

Abstract
Rabbits, mice, rats, non-human primates, sheep and cattle have been used to study the effect of Clostridium perfringens enterotoxin (CPE). CPE produces mostly necrosis of the small intestinal epithelium along with fluid accumulation in rabbits and mice. In the latter, CPE can bind to internal organs such as the liver, which induces lethal potassium levels in blood.
Keywords: animal models, calf, Clostridium perfringens, enterotoxin, lamb, mouse, non-human primate, rabbit, rat
1. Introduction
Clostridium perfringens, a Gram-positive spore forming anaerobic bacterium, is a prolific toxin producer [1]. At least 17 different C. perfringens toxins are reported in the literature, but individual isolates only produce some of these toxins [1]. This variability in toxin production provides the basis for a commonly-used toxin typing classification system where individual strains are assigned to one of five types based upon their production of four typing toxins, i.e., alpha beta, epsilon and iota toxins. All isolates produce alpha toxin, but type B strains also produce beta and epsilon toxins, type C isolates also produce beta toxin, type D strains also produce epsilon toxin and type E isolates also produce iota toxin.
Besides producing one or more of the typing toxins, some C. perfringens strains produce additional toxins. One of those non-typing toxins is C. perfringens enterotoxin (CPE), which can be produced by all types, except type B. Although only ~5% of type A strains produce this enterotoxin [2], those CPE-positive type A strains have exceptional biomedical importance. They cause C. perfringens type A food poisoning, which is the second most common bacterial illness in the USA, where about 1 million cases/year occur [3]. In addition these strains cause ~3-15% of all cases of human non-food borne human gastrointestinal diseases, including antibiotic-associated diarrhea and sporadic diarrhea [4]. Molecular Koch’s postulate analyses strongly support CPE production as the essential factor necessary for CPE-positive type A isolates to cause intestinal pathology in animal models [5].
CPE is encoded by a gene (cpe) that can be located on the chromosome or on large plasmids [6]. When located on large plasmids it is associated with a putative transposon, named Tn5565, which is flanked by IS1470 insertion sequences [7]. In type A isolates, these are two major families of cpe plasmids [8]. The 75.3 kb pCPF5603 cpe plasmid family has an IS1151 insertion sequence near cpe and also carries the gene (cpb2) encoding beta2 toxin. In contrast, the 70.5 kb pCPF4969 cpe plasmid family has an IS1470-like sequence near cpe; it lacks the cpb2 gene but encodes a bacteriocin gene cluster. Both the pCPF5603 and pCPF4969 cpe plasmids carry the tcp locus [8], which has been shown to mediate conjugative transfer for some other C. perfringens plasmids [9]. The tcp region likely also mediates conjugative transfer of the cpe plasmids, since mixed mating studies have shown conjugative transfer of pCPF4969 [10].
CPE is a single 319 amino acid polypeptide of ~35 kDa with a unique sequence [11], except for some limited homology (of still unknown significance) with a nonneurotoxic protein made by C. botulinum [6]. The structure of CPE was recently solved by two groups [12,13], which revealed this toxin belongs to the aerolysin family of pore-forming toxins. Notably, this family also includes two other clostridial toxins, i.e., C. perfringens epsilon toxin and C. septicum alpha toxin. Structurally, the CPE protein consists of two domains, including a C-terminal domain that mediates receptor binding [14] and an N-terminal domain that is responsible for post binding cytotoxic functions [15,16,17], including oligomerization and membrane insertion/pore formation as described below. The D48 and I51 residues of CPE are particularly important for formation of the CPE oligomers described below [16,17]. The transmembrane domain of CPE responsible for mediating insertion of the toxin during pore formation (see below) appears to involve an alpha helical region named TM1, which spans from CPE amino acids 81 to 106 [17].
The first step in CPE action is binding of the toxin to certain members of the claudin family of tight junction proteins [18,19,20]. Claudins are ~22 kDa proteins whose structure consists of four transmembrane domains, two extracellular loops (ECLs) and a cytoplasmic tail [21]. Binding of CPE to claudin receptors involves interactions between tyrosine residues in the 30 C-terminal amino acids of CPE and ECL-2 of receptor claudins [14,22,23,21]. Interestingly, not all claudins can serve as CPE receptors [18,22,21]; recent studies have identified an Asn located in the middle of ECL-2 as being critical for CPE binding [22,23,21]. CPE receptor claudins are present on many epithelial cells, including those found in the intestines [24].
Once bound to a claudin receptor, the toxin becomes localized in a still poorly characterized small complex [25]. This SDS-sensitive complex is ~90 kDa in size and apparently contains both claudin receptors and nonreceptor claudins [26,25]. Small complex formation will occur at 4°C but CPE action proceeds no further at low temperature [27].
At 37°C, CPE in small complexes apparently oligomerizes into an SDS-resistant complex named CH-1 [26]. CH-1 is ~450 kDa in size and contains, besides 6 copies of CPE, both receptor and nonreceptor claudins. Initial formation of CH-1 occurs on the membrane surface; however at 37°C, the CH-1 prepore rapidly inserts into membranes to form an active pore [17].
CPE pore formation results initially in a rapid breakdown of membrane permeability properties for small molecules of <200 Daltons [28]. Of particular importance, formation of the CPE pore causes a Ca2+ influx into CPE-treated cells that activates cell death pathways [29,30]. At low CPE doses there is a modest Ca2+ influx that triggers a classical caspase-3 cell death pathway. In contrast, high CPE doses induce a massive Ca2+ influx that causes cell death by oncosis.
Formation of the CPE pore also results in morphologic damage that exposes the basolateral surface of sensitive host cells [31]. This effect results in formation of CH2, a second SDS-resistant CPE complex of ~600 kDa [26,32]. CH-2 contains, at minimum, 6 copies of CPE, receptor and nonreceptor claudins and a second tight junction protein named occludin. The presence of occludin in CH-2 is most likely due to occludin:claudin interactions since CPE does not bind directly to occludin [32].
