

Abstract
Background
Abnormal endothelial function promotes atherosclerotic vascular disease in diabetes. Experimental studies indicate that disruption of endothelial insulin signaling through the activity of protein kinase C-β (PKCβ) and nuclear factor κB (NFκB) reduces nitric oxide availability. We sought to establish whether similar mechanisms operate in the endothelium in human diabetes mellitus.
Methods and Results
We measured protein expression and insulin response in freshly isolated endothelial cells from patients with Type 2 diabetes mellitus (n=40) and non-diabetic controls (n=36). Unexpectedly, we observed 1.7-fold higher basal endothelial nitric oxide synthase (eNOS) phosphorylation at serine 1177 in patients with diabetes (P=0.007) without a difference in total eNOS expression. Insulin stimulation increased eNOS phosphorylation in non-diabetic subjects but not in diabetic patients (P=0.003) consistent with endothelial insulin resistance. Nitrotyrosine levels were higher in diabetic patients indicating endothelial oxidative stress. PKCβ expression was higher in diabetic patients and was associated with lower flow-mediated dilation (r=−0.541, P=0.02) Inhibition of PKCβ with LY379196 reduced basal eNOS phosphorylation and improved insulin-mediated eNOS activation in patients with diabetes. Endothelial NFκB activation was higher in diabetes and was reduced with PKCβ inhibition.
Conclusions
We provide evidence for the presence of altered eNOS activation, reduced insulin action and inflammatory activation in the endothelium of patients with diabetes. Our findings implicate PKCβ activity in endothelial insulin resistance.
Keywords: endothelium, diabetes, insulin resistance, nitric oxide
Type 2 diabetes mellitus affects a rapidly growing portion of the global population and is associated with high cardiovascular risk.1,2 Abnormal endothelial function contributes to the development of atherosclerosis and clinical vascular events in diabetes.3,4 Novel therapeutic strategies that restore endothelial function hold promise as interventions to lower cardiovascular risk in diabetes.
Experimental studies link the metabolic disturbances in diabetes to altered endothelial phenotype. In cultured endothelial cells, insulin activates endothelial nitric oxide synthase (eNOS) through phosphorylation at serine 1177.5 In animal models relevant to diabetes, endothelial insulin resistance impairs eNOS activation, reduces endothelium-dependent vasodilation, and promotes atherogenesis.6,7 Endothelial inflammatory activation via nuclear factor kappa B (NFκB) may perturb insulin-mediated signaling pathways thereby reducing nitric oxide bioactivity and increasing cytokine release.8,9
Prior studies link increased activity of protein kinase C beta (PKCβ) to endothelial dysregulation in diabetes.10 In the diabetic state, excess nutrients stimulate PKCβ through increased formation of lipid diacylglycerol (DAG), a byproduct of glucose and lipid metabolism.11 In cultured endothelial cells and animal models, PKCβ activation interrupts insulin-induced eNOS stimulation and PKCβ inhibition restores vascular function.12,13 Increased PKCβ activity has also been implicated in endothelial cell inflammatory activation.14
Endothelial dysfunction is a prominent feature of clinical diabetes. Prior studies by our group and others have demonstrated reduced endothelium-dependent vasodilation in patients with diabetes.4,15 The consequences of diabetes on endothelial insulin signaling and endothelial inflammatory activation remain incompletely characterized in the human vasculature. The present study sought to investigate endothelial insulin signaling, inflammation and the contribution of PKCβ activation to altered endothelial phenotype in human diabetes.
Methods
Study Subjects
We enrolled adults with type 2 diabetes mellitus defined as fasting serum glucose ≥126 mg/dL or ongoing treatment for type 2 diabetes at Boston Medical Center and control individuals without diabetes defined as fasting glucose <100mg/dl. All subjects were studied in the fasting state and a blood sample was taken for measurement of lipid levels and glucose levels in the Boston Medical Center Clinical Laboratory. Vasoactive medications were held on the morning of the study (last dose 24 hours prior to testing). The study protocol was approved by the Boston Medical Center Institutional Review Board and all participants provided written informed consent.
Vascular Function Testing
We measured brachial artery flow-mediated dilation as described.16 Briefly, high-resolution ultrasound was used to measure brachial artery diameter before and 1 minute following a 5-minute cuff occlusion of the upper arm (proximal to the imaged portion of the artery) designed to induce a hyperemic response. Doppler flow signals were measured in the resting state and immediately after cuff release.
Peripheral Endothelial Cell Collection
Peripheral venous endothelial cell biopsy was performed as previously described.17,18 Briefly, a 20-gauge intravenous catheter was inserted into a superficial forearm vein under aseptic technique. An 0.018-in J-wire (Arrow International, Reading PA) was introduced through the catheter and endothelial cells were collected by gentle abrasion of the vessel wall. Endothelial cells were recovered from the wire tip by centrifugation in a dissociation buffer and plated on poly-L-lysine coated microscope slides (Sigma). Once plated, cells were either fixed immediately in 4% paraformaldehyde or stimulated with insulin (described below) prior to fixation. Slides were washed in phosphate buffered saline (PBS), dried, and stored at −80°C until further processing.
Insulin-stimulation of Freshly Isolated Human Endothelial Cells
In order to select the optimal time-point for evaluating insulin-mediated eNOS phosphorylation in endothelial cells, we performed a time course evaluation using commercially available cultured human aortic endothelial cells (HAECs, Lonza). After overnight starvation, HAEC’s were treated with 10nM in EGM-2 medium devoid of growth factors at 37°C and fixed at multiple time points up to 60 minutes. With insulin treatment, eNOS phosphorylation increased transiently at 5 minutes and then was sustained at 30 through 60 minutes (Supplemental Figure 1). Thus, for our studies of freshly isolated human cells, we measured insulin-mediated eNOS phosphorylation at 30 minutes.
