

Abstract
Background
In cancer patients, chemo and radiotherapy can cause infertility by damaging spermatogenesis process. This process is based on self-renewal and differentiation of a rare population of the testicular cells called Spermatogonial Stem Cells (SSCs). Scientists have tried to isolate, enrich and culture Human spermatogonial stem cells, hoping to resolve infertility problems in cancer recovered patients in the future.
Methods
Spermatogonial stem cells were isolated and purified from human testicular biopsies sample consisting of at least 500,000 and at most 2,000,000 cells. Two enzymatic digestion steps were performed. Enriching methods, differential plating, and specific culture in serum-free medium with added growth factors: human GDNF, bFGF, EGF and LIF was performed on coated dishes.
Results
Human spermatogonial stem cell clusters were observed after 7 to 10 days in specific culture, then after several passages and successful expanding duration of 52 days, the cells were evaluated by three layer immunocytochemistry test (LSAB) to stain GPR125 protein as a surface marker in human spermatogonial stem cells.
Conclusion
In current study human spermatogonial stem cell were isolated and expanded with the least manipulations in comparison with the other usual isolation methods like florescent or magnetic activated cell sorting. In contrast to the other SSCs isolation and culture methods, this system is based on the testicular biopsies against large samples, thus suggested method in this study is closer to clinical usage in the future.
Keywords: Culture, Enrichment, GPR125, Human spermatogonial stem cell, Infertility, Isolation
Introduction
Today, treating cancer in children has been improved immensely. However, chemotherapy and radiotherapy as the methods of cancer treatment damage different types of cells especially proliferating cells like spermatogonial ones in testis. With the hope of treatment and recovery from cancer in children, one of the major challenges facing these patients after puberty is infertility later in life (1). Since spermatogenesis does not begin in childhood, freezing mature sperm is impossible. Then autotransplant of testis stem cells has been suggested by scientists as a practical clue. On the other hand, the number of spermatogonial stem cells in one testis biopsy is not sufficient to transplant for healing the damages of germinal layer caused by cancer therapy. Hence, before each treatment, the possibility of specific isolation, enrichment, culturing and expanding spermatogonial stem cells as major sperm generators should be provided to autotransplant and recover fertility after cancer treatment.
Different types of human spermatogonia include progenitor Adark-spermatogonia, progenitor Apale-spermatogonia, committed Apale-spermatogonia, and B-spermatogonia. The progenitor Adark-spermatogonia apparently are stems or reserve spermatogonia (2). For specific isolation of spermatogonial stem cells and undifferentiated spermatogonia, scientists have tried to identify specific markers in rodent, monkey and human (3). Recently, GPR125 has been introduced as a marker for mouse spermatogonial stem/progenitor cells (4, 5). Furthermore, GPR125 has been approved as a reliable marker for human SSCs (6, 7). He Z. et al 2010 studied more than six markers that have been identified for SSCs and progenitors in other species to characterize the phenotypes of human spermatogonia and more differentiated germ cells. Then they could obtain ∼3×104 human GPR125-positive spermatogonia from ∼6×106 germ cells after MACS (7). (It implies 2% of total tested cells).
By considering the fact that human spermatogonial stem cells are too rare in testis as mentioned above, for any clinical usage of spermatogonial stem cells, enrichment and culture of these cells will be the first requirement. In culture field, Kanatsu-Shinohara et al cultured mouse SSCs in such a way that these cells propagated themselves on a feeder layer. A special Serum Free Medium (SFM) has been introduced by them that contained special supplements and several growth factors, including GDNF (8).
For the first time, Dirami et al performed differential plating to enrich porcine Type A Spermatogonia to decrease Sertoli and myoid cells (9). Culturing human spermatogonial stem cell had not been reported until 2009 that Sadri et al were able to propagate the Human Spermatogonial Stem Cells In vitro (10). In addition, in recent decades, in culturing systems laminin coated dishes replaced co-culturing system that based on feeder layers.
It should be mentioned that at this stage of studies identifying specific markers, enrichment and isolation undifferentiated spermatogonia from somatic cells and differentiated germ cells were fulfilled by researches. Nevertheless, isolating germ cells from blood-related malignancies needs more investigation before any clinical procedures can take place.