CPE-induced cell death fosters the development of microscopic damage in the intestines. CPE-induced microscopic damage includes villus blunting, epithelial desquamation and epithelial necrosis [24]. This damage appears to trigger the fluid and electrolyte fluxes responsible for diarrhea based upon the fact that only CPE doses causing microscopic damage are capable of causing fluid/electrolyte changes in rabbit ileal loops [33]. In addition, the development of fluid and electrolyte alterations in CPE-challenged rabbit ileal loops correlates closely with the onset of microscopic damage [34].
Several animal models have been used extensively to study the intestinal and systemic activity of CPE [35,36,37,38,39,34]. We review here the information published on animal models to study the pathogenesis of CPE-positive C. perfringens type A infections.
2. Rabbit models
Rabbit intestinal loops have been used for many years to study the effects of CPE in vivo[40,36,41,24]. The effect of CPE has been studied mostly in the small intestine (jejunum and ileum) of rabbits. Purified CPE caused net secretion of fluid and eletrolytes, together with microscopic damage in the rabbit ileum and jejunum, but not in the duodenum [38,37,42]. Thus it appears that, within the rabbit small intestine, the sensitivity to the action of CPE increases moving aborally from the proximal duodenum, with the terminal ileum being the most responsive [38].
Microscopic changes observed in the small intestinal loops of rabbits treated with CPE are dose-dependent and they consist of necrosis of superficial epithelium and lamina propria, villus blunting and fusion, mucosal and submucosal edema and hemorrhage [36,41,24] (Fig. 1). Initial CPE-induced intestinal damage in rabbits begins at villus tips in small intestinal loops [36,34]. This was also confirmed by electron microscopy studies, in which CPE enterocyte damage and heavy cellular blebbing were found predominantly at villus tips of CPE-treated rabbit ileum [42]. Immunolocalization results on rabbit intestinal loops indicated that more CPE binds to villus tips than elsewhere on intestinal villi, which coincides with greater CPE receptor (Claudin-4) density in villus tips [24]. The correlation between claudin-4 density and CPE binding at villus tips also suggests this claudin, which is a known CPE receptor [19,20,26], plays an important role in intestinal CPE binding. However, since certain other claudins can also bind CPE in vitro [18], this correlation does not preclude the possibility that other claudins also contribute to CPE binding in vivo.
Figure 1.
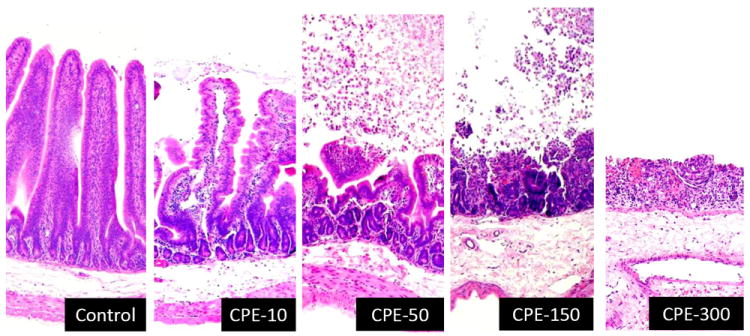
Histology of rabbit small intestinal loops treated with buffer (control), 10 μg of CPE (CPE-10), 50 μg of CPE (CPE-50), 150 μg of CPE (CPE-150) or 300 μg of CPE (CPE-300). After 6 h treatment, loop tissues were formalin fixed, embedded in paraffin and processed for the production of 4 um thick, hematoxylin and eosin stained sections. Final magnification: X200.
Although several studies [43,34] have supported a linkage between CPE-induced cytotoxicity, intestinal microscopic damage and the onset of intestinal fluid transport alterations, more recent studies [19,20,44,45] reported that 4-6 h treatment with noncytotoxic, but binding-capable, C-terminal CPE fragments can induce claudin internalization and alter transepithelial resistance in cultured cells. In addition, those Cterminal CPE fragments were unable to induce microscopic damage in the ileum and alter transepithelial resistance and dextran absorption in ileal loops [44]. Those observations could have indicated that C-terminal CPE fragments can, independently of cytotoxicity or microscopic changes, induce paracellular permeabilty changes that contribute to intestinal fluid transport changes. However, no fluid accumulation was measured in the intestine in the study referred to [44]. To address this, the in vivo effects of CPE versus two rCPE variants that are binding-capable, yet non-cytotoxic in vitro; i.e. rCPE168-319 and rCPE D48A [46,16] were compared [24]. In that study, CPE (which is cytotoxic) caused both intestinal fluid accumulation and microscopic damage in rabbit ileal loops [24]. However, neither of those effects was observed when small intestinal loops were treated with similar pathophysiologically-relevant (up to 100 μg/ml) doses of the rCPE168-319 or rCPE D48A non-cytotoxic variants of CPE, even though both those rCPE variants were demonstrated to bind to the small intestinal epithelium (Figs. 2 and 3). These findings support cytotoxic activity as being important for the development of both the microscopic and intestinal fluid transport effects of CPE in this model.
Figure 2.
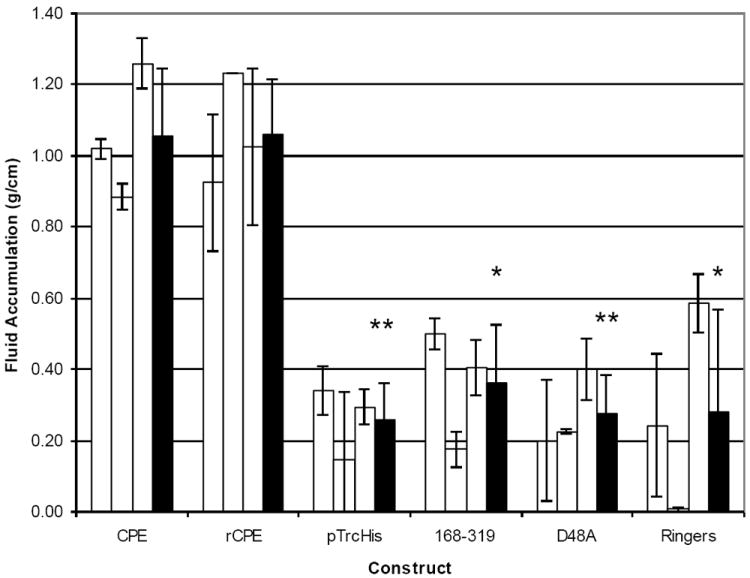
Fluid accumulation in rabbit small intestinal loops inoculated with 300 μg of CPE, rCPE, two non-cytotoxic CPE variants (168-319 and D48A) or two different buffers (negative controls) and thenincubated for 6 h while rabbits were under anesthesia. At the conclusion of the experiment, loop fluid content was measured and the amount of accumulated fluid was related inversely to the length of each loop (g/cm). White bars for each inoculum preparation represent the average of duplicate ileal loop samples in a single rabbit, while black bars represent the combined average results for three rabbits. Error bars represent the standard deviation from the mean. Statistical significance of <0.005 between a construct and rCPE is indicated with a ‘**’, whereas ‘*’ similarly indicates statistical significance of <0.05. Copyright © American Society for Microbiology, Infection and Immunity, 76, 2008, 3793-3800, DOI: 10.1128/IAI.00460-08. Used with permission.