Freshly isolated endothelial cell samples were split immediately after isolation and prior to fixation with paraformaldahyde and incubated with either 0 or 10 nM insulin in EGM-2 Bullet Kit medium (Lonza Inc, Walkersville, MD), devoid of growth factors, at 37°C for 30 min. Cells were then washed in PBS, fixed, dried, and frozen as above. To test the hypothesis that PKCβ inhibition improves endothelial insulin signaling in diabetes, cells were similarly incubated with 0 or 10 nM insulin concurrently with LY379196, a PKCβ-specific inhibitor (kindly supplied by Eli Lilly and Company, Indianapolis, IN), at a concentration of 0 or 30 nM for 30 min. The dose of LY379196 was based on prior reports evaluating commercially available cultured endothelial cells.19 Cells were processed as described above.
Assessment of Protein Expression by Quantitative Immunofluorescence
Fixed samples were thawed and rehydrated with PBS containing 50mM glycine (Sigma) for 10 minutes. Cells were permeabilized with 0.1% Triton X-100 and non-specific binding sites were blocked with 0.5% bovine serum albumin (BSA). Slides were incubated for 1 hour at 37°C with primary antibodies against one of the following targets: eNOS (1:100 dilution, BD Biosciences, San Diego, CA), phosphorylated eNOS at serine 1177 (1:200 dilution, Millipore, Billerica, MA) and threonine 495 (1:100 dilution, Cell Signaling, Danvers, MA), Akt (1:100 dilution, Cell Signaling), PKCβ (1:100 dilution, Abcam, Cambridge, MA), phosphatase and tensin homolog (PTEN) (1:100, Santa Cruz Biotechnology, Santa Cruz, CA), nitrotyrosine (1:1000 dilution, Millipore, Billerica, MA), ICAM (1:150 dilution, Santa Cruz Biotechnology), IκBα (dilution, Novus, Littleton, CO), or p65 (1:375 dilution, Novus). All cells were also stained with an anti-von Willebrand Factor (vWF) antibody (1:300 dilution, Dako, Carpinteria, CA) to aid in endothelial cell identification. After incubation with the primary antibodies, the slides were washed and incubated with corresponding Alexa Fluor-488 and Alexa Fluor-594 antibodies (1:200 dilution, Invitrogen, Carlsbad, CA) and mounted under glass coverslips with Vectashield containing DAPI for nuclear identification (Vector Laboratories, Burlingame, CA). For each batch of patient-derived cells, we stained a control slide of cultured HAECs,, maintained in EGM-2 Bullet Kit medium (Lonza) at 37°C with 5% CO2 and taken from a single index passage.
Slides were imaged with a fluorescence microscope (Nikon Eclipse TE2000-E) at 20x magnification and digital images of the cells were captured using a Photometric CoolSnap HQ2 Camera (Photometrics, Tucson, AZ). Exposure time was held constant and image intensity was corrected for background fluorescence. Fluorescent intensity was quantified by NIS Elements AR Software (Nikon Instruments Inc, Melville, NY). For each protein of interest, fluorescent intensity was quantified in 20 cells from each patient and averaged. To minimize batch-to-batch variability in staining intensity, fluorescence intensity for each patient sample was normalized to the intensity of HAEC staining performed simultaneously. Intensity is expressed in arbitrary units (au) calculated by dividing the average fluorescence intensity from the patient sample by the average fluorescence intensity of the HAEC sample and multiplying by 100. Intensity quantification was performed blinded to subject identity and diabetes status, and each batch included patients with and without diabetes.
Statistical Analyses
Statistical analyses were performed using SPSS version 19.0. The distribution of continuous clinical characteristics and vascular function measures were evaluated by examining a histogram and the Shapiro-Wilk test and compared between diabetic patients and non-diabetic controls using the independent samples t-test or Mann-Whitney U test as appropriate. Categorical clinical characteristics were compared using chi-square testing. Baseline endothelial cell protein expression, and change in eNOS phosphorylation with insulin were compared between diabetic patients and non-diabetic controls using Mann-Whitney U test given the sample size. We compared flow-mediated dilation between patients with diabetes and non-diabetic controls adjusting for race, body-mass index, total cholesterol, HDL cholesterol, systolic blood pressure using a general linear model. We evaluated the effect of LY379196 on eNOS phosphorylation, the response to insulin, and IκBα expression with related samples Wilcoxon Signed Rank Test. We compared clinical characteristics, vascular measures and eNOS phosphorylation responses by evaluating correlation coefficients. A two-sided P-value <0.05 was considered statistically significant.
Results
Study Subjects and Vascular Function
We enrolled 40 patients with diabetes and 36 non-diabetic control participants. As shown in Table 1, patients with diabetes had metabolic abnormalities including higher body mass index, systolic blood pressure, fasting serum glucose, and glycosylated hemoglobin A1c, and lower HDL cholesterol. Patients with diabetes had lower LDL cholesterol, likely related to concomitant lipid-lowering therapies. Flow-mediated dilation of the brachial artery was lower in the patients with diabetes as compared to the non-diabetic controls, consistent with the presence of endothelial dysfunction. In a multivariable model adjusting for race, total cholesterol, HDL cholesterol, and systolic blood pressure, patients with diabetes had lower flow-mediated dilation than non-diabetic controls (least square mean±standard error, 6.4±0.7 vs. 9.7±0.7, P=0.003). Nitroglycerin-mediated dilation was lower in the patients with diabetes suggesting the presence of smooth muscle dysfunction or inactivation of nitric oxide by reactive oxygen species as well as impaired endothelium-dependent vasodilation. There were no differences in arterial diameter or resting or hyperemic flow.
Table 1.