In the current study, we tried to isolate, enrich and specifically culture spermatogonial stem cells derived from tiny biopsy samples. In the next step, after expanding human spermatogonial stem cells, we evaluate our culture system by immunocytochemistry test to stain GPR125 protein as surface marker in human spermatogonial stem cells.
Materials and Methods
Sampling and digestion
This study is based on 12 biopsy samples taken from different azoospermic patients referred to Avicenna infertility clinic center for operating TESE procedure (testicular sperm extraction) (11). After performing clinical procedure and confirming active spermatogenesis, and obtaining verbal informed consent from the patients, surplus testis samples were donated to the research laboratory. Two enzymatic digestion steps were performed by 2.5 mg/ml collagenase IV (Sigma-aldrich) for 15 min and then trypsin-EDTA (0.25%-1 mM) for 5-7 minutes on each biopsy in 37°C temperature (9). Biopsy samples consisted of at least 500,000 and at most 2,000,000 cells.
SSCs culture technique
In the cases which the biopsies contained more than 106 cells, cells were suspended in DMEM-F12 (Sigma) with 10% FBS (Gibco) for 3 hr or for an overnight (in different cases) in uncoated plates to enrich spermatogonial cells and decrease the number of myoid and sertoli cells. During this time, myoid and sertoli cells make opportunities to attach culture dishes. Then, suspended cells were collected and cultured in SFM with some supplements and growth factors: 10 ng/ml human GDNF (Sigma), 10 ng/ml bFGF (Sigma), 20 ng/ml EGF and 103 U/ml LIF (Chemicon) in 20 µl/ml laminin (Sigma) coated or 0.2% gelatin (Sigma) coated dishes at 37°C in an atmosphere of 5% CO2 (8, 10) (base medium was DMEM-F12).
In the cases that biopsies contained less than 106 cells, we bypassed the differential plating method that was mentioned above. It was preferred to save myoid and sertoli cells in these cases as feeder cells, because the primary cell number was so few. Thus, cell suspended in DMEM-F12 with 10% FBS for overnight at a density of 30,000/cm 2 in 20 µl/ ml laminin (10) coated or 0.2% gelatin (8) coated plates, the following day, DMEM-F12 with 10% FBS media was changed to SFM contained specific growth factors within the previous plates. Spermatogonial stem cells clusters were passaged every 7-10 days. The cells were cultured and passaged 6 times during 52 days, in this study.
Immunochemistry
Culture of human spermatogonial stem cells was evaluated by immunocytochemistry test. After 52 days of culture, the cells were trypsinated and cytospind onto slides for DAB staining GPR125 protein as surface marker on these cells. Slides were fixed in NBF for 10 min. Serum blocking was performed by 10% sheep normal serum for 30 min at room temperature and the endogenous peroxidase activity was quenched with 3% hydrogen peroxide in dark. The slides were incubated with primary antibodies (AB), including rabbit polyclonal AB to GPR125 (Ab5175, Abcam Inc., Cambridge, MA) at a 1:100 dilution at 4°C overnight (7). After 3 washes with TBS-1% BSA, the slides were incubated by secondary antibody, biothinylated sheep anti rabbit at 2.5 µg/ml for 45 min at room temperature and washed 3 times. Next, streptavidin-HRP complex (Invitrogen) was added to the cells for 30 min and treated by diaminobenzidine (DAB) as the chromogen and hematoxyline as counterstian. Then, dehydration was performed through a series of graded alcohols. As negative control, somatic cells derived from an 86-year-old man testis, were used, after several hours differential plating.
For this purpose, frozen 86-year-old man testis, was thawed. After digestion, the cell suspension placed in DMEM-F12 with 10% FBS, then float cells (include germ cells and floated somatic cells) were removed after several hours. The resistant attached spindle cells at the bottom of plate, were cultured in DMEM-F12 with 10% FBS for 3 weeks, these attached somatic cells trypsinated after 2 passages and were used as negative control. Hela cell line was used as positive control (12). In the end, slides were examined under microscope (OLYMPUS BX51).