Figure 3.
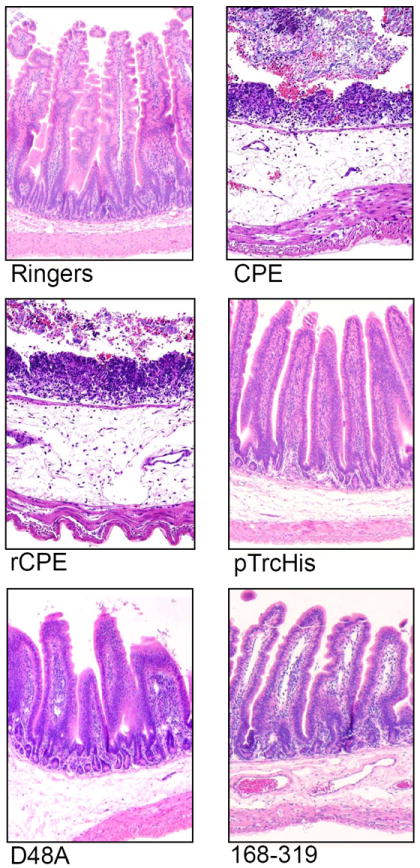
Histopathological changes induced by the toxin preparations described in Figure 2, in rabbit small intestinal loops. After the 6 h fluid accumulation experiments, rabbit small intestinal loops selected for histological examination were formalin-fixed and embedded in paraffin. Hematoxylin and eosin was used to stain 4 μm thick sections of intestinal tissue treated with the indicated construct. The preparation used to treat each loop is noted at the bottom of each panel. Final magnification: 200×. Copyright © American Society for Microbiology, Infection and Immunity, 76, 2008, 3793-3800, DOI: 10.1128/IAI.00460-08. Used with permission.
Results of the study referred to above [24] suggested the following model for CPE enterotoxicity. Once produced in the small intestine during sporulation, CPE first binds to certain claudin receptors at the villus tips because cells in that region have more abundant and probably more exposed (due to normal cellular extrusion processes) enterotoxin receptors. This CPE binding results in small complex formation, followed by formation of hexameric CH-1 pores on the enterocyte surface. These prepore CH-1 complexes then penetrate the plasma membrane to form a pore that permits a Ca2+ influx to drive death of villus tip cells, which are already primed for apoptosis. The resultant death of villus tip enterocytes exposes the basolateral membranes of adjacent enterocytes, allowing the process to repeat such that epithelial desquamation developing at the villus tips impairs intestinal absorption. As the process continues, intestinal villi become increasingly blunted and denuded, causing net fluid secretion in the small intestine. These effects then manifest clinically as diarrhea and abdominal cramps.
Information about the action of CPE in the colon is scant and sometimes contradictory. Because the colon is involved primarily in fluid reabsorption, it is thought that the diarrhea characteristic of the human disease could be caused in part by inhibition of fluid reabsorption or by enhancement of fluid secretion by the colon [38]. In a study of the effect of CPE on small and large human intestine ex-vivo, the toxin bound to ileal epithelium and induces microscopical damage concurrently with reduced short-circuit current, transepithelial resistance and net water resporption [47]. In the same study, it was also found that CPE binds to the human colon but causes only slight morphological and transport changes. Rabbit colonic loops inoculated with CPE had no apparent response to the CPE in the transport of fluid and electrolytes, or microscopic damage [38]. This seems to be in disagreement with human cases of fatal necrotizing colitis that occurred in people consuming turkey contaminated with enterotoxigenic C. perfringens [48]. However, colonic epithelial cells bound 125I-labeled CPE at levels even greater than those observed in the ileum. Based on these results, Mc Donel and Demers [38] concluded that although binding of CPE is necessary for its biological activity, biological activity does not necessarily follow binding to specific receptors.
Rabbits have also been used to study the binding of CPE to tissues other than the intestine [35]. 125I-labeled CPE was incubated with homogenates of rabbit liver, kidney and brain. CPE bound in a specific manner to tissue homogenates from liver and kidney but not brain. The amount of CPE molecules that bound was similar in intestine, liver and kidney. This binding was demonstrated to be heat-labile, as the binding ability was lost after CPE was heated for 10 minutes at 60°C [35]. These results, coupled with similar additional studies in mice [49], suggest that CPE absorbed from the intestine can be responsible for systemic alterations, which may help explain the lethality observed in some cases of experimental animals and human patients [48].
3-Mouse models
Mice have been used, although less extensively than rabbits, to study the pathogenesis of CPE intoxication [50,49]. In mouse models, both the intestinal and systemic effects of CPE were studied.
Sakaguchi et al [51] found that the lethal CPE dose to a mouse by i.v. injection is about 2 μg. Death of animals usually occurred within 30 minutes after i.v. injection of CPE and animals that survived for 30 minutes rarely died later [51,52]. When anesthetized mice received an i.v. inoculation with purified CPE, a 50 μg/kg dose was lethal [53]. Death in these mice was preceded by changes of the ECG, rapid fall of blood pressure and transient hyperpnea followed by respiratory depression. Elevated levels of plasma potassium were detected in these mice, suggesting that the ECG alterations and death were produced by hyperkalemia. This was supported by the fact that even large doses of CPE (up to 100 μg) failed to produce significant cardiotoxicity on the isolated heart.