Clinical Characteristics and Vascular Function
| Non-Diabetic (n=36) | Diabetic (n=40) | |
|---|---|---|
| Clinical Characteristics | ||
| Age, years | 51±10 | 55±7 |
| Female Sex, % | 33 | 45 |
| Black Race, % | 36 | 57 |
| Body Mass Index, kg/m2 | 27.4±4.8 | 33.5±7.7* |
| Total Cholesterol, mg/dl | 198±35 | 172±48* |
| LDL cholesterol, mg/dl | 121±26 | 96±34* |
| HDL cholesterol, mg/dl | 54±18 | 45±13* |
| Triglycerides, mg/dl | 118±77 | 163±125 |
| Fasting Glucose, mg/dl | 89±11 | 147±77* |
| Hemoglobin A1c, % | 5.4±0.5 | 8.9±5.5* |
| Systolic Blood Pressure, mmHg | 126±12 | 138±20* |
| Diastolic Blood Pressure, mmHg | 73±7 | 77±12 |
| Antiplatelet Therapy, % | 3 | 41* |
| Lipid Lowering Therapy, % | 8 | 49* |
| ACE inhibitor or ARB therapy, % | 3 | 61* |
| Metformin, % | 0 | 67* |
| Sulfonylureas, % | 0 | 18* |
| Thiazolidinediones, % | 0 | 5* |
| Insulin Therapy, % | 0 | 43* |
| Vascular Function | ||
| Baseline Diameter, mm | 4.19±0.72 | 4.33±0.77 |
| Baseline Flow, ml/min | 119±68 | 133±75 |
| Hyperemic Flow, ml/min | 1018±438 | 892±469 |
| Flow-mediated dilation, % | 10.0±4.0 | 6.5±4.3* |
| Flow-mediated dilation, mm* | 0.40±0.13 | 0.26±0.16* |
| Nitroglycerin-mediated dilation† | 13.6±5.2 | 10.1±4.6* |
Data expressed as Mean±SD
P<0.05
n=18, 26 non-diabetic, diabetic
Endothelial Nitric Oxide Synthase Activation and Response to Insulin in Freshly Isolated Endothelial Cells
To evaluate activation of eNOS, we quantified levels of eNOS phosphorylation at serine 1177 and threonine 495 in diabetic individuals. Notably, diabetic individuals had higher basal levels of activated eNOS at serine 1177 compared to controls (Figure 1, Supplemental Figure 2). To measure insulin-induced eNOS phosphorylation, we exposed cells from diabetic and non-diabetic subjects to 0 or 10 nM insulin for 30 min as shown in Figure 1, Supplemental Figure 2. With insulin stimulation, eNOS phosphorylation at serine 1177 increased 38±30% in cells from non-diabetic subjects (n=9, P=0.04). In contrast, eNOS phosphorylation at serine 1177 decreased 10±9% with insulin stimulation in cells from diabetic patients (n=12, P=0.006). The difference in overall insulin-induced eNOS phosphorylation at serine 1177 between the diabetic and non-diabetic groups was highly significant (P=0.003). Among the patients with diabetes, there were no differences in basal or insulin-stimulated eNOS phosphorylation at serine 1177 based on use of statins, ACE inhibitor or ARB, metformin, or insulin (Supplemental Table 1).
Figure 1.

Diabetes mellitus is associated with altered eNOS activation and insulin resistance in endothelial cells. As described in Methods, freshly isolated endothelial cells identified by vWF staining (green) and nuclear morphology (DAPI in blue) were obtained from patients with diabetes (n=12) and non-diabetic controls (n=9). Cells were incubated with 0nM or 10nM insulin for 30 minutes and then fixed. eNOS activation was evaluated as eNOS phosphorylation at serine 1177 (red). Protein levels were quantified by evaluating 20 cells for each patient under each condition. A. Representative cells from a patient with diabetes (right) show higher basal eNOS phosphorylation at serine 1177 (top, red) compared to the non-diabetic control (left top, red). Insulin increased eNOS phosphorylation at serine 1177 in the non-diabetic control (left top, red vs. left bottom, red) but not in the diabetic patient (right top, red vs. right bottom, red). B. Pooled data show that basal eNOS phosphorylation at serine 1177 was 67% higher in endothelial cells from patients with diabetes compared to non-diabetic controls (*P<0.01). C. Pooled data demonstrate that insulin increased eNOS phosphorylation at serine 1177 in endothelial cells from non-diabetic controls but not in endothelial cells from patients with diabetes (*P=0.003).
There was no difference in basal eNOS phosphorylation at the inhibitory site threonine 495 (Figure 2, Supplemental Figure 2). In response to insulin stimulation, the change in inhibitory eNOS phosphorylation at threonine 495 differed between diabetic patients (18±9%) and non-diabetic controls (−18±5%) (Figure 2, P=0.002).
Figure 2.

Diabetes mellitus and inhibitory eNOS phosphorylation. As described in Methods, freshly isolated endothelial cells identified by vWF staining (green) and nuclear morphology (DAPI in blue) were obtained from patients with diabetes (n=9) and non-diabetic controls (n=6). Cells were incubated with 0nM or 10nM insulin for 30 minutes and then fixed. eNOS phosphorylation at threonine 495 (red) was quantified by evaluating 20 cells for each patient under each condition. A. Representative cells from a patient with diabetes (right) show similar basal eNOS phosphorylation at threonine 495 (top, red) compared to the non-diabetic control (left top, red). Insulin decreased eNOS phosphorylation at threonine 495 in the non-diabetic control (left top, red vs. left bottom, red) and increased phosphorylation at this site in the diabetic patient (right top, red vs. right bottom, red). B. Pooled data show that basal eNOS phosphorylation at threonine 495 was similar in endothelial cells from patients with diabetes compared to non-diabetic controls (P=0.95). C. Pooled data demonstrate that insulin decreased eNOS phosphorylation at threonine 495 in endothelial cells from non-diabetic controls but increased in endothelial cells from patients with diabetes (*P=0.009).