Results
Differential plating was suggested to enrich spermaogonia in cell suspension derived from testis sample after enzymatic digestion by some articles (9, 10). In the current study, after investigation on 12 testicular biopsies, we observed that decrease in the number of somatic cells (like myoid and sertoli cells) by differential plating was unsuccessful to maintain spermatogonia and early cell death was observed in culture conditions when the number of cells in testicular biopsies was less than 106. Thus, when primary count was less than 106 cells, instead of differential plating, cells should be suspended in DMEM-F12 with 10% FBS for overnight in 20 µl/ml laminin coated or 0.2% gelatin coated plates. A day later, DMEM-F12 with 10% FBS media should be changed to SFM contained specific human spermatogonial stem cells growth factors in the prior plates. In this way, we could prevent excessive loss of cells and restrict proliferation somatic cells by decreasing serum in culture condition at last.
On the other hand, we observed that when primary cell count was more than 106 cells in digested cell suspension, differential plating, was beneficial to enrich human spermatogonia and decrease disturbing of somatic cells before transferring the cells to SFM. To obtain the appropriate time for primary culture, two culture times of 3 hr primary culture and overnight culture in DMEM-F12 media with 10% FBS were compared to each other. The bottom of each dish was examined by microscope (OLYMPUS CK41) after collecting the suspended cells. The plate that related to 3 hr primary culture showed little cell attachment. This means that many testicular somatic cells were not resistant to be attached to plastic dishes during 3 hr (Figure 1A). Then, major of somatic cells will be suspended and transferred to laminin coated dish that was prepared for specific culture. Nevertheless, the plate that related to overnight primary culture showed appropriate somatic cells attachment during this period (Figure 1B). Thus, enriching human spermatogonia in digested cell suspension was fulfilled as the aim of differential plating. Collection and centrifuge of overnight cell suspension will help remove blood cells and disturbing particles from culture system too.
Figure 1.
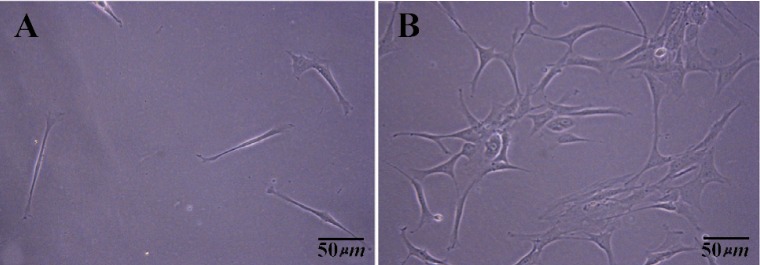
Somatic cells attached at the bottom of plastic dish in primary culture of testis cells after collecting the floating media and suspended cells. A) 3 hr differential plating; B) An overnight differential plating
Based on morphology, human spermatogonial stem cells cluster were appeared between 7 to 10 days of culture in SFM media included mentioned growth factors with compact and raised appearance. Primary spermatogonial germ cells cluster was near or on the somatic cells (Figure 2).
Figure 2.
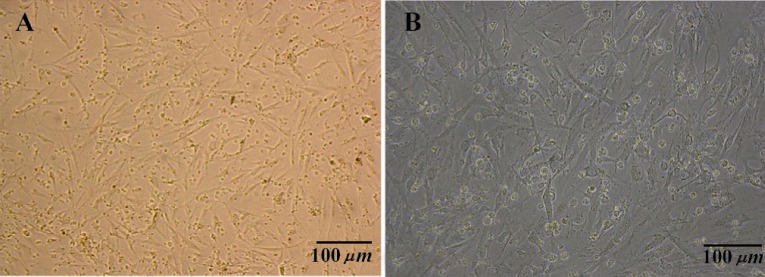
Human germ cell clusters to gather somatic cells at the 7th day of testis cells culture in SFM media. A) 10×10X; B) 10×20X
After the first passage, the clusters got more typical appearance. Despite the fact whether the cells compact reach 80% confluency or not between 7 to 10 days, the morphologic appearance of undifferentiated spermatogonia cluster started to change and their bounder lines began to become indistinguishable after several days in culture. The appropriate time to passage the undifferentiated spermatogonia was around the time when morphological changes were taking place. Human spermatogonial stem cells clusters propagated by expanding the surface area culture after each passage, but the number of fibroblastic cells did not increase as much. Even the somatic cells aged and declined in SFM media (Figure 3).
Figure 3.