Experimental inoculation of CPE i.v. [53] provided valuable information on the pathogenesis of the systemic effects of CPE. However, i.v. administration does not closely mimic natural CPE action, where CPE is produced in the intestines. To overcome this problem, intestinal loop models were developed in mice [50,49]. When CPE was inoculated into ligated small intestinal loops of mice, similar intestinal effects to those seen in rabbits were observed [50,49]. CPE caused marked fluid accumulation; the effect being proportional to the applied CPE dose and occurring as early as 10 minutes after toxin inoculation [50]. This effect, however, was arrested by washing the loop with saline or by injection of anti-CPE antibodies into the loop as late as 30 minutes after inoculation of CPE. Based on these results, the authors suggested that CPE is neither bound firmly to the mucosal membrane nor does it permeate into the cells of the intestinal wall [50]. These results are in contrast with the irreversible nature of CPE binding to intestinal cells observed in several other studies [35]. This discrepancy might suggest that the rapid onset of fluid accumulation observed in one mouse intestinal loop study [50] involved the presence of another factor in addition to CPE.
Mouse intestinal loop models have also been used to study the systemic effects of CPE in vivo [49], something which cannot be evaluated in rabbit intestinal loops mainly because, in rabbit studies, multiple intestinal loops are typically constructed in a single animal to test different inocula. In addition, use of multiple loops in one rabbit is not conductive to time course studies since the rabbit is euthanized when the first time-point is reached. In the mentioned study [49], significant lethality occurred when mouse intestinal loops were treated with 100 or 200 μg CPE doses for several hours (Fig. 4). Those CPE doses rarely cause lethality in rabbit small intestinal loop models, even after 6 h of incubation. It was then suggested that CPE absorbed from the intestine into the circulation could have been responsible for the lethality observed with the mouse intestinal loop model. This suggestion was based on the fact that seroprotection against CPE-induced lethality was observed in the mouse intestinal loop model after i.v. injection of CPE antibody, and the presence of CPE was detected in sera of these mice. The difference in the size of mice and rabbits (and therefore, a difference in doses per Kg of body weight) was also suggested as a possible explanation for the difference in response between rabbits and mice since similar doses were used in both species [49].
Figure 4.
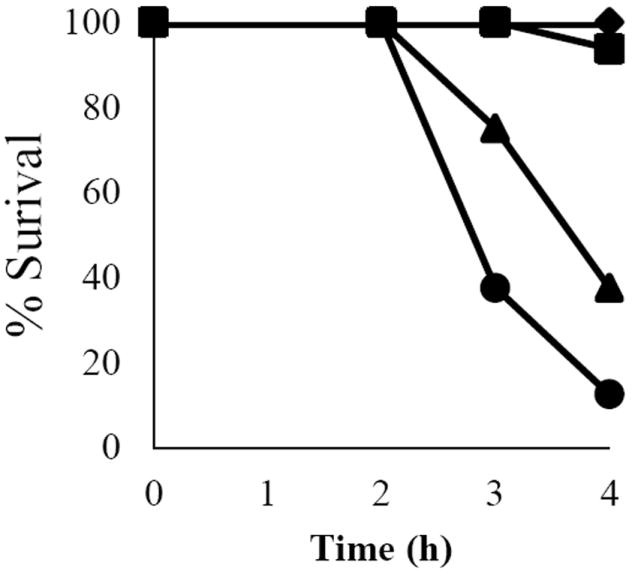
Dose- and time-dependent representation of mouse survival after intraduodenal CPE challenge. The percent survival of mice was calculated and plotted against the time of treatment (in hours, x-axis) for each dose of CPE at 1, 2, 3, and 4 h. Buffer control animals (-+JmY--), 50 +A7w-g treatment (-+JaA--), 100 +A7w-g treatment (-+JbI--), 200 +A7w-g treatment (-+Jc8--). Survival was calculated from 8 animals for each CPE dose. Copyright +AKk- American Society for Microbiology, Infection and Immunity, 79, 2011, 3020-3027, DOI: 10.1128/IAI.01342-10. Used with permission.
CPE treated mouse intestinal loops showed dose- and time-dependent intestinal histopathologic damage mimicking that observed in rabbit ileal loops, thus validating the usefulness of mouse intestinal loops for studying CPE action. However, no luminal fluid accumulation was observed in the intestinal loops of CPE-treated mice, probably due to the short (up to 4 h) experimental challenge of mice used in the model [49], compared to the 6 or more h incubation period usually used in rabbits. Experiments using longer treatment times are required to clarify this issue.
When potassium levels in sera were measured in mice whose intestinal loops were treated for 2 h with 200 μg of CPE, a lethal (~14 mM) concentration of potassium was detected (Fig. 5). The mouse lethality results in the intestinal loop model described by Caserta et al [49], appears to reproduce results by Sakaguchi et al [51] who detected hyperkalemia in mice inoculated i.v. with CPE. These results may also help to explain the unusually high lethality associated with the recent C. perfringens type A food poisoning outbreaks in mental institutions [48]. Specifically, the study by Caserta et al [49] has found that CPE can be absorbed from the intestines into the circulation and this absorbed CPE can have lethal consequences. The atypical lethality associated with CPE in psychiatric patients referred to before in Oklahoma was thought to involve drugs that cause constipation as a side effect [48]. Constipation would prevent diarrhea from flushing CPE out of the intestines and thus prolong intestinal exposure to CPE (and perhaps other C. perfringens toxins). The mouse ligated intestinal loop model developed by Caserta et al [49] mimics somewhat the situation of constipated human patients. The mechanism by which CPE gains access to the bloodstream is currently unknown. CPE might gain access to the blood simply due to the overwhelming tissue destruction in the CPE-treated intestine, which could expose the underlying microvasculature. However, in the study by Caserta et al [49], small amounts of CPE were still detected in the blood of mice inoculated with 50 μg of CPE that had minimal intestinal damage. This observation suggests that substantial gross or microscopic damage may not be essential for CPE to gain access to the circulation.
Figure 5.