Overall, a greater increase in eNOS phosphorylation with insulin stimulation was associated with higher flow-mediated dilation (r=0.523, P=0.04, Supplemental Figure 3). Among the diabetic patients, higher body mass index was associated with lower insulin-stimulated eNOS phosphorylation (r=−0.600, P=0.04). Fasting glucose and hemoglobin A1c were not associated with insulin-stimulated eNOS phosphorylation.
As shown in Figure 3, total eNOS and Akt expression were similar in endothelial cells from patients with diabetes compared to control subjects. Expression of PTEN, a phosphatase that opposes PI3 kinase action and inhibits Akt activation, was similar in diabetic patients and non-diabetic controls (Figure 3, Supplemental Figure 4). These results suggest alterations in eNOS activation with comparable eNOS expression in patients with diabetes. As shown in Figure 3, expression of nitrotyrosine, a marker of oxidative stress, was higher in patients with diabetes than in non-diabetic controls. Basal eNOS phosphorylation at serine 1177, total eNOS expression, and nitrotyrosine level were not associated significantly with flow-mediated dilation.
Figure 3.

Abnormal eNOS activation is not attributable to differential eNOS, Akt or PTEN expression in diabetes. Diabetes is associated with oxidative stress in endothelial cells. Freshly isolated endothelial cells collected from patients with diabetes and non-diabetic controls were fixed and stained (20 cells per patient for each protein). A. Representative cell from a diabetic patient (right) shows similar eNOS expression (red) compared with a cell from a non-diabetic control (left, red). Pooled data show that eNOS levels were comparable in the non-diabetic controls (n=8) and diabetics (n=10, P=0.46). B. Representative cell from a diabetic patient (right) shows similar Akt expression (red) compared with a cell from a non-diabetic control (left, red). Pooled data show that Akt levels were comparable in the non-diabetic controls (n=6) and diabetics (n=10, P=0.79). C. Representative cell from a diabetic patient (right) shows similar PTEN expression (red) compared with a cell from a non-diabetic control (left, red). Pooled data show that PTEN levels were comparable in the non-diabetic controls (n=8) and diabetics (n=10, P=1.0). D. Representative cell from a diabetic patient (right) shows higher nitrotyrosine expression (red) compared with a cell from a non-diabetic control (left, red). Pooled data show that nitrotyrosine levels were higher diabetic patients (n=5) compared to the non-diabetic controls (n=5, *P=0.03).
PKCβ Inhibition Restores Endothelial Nitric Oxide Synthase Activation and Insulin Signaling in Diabetes
To investigate PKCβ activation in the diabetic endothelium, we measured total PKCβ expression. As shown in Figure 4 and Supplemental Figure 5, PKCβ expression was higher in patients with diabetes compared to non-diabetic controls. Higher PKCβ expression was associated with lower flow-mediated dilation of the brachial artery in the overall sample (Figure 4) and among the patients with diabetes (r=−0.674, P=0.03).
Figure 4.
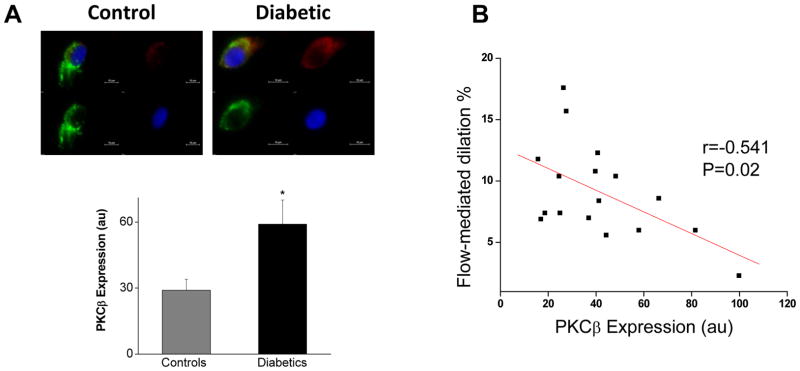
PKCβ and Endothelial Function. A. Representative cell from a diabetic patient (right) shows higher PKCβ expression (red) compared with a cell from a non-diabetic control (left, red). Pooled data (20 cells analyzed per patient) show that PKCβ levels were higher in diabetic patients (n=11) compared to the non-diabetic controls (n=7, *P=0.03). B. Higher endothelial PKCβ expression levels were associated with lower flow-mediated dilation.
To evaluate the role of PKCβ activation in the impairment of eNOS signaling in diabetes, we treated freshly isolated endothelial cells with LY379196, a pharmacologic inhibitor of PKCβ.19 As shown in Figure 5 and Supplemental Figure 6, PKCβ inhibition decreased the basal levels of eNOS phosphorylation at serine 1177 in endothelial cells from patients with diabetes. The change in basal eNOS phosphorylation with PKCβ inhibition was not significantly correlated with flow-mediated dilation.
Figure 5.

Inhibition of PKCβ alters eNOS phosphorylation and insulin response in endothelial cells from diabetic patients. As described in Methods, freshly isolated endothelial cells from patients with diabetes (n=7) were treated with 0 or 30nM LY379196, a pharmacologic inhibitor of PKCβ, along with 0 or 10nM insulin for 30 minutes and eNOS phosphorylation at serine 1177 was measured (20 cells per patient in each condition). A. Representative cells from a patient with diabetes treated with LY379196 (right) shows lower basal eNOS phosphorylation at serine 1177 (top, red) compared to control condition (left top, red). Treatment with LY379196 improved the insulin-mediated change in eNOS phosphorylation at serine 1177 (right top, red vs. right bottom, red) compared to control condition (left top, red vs. left bottom, red). B. Basal eNOS phosphorylation at serine 1177 was reduced in endothelial cells from patients with diabetes treated with LY379196 (*P=0.02). C. Treatment with LY379196 improved insulin-mediated eNOS phosphorylation at serine 1177 in endothelial cells from patients with diabetes (*P=0.04).
