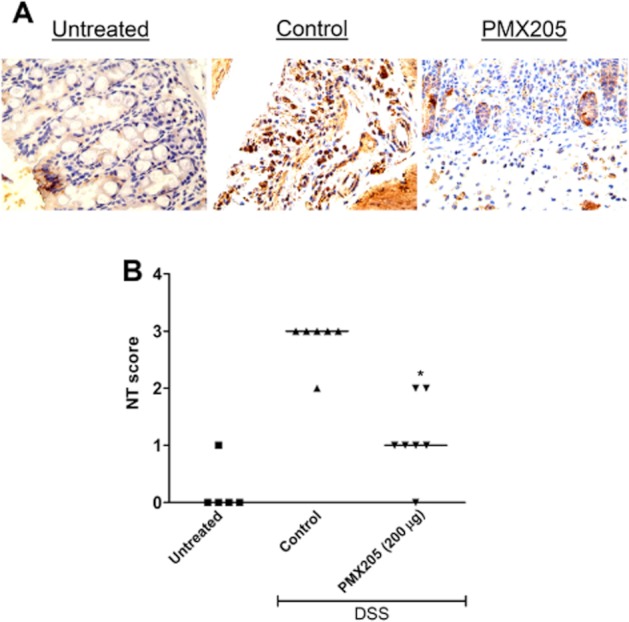
Effect of PMX205 on NO activity estimated by NT staining of colon sections. (A) Representative examples of immunohistochemical staining for NT. (B) Quantitation of the staining signal identifies a significant reduction in the intensity of NT in the PMX205 group. An isotype control antibody did not show any visible staining (data not shown). *P < 0.05 versus control.
PMX205 does not protect from DSS induced colitis in CD88−/− mice
Johswich and co-workers showed that CD88–/– mice are not protected from colon inflammation due to DSS (Johswich et al., 2009). To confirm whether PMX205-mediated protection is due to its interaction with CD88 and not because of off-target effects, we used it prophylactically at a dose of 200 μg day−1 in DSS-inflamed BALB/c mice lacking CD88. Figure 7A shows that the average colon lengths of the two groups were not significantly different. Microscopically, DSS administration to CD88–/– animals resulted in cellular infiltration, oedema, ulcers and extensive crypt loss, and PMX205 treatment did not result in any significant improvement in this histopathology (Figure 7B,C), supporting that PMX205 is specific for CD88.
Figure 7.
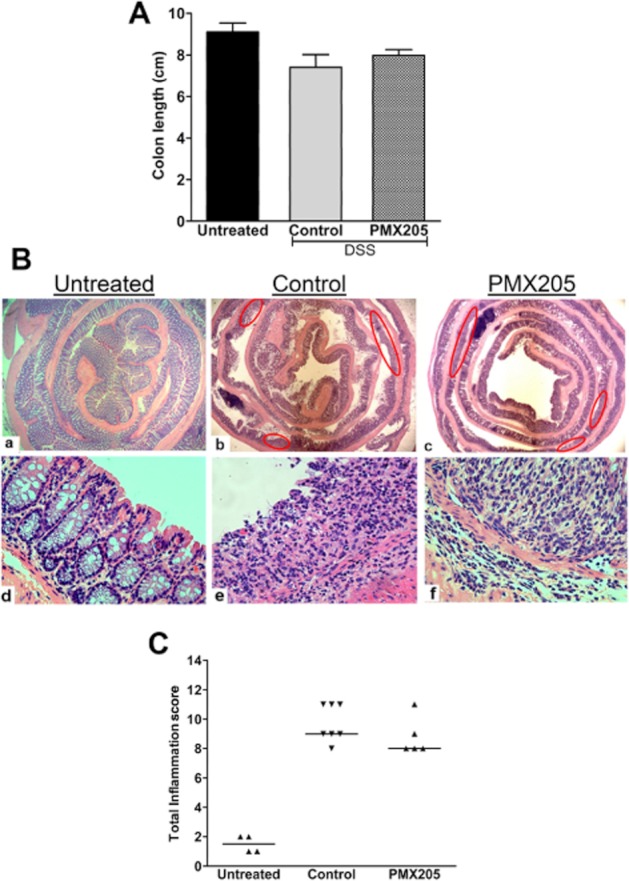
PMX205 did not abrogate DSS-induced damage in mice lacking CD88. PMX205 was used in the prophylactic regime in CD88−/− mice to test the drug specificity for CD88. (A) PMX205 had no protective effect on colon shortening due to the DSS. Shown are the mean colon lengths ± SEM (n = 5–7 mice per group). (B) Representative sections of the colon of inflamed CD88−/− mice. Red ovals identify ulcers. (C) Inflammation scores derived from the histology; each dot represents a single mouse and the line represents the median score.