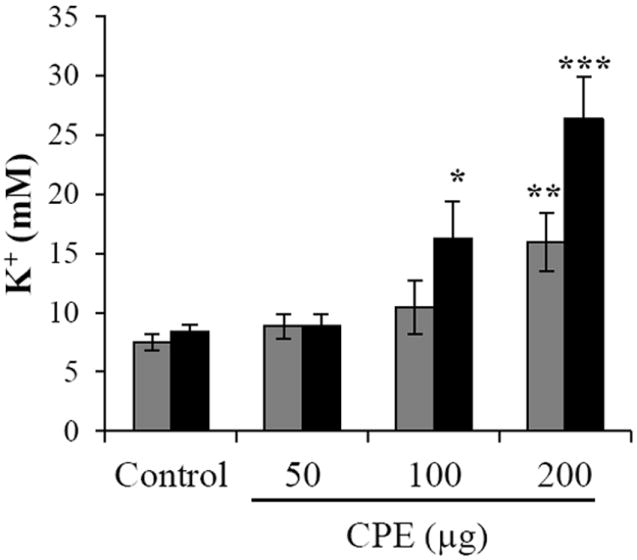
Serum potassium levels after CPE intraduodenal challenge of mice. After 2 h (gray bars) or 3 h (black bars) of intestinal loop treatment with buffer control or buffer containing 50 μg, 100 μg, or 200 μg of CPE, sera were collected from mice. Those sera were analyzed by utilizing inductively coupled argon plasma emission spectrometry. After precipitation of proteins, the protein-free supernatant of each sample was analyzed for potassium levels. Accuracy of results was measured by analyzing quality assurance sera obtained from the Veterinary Laboratory Association Quality Assurance Program. Data were accepted if analyzed quality assurance serum values were within 2 standard deviations of the reference values. Data shown are mean values obtained using 8 mice. Error bars represent the standard error of the mean. *p<0.02, **p<0.01, or ***p<0.0002 compared to control. Copyright © American Society for Microbiology, Infection and Immunity, 79, 2011, 3020-3027, DOI: 10.1128/IAI.01342-10.
Early studies determined that, when i.v.-injected into mice, most CPE distributes to the liver and, to a lesser extent, the kidney [54]. More recently liver and kidney homogenates were prepared from mice whose intestinal loops had been challenged with 200 μg of CPE. Analysis of those homogenates by Western blotting with an anti-CPE antibody showed that CPE had bound and formed a CH-1-like complex in both the liver and kidney. However, no CPE binding or complex formation was observed in homogenates prepared from the livers and kidneys of mice whose loops had received 50 μg of CPE. In addition, no CH-2-like complex was detected in homogenates prepared from liver or kidney of mice intestinally challenged with either the 50 or 200 μg CPE dose. The study referred to [49] also showed that CPE can bind and form complexes resembling CH-1 in internal organs, including the liver and kidney. The ability of CPE to bind to liver and kidney is consistent with the strong expression of claudins, including some known claudin CPE receptors, in these two organs [55]. As the portal vein system connects the GI tract directly to the liver, this might explain the abundance of CPE binding to this organ.
Administration of CPE by the i.v. route also caused elevated levels of blood potassium, which resulted in cardiac dysfunction and, eventually, respiratory depression and death [54,53]. This is consistent with results of CPE treatment of liver ex vivo shown to cause a massive potassium loss, suggesting the liver as a major source of the lethal blood potassium levels measured in mice receiving an i.v. CPE injection [54]. In mice receiving a CPE injection into their intestinal loops, both a CPE dose- and time-dependent increase in serum potassium were found and this increase exceeded lethal levels. Furthermore, these increased serum potassium levels slightly preceded the onset of lethality in mice that were intestinally-challenged with CPE, consistent with a cause and effect relationship. These results suggest that formation of a CH-1-like pore in liver may be responsible, at least in part, for the observed potassium release and subsequent lethality observed in some intestinally-challenged mice.
Interestingly, formation of a CH-2-like complex was not observed in mouse intestinal tissue that had been CPE-treated in vivo or in mouse intestinal homogenates treated with CPE in vitro. Since Western blot analysis showed that virtually all of the CPE bound to intestinal tissue had localized in CH-1 and no CH-2 formation was detected in vivo, formation of a CH-1-like CPE complex appears to be sufficient for CPE intestinal action in vivo. Similarly, no CH-2-like complex was detected in kidney or liver tissue from mice whose intestinal loops had been CPE-treated or in homogenates of those organs treated in vitro with CPE.
4-Rat Models
Rats have also been used, albeit less extensively than mice and rabbits, to study the in vivo effects of CPE [53]. Similarly to what was observed in mice (see above), when anesthetized rats were inoculated i.v. with purified CPE, 50 μg/kg of this toxin were enough to produce lethality. Death in these rats was preceded by changes of the ECG, rapid fall of blood pressure and transient hyperpnea followed by respiratory depression. Elevated levels of plasma potassium were also detected in these rats, suggesting that the ECG alterations and death were produced by hyperkalemia. This was supported by the fact that even large doses of CPE (up to 100 μg) failed to produce significant cardiotoxicity on the isolated heart of rats. By perfusing isolated rat organs with CPE, the same authors [53] demonstrated that there was a marked increase of potassium and cytoplasmic enzymes (GPT, GOT and LDH) in the eluent of the liver (but not lungs or lower extremities), indicating that the elevated potassium found in plasma originated, at least partly, in the liver. Based on these results, Sugimoto et al [53] concluded that hyperkalemia elicited by the cytotoxic action of CPE on hepatocytes caused cardiac failure leading to death. Binding to liver cells was demonstrated to be specific on hepatocytes rather than on stromal cells and on these specific for the plasma membrane [56].
5. Other animal models
To the best of our knowledge, only one report has been published describing the use of non-human primates to study the effects of CPE. Uemura et al [57] fed cynomolgus monkeys with 5 mg of purified CPE, causing vomiting and diarrhea only when the normally low gastric pH had been neutralized by giving the animals sodium bicarbonate. Only diarrhea was consistently observed in all the monkeys dosed orally with CPE positive C. perfringens type A and sodium bicarbonate. No lethality was observed and no CPE antibodies were detected in any of the monkeys up to 21 days after the challenge. Based on these results, the authors concluded that no significant amount of CPE is absorbed from the intestine (it should be noted, however, that the development of serum antibodies against CPE has been detected in human food poisoning victims [58). Unfortunately no post mortem examinations were performed on these animals and no information on gross or histopathological changes of non-human primates intoxicated with CPE is therefore available.