Treatment with LY379196 improved the insulin-mediated change in eNOS phosphorylation at serine 1177 in endothelial cells from diabetic patients, suggesting that PKCβ activity impedes insulin-mediated activation of eNOS in diabetes (Figure 5). PKCβ inhibition did not change in basal eNOS phosphorylation at threonine 495 in patients with diabetes (Supplementary Figure 7). Treatment with LY379196 did not change significantly the insulin-mediated levels of eNOS phosphorylation at threonine 495 in patients with diabetes (Supplementary Figure 7).
NFκB Activation in Freshly Isolated Endothelial Cells in Diabetes
To evaluate endothelial inflammatory activation, we examined the activity of NFκB by quantification of subunit p65, the inhibitory regulator IκBα, and ICAM-1. (Figure 6, Supplemental Figure 5).20 Endothelial cells from diabetic patients showed a trend toward higher levels of p65 when compared with non-diabetic controls. Expression of the negative regulator of NFκB, IκBα, was lower in endothelial cells from diabetic patients than in non-diabetic controls. Further, expression of ICAM-1, a protein regulated by NFκB, trended toward higher in endothelial cells from patients with diabetes than in non-diabetic controls. Endothelial p65, ICAM-1, and IκBα expression levels were not associated significantly with flow-mediated dilation.
Figure 6.

PKCβ and endothelial inflammatory activation in diabetes. A. There was a trend for higher expression of NFκB subunit p65 in endothelial cells from patients with diabetes (n=4 diabetic and 5 non-diabetic, *P=0.06). B. There was a trend for higher expression of ICAM-1, a cellular adhesion molecule regulated by NFκB, in endothelial cells from patients with diabetes (n=4 diabetic and 5 non-diabetic, *P=0.06). C. Expression of IκBα, an inhibitor of NFκB, was lower in endothelial cells from patients with diabetes (n=6 diabetic and 5 non-diabetic, *P=0.004). Treatment with LY379196 for 30 minutes increased IκBα expression in patients with diabetes (**P=0.03).
To evaluate the role of PKCβ in the NFκB activation in diabetes, we measured IκBα expression before and after treatment with LY379196 for 30 minutes. In endothelial cells from diabetic patients, inhibition of PKCβ increased IκBα expression suggesting a link between PKCβ activity and activation of NFκB (Figure 6).
Discussion
In the present study, we provide direct evidence that signaling pathways responsible for eNOS activation are altered in endothelial cells from patients with diabetes. Interestingly, we found elevated levels of activated eNOS in conjunction with reduced flow-mediated vasodilation in diabetic patients compared to control individuals. Basal expression of total eNOS, Akt and PTEN were similar in diabetes suggesting that abnormal endothelial function is not attributable to altered levels of these proteins. Activation of eNOS by insulin was severely blunted in endothelial cells from diabetic patients indicating the presence of endothelial insulin resistance. Inhibitory eNOS phosphorylation was increased by insulin in patients with diabetes but decreased in non-diabetic controls. In addition, we observed endothelial inflammatory activation and oxidative stress evidenced by increased markers of NFκB activity and increased nitrotyrosine expression in endothelial cells from diabetic patients.
Inhibition of PKCβ with LY379196 blunted the diabetes-associated endothelial abnormalities in basal and insulin-stimulated eNOS phosphorylation state. Further, PKCβ inhibition increased IκBα expression suggesting that PKCβ inhibition reduced NFκB activation in endothelial cells from patients with diabetes. Taken together, our findings suggest that PKCβ activity contributes to the abnormal endothelial phenotype characterized by impaired insulin-mediated eNOS activation in diabetes.
Experimental studies have identified insulin as a regulator of endothelial nitric oxide production. In commercially-available cultured endothelial cells, insulin enhances eNOS activity through phosphatidylinositol-3 kinase (PI3K)–Akt mediated phosphorylation at serine 1177.5 Relevant to our study of flow-mediated dilation, shear stress also activates eNOS via the PI3K-Akt signaling axis.21 In animal models, diminished vascular insulin signaling is associated with reduced nitric oxide bioavailability.22,23 Further, endothelial-specific elimination of the insulin receptor leads to reduced insulin-mediated eNOS activation, endothelial vasomotor dysfunction, inflammatory activation and accelerated plaque formation.7 These studies support the concept that endothelial insulin resistance contributes to the development of endothelial dysfunction and atherosclerosis; however, the relevance to human diabetes has not been established previously.
Our data provide insight into the consequences of human diabetes on pathways that modulate eNOS activity under basal and stimulated conditions. We observed excessive basal eNOS activation and reduced flow-mediated dilation in diabetic patients. The differences in resting eNOS phosphorylation are concordant with a previous report describing high basal eNOS phosphorylation in obese subjects.24 Complex processes determine nitric oxide bioavailability, including the amount of nitric oxide synthesized and the extent of nitric oxide destruction by reactive oxygen species.25 Higher eNOS activation may represent an adaptive response to high oxidative stress in diabetes.26 Consistent with this possibility, we observed higher nitrotyrosine levels in the endothelial cells from diabetic patients.27,28 Under pathologic conditions including diabetes, eNOS may become uncoupled and produce superoxide, rather than NO.29 It is plausible that elevated eNOS phosphorylation in diabetes reflects a compensatory alteration to chronically reduced nitric oxide levels. The higher basal Ser1177 phosphorylation in endothelial cells from diabetic subjects raises the possibility that the blunted response to insulin stimulation represents a “ceiling” effect that limits the amount of additional phosphorylation that can occur.