PMX205 ameliorates DSS-induced colitis in C57BL/6 mice
C57BL/6 mice are highly susceptible to DSS and their colitis may develop into a chronic disease (Melgar et al., 2005), implying that mechanisms different from the BALB/c strain may be occurring. Therefore, to test whether the benefit behind CD88 blockade is strain dependent, we used PMX205 in C57BL/6 mice. We used 3% DSS in the drinking water in the protocol, a dose that in our experience results in colitis of a similar magnitude to 5% DSS in BALB/c mice. DSS-inflamed C57BL/6 mice showed body weight loss and colon shortening. Similar to the BALB/c mice, prophylactic oral treatment with PMX205 abrogated this body weight loss (Figure 8A) and prevented colon shortening (Figure 8B). The colon inflammation scores confirmed a significant improvement in the PMX205-treated C57BL/6 mice (Figure 8D).
Figure 8.
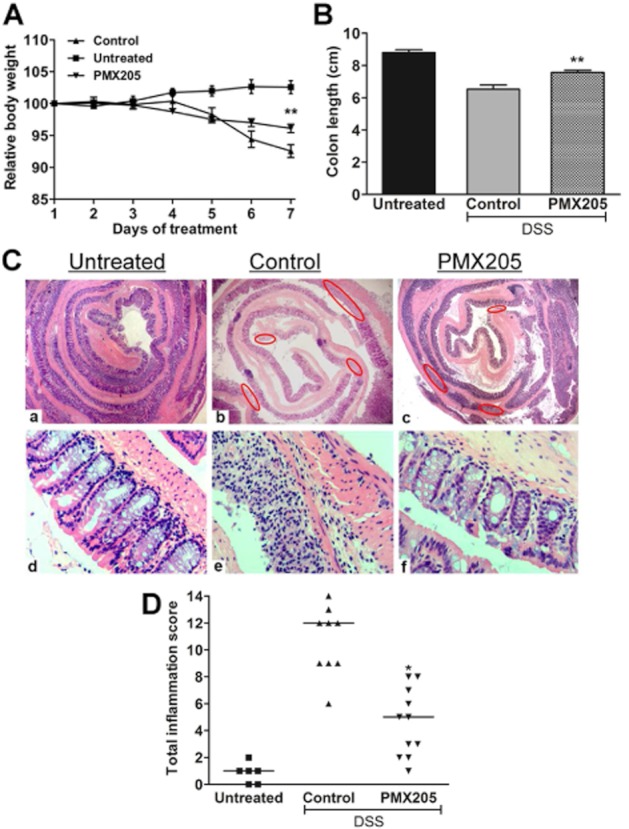
PMX205 ameliorates DSS induced body weight loss, colon shortening and histopathological changes in C57BL/6 mice. C57BL/6 mice had 3% DSS added to their water for 5 days. Untreated mice were kept on facility water until day 7. PMX205 or water (control) was administered orally beginning 1 day prior to the DSS start (day 0). PMX205 significantly prevented the (A) body weight and (B) colon shortening due to the DSS exposure. Results are presented as mean ± SEM (n = 5–14 mice per group). (C) Haematoxylin and eosin-stained colons from (a,d) untreated, (b,e) control or (c,f) PMX205-treated group. (D) Inflammation scores, where each dot represents a single mouse and the line represents median score. *P < 0.05 and **P < 0.01.
Therapeutic effect of PMX205
To ascertain whether blocking CD88 is efficacious in a therapeutic protocol, BALB/c mice were treated with PMX205 (200 μg per mouse) starting from Day 3 (48 h after starting DSS) through Day 6. The PMX205 group had significantly less body weight loss on day 7, less colon shortening, and showed significant reductions in clinical illness and inflammation (Figure 9), affirming that this approach to treating colitis has potential to be effective when used therapeutically.