Niilo [59,60] reported that i.v. administration of crude cell extracts from sporulating cells of enterotoxigenic strains of C. perfringens type A into calves caused partial loss of villus epithelium associated with diarrhea and other systemic reactions. He also reported variable damage in ligated loops of lambs when CPE was placed in the intestinal lumen. Hauschild et al [61] reported no significant lesions in lambs challenged with cells or culture filtrates of enterotoxigenic C. perfringens strains.
6. Conclusions
Although significant progress has been made over the last few decades towards the understanding of CPE-positive C. perfringens type A infections, new insights into the pathogenesis of these infections, including the mechanism of action of CPE, have come to light using animal models. Several examples showed that CPE has different effects depending on the physiologic condition of the patient. An example of this is the necrotizing colitis presented by psychiatric patients medicated with drugs that produced constipation; an effect not usually observed in other patients. It is also important to point out that different animal species provide tools to study different aspects of CPE effects. For instance, although the rabbit intestinal loop model has been very useful to study the intestinal effect of CPE, that model could not be used to study the systemic effects of this toxin. The latter could only be at least partly understood when the new mouse model was used.
It is also worth mentioning that most in vivo research performed to date was done using CPE. To the best of our knowledge no animal model has been developed based on CPE-positive C. perfringens infections because it is difficult to obtain good levels of in vivo sporulation, as needed for CPE production, in small animals. Such a model would be very useful to fully understand the pathogenesis of these infections. This aspect of CPE disease might also benefit from the development of non-human primates model/s.
Acknowledgments
This work was supported by Public Health Service grant R37 AI019844-29 (BAM) from the National Institute of Allergy and Infectious Diseases. We thank Ms. S. Fitisemanu and N. Harrigan for excellent secretarial help.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McClane BA, Uzal FA, Miyakawa MF, Lyerly D, Wilkins T. The Enterotoxic Clostridia, p. In: Dworkin M, Falkow S, Rosenburg E, Schleifer H, Stackebrandt E, editors. The Prokaryotes. 3. Springer NY; New York: 2006. pp. 688–752. [Google Scholar]
- 2.Kokai-Kun JF, Songer JG, Czeczulin JR, Chen F, McClane BA. Comparison of Western immunoblots and gene detection assays for identification of potentially enterotoxigenic isolates of Clostridium perfringens. J Clin Microbiol. 1994;32:2533–2539. doi: 10.1128/jcm.32.10.2533-2539.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M, Roy S, Jones JL, Griffin PM. Foodborne illness acquired in the United States-major pathogens. Emer Infect Dis. 2011;17:7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carman RJ. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev Med Microbiol. 1997;8(supplement 1):S43–S45. [Google Scholar]
- 5.Sarker MR, Carman RJ, McClane BA. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Molec Microbiol. 1999;33:946–958. doi: 10.1046/j.1365-2958.1999.01534.x. [DOI] [PubMed] [Google Scholar]
- 6.McClane BA. Clostridium perfringens. In: Doyle MP, Beuchat LR, editors. Food Microbiology. Third Edition. ASM press; Washington D.C: 2007. pp. 423–444. [Google Scholar]
- 7.Brynestad S, Synstad B, Granum PE. The Clostridium perfringens enterotoxin gene is on a transposable element in type A human food poisoning strains. Microb. 1997;143:2109–2115. doi: 10.1099/00221287-143-7-2109. [DOI] [PubMed] [Google Scholar]
- 8.Miyamoto K, Fisher DJ, Li J, Sayeed S, Akimoto S, McClane BA. Complete sequencing and diversity analysis of the enterotoxin-encoding plasmids in Clostridium perfringens type A non-food-borne human gastrointestinal disease isolates. J Bacteriol. 2006;188:1585–1598. doi: 10.1128/JB.188.4.1585-1598.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bannam TL, Teng WL, Bulach D, Lyras D, Rood JI. Functional identification of conjugation and replication regions of the tetracycline resistance plasmid pCW3 from Clostridium perfringens. J Bacteriol. 2006;188:4942–51. doi: 10.1128/JB.00298-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brynestad S, Sarker MR, McClane BA, Granum PE, Rood JI. The enterotoxin (CPE) plasmid from Clostridium perfringens is conjugative. Infect Immun. 2001;69:3483–3487. doi: 10.1128/IAI.69.5.3483-3487.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Czeczulin JR, Hanna PC, McClane BA. Cloning, nucleotide sequencing and expression of the Clostridium perfringens enterotoxin gene in Escherichia coli. Infect Immun. 1993;61:3429–3439. doi: 10.1128/iai.61.8.3429-3439.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Briggs DC, Naylor CE, Smedley JG, 3rd, Lukoyanova N, Robertson S, Moss DS, McClane BA, Basak AK. Structure of the food-poisoning Clostridium perfringens enterotoxin reveals similarity to the aerolysin-like pore-forming toxins. J Mol Biol. 2011;413:138–149. doi: 10.1016/j.jmb.2011.07.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitadokoro K, Nishimura K, Kamitani S, Fukui-Miyazaki A, Toshima H, Abe H, Kamata Y, Sugita-Konishi Y, Yamamoto S, Karatani H, Horiguchi Y. Crystal structure of Clostridium perfringens enterotoxin displays features of beta-pore-forming toxins. J Biol Chem. 2011;286:19549–19555. doi: 10.1074/jbc.M111.228478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanna PC, Wnek AP, McClane BA. Molecular cloning of the 3’ half of the Clostridium perfringens enterotoxin gene and demonstration that this region encodes receptor-binding activity. J Bacteriol. 1989;171:6815–6820. doi: 10.1128/jb.171.12.6815-6820.