Our study extends our understanding of nitric oxide regulation in diabetic endothelial cells by examining the functional response to insulin. We demonstrated that insulin promotes eNOS activity by increasing activating (Ser1177) and decreasing inhibitory (Thr495) phosphorylation in endothelial cells from non-diabetic control individuals. In contrast, endothelial cells isolated from diabetic patients displayed a marked abnormality of endothelial insulin action. These findings suggest that modulation of eNOS activation through the PI3K-Akt pathway is disrupted in human diabetes. Further, insulin augments inhibitory phosphorylation of eNOS that may contribute to uncoupling and oxidative stress.30 The inability to upregulate eNOS activity in response to acute insulin administration was accompanied by abnormal shear-stress mediated vasodilator responses. Our findings support the possibility that insulin resistance in the endothelium contributes to endothelial dysfunction in humans with diabetes.
The present study suggests that PKCβ activity is an important determinant of abnormal eNOS activation in diabetes. In animal models of diabetes, PKCβ inhibition improved insulin-mediated PI3K/Akt signaling in parallel with endothelium-mediated vasodilation.12 In human subjects, PKCβ inhibition with ruboxistaurin treatment prevented the development of endothelial dysfunction induced by acute hyperglycemia and improved flow-mediated dilation in diabetic patients.31,32 Consistent with these studies, we observed higher PKCβ levels in endothelial cells from patients with diabetes. Higher PKCβ expression was associated with reduced endothelial function evidenced by lower flow-mediated dilation. Inhibiting PKCβ activity improved the responsiveness to insulin in endothelial cells and reduced basal eNOS phosphorylation in diabetes. In cell culture, PKCβ has been described as reducing eNOS activity through phosphorylation at the inhibitory site Thr495. We did not observe differences in basal or insulin-mediated eNOS phosphorylation at threonine 495 with PKCβ inhibition. Collectively, our findings support the concept that PKCβ activity impedes regulation of eNOS activation in diabetes.
Prior investigations of eNOS expression in diabetic conditions have yielded conflicting results. In cultured endothelial cells, hyperglycemia has been shown to decrease total eNOS protein levels; however, similar glucose conditions were reported to increase eNOS expression along with increased superoxide production and reduced NO bioactivity.33,34 Increased vascular eNOS expression has also been observed in the aortas of insulin resistant rats and eNOS expression was normalized after treatment with a PKC inhibitor.12 In patients undergoing coronary artery bypass grafting, expression of eNOS was lower in the internal mammary arteries in the presence of diabetes.35 However, eNOS levels were similar in freshly isolated endothelial cells from obese and lean individuals.24 In the current study, endothelial eNOS expression was comparable in diabetic patients and controls suggesting that eNOS deficiency is not an important mechanism for endothelial dysfunction in diabetes.
Inflammatory activation may contribute to endothelial dysfunction in diabetes. Experimental data links NFκB activity to vascular insulin resistance and endothelial dysfunction.36 Prior human data in older and obese individuals supports a pro-inflammatory phenotype in endothelial cells characterized by increased NFκB activation.24,37 Similarly, we found evidence of higher NFκB activation in endothelial cells of patients with diabetes in conjunction with abnormal eNOS signaling. Our findings suggest the possibility that endothelial inflammatory activation through NFκB contributes to vascular insulin resistance. PKCβ inhibition increased IκBα expression in endothelial cells from patients with diabetes consistent with a link between PKCβ activity and endothelial inflammation.14 Additional studies are needed to evaluate whether NFκB inhibitors have a beneficial effect on endothelial signaling in human diabetes.
Several limitations of our study should be noted. The studies were performed using venous and not arterial endothelial cells that may be more directly relevant to atherogenesis. However, venous endothelial cells are exposed to the systemic metabolic disturbances present in diabetes. Further, previous reports indicate correlations between protein expression in venous and arterial endothelial cells obtained by similar biopsy methodology.18,38 The relatively modest sample size for the endothelial cell evaluations precludes multivariable adjustment for confounders. Dysregulation of eNOS activation in diabetes is likely complex and multifactorial.5 Based on animal studies, we prospectively sought to investigate the contribution of PKCβ activity.10 Further studies are warranted to evaluate additional mechanisms for altered eNOS signaling in diabetes. Additional phosphorylation sites have been described that regulate eNOS activity that we have not evaluated in the present study.25 As has been described previously, we observed lower flow-mediated and nitroglycerin-mediated vasodilation in patients with diabetes indicating multilevel abnormalities in vasomotor function.39–42 These limitations are counterbalanced by the novel functional demonstration of insulin action in endothelial cells from patients and evidence supporting a role for PKCβ in abnormal endothelial signaling in humans with diabetes.
In summary, we have demonstrated the presence of endothelial insulin resistance in diabetic patients with endothelial dysfunction that is alleviated by PKCβ inhibition. The emergent diabetes epidemic necessitates the identification of novel therapeutic interventions for cardiovascular disease reduction. Pharmaceutical agents that reduce PKCβ activity have shown potential in selected clinical trials to reduce microvascular complications in diabetes.10 Our findings provide support for developing interventions that restore insulin action in the endothelium, potentially by inhibiting PKCβ, as a strategy to lower vascular disease burden in patients with diabetes.
Supplementary Material
Atherosclerotic vascular disease in patients with type 2 diabetes mellitus is a pressing health problem. Abnormal endothelial function contributes to cardiovascular disease development and manifestation in diabetic patients. Experimental studies link insulin resistance and endothelial dysfunction. We demonstrated that endothelial cells collected directly from patients with diabetes have impaired activation of endothelial nitric oxide synthase in response to insulin. Further, we observed evidence of increased oxidative stress and inflammatory activation in the endothelial cells from diabetic patients. We found evidence of higher levels of PKCβ in the endothelial cells from diabetic patients that were associated with lower endothelial function measured by brachial artery flow-mediated dilation. Treatment with a PKCβ inhibitor improved insulin signaling and reduced inflammatory activation in the endothelial cells from patients with diabetes. This study provides evidence that patients with diabetes have endothelial insulin resistance and supports the possibility that treatments aimed at restoring insulin action in the endothelium may be a novel strategy to reduce vascular disease burden in this high risk group.