Figure 9.
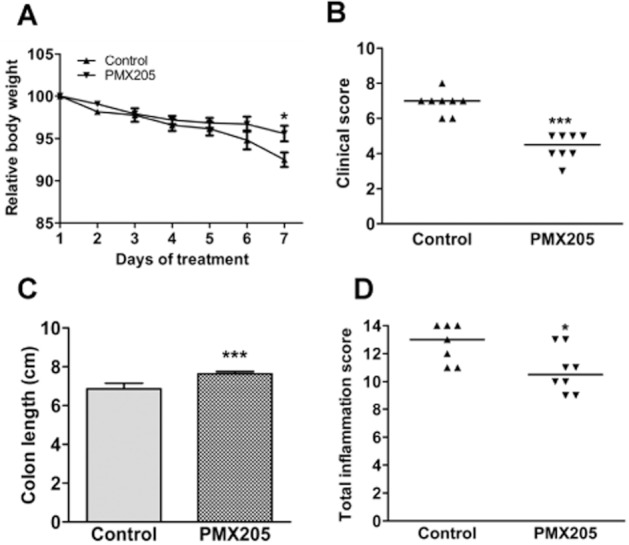
Effects of therapeutic PMX205 on measures of DSS-induced colitis in BALB/c mice. Mice were fed 5% DSS in drinking water from day 1 until day 6. PMX205 or water (control) was orally administered starting from day 3 through day 6. PMX205 significantly reduced measures of colitis including (A) body weight loss, (B) colon shortening, (C) clinical illness and (D) colon inflammation scores. Results are presented as mean ± SEM (n = 6–8 mice/group). *P < 0.05 and ***P < 0.001 versus DSS controls.
Discussion and conclusions
Complement is an arm of the innate immune system that plays a key role in the mammalian host's defence against microorganisms. However, excessive activation of complement can occur under diseased conditions and contribute to the pathology of inflammatory diseases (Ricklin and Lambris, 2007; Ehrnthaller et al., 2011). Of the split complement proteins, C5a is pathogenic in various diseases including sepsis (Czermak et al., 1999), asthma (Baelder et al., 2005) and ischaemia/reperfusion injury (Arumugam et al., 2004). As DSS activates the innate immune system (Wirtz et al., 2007) and mice show split complement protein deposition early on the mucosa (Lin et al., 2004), the model is highly suitable to determining the effects of pharmacological blockade of complement factors early in colitis. Complement pathways are also required for protecting the host, so rather than block an activation event upstream of C5 cleavage, our strategy was to specifically block CD88.
We first confirmed that there is increased mucosal generation of anaphylatoxins in the DSS-inflamed mouse, and colonic C3a and C5a concentrations were indeed elevated. Next, we confirmed that C5a is pathogenic by showing protection from DSS colitis using PMX205 in both prophylactic and therapeutic regimes; an outcome consistent with the effect of the PMX compounds on TNBS-induced colitis in rats (Woodruff et al., 2003; 2005). Mice show strain-related susceptibilities to DSS (Melgar et al., 2005) and we aimed to ensure that the benefits of PMX205 are not restricted by genetic background in complement competent mice. Consequently, we show that PMX205 administration also dramatically inhibited clinical disease and histopathological damage in the colons of C57BL/6 mice. Preventing mucosal damage is highly desirable as mucosal healing is one of the treatment goals for IBD. In contrast to our results using complement competent mice, PMX205 had no effect on DSS inflammation in mice lacking CD88, which we used as a specificity control for the drug. It seems paradoxical that CD88–/– mice should not be protected from DSS colitis, but others reported that CD88–/– mice on the C57BL/6 genetic background had similar colon pathology as control mice by the end of an 8 day DSS treatment period, despite less clinical disease (Johswich et al., 2009). It is unlikely that the difference in protocol (8 days vs. 5 days) affects these disparate outcomes, as we also observe that after 5 days of DSS, CD88–/– mice (BALB/c background) are not protected from colitis. Contradictory results between inhibition and deficiency have also been observed in the case of TNF in animal models of colitis (Myers et al., 2003; Naito et al., 2003), as have several other mediators. Collectively, these reports indicate that gene knockout strains do not necessarily recapitulate the outcomes of wild-type animals responding to pharmacological blockade of the gene product. Notwithstanding unknown model specific susceptibilities to DSS, we argue that these knockout mice affirm the specificity of PMX205 for CD88.