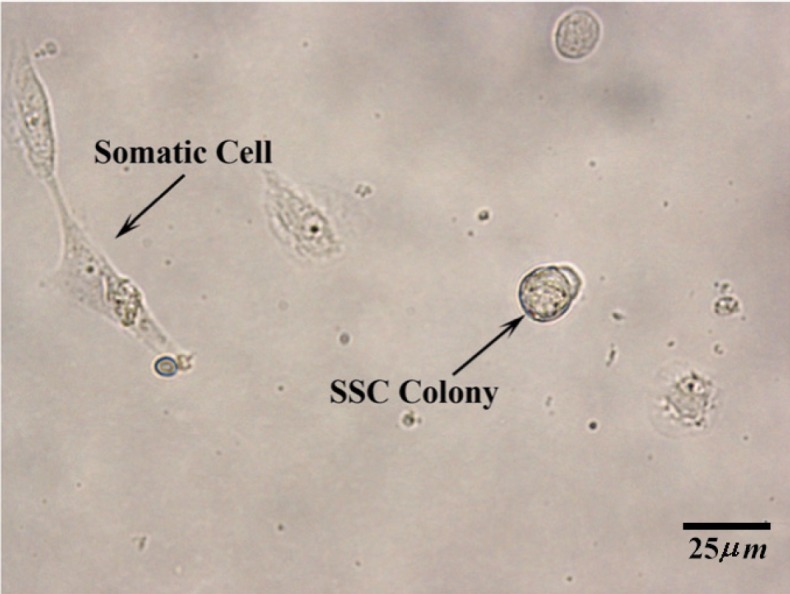
Two weeks after specific culture of human spermatogonial stem cells in SFM. At the left, a somatic cell and at the right, a human germ cell colony are shown. (10×40X)
So far, human spermatogonial germ cells cluster attached to gelatine or laminin coated plates with the minimum somatic cells accompaniment after several passages (Figure 4).
Figure 4.
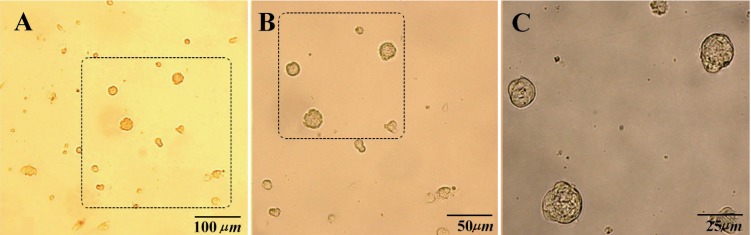
Human germ cells colonies were attached to laminin coated plate without fibroblast accompaniment after the second passage. A) 10×10X; B) 10×20X; C) 10×40X
The colonies condition was not mentioned for different cases on gelatin and laminin coated plated when the primary count was less than 106 cells in biopsy samples.
The specific culture of human spermatogonial stem cells was evaluated and approved by collecting the cultured cells after 52 days and DAB staining GPR125 as specific surface marker on these cells. The majority of the cells on each field were positive. By considering spermatogonial stem cell number Hela cells as positive control and testis somatic cells derived from an 86-year-old man as negative control are shown in Figure 5).
Figure 5.
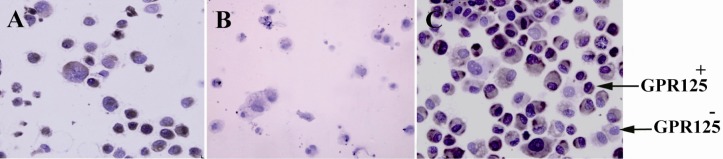
GPR125 immunochemistry staining. A) Hela cells as positive control; B) testis somatic cells derived from 86-year-old man as negative control; C) GPR125+ cells in specific culture of human spermatogonial stem cells after 52 days
Discussion
Reproductive potential of an organism can be prolonged indefinitely by using germ-line stem cells (13). In rodent, only 0.03% of all germ cells are spermatogonial stem cells. They are the only cell type that can repopulate and restore fertility to congenitally infertile recipient mice following transplantation (13, 14). In recent years, three major requirements needed to advance spermatogonial stem cell research were (i) an in vivo assay for stem cell function like methods for spermatogonial transplantation, (ii) knowledge of stem cell markers, and (iii) a method to maintain the stem cells continuously in culture (15).