Acknowledgments
We gratefully acknowledge Eli Lilly and Company for providing the experimental reagent LY379196.
Funding Sources: The project was supported by HL102299 and HL109790 from the National Heart, Lung and Blood Institute. Drs Tabit, Hamburg, and Vita received support from the National Institutes of Health (NIH)–sponsored Boston University Medical Center Leadership Program in Vascular Medicine (K12 HL083781). Drs. Fetterman, Dohadwala, and Farb received support from T32 HL007224. Dr Vita is supported by NIH grants HL083801, HL081587, HL083269, and HL75795.
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Lin JK, Farzadfar F, Khang YH, Stevens GA, Rao M, Ali MK, Riley LM, Robinson CA, Ezzati M. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011;378:31–40. doi: 10.1016/S0140-6736(11)60679-X. [DOI] [PubMed] [Google Scholar]
- 2.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, de Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Executive summary: heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:948–54. doi: 10.1161/CIRCULATIONAHA.109.192666. [DOI] [PubMed] [Google Scholar]
- 3.Tabit CE, Chung WB, Hamburg NM, Vita JA. Endothelial dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev Endocr Metab Disord. 2010;11:61–74. doi: 10.1007/s11154-010-9134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Creager MA, Luscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation. 2003;108:1527–32. doi: 10.1161/01.CIR.0000091257.27563.32. [DOI] [PubMed] [Google Scholar]
- 5.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 6.Kearney MT, Duncan ER, Kahn M, Wheatcroft SB. Insulin resistance and endothelial cell dysfunction: studies in mammalian models. Exp Physiol. 2008;93:158–63. doi: 10.1113/expphysiol.2007.039172. [DOI] [PubMed] [Google Scholar]
- 7.Rask-Madsen C, Li Q, Freund B, Feather D, Abramov R, Wu IH, Chen K, Yamamoto-Hiraoka J, Goldenbogen J, Sotiropoulos KB, Clermont A, Geraldes P, Dall’Osso C, Wagers AJ, Huang PL, Rekhter M, Scalia R, Kahn CR, King GL. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein E null mice. Cell Metab. 2010;11:379–89. doi: 10.1016/j.cmet.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Read MA, Whitley MZ, Williams AJ, Collins T. NF-kappa B and I kappa B alpha: an inducible regulatory system in endothelial activation. J Exp Med. 1994;179:503–12. doi: 10.1084/jem.179.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A, Schwartz MW. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28:1982–8. doi: 10.1161/ATVBAHA.108.169722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–31. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rask-Madsen C, King GL. Proatherosclerotic mechanisms involving protein kinase C in diabetes and insulin resistance. Arterioscler Thromb Vasc Biol. 2005;25:487–96. doi: 10.1161/01.ATV.0000155325.41507.e0. [DOI] [PubMed] [Google Scholar]
- 12.Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K, Way KJ, Jacobs JR, Clermont AC, Ueki K, Ohshiro Y, Zhang J, Goldfine AB, King GL. Activation of vascular protein kinase C-beta inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes. 2006;55:691–8. doi: 10.2337/diabetes.55.03.06.db05-0771. [DOI] [PubMed] [Google Scholar]
- 13.Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A, Bursell SE, Kern TS, Ballas LM, Heath WF, Stramm LE, Feener EP, King GL. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science. 1996;272:728–31. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 14.Kouroedov A, Eto M, Joch H, Volpe M, Luscher TF, Cosentino F. Selective inhibition of protein kinase Cbeta2 prevents acute effects of high glucose on vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 2004;110:91–6. doi: 10.1161/01.CIR.0000133384.38551.A8. [DOI] [PubMed] [Google Scholar]
- 15.Hamburg NM, Palmisano J, Larson MG, Sullivan LM, Lehman BT, Vasan RS, Levy D, Mitchell GF, Vita JA, Benjamin EJ. Relation of brachial and digital measures of vascular function in the community: the Framingham heart study. Hypertension. 2011;57:390–6. doi: 10.1161/HYPERTENSIONAHA.110.160812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McMackin CJ, Vita JA. Update on nitric oxide-dependent vasodilation in human subjects. Methods Enzymol. 2005;396:541–53. doi: 10.1016/S0076-6879(05)960-16-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess MA, Levit A, Kim B, Hartman ML, Joseph L, Shirihai OS, Vita JA. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444–53. doi: 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colombo PC, Ashton AW, Celaj S, Talreja A, Banchs JE, Dubois NB, Marinaccio M, Malla S, Lachmann J, Ware JA, Le Jemtel TH. Biopsy coupled to quantitative immunofluorescence: a new method to study the human vascular endothelium. J Appl Physiol. 2002;92:1331–8. doi: 10.1152/japplphysiol.00680.2001. [DOI] [PubMed] [Google Scholar]
- 19.Quagliaro L, Piconi L, Assaloni R, Da Ros R, Maier A, Zuodar G, Ceriello A. Intermittent high glucose enhances ICAM-1, VCAM-1 and E-selectin expression in human umbilical vein endothelial cells in culture: the distinct role of protein kinase C and mitochondrial superoxide production. Atherosclerosis. 2005;183:259–67. doi: 10.1016/j.atherosclerosis.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 20.Pierce JW, Read MA, Ding H, Luscinskas FW, Collins T. Salicylates inhibit I kappa B-alpha phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. J Immunol. 1996;156:3961–9. [PubMed] [Google Scholar]