Mediator profiling in DSS colitis has shown significantly increased production of multiple Th1 and Th17 cytokines (Yan et al., 2009), and C5a acts on various cell types to induce the production of Th1 cytokines (Riedemann et al., 2002; Monsinjon et al., 2003). Blocking CD88 with PMX205 resulted in significant reductions in IL-1β, IL-6 and TNF, while IL-12, IL-17 and CXCL2 were unaffected. Treatment with PMX205, however, enhanced levels of the Th2 cytokines, IL-4 and IL-10. This observation is consistent with the report showing that blocking C5a in TNBS colitis resulted in a shift in the cytokine balance (Chen et al., 2011). IL-10 in particular has been shown to be protective in colitis. Apart from IL-10–/– mice spontaneously developing colitis (Kühn et al., 1993), injecting IL-10 was shown to be protective against DSS colitis (Tomoyose et al., 1998) and adenoviral vector-encoded IL-10 prevented damage in TNBS colitis (Lindsay et al., 2002). Regarding how these cytokines become elevated, C3a is one possibility as it reportedly stimulates a Th2-like response in the absence of C5a (Hawlisch et al., 2004). In fact, very little is known about the impact of C3a in colitis, yet mucosal levels are increased and are consistently greater than levels of C5a, which warrant further exploration. Multiple leukocyte types have been implicated in the pathogenesis of DSS colitis and thus we looked at the effect of CD88 inhibition on the granulocyte infiltrate. We found that neutrophil numbers were significantly reduced in the mucosa of PMX205-treated group, concurrent with less crypt loss and fewer ulcers. Statistical significance could not be reached in the submucosal population of neutrophils, although the trend was higher in the DSS control group. With respect to macrophages, it has been shown that C5a promotes the development of M1 (inflammatory) macrophages (Langer et al., 2010) which mediate tissue inflammation mainly by producing NO (Munder, 2009). On the other hand, the development of M2 (anti-inflammatory) macrophages is promoted by anti-inflammatory cytokines (IL-4 or IL-13), and this subtype, in turn, plays a protective role in inflammation primarily by secreting IL-10 (Gordon, 2003). Because in our study the macrophage numbers were similar in the two groups but the cytokines IL-10 and IL-4 were increased in the PMX205 group, we wondered if there was a relative increase in the anti-inflammatory activity of macrophages due to C5aR blockade. NO, which is generated by NOS, further leads to the formation of NT, which serves as an indirect marker for M1. Another enzyme, arginase I, competes with NOS for the same L-arginine substrate, where arginase-I-mediated consumption of L-arginine results in the anti-inflammatory phenotype of the cells (Munder, 2009). NT was significantly less in the PMX205 group whereas arginase-positive cell numbers were not different between the groups. Reduced NT with unchanged arginase levels implies less inflammatory activity of M1 macrophages without an increase in the M2 population. Alternatively, less NO production might reflect higher arginase I activity. Another important implication of reduced NT is the implied reduction in the levels of reactive nitrogen species. Reactive nitrogen species have been implicated in the pathogenesis of colitis and C5a is also known to induce the generation of reactive nitrogen radicals from various cell types (DiScipio et al., 2006). Given these results, we conclude that PMX205-mediated protection is likely mainly due to a reduction in the inflammatory characteristics of the cells in the mucosa.
In summary, data from this study show that acute colitis was ameliorated in both preventive and therapeutic models by oral treatment with PMX205, and was associated with lower inflammatory markers (cytokines, NO production) and up-regulated anti-inflammatory cytokines in the prophylactic regime. In considering the translation of these results and complement therapies for IBD, there are many axis of the cascade that could be blocked. One example of an effective anti-complement therapy is eculizamab, a monoclonal antibody which prevents the cleavage of C5, used to treat paroxysmal nocturnal haemoglobinuria (McKeage, 2011). Yet specifically blocking CD88 leaves other antimicrobial activities of the complement cascade intact. Finally, oral administration of PMX205 is less likely to be associated with side effects or systemic effects of a parenteral route of administration. In fact, an analogue, PMX53, has successfully undergone three Phase Ia clinical safety trials in humans, demonstrating the safe oral use of this class of compounds (Woodruff et al., 2011), but not yet in colitis patients.