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kokai-Kun JF, Benton K, Wieckowski EU, McClane BA. Identification of a Clostridium perfringens enterotoxin region required for large complex formation and cytotoxicity by random mutagenesis. Infect Immun. 1999;67:6534–6541. doi: 10.1128/iai.67.11.5634-5641.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smedley JG, III, McClane BA. Fine-mapping of the N-terminal cytotoxicity region of Clostridium perfringens enterotoxin by site-directed mutagenesis. Infect Immun. 2004;72:6914–6923. doi: 10.1128/IAI.72.12.6914-6923.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smedley JG, 3rd, Uzal FA, McClane BA. Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect Immun. 2007;75:2381–2390. doi: 10.1128/IAI.01737-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita K, Katahira J, Horiguchi Y, Sonoda N, Furuse M, Tskuita S. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction membrane protein. FEBS Letters. 2000;476:258–261. doi: 10.1016/s0014-5793(00)01744-0. [DOI] [PubMed] [Google Scholar]
- 19.Katahira J, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J Cell Biol. 1997;136:1239–1247. doi: 10.1083/jcb.136.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katahira J, Sugiyama H, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J Biol Chem. 1997;272:26652–26658. doi: 10.1074/jbc.272.42.26652. [DOI] [PubMed] [Google Scholar]
- 21.Veshnyakova A, Protze J, Rossa J, Blasig IE, Krause G, Piontek J. On the interaction of Clostridium perfringens enterotoxin with claudins. Toxins. 2010;2:1336–1356. doi: 10.3390/toxins2061336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitchell LA, Koval M. Specificity of interactions between Clostridium perfringens enteortoxin and claudin-family tight junction proteins. Toxins. 2010;2:1595–1611. doi: 10.3390/toxins2071595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robertson S, Smedley JG, III, McClane BA. Identification of a claudin-4 residue important for mediating the host cell binding and action of Clostridium perfringens enterotoxin. Infect Immun. 2010;78:505–517. doi: 10.1128/IAI.00778-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smedley JG, 3rd, Saputo J, Parker JC, Fernandez-Miyakawa ME, Robertson SL, McClane BA, Uzal FA. Noncytotoxic Clostridium perfringens enterotoxin (CPE) variants localize CPE intestinal binding and demonstrate a relationship between CPE-induced cytotoxicity and enterotoxicity. Infect Immun. 2008;76:3793–800. doi: 10.1128/IAI.00460-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wieckowski EU, Wnek AP, McClane BA. Evidence that an ~50kDa mammalian plasma membrane protein with receptor-like properties mediates the amphiphilicity of specifically-bound Clostridium perfringens enterotoxin. J Biol Chem. 1994;269:10838–10848. [PubMed] [Google Scholar]
- 26.Robertson SL, Smedley JG, 3rd, Singh U, Chakrabarti G, Van Itallie CM, Anderson JM, McClane BA. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell Microbiol. 2007;9:2734–2755. doi: 10.1111/j.1462-5822.2007.00994.x. [DOI] [PubMed] [Google Scholar]
- 27.McClane BA, Wnek AP. Studies of Clostridium perfringens enterotoxin action at different temperatures demonstrate a correlation between complex formation and cytotoxicity. Infect Immun. 1990;58:3109–3115. doi: 10.1128/iai.58.9.3109-3115.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McClane BA. Clostridium perfringens enterotoxin acts by producing small molecule permeability alterations in plasma membranes. Toxicol. 1994;87:43–67. doi: 10.1016/0300-483x(94)90154-6. [DOI] [PubMed] [Google Scholar]
- 29.Chakrabarti G, McClane BA. The importance of calcium influx, calpain and calmodulin for the activation of CaCo-2 cell death pathways by Clostridium perfringens enterotoxin. Cell Microbiol. 2005;7:129–146. doi: 10.1111/j.1462-5822.2004.00442.x. [DOI] [PubMed] [Google Scholar]
- 30.Chakrabarti G, Zhou X, McClane BA. Death pathways activated in CaCo-2 cells by Clostridium perfringens enterotoxin. Infect Immun. 2003;71:4260–4270. doi: 10.1128/IAI.71.8.4260-4270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh U, Mitic LL, Wieckowski E, Anderson JM, McClane BA. Comparative biochemical and immunochemical studies reveal differences in the effects of Clostridium perfringens enterotoxin on polarized CaCo-2 cells versus Vero cells. J Biol Chem. 2001;276:33402–33412. doi: 10.1074/jbc.M104200200. [DOI] [PubMed] [Google Scholar]
- 32.Singh U, Van Itallie CM, Mitic LL, Anderson JM, McClane BA. CaCo-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J Biol Chem. 2000;275:18407–18417. doi: 10.1074/jbc.M001530200. [DOI] [PubMed] [Google Scholar]
- 33.McDonel JL. Toxins of Clostridium perfringens types A, B, C, D and E. In: Dorner F, Drews H, editors. Pharmacology of Bacterial Toxins. Pergamon Press; Oxford: 1986. pp. 477–517. [Google Scholar]
- 34.Sherman S, Klein E, McClane BA. Clostridium perfringens type A enterotoxin induces concurrent development of tissue damage and fluid accumulation in the rabbit ileum. J Diar Dis Res. 1994;12:200–207. [PubMed] [Google Scholar]
- 35.McDonel JL. Binding of Clostridium perfringens [125I]enterotoxin to rabbit intestinal cells. Biochemistry. 1980;19:4801–4807. doi: 10.1021/bi00562a014. [DOI] [PubMed] [Google Scholar]
- 36.McDonel JL, Duncan CL. Histopathological effect of Clostridium perfringens enterotoxin in the rabbit ileum. Infect Immun. 1975;12:1214–1218. doi: 10.1128/iai.12.5.1214-1218.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonel JL, Duncan CL. Regional localization of activity of Clostridium perfringens type A enterotoxin in the rabbit ileum, jejunum and duodenum. J Infect Dis. 1977;136:661–666. doi: 10.1093/infdis/136.5.661. [DOI] [PubMed] [Google Scholar]
- 38.McDonel JL, Demers GW. In vivo effects of enterotoxin from Clostridium perfringens type A in the rabbit colon: Binding vs. biologic activity. J Infect Dis. 1982;145:490–494. doi: 10.1093/infdis/145.4.490. [DOI] [PubMed] [Google Scholar]