- 21.Dimmeler S, Assmus B, Hermann C, Haendeler J, Zeiher AM. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ Res. 1998;83:334–41. doi: 10.1161/01.res.83.3.334. [DOI] [PubMed] [Google Scholar]
- 22.Jiang ZY, Lin YW, Clemont A, Feener EP, Hein KD, Igarashi M, Yamauchi T, White MF, King GL. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest. 1999;104:447–57. doi: 10.1172/JCI5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wheatcroft SB, Shah AM, Li JM, Duncan E, Noronha BT, Crossey PA, Kearney MT. Preserved glucoregulation but attenuation of the vascular actions of insulin in mice heterozygous for knockout of the insulin receptor. Diabetes. 2004;53:2645–52. doi: 10.2337/diabetes.53.10.2645. [DOI] [PubMed] [Google Scholar]
- 24.Silver AE, Beske SD, Christou DD, Donato AJ, Moreau KL, Eskurza I, Gates PE, Seals DR. Overweight and obese humans demonstrate increased vascular endothelial NAD(P)H oxidase-p47(phox) expression and evidence of endothelial oxidative stress. Circulation. 2007;115:627–37. doi: 10.1161/CIRCULATIONAHA.106.657486. [DOI] [PubMed] [Google Scholar]
- 25.Hamburg NM, Vita JA. Endothelial dysfunction in atherosclerosis: Mechanisms of impaired nitric oxide bioactivity. In: Loscalzo J, editor. Molecular mechanisms of atherosclerosis. London: Taylor & Francis; 2006. pp. 95–110. [Google Scholar]
- 26.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–62. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 27.Jelic S, Lederer DJ, Adams T, Padeletti M, Colombo PC, Factor PH, Le Jemtel TH. Vascular inflammation in obesity and sleep apnea. Circulation. 2010;121:1014–21. doi: 10.1161/CIRCULATIONAHA.109.900357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shishehbor MH, Aviles RJ, Brennan ML, Fu X, Goormastic M, Pearce GL, Gokce N, Keaney JF, Jr, Penn MS, Sprecher DL, Vita JA, Hazen SL. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA. 2003;289:1675–80. doi: 10.1001/jama.289.13.1675. [DOI] [PubMed] [Google Scholar]
- 29.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KAJ. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95:9220–5. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin MI, Fulton D, Babbitt R, Fleming I, Busse R, Pritchard KA, Jr, Sessa WC. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J Biol Chem. 2003;278:44719–26. doi: 10.1074/jbc.M302836200. [DOI] [PubMed] [Google Scholar]
- 31.Beckman JA, Goldfine AB, Gordon MB, Garrett LA, Creager MA. Inhibition of protein kinase C beta prevents impaired endothelium- dependent vasodilation caused by hyperglycemia in humans. Circ Res. 2002;90:107–11. doi: 10.1161/hh0102.102359. [DOI] [PubMed] [Google Scholar]
- 32.Mehta NN, Sheetz M, Price K, Comiskey L, Amrutia S, Iqbal N, Mohler ER, Reilly MP. Selective PKC beta inhibition with ruboxistaurin and endothelial function in type-2 diabetes mellitus. Cardiovasc Drugs Ther. 2009;23:17–24. doi: 10.1007/s10557-008-6144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding Y, Vaziri ND, Coulson R, Kamanna VS, Roh DD. Effects of simulated hyperglycemia, insulin, and glucagon on endothelial nitric oxide synthase expression. Am J Physiol Endocrinol Metab. 2000;279:E11–E17. doi: 10.1152/ajpendo.2000.279.1.E11. [DOI] [PubMed] [Google Scholar]
- 34.Cosentino F, Hishikawa K, Katusic ZS, Luscher TF. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation. 1997;96:25–8. doi: 10.1161/01.cir.96.1.25. [DOI] [PubMed] [Google Scholar]
- 35.Okon EB, Chung AW, Rauniyar P, Padilla E, Tejerina T, McManus BM, Luo H, van Breemen C. Compromised arterial function in human type 2 diabetic patients. Diabetes. 2005;54:2415–23. doi: 10.2337/diabetes.54.8.2415. [DOI] [PubMed] [Google Scholar]
- 36.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pierce GL, Lesniewski LA, Lawson BR, Beske SD, Seals DR. Nuclear factor-{kappa}B activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation. 2009;119:1284–92. doi: 10.1161/CIRCULATIONAHA.108.804294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silver AE, Christou DD, Donato AJ, Beske SD, Moreau KL, Magerko KA, Seals DR. Protein expression in vascular endothelial cells obtained from human peripheral arteries and veins. J Vasc Res. 2010;47:1–8. doi: 10.1159/000231715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ayer JG, Harmer JA, David C, Steinbeck S, Seale JP, Celermajer DS. Severe obesity is associated with impaired arterial smooth muscle function in young adults. Obesity. 2011;19:54–60. doi: 10.1038/oby.2010.114. [DOI] [PubMed] [Google Scholar]
- 40.Adams MR, Robinson J, McCredie R, Seale JP, Sorensen KE, Deanfield JE, Celermajer DS. Smooth muscle dysfunction occurs independently of impaired endothelium-dependent dilation in adults at risk of atherosclerosis. J Am Coll Cardiol. 1998;32:123–7. doi: 10.1016/s0735-1097(98)00206-x. [DOI] [PubMed] [Google Scholar]
- 41.Clarkson P, Celermajer DS, Donald AE, Sampson M, Sorensen KE, Adams M, Yue DK, Betteridge DJ, Deanfield JE. Impaired vascular reactivity in insulin-dependent diabetes mellitus is related to disease duration and low density lipoprotein cholesterol levels. J Am Coll Cardiol. 1996;28:573–9. doi: 10.1016/0735-1097(96)82380-1. [DOI] [PubMed] [Google Scholar]
- 42.Sorensen VR, Mathiesen ER, Clausen P, Flyvbjerg A, Feldt-Rasmussen B. Impaired vascular function during short-term poor glycaemic control in Type 1 diabetic patients. Diabet Med. 2005;22:871–6. doi: 10.1111/j.1464-5491.2005.01543.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.