Acknowledgments
We thank Hana James for expert technical assistance on the project. The project was funded by a partnership grant from the Canadian Institutes of Health Research, the Nova Scotia Health Research Foundation, and the Crohn's and Colitis Foundation.
Glossary
- CD
Crohn's disease
- DSS
dextran sulphate sodium
- IBD
inflammatory bowel disease
- NT
nitrotyrosine
- TNBS
trinitrobenzenesulfonic acid
- UC
ulcerative colitis
Conflict of interest
The authors have no financial conflicts of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam TV, Woodruff TM, Stocks SZ, Proctor LM, Pollitt S, Shiels IA, et al. Protective effect of a human C5a receptor antagonist against hepatic ischaemia-reperfusion injury in rats. J Hepatol. 2004;40:934–941. doi: 10.1016/j.jhep.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Baelder R, Fuchs B, Bautsch W, Zwirner J, Köhl J, Hoymann HG, et al. Pharmacological targeting of anaphylatoxin receptors during the effector phase of allergic asthma suppresses airway hyperresponsiveness and airway inflammation. J Immunol. 2005;174:783–789. doi: 10.4049/jimmunol.174.2.783. [DOI] [PubMed] [Google Scholar]
- Chen G, Yang Y, Gao X, Dou Y, Wang H, Han G, et al. Blockade of complement activation product C5a activity using specific antibody attenuates intestinal damage in trinitrobenzene sulfonic acid induced model of colitis. Lab Invest. 2011;91:472–483. doi: 10.1038/labinvest.2010.183. [DOI] [PubMed] [Google Scholar]
- Czermak BJ, Sarma V, Pierson CL, Warner RL, Huber-Lang M, Bless NM, et al. Protective effects of C5a blockade in sepsis. Nat Med. 1999;5:788–792. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–1652. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- DiScipio RG, Schraufstatter IU, Sikora L, Zuraw BL, Sriramarao P. C5a mediates secretion and activation of matrix metalloproteinase 9 from human eosinophils and neutrophils. Int Immunopharmacol. 2006;6:1109–1118. doi: 10.1016/j.intimp.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Ehrnthaller C, Ignatius A, Gebhard F, Huber-Lang M. New insights of an old defense system: structure, function, and clinical relevance of the complement system. Mol Med. 2011;17:317–329. doi: 10.2119/molmed.2010.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer's disease. J Immunol. 2009;183:1375–1383. doi: 10.4049/jimmunol.0901005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Halstensen TS, Brandtzaeg P. Local complement activation in inflammatory bowel disease. Immunol Res. 1991;10:485–492. doi: 10.1007/BF02919746. [DOI] [PubMed] [Google Scholar]
- Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12:S3–S9. doi: 10.1097/01.mib.0000195385.19268.68. [DOI] [PubMed] [Google Scholar]
- Hawlisch H, Wills-Karp M, Karp CL, Köhl J. The anaphylatoxins bridge innate and adaptive immune responses in allergic asthma. Mol Immunol. 2004;41:123–131. doi: 10.1016/j.molimm.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Hunter MM, Wang A, Parhar KS, Johnston MJ, Van Rooijen M, Beck PL, et al. In vitro-derived alternatively activated macrophages reduce colonic inflammation in mice. Gastroenterology. 2010;138:1395–1405. doi: 10.1053/j.gastro.2009.12.041. [DOI] [PubMed] [Google Scholar]
- Johswich K, Martin M, Bleich A, Kracht M, Dittrich-Breiholz O, Gessner JE, et al. Role of the C5a receptor (C5aR) in acute and chronic dextran sulfate-induced models of inflammatory bowel disease. Inflamm Bowel Dis. 2009;15:1812–1823. doi: 10.1002/ibd.21012. [DOI] [PubMed] [Google Scholar]
- Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Langer HF, Chung KJ, Orlova VV, Choi EY, Kaul S, Kruhlak MJ, et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood. 2010;116:4395–4403. doi: 10.1182/blood-2010-01-261503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Spencer D, Hatala DA, Levine AD, Medof ME. Decay-accelerating factor deficiency increases susceptibility to dextran sulfate sodium-induced colitis: role for complement in inflammatory bowel disease. J Immunol. 2004;172:3836–3841. doi: 10.4049/jimmunol.172.6.3836. [DOI] [PubMed] [Google Scholar]