- 39.McDonel JL, Asano T. Analysis of unidirectional fluxes of sodium during diarrhea induced by Clostridium perfringens enterotoxin in the rat terminal ileum. Infect Immun. 1975;11:526–529. doi: 10.1128/iai.11.3.526-529.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duncan CL, Strong DH. Ileal loop fluid accumulation and production of diarrhea in rabbits by cell-free products of Clostridium perfringens. J Bacteriol. 1969;100:86–94. doi: 10.1128/jb.100.1.86-94.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duncan CL, Sugiyama H, Strong DH. Rabbit ileal loop responseto strains of Clostridium perfringens. J Bacteriol. 1968;95:1560–1566. doi: 10.1128/jb.95.5.1560-1566.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McDonel JL, Chang LW, Pounds JG, Duncan CL. The effects of Clostridium perfringens enterotoxin on rat and rabbit ileum: an electron microscopic study. Lab Invest. 1978;39:210–218. [PubMed] [Google Scholar]
- 43.McDonel JL, McClane BA. Production, purification and assay of Clostridium perfringens enterotoxin. Methods Enzymol. 1988;165:94–103. doi: 10.1016/s0076-6879(88)65018-x. [DOI] [PubMed] [Google Scholar]
- 44.Masuyama A, Kondoh M, Seguchi H, Takahashi A, Harada M, Fujii M, Mizuguchi H, Horiguchi Y, Watanabe Y. Role of N-terminal amino acids in the absorption-enhancing effects of the C-terminal fragment of Clostridium perfringens enterotoxin. J Pharmacol Exp Ther. 2005;314:789–795. doi: 10.1124/jpet.105.085399. [DOI] [PubMed] [Google Scholar]
- 45.Sonoda N, Furuse M, Sasaki H, Yonemura S, Katahira J, Horiguchi Y, Tsukita S. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: Evidence for direct involvement of claudins in tight junction barrier. J Cell Biol. 1999;147:195–204. doi: 10.1083/jcb.147.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kokai-kun JF, McClane BA. Determination of functional regions of Clostridium perfringens enterotoxin through deletion analysis. Clin Infect Dis. 1997;25:S165–S167. doi: 10.1086/516246. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez Miyakawa ME, Pistone Creydt V, Uzal FA, McClane BA, Ibarra C. Clostridium perfringens enterotoxin damages the human intestine in vitro. Infect Immun. 2005;73:8407–8410. doi: 10.1128/IAI.73.12.8407-8410.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bos J, Smithee L, McClane B, Distefano RF, Uzal F, Songer JG, Mallonee S, Crutcher JM. Fatal necrotizing colitis following a foodborne outbreak of enterotoxigenic Clostridium perfringens type A infection. Clin Infect Dis. 2005;40:78–83. doi: 10.1086/429829. [DOI] [PubMed] [Google Scholar]
- 49.Caserta JA, Robertson SL, Saputo J, Shrestha A, McClane BA, Uzal FA. Development and application of a mouse intestinal loop model to study the in vivo action of Clostridium perfringens enterotoxin. Infect Immun. 2011;79:3020–3027. doi: 10.1128/IAI.01342-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamamoto K, Ohishi I, Sakaguchi G. Fluid accumulation in mouse ligated intestine inoculated with Clostridium perfringens enterotoxin. Appl Environ Microbiol. 1979;37:181–186. doi: 10.1128/aem.37.2.181-186.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakaguchi G, Uemura T, Riemann HP. Simplified method for purification of Clostridium perfringens type A enterotoxin. Appl Microbiol. 1973;26:762–767. doi: 10.1128/am.26.5.762-767.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hauschild AHW, Hilcheimer R. Purification and characteristics of the enterotoxin of Clostridium perfringens type A. Can J Microbiol. 1971;17:1425–1433. doi: 10.1139/m71-227. [DOI] [PubMed] [Google Scholar]
- 53.Sugimoto N, Chen YM, Lee SY, Matsuda M, Lee CY. Pathodynamics of intoxication in rats and mice by enterotoxin of Clostridium perfringens type A. Toxicon. 1991;29:751–759. doi: 10.1016/0041-0101(91)90067-2. [DOI] [PubMed] [Google Scholar]
- 54.Skjelkvale R, Tolleshaug H, Jarmund T. Binding of enterotoxin from Clostridium perfringens type A to liver cells in vivo and in vitro. The enterotoxin causes membrane leakage. Acta Pathol Microbiol Scand B. 1980;88:95–102. doi: 10.1111/j.1699-0463.1980.tb02612.x. [DOI] [PubMed] [Google Scholar]
- 55.D’Souza T, Sherman-Baust CA, Poosala S, Mullin JM, Morin PJ. Age-related changes of claudin expression in mouse liver, kidney and pancreas. J Gerontol A Biol Sci Med Sci. 2009;64:1146–1153. doi: 10.1093/gerona/glp118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tolleshaug H, Skjelkvale R, Berg T. Quantitation of binding and subcellular distribution of Clostridium perfringens enterotoxin in rat liver cells. Infect Immun. 1982;37:486–491. doi: 10.1128/iai.37.2.486-491.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uemura T, Sakaguchi G, Ito T, Okazawa K, Sakai S. Experimental diarrhea in cynomogus monkeys by oral administration with Clostridium perfringens type A viable cells or enterotoxin. Jpn J Med Sci Biol. 1975;28:165–177. doi: 10.7883/yoken1952.28.165. [DOI] [PubMed] [Google Scholar]
- 58.Birkhead G, Vogt RL, Heun EM, Snyder JT, McClane BA. Characterization of an outbreak of Clostridium perfringens food poisoning by quantitative fecal culture and fecal enterotoxin measurement. J Clin Microbiol. 1988;26:471–474. doi: 10.1128/jcm.26.3.471-474.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niilo L. Mechanism of action of the enteropathogenic factor of Clostridium perfringens type A. Infect Immun. 1970;3:100–106. doi: 10.1128/iai.3.1.100-106.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Niilo L. Effect on calves of the intravenous injection of the enterotoxin of Clostridium welchii type A. J Comp Path. 1973;83:265–269. doi: 10.1016/0021-9975(73)90051-0. [DOI] [PubMed] [Google Scholar]
- 61.Hauschild AHW, Hilsheimer R, Rogers CG. Experimental enteritis with food poisoning and classical strains of Clostridium perfringens type A in lambs. J Infect Dis. 1967;117:379–386. doi: 10.1093/infdis/117.5.379. [DOI] [PubMed] [Google Scholar]