Kanatsu-Shinohara et al cultured SSCs in such a way that these cells propagated themselves, while retaining their capacity to repopulate a recipient mouse testis upon transplantation (8). Differential plating to enrich Porcine Type A Spermatogonia was performed by Dirami et al Digested cell suspension was incubated in DMEM/F12 containing 5% hr serum for 4 hr at 34°C. Sertoli and myoid cells attached to the culture plates. The spermatogonial cells, which remained in suspension, were collected before plating (9).
Sadri et al cultured human testicular cells overnight in uncoated dishes in supplemented MEM containing 10% FCS at 37°C and 5% CO2. After overnight incubation, floating cells were collected and cultured in uncoated dishes with supplemented StemPro-34 with human growth factors. In the case of overgrowth of flat cells with long extensions which were interpreted as somatic cells, only germ cells and round dividing cells were differentially passed to fresh dishes coated with human placenta laminin by vigorous pipetting (10).
In the current study, we have worked on human testicular biopsies that included at least 500,000 and at most 2,000,000 cells. We have also studied differential plating to enrich human spermatogonia on digested cell suspension in two aspects. First, was the common method of differential plating helpful to enrich human spermatogonia for all tiny biopsy samples. And also, when cell count was more than 106 cells in digested cell suspension, primary culture in DMEM-F12 with 10% FBS for overnight and transfer the suspended cells to specific human spermatogonial stem cells culture system, prevented disturbing influences of somatic cells over growth in SFM media.
Previous studies have reported different times for primary culture, 4 hours in Porcine, overnight in bovine and 3 hr in human (7, 9, 16). The second question asks the appropriate time to gain successful attachment of human sertoli and myoid cells to plastic dishes. After 3 hours examination, we observed many cells that had suspended and the majority of cells transferred with spermatogonia to SFM. However, after overnight examination we observed human fibroblast attached plastic dishes successfully.
When the cell number in primary count of testis biopsy was less than 106 cells, enriching the spermatogonia was unsuccessful by differential plating. Decreasing the attaching cells during differential plating makes the confluency of cells in vitro condition too little. Then it is likely, that the cells could not interact with each other and early cell death would be observed. In this condition, (primary count of testis biopsy was less than 106 cells) we do not suggest the primary culture in plastic dishes. So that we tried to support all the digested cells on gelatin and laminin coated dishes in DMEM-F12 with 10% FBS media for the first day and then changed the media with serum to the SFM the next day. In this way, we could culture human spermatogonial stem cells clusters derived from tiny testis biopsy.
Gelatin has often been used to coat plastic surfaces and enhance attachment of various cell type experiments in vitro culture. Gelatin is a derivative of matrix constituency and assists in attachment of cells to a surface and allows them to proliferate (17).
A previous study on mouse identified α6- and β1-integrins as surface markers on spermatogonial stem cells (18). These integrins act as a laminin receptor and mediate cell attachment to laminin (19).
In this study, testicular biopsies cells were cultured (with less than 106 cells) in 20 µl/ml laminin coated and 0.2% gelatin coated plates. Fibroblastic and myoid cell divided were controlled by decreasing amount of serum in SFM, but they supported spermatogonial stem cells clusters for several weeks. Just by morphology, human spermatogonial stem cell clusters condition was not so different on gelatin or laminin during several passages. It should be mentioned that we collected the colonies after 52 days to evaluate the culture system by immonochemistry test. For a long-term culture, spermatogonial stem cells cluster could not be supported by gelatin coated dish alone probably after aging fibroblastic cells. The accurate comparison between spermatogonial stem cell clusters condition on laminin or gelatin coated plates would be reliable by following the fate of the cells after transplantation.
Seandel et al, have identified GPR125 as a surface marker for self renewing clonogenic spermatogonial progenitor cells in mouse (5). He et al, isolated GPR125-positive spermatogonia from adult human testis, followed by magnetic-activated cell sorting. Results indicate that GPR125-positive spermatogonia are phenotypically putative human spermatogonial stem cells and retain an undifferentiated status in vitro. Then they could obtain ∼3×104 human GPR125-positive spermatogonia from ∼6×106 germ cells after MACS (7) (It implies 2% of total tested cells).