- Lindsay J, Van Montfrans C, Brennan F, Van Deventer S, Drillenburg P, Hodgson H, et al. IL-10 gene therapy prevents TNBS-induced colitis. Gene Ther. 2002;9:1715–1721. doi: 10.1038/sj.gt.3301841. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeage K. Eculizumab: a review of its use in paroxysmal nocturnal haemoglobinuria. Drugs. 2011;71:2327–2345. doi: 10.2165/11208300-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Mañé J, Lorén V, Pedrosa E, Ojanguren I, Xaus J, Cabré E, et al. Lactobacillus fermentum CECT 5716 prevents and reverts intestinal damage on TNBS-induced colitis in mice. Inflamm Bowel Dis. 2009;15:1155–1163. doi: 10.1002/ibd.20908. [DOI] [PubMed] [Google Scholar]
- Melgar S, Karlsson A, Michaelsson E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1328–G1338. doi: 10.1152/ajpgi.00467.2004. [DOI] [PubMed] [Google Scholar]
- Melgar S, Karlsson L, Rehnstrom E, Arlsson A, Utkovic H, Jansson L, et al. Validation of murine dextran sulfate sodium-induced colitis using four therapeutic agents for human inflammatory bowel disease. Int Immunopharmacol. 2008;8:836–844. doi: 10.1016/j.intimp.2008.01.036. [DOI] [PubMed] [Google Scholar]
- Molodecky NA, Kaplan GG. Environmental risk factors for inflammatory bowel disease. Gastroenterol Hepatol (N Y) 2010;6:339–346. [PMC free article] [PubMed] [Google Scholar]
- Monk PN, Scola AM, Madala P, Fairlie DP. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol. 2007;152:429–448. doi: 10.1038/sj.bjp.0707332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsinjon T, Gasque P, Chan P, Ischenko A, Brady JJ, Fontaine MC. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 2003;17:1003–1014. doi: 10.1096/fj.02-0737com. [DOI] [PubMed] [Google Scholar]
- Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol. 2009;158:638–651. doi: 10.1111/j.1476-5381.2009.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KJ, Murthy S, Flanigan A, Witchell DR, Butler M, Murray S, et al. Antisense oligonucleotide blockade of tumor necrosis factor-alpha in two murine models of colitis. J Pharmacol Exp Ther. 2003;304:411–424. doi: 10.1124/jpet.102.040329. [DOI] [PubMed] [Google Scholar]
- Naito Y, Yakagi T, Handa O, Ishikawa T, Nakagawa S, Yamaguchi T, et al. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J Gastroenterol Hepatol. 2003;18:560–569. doi: 10.1046/j.1440-1746.2003.03034.x. [DOI] [PubMed] [Google Scholar]
- Pavlovski D, Thundyil J, Monk PN, Wetsel RA, Taylor SM, Woodruff TM. Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. FASEB J. 2012;9:3680–3690. doi: 10.1096/fj.11-202382. [DOI] [PubMed] [Google Scholar]
- Perianayagam MC, Balakrishnan VS, King AJ, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int. 2002;61:456–463. doi: 10.1046/j.1523-1755.2002.00139.x. [DOI] [PubMed] [Google Scholar]
- Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25:1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedemann NC, Guo RF, Sarma VJ, Laudes IJ, Huber-Lang M, Warner RL, et al. Expression and function of the C5a receptor in rat alveolar epithelial cells. J Immunol. 2002;168:1919–1925. doi: 10.4049/jimmunol.168.4.1919. [DOI] [PubMed] [Google Scholar]
- Satsangi J, Morecroft J, Shah NB, Nimmo E. Genetics of inflammatory bowel disease: scientific and clinical implications. Best Pract Res Clin Gastroenterol. 2003;17:3–18. doi: 10.1053/bega.2002.0349. [DOI] [PubMed] [Google Scholar]
- Sivaram G, Tiwari SK, Bardia A, Manoj G, Santhosh B, Saikant R, et al. Association of genetic variants of mannan-binding (MBL) lectin-2 gene, MBL levels and function in ulcerative colitis and Crohn's disease. Innate Immun. 2011;17:526–531. doi: 10.1177/1753425910384531. [DOI] [PubMed] [Google Scholar]
- Snyderman R, Phillips J, Mergenhagen SE. Polymorphonuclear leukocyte chemotactic activity in rabbit serum and Guinea pig serum treated with immune complexes: evidence for C5a as the major chemotactic factor. Infect Immun. 1970;1:521–525. doi: 10.1128/iai.1.6.521-525.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyderman R, Pike M, McCarley D, Lang L. Quantification of mouse macrophage chemotaxis in vitro: role of C5 for the production of chemotactic activity. Infect Immun. 1975;11:488–492. doi: 10.1128/iai.11.3.488-492.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon L, Mansor S, Mallon P, Donnelly E, Hoper M, Loughrey M, et al. The dextran sulphate sodium (DSS) model of colitis: an overview. Comp Clin Pathol. 2010;19:235–239. [Google Scholar]
- Stallmach A, Hagel S, Bruns T. Adverse effects of biologics used for treating IBD. Best Pract Res Clin Gastroenterol. 2010;24:167–182. doi: 10.1016/j.bpg.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Stevceva L, Pavli P, Husband A, Doe W. The inflammatory infiltrate in the acute stage of the dextran sulphate sodium induced colitis: B cell response differs depending on the percentage of DSS used to induce it. BMC Clin Pathol. 2001;1:3. doi: 10.1186/1472-6890-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillie R, Stadnyk AW. Role of TNF receptors, TNFR1 and TNFR2, in dextran sodium sulfate-induced colitis. Inflamm Bowel Dis. 2009;15:1515–1525. doi: 10.1002/ibd.20951. [DOI] [PubMed] [Google Scholar]
- Tomoyose M, Mitsuyama K, Ishida H, Toyonaga A, Tanikawa K. Role of interleukin-10 in a murine model of dextran sulfate sodium-induced colitis. Scand J Gastroenterol. 1998;33:435–440. doi: 10.1080/00365529850171080. [DOI] [PubMed] [Google Scholar]
- Tsuji RF, Kawikova I, Ramabhadran R, Akahira-Azuma M, Taub D, Hugli TE, et al. Early local generation of C5a initiates the elicitation of contact sensitivity by leading to early T cell recruitment. J Immunol. 2000;165:1588–1598. doi: 10.4049/jimmunol.165.3.1588. [DOI] [PubMed] [Google Scholar]
- Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Arumugam TV, Shiels IA, Reid RC, Fairlie DP, Taylor SM. A potent human C5a receptor antagonist protects against disease pathology in a rat model of inflammatory bowel disease. J Immunol. 2003;171:5514–5520. doi: 10.4049/jimmunol.171.10.5514. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Pollitt S, Proctor LM, Stocks SZ, Manthey HD, Williams HM, et al. Increased potency of a novel complement factor 5a receptor antagonist in a rat model of inflammatory bowel disease. J Pharmacol Exp Ther. 2005;314:811–817. doi: 10.1124/jpet.105.086835. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Crane JW, Proctor LM, Buller KM, Shek AB, de Vos K, et al. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 2006;2006:1407–1417. doi: 10.1096/fj.05-5814com. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Costantini KJ, Crane JW, Atkin JD, Monk PN, Taylor SM, et al. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol. 2008;181:8727–8734. doi: 10.4049/jimmunol.181.12.8727. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5-C5a receptor axis. Mol Immunol. 2011;48:1631–1642. doi: 10.1016/j.molimm.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Yan Y, Kolachala V, Dalmasso G, Nguyen H, Laroui H, Sitaraman SV, et al. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. Plos ONE. 2009;4:e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YJ, Macneil AJ, Junkins R, Carrigan SO, Tang JT, Forward N, et al. Regulator of calcineurin 1 (Rcan1) is required for the development of pulmonary eosinophilia in allergic inflammation in mice. Am J Pathol. 2011;179:1199–1210. doi: 10.1016/j.ajpath.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]