In this study, the culture process was evaluated after 52 days of specific human spermatogonial stem cells culture by GPR125 staining. The majority of cells on each field (more than 70%) were GPR125 positive. While based on previous human study that has been mentioned above, about 2% of germ cells reported GPR125-positive spermatogonia before culture. So far, we have been able to culture and proliferate human spermatogonial stem cells in human testicular biopsies successfully by composing some methods such as differential plating, using gelatin and laminin coated plates and SFM that consist human specific growth factors. Expression of spermatogonial stem cells markers on the surface of these cells is usually weak. For Immunochemistry in those cases, it has been suggested to replace three layers staining instead of two layers staining (20). Thus, we improved sensitivity of the test by using Labeled Streptavidin Biotin (LSAB) method.
Cell sorting methods like MACS and FACS were used to enrich spermatogonia from mouse, hamster, and monkey testicular tissue (7, 21–23). In general, when spermatogonial stem cells have been separated by conjugated antibodies, a high degree of cell purity was obtained, but a low number of cells resulted. After reviewing previous experiments using these technique on testicular tissue, the effect of viability on subsequent experiments is still questionable since viability was not often reported (24).
This study has introduced a method to isolate and enrich human spermatogonial stem cells without common manipulations of the cells that is usual in cell sorting methods. Also, spermatogonial stem cells derived from testicular biopsies. It means a huge volume of testis sample with large amount of cells is not needed to isolate human spermatogonial stem cells. In this way, we get closer to the clinical purpose of spermatogonial stem cell in fertility treatment in the future.
Conclusion
Isolation and culture of human spermatogonial stem cells was fulfilled from TESE positive testicular biopsies with at least 500,000 to at most 2,000,000 cells. Differential plating performed to enrich human spermatogonial stem cells. Isolated cells proliferated in DMEM-F12 with GDNF, bFGF, EGF, LIF growth factors. Immunocytochemistry test showed that the majority of cells were GPR125 positive after 52 days of specific culture. Before any clinical usage of spermatogonial stem cells, more investigation is required to achieve isolated cancer cells from spermatogonial stem cells. In addition, improved freezing methods is required to maintain the cells for a long period of time.
Acknowledgement
This work was financially supported by the Avicenna research institute. We would like to thank all of the individuals who kindly accepted to participate in this study. We are also deeply indebted to the personnel of the Avicenna Reproductive Biotechnology and Nanobiotechnology departments.
References
- 1.Woodruff TK, Snyder KA. Oncofertility fertility preservation for cancer survivors. New York: Springer Verlag; 2007. [Google Scholar]
- 2.Clermont Y. Kinetics of spermatogenesis in mammals: seminiferous epithelium cycle and spermatogonial renewal. Physiol Rev. 1972;52(1):198–236. doi: 10.1152/physrev.1972.52.1.198. [DOI] [PubMed] [Google Scholar]
- 3.Dym M, Kokkinaki M, He Z. Spermatogonial stem cells: mouse and human comparisons. Birth Defects Res C Embryo Today. 2009;87(1):27–43. doi: 10.1002/bdrc.20141. [DOI] [PubMed] [Google Scholar]
- 4.Seandel M, James D, Shmelkov SV, Falciatori I, Kim J, Chavala S, et al. Generation of functional multipotent adult stem cells from GPR125+ germ-line progenitors. Nature. 2007;449(7160):346–350. doi: 10.1038/nature06129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golestaneh N, Kokkinaki M, Pant D, Jiang J, De-Stefano D, Fernandez-Bueno C, et al. Pluripotent stem cells derived from adult human testes. Stem Cells Dev. 2009;18(8):1115–1126. doi: 10.1089/scd.2008.0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izadyar F, Wong J, Maki C, Pacchiarotti J, Ramos T, Howerton K, Yuen C, et al. Identification and characterization of repopulating spermatogonial stem cells from the adult human testis. Hum Reprod. 2011;26(6):1296–1306. doi: 10.1093/humrep/der026. [DOI] [PubMed] [Google Scholar]
- 7.He Z, Kokkinaki M, Jiang J, Dobrinski I, Dym M. Isolation, characterization, and culture of human spermatogonia. Biol Reprod. 2009;82(2):363–372. doi: 10.1095/biolreprod.109.078550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanatsu-Shinohara M, Ogonuki N, Inoue K, Miki H, Ogura A, Toyokuni S, et al. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol Reprod. 2003;69(2):612–616. doi: 10.1095/biolreprod.103.017012. [DOI] [PubMed] [Google Scholar]
- 9.Dirami G, Ravindranath N, Pursel V, Dym M. Effects of stem cells factor and granulocyte macrophage-colony stimulating factor on survival of porcine type A spermatogonia cultured in KSOM. Biol Reprod. 1999;61(1):225–230. doi: 10.1095/biolreprod61.1.225. [DOI] [PubMed] [Google Scholar]
- 10.Sadri-Ardekani H, Mizrak SC, van Daalen SK, Kor-ver CM, Roepers-Gajadien HL, Koruji M, et al. Propagation of human spermatogonial stem cells in vitro. JAMA. 2009;302(19):2127–2134. doi: 10.1001/jama.2009.1689. [DOI] [PubMed] [Google Scholar]
- 11.Amirjannati N, Heidari-Vala H, Akhondi MA, Hos-seini Jadda SH, Kamali K, Sadeghi MR. Comparison of intracytoplasmic sperm injection outcomes between spermatozoa retrieved from testicular biopsy and from ejaculation in cryptozoospermic men. Andrologia. 2011;44(Suppl 1):704–709. doi: 10.1111/j.1439-0272.2011.01253.x. [DOI] [PubMed] [Google Scholar]
- 12.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, et al. Towards a knowledge-based human protein atlas. Nat Biotechnol. 2010;28(12):1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 13.Brinster RL, Zimmermann JW. Spermatogenesis following male germ-cell transplantation. Proc Natl Acad Sci USA. 1994;91(24):11298–11302. doi: 10.1073/pnas.91.24.11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avarbock MR, Brinster CJ, Brinster RL. Reconstitution of spermatogenesis from frozen spermatogonial stem cells. Nat Med. 1996;2(6):693–696. doi: 10.1038/nm0696-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matzuk MM. Germ-line immortality. PNAS. 2004;101(47):16395–16396. doi: 10.1073/pnas.0407344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Izadyar F, Spierenberg GT, Creemers LB, den Ou-den K, de Rooij DG. Isolation and purification of type A spermatogonia from the bovine testis. Reproduction. 2002;124(1):85–94. [PubMed] [Google Scholar]
- 17.Folkman J, Haudenschild CC, Zetter BR. Long-term culture of capillary endothelial cells. Proc Natl Acad Sci USA. 1979;76(10):5217–5221. doi: 10.1073/pnas.76.10.5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shinohara T, Avarbock MR, Brinster RL. Beta 1- and alpha 6-integrin are surface markers on mouse spermatogonial stem cells. Proc Natl Acad Sci USA. 1999;96(10):5504–5509. doi: 10.1073/pnas.96.10.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69(1):11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 20.Howard GC, Kaser MR. Making and using antibodies: A practical handbook. In: Ramos Vara JA, Saettele JA, editors. Immunohistochemical methods. United Kingdom: Taylor & Francis; 2006. pp. 273–314. [Google Scholar]
- 21.Kubota H, Avarbock MR, Brinster RL. Culture conditions and single growth factors affect fate determination of mouse spermatogonial stem cells. Biol Reprod. 2004;71(3):722–731. doi: 10.1095/biolreprod.104.029207. [DOI] [PubMed] [Google Scholar]
- 22.Hofmann MC, Braydich-Stolle L, Dym M. Isolation of male germ-line stem cells; influence of GDNF. Dev Biol. 2005;279(1):114–124. doi: 10.1016/j.ydbio.2004.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shinohara T, Brinster RL. Brinster, enrichment and transplantation of spermatogonial stem cells. Int J Androl. 2000;23(Suppl 2):89–91. doi: 10.1046/j.1365-2605.2000.00025.x. [DOI] [PubMed] [Google Scholar]
- 24.Miller SR. Assessment of nycodenz gradient on enrichment and culture of perinatal porcine spermatogonial stem cells; [NCSU libraries]: NC State University; 2006. p. 155. [master's thesis] [Google Scholar]